

SUMMARY
Nuclear receptors induce both transcriptional activation and repression programs responsible for development, homeostasis and disease. Here, we report a previously overlooked enhancer decommissioning strategy underlying a large estrogen receptor alpha (ERα)-dependent transcriptional repression program. The unexpected signature for this E2-induced program resides in indirect recruitment of ERα to a large cohort of pioneer factor basally-active FOXA1-bound enhancers that lack cognate ERα DNA binding elements. Surprisingly, these basally-active estrogen-repressed (BAER) enhancers are decommissioned by ERα-dependent recruitment of the histone demethylase, KDM2A, functioning independently of its demethylase activity. Rather, KDM2A tethers the E3 ubiquitin-protein ligase, NEDD4, to ubiquitylate/dismiss Pol II to abrogate eRNA transcription, with consequent target gene down-regulation. Thus, our data reveal that Pol II ubiquitylation/dismissal may serve as a potentially broad strategy utilized by indirectly bound nuclear receptors to abrogate large programs of pioneer factor-mediated, eRNA-producing enhancers.
eTOC Blurb
Tan et al. discover a mechanism underlying repression of a large Basally Active Estrogen Repressed (BAER) enhancer program. The expression of these basally-active genes in human breast cancer cells is repressed by estrogen, and the indirectly-bound ERα (in trans) at BAER enhancers dismisses RNA Pol II to abrogate large, eRNA-producing enhancer programs.
Graphical Abstract
INTRODUCTION
Functional specialization of distinct cell types requires cells to elicit unique responses to developmental and homeostatic signaling by generating specific transcriptional programs despite harboring identical genetic material. These programs are primarily dictated by enhancers, which were initially discovered more than 35 years ago and defined as DNA elements that act at “long” distances to positively regulate target genes expression (Banerji et al., 1981). The precise pattern of activation of specific cohorts of enhancers is critical for development and cell lineage determination, as well as cellular responses to stimuli (Fukaya et al., 2016; Shen et al., 2016).
An intriguing property of active enhancers is their ability to recruit Pol II to initiate transcription, and recruitment of Pol II to enhancers actually temporally precedes transcriptional induction of target gene promoters (Arner et al., 2015; Kim et al., 2015). Enhancers can even function as alternative promoters to active target gene transcription when their promoters are deleted (Kowalczyk et al., 2012), which further suggested that the presence of Pol II at enhancers plays a critical role in target gene expression. We have reasonably clear ideas of the mechanisms underlying enhancer activation, which is usually correlated with an increase in enhancer RNAs (eRNAs) transcription (Li et al., 2016), and eRNA-producing enhancers are preferentially engaged in interactions with the proximal promoters (Ghavi-Helm et al., 2014; Li et al., 2013). However, the mechanisms by which most ligand- or signal-dependent transcription factors mediate equivalently large programs of transcriptional repression remain poorly understood.
17β-estradiol (E2)-bound estrogen receptor α (ERα), a ligand-dependent sex steroid-regulated transcription factor and a well-established regulator of breast epithelial proliferation (Khan et al., 1998; Malik et al., 2010), mediates most of the biological effects of estrogens, primarily at the level of transcription (Li et al., 2013). ERα binds to estrogen response elements (EREs) (Schwabe et al., 1993) in human breast cancer MCF7 cells, a large subset of which harbor the marks of enhancers, and recruits co-factors such as p300, Mediator, cohesin, condensin, and Mega-Trans complex to induce enhancer activation (Li et al., 2016). While several strategies that critically impose repressive programs acting at individual gene promoters have been elucidated (Malik et al., 2010), genome-wide data actually showed that most ERα binding sites are located on potential enhancers (Li et al., 2013). Therefore, we wished to use a genomic approach to investigate the mechanistic basis underlying ERα mediated enhancer based repression programs.
Here, we employ GRO-seq, ChIP-seq, 3C/4C/Hi-C assays and CRISPR-Cas9 genome editing to probe E2-mediated transcriptional repression programs at a genome-wide scale. These studies reveal that ERα mediates enhancer-dependent transcriptional repression is based on its indirect binding to a large cohort of Basally-Active Estrogen Repressed (BAER) enhancers in the absence of an estrogen response element (ERE). While indirectly bound ERα recruits coactivators to these enhancers in a ligand-dependent fashion, the repression events, unexpectedly, reflect the availability of the ERα DNA binding domain (DBD) to interact with the histone demethylase, KDM2A, which, even in the absence of its demethylase function, serves to recruit the ubiquitin ligase NEDD4 to these enhancers, resulting ubiquitylation and dismissal of Pol II and consequent inhibition of these enhancer activities. Therefore, our data reveal a previously unappreciated mechanism underling global repression programs, based on indirect recruitment of estrogen receptors to the large cohort of basally active, eRNA-producing enhancers, eventuating in their decommissioning by abrogating enhancer RNA Polymerase II transcription.
RESULTS
17β-estradiol induces decommissioning of a basally active enhancer program
To investigate whether the ligand-dependent binding of ERα to active enhancers might lead to their decommissioning, we examined eRNA transcription by global run-on sequencing (GRO-seq) (Core et al., 2008), which allows us to detect the transcriptional changes in both enhancers and their cognate target genes, assessing those enhancers marked by active histone marks (H3K27ac and H3K4me1) and ERα in MCF7 breast cancer cells. In combination with our previous GRO-seq datasets (GSM1115995/6) (Li et al., 2013) and our current GRO-seq data, we assessed the E2-mediated transcriptional programs, and we found that 820 active enhancers were repressed (referred to as the BAER enhancers) and 1,206 enhancers were activated (referred as E2 activated enhancers) following 1hr E2 treatment (Figure 1A). This was associated with 1,205 down-regulated and 1,573 up-regulated coding target genes (Figure S1A and Table S1). Further analysis showed that 70.5% of the down-regulated transcription units corresponded to those reported by other studies (Hah et al., 2011; Li et al., 2013).
Figure 1. BAER enhancers decommissioned by ERα and interact with E2 repressed gene promoters.

A, MAplots to show GRO-seq tags at ERα-bound enhancers in MCF7 cells under 1 hr treatment of EtOH (Veh) or E2, respectively. Red colors indicate whether genes scored as statistically different (FDR<0.01, Fold Change>1.5) between these two conditions. B-C, Genomic loci for Plekhf2 (B) and Sytl2 (C) showing indicated tag counts for GRO-seq at Veh and E2 condition. H3K27ac serves as an enhancer marker in the intergenic region. D-E, In situ Hi-C showing the interactions surrounding Plekhf2 (D) and Sytl2 (E) loci in MCF7 cells under vehicle condition. Contact domains (marked by yellow square) and loop domains (marked by cyan blocks) are generated from the same regions in GM12878 lymphoblastoid cells with highly deep sequencing (GSE63525). The enhancer and promoter region of Plekhf2 and Sytl2 are marked with light blue box. F, The percentage of E2-dependent enhancers surrounding E2 repressed gene promoters are presented.
Interestingly, BAER enhancers are usually adjacent to the down-regulated gene promoters, as exemplified by the Plekhf2 (Figure 1B), Sytl2 (Figure 1C) and Ncam2 (Figure S1B) loci. We also observed that the enhancers and promoters in Plekhf2 (Figure 1D), Sytl2 (Figure 1E) loci are located in the same contact domain, which is detected by ultra-deep in situ Hi-C and conserved in mammalian cells (Rao et al., 2014), indicating that BAER enhancers have high probability of interacting with their neighboring, down regulated, gene promoters. Indeed, about 80% of ERα regulated enhancers within 100 kilobases (kb) of the down-regulated coding gene TSSs are repressed upon E2 treatment (Figure 1F). To ask whether there have been previous data suggesting interactions between BAER enhancers and E2 down-regulated gene promoters, we analyzed published Pol II ChIA-PET data, which allowed us to profile the interactome between Pol II-enriched regions in normal MCF7 cells (Li et al., 2012). We found that 392 of the BAER enhancers were detected in Pol II-enriched regions in MCF7 cells that had been cultured under complete growth medium. These enhancers exhibited a relatively higher enrichment of Pol II than other BAER enhancers under vehicle conditions (Figure S1C), and could interact with E2-repressed gene promoters exemplified by Plekhf2 and Sytl2 loci (Figure S1D). 3C PCR assays were also employed, showing that Sytl2-enhancer looped with Sytl2-promoter (Figure S1E) with the confirmation by traditional Sanger sequencing (Figure S1F).
BAER enhancers modulate the expression of ERα-mediated down-regulated genes under basal conditions
To examine whether BAER enhancers control the transcription of E2 repressed genes, we employed CRISPR-Cas9, using different pairs of appropriate guide RNAs, to evaluate if their robust expression was abrogated by removal of presumed regulatory enhancers (Figure 2A). Deletion of the Plekhf2 enhancer was assessed by traditional PCR (Figure S2A), Sanger sequencing (Figure S2B) and droplet digital™ polymerase chain reaction (ddPCR) (Figure 2B) to ensure homologous deletion. Our data using these gene-edited clonal lines clearly showed that the expression of E2 repressed genes was decreased by knockout (KO) of the neighboring BAER enhancer under basal culture conditions (Figure 2C). We also observed similar results for the Sytl2-enhancer (Figures 2D, S2C and S2D), and the Ncam2-enhancer (Figures 2E, S2E and S2F) knockout cells. To further support the specificity of enhancer KO effects in MCF7 cells, we investigated the expression of randomly chosen transcription units, such as Sumo2, in these KO cells, finding that the expression of control transcription units such as Sumo2 was not changed (Figure S2G). Consistent with these results, BAER enhancers were capable of up-regulating a promoter-driven downstream luciferase reporter gene (Figure S2H). Together, our data revealed that there is a large cohort active enhancers which are responsible for the expression of E2 down-regulated genes at basal conditions are decommissioned that is functionally initiated by ERα recruitment.
Figure 2. BAER enhancers dictate the expression of E2 repressed gene at vehicle condition in MCF7 cells.

A, Schematic of Lenti-CRISPR vector for U6 promoter-driven sgRNA expression with a puromycin selection marker used for all subsequent experiments. B, Droplet digital polymerase chain reaction (ddPCR) showed one-dimensional scatterplots of event number (droplets) vs. fluorescence amplitude for an ideal assay with a clear separation of positive (above the purple line) and negative (under the purple line) droplets. HEX fluorescence is shown in green (outside of Plekhf2 enhancer region, up panel) and FAM fluorescence is shown in blue (inner of enhancer region, bottom panel). In outside of Plekhf2 enhancer region, all clones displayed positive signal, but in the center of the enhancer region, only wild type (WT) clone showed the positive signal, the knock out (KO) cell showed a negative signal, which means the knockout clones were homozygous. C-E, Analysis of the expression level of E2 down-regulated genes such as Plekhf2 (C), Sytl2 (D) and Ncam2 (E) in wild type and BAER enhancer knockout cells with 2 different pairs of sgRNAs by RT-qPCR. Plekhf2 and Sytl2 enhancer knockout was carried with 2 different pairs of sgRNAs; and the 2nd (pair 2) sgRNA for Ncam2 enhancer knockout was carried with nickase Cas9 strategy to preclude the off-target effects.
ERα binds indirectly to the BAER enhancers
To delineate the potential differential features that underlie the BAER vs. E2 activated enhancers, we explored the predicted factor binding motifs enriched in these enhancers and found that BAER enhancers were enriched for FOXA1 motifs (Figure 3A); however, there was little representation of ERE motifs (Schwabe et al., 1990), which, in contrast, is characteristically the most enriched motif for activated enhancers (Liu et al., 2014). Therefore, we tested the possibility the ERα might be recruited to these enhancers indirectly.
Figure 3. ERα binds indirectly to BAER enhancers.

A, Sequence logos are shown for the most highly enriched sequence motifs in BAER enhancers. The fraction of peaks containing at least one instance of each motif within 200 bp of the peak center is given to the right of the motif with the expected frequency of the motif in random genome regions with the same GC-content given in parentheses. B, Average enrichment profile (significant counts) of wild type ERα and Pbox mutated biotin ligase recognition peptide (BLRP)-and HA-tagged ERα relative to the center of E2 activated enhancer and BAER enhancer under E2 condition. The x-axis represents the distance to the center of selected ChIP regions. The y-axis represents average ChIP-seq tag count numbers. C, RT-qPCR showing gene expression levels of E2 active gene-Pgr, E2 repressed gene-Sytl2 and Plekhf2 in response of E2 treatment when FoxA1 was knockdown by siRNAs. Data are presented as mean ± s.d. P values denote differences between siControl (siCTL) and siFoxA1 are shown (N = 4; unpaired t test). D, The significant peaks of ERα spanning ±3kb region from the center of BAER enhancers under siCTL and siFoxA1 condition. E, Box-and-whisker plots showing ERα ChIP-seq signals (tag counts) in siCTL or FoxA1 knockdown MCF7 cells before and after E2 treatment (1 h) at BAER enhancers. P values denote statistical differences between treatment conditions. Centerlines show the medians, box limits indicate the 25th and 75th percentiles, and whiskers extend 1.5× the interquartile range from the 25th and 75th percentiles. F, The “openness” of the enhancer by FAIRE-seq (E-MTAB-223) at E2 active and BAER enhancer under siCTL or siFoxA1 conditions are represented as histographs.
Using MCF7 cell lines that express proximal box (P-box) mutant ERα that dramatically reduces, but does not entirely eliminate, its ability to bind to its cognate DNA binding sites (Stender et al., 2010), we evaluated their binding to E2 activated and BAER enhancers by chromatin immunoprecipitation sequencing (ChIP-seq). Consistent with the markedly reduced ability of this mutant receptor to bind to its cognate DNA recognition element, the recruitment of the P-box mutant ERα was highly compromised for binding to active enhancers, exemplified by the Tff1 enhancer (Figure S3A), which harbor the ERE motif. In contrast, the E2-dependent recruitment of ERα was observed to be quantitatively equivalent when comparing wild type and P-box mutant ERα on BAER enhancers (Figure 3B), as illustrated by a browser image of the Plekhf2-enhancer (Figure S3B). These data support the model that ERα is directly recruited to the activated enhancers based on the cognate binding site EREs (referred to as “direct binding”), but indirectly to the BAER enhancers (referred to as “indirect binding”), which do not harbor EREs, dictating their opposing activated vs deactivated responses to E2.
Based on the well-established interactions between ERα and its pioneer protein, FOXA1 on activated enhancers (Hurtado et al., 2011), we tested the effects of knockdown of FoxA1 by siRNAs on the recruitment of ERα on BAER enhancers and the expression of their coding target genes. Examining whether FOXA1 was itself functionally important for the recruitment of ERα to these BAER enhancers, we found that FoxA1 knockdown decreased ERα recruitment on the enhancers, such as the Plekhf2-enhancer (Figure S3C) and the Sytl2-enhancer (Figure S3D), similar to what is observed for activated enhancers. Consistent with previous microarray data (Hurtado et al., 2011), FoxA1 knockdown caused dramatic decrease of Plekhf2 and Sytl2 gene basal level expression (Figure 3C), suggesting the FOXA1 is one of the major proteins to maintain the activation of these genes. Moreover, after exposure of these knock-down enhancers to E2, Plekhf2 and Sytl2 gene are not inhibited further (Figure 3C), exhibiting decreased recruitment of ERα at these BAER enhancers
To evaluate the global effects of ERα binding to BAER enhancers following knockdown of FoxA1, we examined ERα ChIP-seq and Formaldehyde Assisted Isolation of Regulatory Elements Sequencing (FAIRE-seq) data in MCF7 cells that were treated with siFoxA1 (Hurtado et al., 2011), finding that siFoxA1 caused a global decrease of ERα recruitment (Figures 3D and 3E) and also a global decrease of chromatin “openness’ by FAIRE-seq on BAER enhancers, which is similar to E2-activated enhancers (Figure 3F). To ask whether ERβ or ERα/β heterodimers could displace of pre-existing ERα (Drouin et al., 1993), we knocked-down ERβ with siRNAs and found that ERβ is not involved in the repression strategy in MCF7 cells (Figure S3E). Therefore, FOXA1 serves as a “pioneer” for our BAER enhancers, analogous to its role on activated enhancers, increasing the chromatin openness, apparently permitting ERα binding to these enhancers upon E2 treatment.
Pol II dismissal from BAER enhancers in response to E2
To explore why indirectly-bound ERα causes BAER enhancer decommissioning, we analyzed the binding of coactivators and Pol II at these enhancers (Figure S4A). Rather unexpectedly, we observed that E2 actually elicited a further increase in recruitment of coactivators, including CBP, p300, SRC3 on the BAER enhancers (Figure 4A), as illustrated by the genome browser of Plekhf2-enhancer (Figure S4B). This is similar to known events on E2-activated Tff1-enhancer (Figure S4C). Pol II levels on these enhancers were also decreased with E2 treatment (Figures 4B and S4D), but were not altered on randomly selected genomic regions (Figure S4E), indicating that the dismissal of Pol II on these enhancers might lead to their decommissioning. Further, knockdown of the endogenous ERα in MCF7 cells that express HA-tagged P-box mutant ERα (Figure S4F) resulted in the dismissal of Pol II at Plekhf2-enhancer and Sytl2-enhancer (Figure 4C), while the recruitment of HA-tagged ERα at these BAER enhancers was not affected (Figure S4G). We next investigated the potential importance of Pol II transcripts on BAER enhancer, eRNAs, in the regulation of gene expression. Transfection of specific antisense oligonucleotides (ASOs) targeting the Sytl2 or Plekhf2 eRNAs, down-regulated the cognate Sytl2 or Plekhf2 mRNAs expression (Figure 4D). To further show the critical function of BAER enhancer transcription on cognate target gene mRNA expression, we investigated the ability of CRISPRi (dCas9 fused with KRAB protein) (Gilbert et al., 2013) targeted to BAER enhancers. We found that the CRISPRi inhibited BAER enhancers eRNAs and cognate target gene expression (Figure 4E). An independent approach to support this result was achieved using an enhancer-dependent reporter assay, for which a Pol II transcriptional terminator (Nojima et al., 2013) was inserted into the Sytl2 or Plekhf2 enhancers, causing a decrease in the reporter gene expression (Figure 4F), but the insertion of a sequence of comparable length without terminators did not inhibit reporter gene expression (Figure S4H).
Figure 4. Indirectly-bound ERα recruits co-activators, but Pol II is dismissed from the enhancers upon E2 treatment.

A, The enrichment of CBP (E-TABM-785), p300 (E-TABM-785) and SRC3 (E-TABM-785) spanning ±3kb region from the center of BAER enhancer under Veh and E2 conditions, represented as heatmaps. B, The enrichment of Pol II spanning ±3kb region from the center of E2 active and BAER enhancer under Veh and E2 conditions were represented as heatmaps. C, The enrichment of Pol II at selected enhancers was measured by RT-qPCR when endogenous ERα was knocked-down using two different siRNAs targeting the 3’UTR of ERα in HA-tagged P-box mutant ERα stable cells. Data are presented as mean ± s.d. (N = 3; unpaired t test). D, Plekhf2 and Sytl2 eRNAs were knocked-down using two different ASOs. The expression level of Plekhf2 and Sytl2 mRNA and eRNAs are measured by RT-qPCR. Data are presented as mean ± s.d. P values denoting differences between between control sgRNA and sgRNAs target to Plekhf2- or Sytl2-enhancers. (N = 3; unpaired t test). E, Plekhf2 and Sytl2 enhancers were knockeddown by dCas9-KRAB with 2 different pairs of sgRNAs. The expression level of Plekhf2 and Sytl2 mRNA and eRNAs were measured by RT-qPCR. Data are presented as mean ± s.d. P values denoting differences between control sgRNA and sgRNAs target to Plekhf2- or Sytl2-enhancers. (N = 3; unpaired t test). F, Insertion of Pol II transcription terminators to Plekhf2 and Sytl2 enhancers could decrease downstream target reporter gene (luciferase) expression in HEK-293T cells. Plekhf2 and Sytl2 enhancers with ~500bp Pol II transcription terminators were cloned into pGL4.23 and transfected into HEK-293T cells, the luciferase value was detected by Dual-Luciferase® Reporter Assay System (Promega). G, 4C-seq under vehicle or 1hr E2 treatment in MCF7 cells is shown. The black line represents the interaction frequency based on liner mean in 200kb window. Gray cloud indicates 20th and 80th percentiles of confidence intervals of the main trend on the curve (black line). Domainogram (heatmap) represents the normalized contact profile, which resolution was ranked from 2kb on the top to 50kb on the bottom. Coordinate of enhancers are indicated with red box, while the promoters are indicated with blue box. H, 3CPCR assays showing the changes of the interaction between enhancer and promoter at Plekhf2 and Sytl2 loci upon vehicle or E2 treatment.
To ascertain whether eRNAs expressed under basal conditions on BAER enhancers were correlated with interactions to their cognate target gene promoters, we performed chromosome conformation capture combined with high-throughput sequencing (4C-seq) assay (van de Werken et al., 2012) on the E2 repressed genome loci, and we detected decreased interactions between BAER enhancers and E2 repressed gene promoters (Figure 4G). Quantitative PCR to evaluate the changes between Veh and E2 conditions by 3C-qPCR for the interactions of gene promoters and enhancers yielded similar results (Figure 4H). Thus, these data show that inhibition of eRNA transcription at BAER enhancers by diverse strategies leads to decreased interactions between BAER enhancers and their cognate target gene promoters, causing deactivation on their cognate target genes.
KDM2A mediates enhancer decommissioning independent on its demethylase function by interacting with the ERα DNA binding domain
To begin to explore potential mechanisms underlying these observations, we performed a mini-siRNA screen designed to target a series of potential corepressors, including NCoR, SMRT, LCoR, GRIP1, SHP, HDAC1/2/3/4/7 (Figure S5A), focusing on the effects of these siRNAs on a subset of target loci, including Sytl2, Plekhf2 Ncoa3, Rnf43, Cdkn1b and Txnip. In this pilot screen, knockdown of suspected corepressors had little or no impact on the E2-induced repression events (Figure 5A). Because histone methylation levels play important roles in transcription, and targeting of histone demethylases (KDMs) are emerging as promising therapeutic agents against cancer (Hojfeldt et al., 2013), we also investigated whether KDMs might be involved in the E2 mediated decrease in BAER enhancer transcription. To this end, we tested siRNAs against Kdm1a/b; Kdm2a/b; Kdm3a/b/c; Kdm4a/b/c/d and found that only the knockdown of Kdm2a, which is a known histone H3 lysine 36 (H3K36me2) demethylase (Tsukada et al., 2006), altered the E2 mediated down-regulation of eRNAs and target coding gene expression (Figure 5B).
Figure 5. KDM2A is involved in ERα induced enhancer decommissioning.

A-B, RNAi screen showed that traditional corepressors (A) and histone demethylases (B) are less likely candidates, but that KDM2A is involved in the ERα mediated transcription repression in MCF7 cells. The ratios of the expression levels for the indicated eRNAs or coding genes between E2 and Veh conditions are showed by heatmaps. C, Heatmap showing that eRNA transcription level detected by PRO-seq was decreased by E2 in siCTL condition, and it is reversed by the knockdown of Kdm2a. The relative fold-change (E2/Veh-1) of each enhancer is shown. D, Boxand-whisker plots showing Pol II ChIP-seq signals (tag counts) in siCTL or Kdm2a knockdown MCF7 cells before and after E2 treatment (1 h) at E2 activated and BAER enhancers. P values denote statistical differences between treatment conditions. Centerlines show the medians, box limits indicate the 25th and 75th percentiles, and whiskers extend 1.5× the interquartile range from the 25th and 75th percentiles. E, Venn diagram showing the genome-wide overlap of ChIP-seq peaks of HA-tagged KDM2A with BAER enhancers in MCF7 cells upon E2 treatment. F, The enrichment of HA-tagged KDM2A spanning ±3kb region from the center of BAER and activated enhancer under Veh and E2 conditions, represented as heatmaps.
To determine whether KDM2A is globally required for the E2-mediated repression program, we employed the Precision nuclear Run-On and sequencing assay (PRO-seq), which maps the location of active RNA polymerases genome-wide with high resolution (Kwak et al., 2013). We observed that 716 enhancers exhibited a decrease of eRNA tags upon E2 treatment. Notably, in the absence of KDM2A, deactivation on the vast majority of BAER enhancers was impaired (Figure 5C), suggesting that KDM2A is a crucial component of this enhancer decommissioning strategy in MCF7 cells. To further confirm this result, we performed Pol II ChIP-seq in MCF7 cells, which revealed that Kdm2a knockdown reversed the decrease of Pol II on BAER enhancers, while having almost no effect on E2 activated enhancers (Figures 5D and S5B). Furthermore, using doxycycline-induced KDM2A-HA tagged stable MCF7 cell line for ChIP-seq, we found that at least 643 ERα-marked BAER enhancers overlapped with very highly reproducible KDM2A peaks (Figures 5E and S5C), which were calculated by HOMER peak-calling software and using ENCODE’s irreproducible discovery rate (IDR) analyses, while only very few KDM2A peaks could be observed on E2 activated enhancers (Figure 5F).
We therefore investigated whether KDM2A could interact with ERα by using a specific α-KDM2A antibody to pull-down nuclear extracts from MCF7 cells. We detected clear ERα: KDM2A interactions in MCF7 cells (Figure 6A). Furthermore, mapping HA-tagged ERα domain interactions with KDM2A indicated that KDM2A could associate with the DNA-binding domain (DBD) of ERα (Figure 6B). Furthermore, the interaction between KDM2A and purified ERα DBD (Figure S6A) was quantitatively competed by DNA oligonucleotides containing ERE motifs (Figure 6C), but not by randomized DNA oligonucleotides (Figure S6B), consistent with the observation that KDM2A was enriched at BAER enhancers, where ERα is indirectly recruited and its DBD is therefore accessible.
Figure 6. KDM2A mediates enhancer decommissioning through its interaction with ERα DNA binding domain.

A, KDM2A interactions with ERα and NEDD4 are revealed by anti-KDM2A antibody (Santa Cruz, H120) co-immunoprecipitation in MCF7 cells under E2 treatment. B, The interaction of ERα with KDM2A is dependent on its DNA-binding domain (DBD), as shown by coimmunoprecipitation using HA-tagged ERα fragments. KDM2A interacts with full length of ERα (ER-FL) and ERα DNA-binding domain (ER-DBD), but not with ERα AF1 domain (ER-AF1). C, In vitro immunoprecipitation of KDM2A with Flag-tagged ERα DBD (Flag-ER DBD) protein, in presence of 0, 0.5, 5 and 50 μl 2xERE oligonucleotides (100 μM) as competitor. The interaction between KDM2A and ERα DBD was quantitatively decreased by DNA oligos encompassing an ERE motif. Empty vector (EV), which does not express Flag-tagged ERα DBD protein, served as a control. D, RT-qPCR showing expression levels of Sytl2-enhancer, Plekhf2-enhancer and their target gene repression by E2 treatment can be rescued by both wild type KDM2A and H212A mutated KDM2A (eS: eRNA sense chain; eAS: eRNA anti-sense chain). Data are presented as mean ± s.d. P values denote differences between each group upon E2 treatment are shown (N = 4; unpaired t test). E, Heatmap showing that eRNA expression levels were decreased by E2 in siCTL condition, and this was reversed by the knockdown of Kdm2a (by siKdm2a5utr); the effect of siKdm2a5utr could be rescued by either wild type or H212A mutant HA-tagged KDM2A in MCF7 cells. The relative fold-changes (E2/Veh-1) of each enhancer are shown.
To ask whether H3K36me2 demethylase activity was required for this enhancer decommissioning function of KDM2A, we performed H3K36me2 ChIP-qPCR on several enhancer loci, and we found that the enrichment of H3K36me2 was very low in these enhancer regions, which is consistent with previous reports showing that H3K36me2 is generally not enriched at enhancers (Kuo et al., 2011), and that knock-down of Kdm2a did not affect H3K36me2 at these regions (Figures S6C). We then explored the ability to rescue function using an enzymatic activity-deficient KDM2A demethylase mutant (H212A in the JmjC domain) (Tanaka et al., 2010) (Figures S6D and S6E). Using the Plekhf2 and Sytl2 enhancers as models, we knocked down the endogenous KDM2A with siRNA targeting the 5’UTR of the Kdm2a gene in HA-tagged wild type (wt) KDM2A or H212A-KDM2A stable cell lines. After verifying the relatively equal expression levels of HA-tagged KDM2A with endogenous KDM2A (Figure S6F), the ability of WT and mutant KDM2A to rescue the inhibitory effects of ERα was then tested. We found that both WT and mutant KDM2A were equivalently effective in re-imposing E2-dependent decrease of eRNA transcripts and cognate target gene expression (Figures 6D and 6E). These data indicated that KDM2A actions in BAER enhancer decommissioning were independent of its demethylase function.
We therefore asked whether KDM2A could directly regulate Pol II on the enhancers. Overexpression of either KDM2A or the enzymatically-inactive KDM2A-H212A mutant in mammalian cells led to a global decrease in Ser2-phosphorylated RNA polymerase II (PolIISer2P), which serves as a signature of transcription elongation (Brookes and Pombo, 2009), linked to enhancer activation (Basnet et al., 2014) (Figure S7A). Such a decrease in protein levels of PolIISer2P is usually due to its ubiquitylation and subsequent degradation, leading to transcriptional repression (Somesh et al., 2005; Sun et al., 2013). Consistent with these observations, when cells were treated with the proteasome inhibitor MG132, we detected an increase of PolIISer2P protein (Figure S7B). To determine whether KDM2A is involved in PolIISer2P ubiquitylation, KDM2A was knocked down by specific siRNAs, and UV-radiation was employed to enhance detection of PolIISer2P ubiquitylation and degradation, finding that Kdm2a knockdown by siRNAs could block these processes (Figure S7C). Then, to test whether KDM2A directly contributed to ubiquitylation-based mechanisms, we purified KDM2A containing complexes from MCF7 cells, and performed in vitro ubiquitylation assays using KDM2A containing complexes purified from MCF7 cells without adding additional E3 ligase, as a result, the characteristic “smeared” ubiquitylation signal was observed above the major band, and this was confirmed as ubiquitylated protein by western blot analysis using a specific anti-ubiquitin antibody (Figure S7D), indicating that KDM2A complexes possess ubiquitin E3 ligase activity.
To ask whether Pol II ubiquitylation underlies the enhancer decommissioning events, we investigated Pol II binding at the BAER enhancers and target gene expression after treating MCF7 cell with MG-132 to block the degradation of ubiquitylated protein. We found that the decrease of Pol II upon E2 treatment at BAER enhancers, such as the Plekhf2 and Sytl2 enhancers, was arrested by MG132 treatment, while the increase of Pol II at E2-activated enhancers, such as Greb1 enhancer, was not affected (Figure 7A). As expected from these findings, the repression of Plekhf2 and Sytl2 genes expression in response to E2 treatment was rescued by MG132 treatment (Figure 7B). Further, we overexpressed HA tagged ubiquitin in MCF7 cells (Figure S7E), and our results showed that MG132 reverses ubiquitylation at BAER enhancers, such as the Plekhf2 and Sytl2 enhancers, in E2-dependent manner, but not on the control gene genome regions, such as Klk3mid (Figure S7F). These data indicated that ubiquitylation of Pol II at BAER enhancers is functionally associated with E2 dependent target gene down-regulation.
Figure 7. KDM2A recruits NEDD4 complex to dismiss Pol II at BAER enhancers.

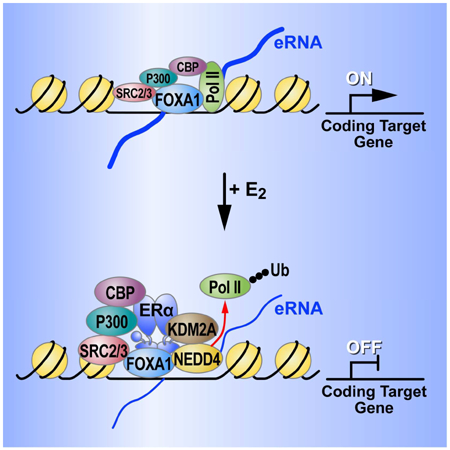
A, E2-mediated decrease of Pol II at the Plekhf2 and Sytl2 enhancers is abolished by 10 M MG132 treatment for 3 h, as shown by Pol II ChIP-qPCR. ChIP signals are presented as percentage of input. Data are mean ± s.d. P values denoting differences between Veh and E2 condition are shown. B, RT-qPCR showing gene expression levels of E2 repressed gene-Sytl2 and Plekhf2 in response of E2 when MCF7 cells was treated with 10 M MG132. Data are presented as mean ± s.d. P values denote differences between +/− E2 treatment are shown (N = 3; unpaired t test). C, The enrichment of NEDD4 spanning ±3kb region from the center of BAER or activated enhancers in Veh and E2 conditions is represented as heatmaps. D, Box-and-whisker plots of NEDD4 ChIP-seq signals (tag counts) in siCTL or Kdm2a knockdown MCF7 cells before and after E2 treatment at BAER enhancers are shown. P values denote statistical differences between treatment conditions. E, Box-and-whisker plots of Pol II ChIP-seq signals (tag counts) in siCTL or Nedd4 knockdown MCF7 cells before and after E2 treatment at BAER enhancers, but not at E2 activated enhancers, are shown. P values denote statistical differences between treatment conditions. Centerlines show the medians, box limits indicate the 25th and 75th percentiles, and whiskers extend 1.5× the interquartile range from the 25th and 75th percentiles. F, Plekhf2 and Sytl2 enhancers were repressed by dCas9 fused with KDM2A or truncated NEDD4 (containing C terminal HECT domain) proteins generated using two different groups of sgRNAs. The expression level of Plekhf2 and Sytl2 mRNA and eRNAs are measured by RT-qPCR. Data are presented as mean ± s.d. P values denoting differences between control sgRNA and sgRNAs target to Plekhf2- or Sytl2-enhancers. (N = 3; unpaired t test; * = p< 0.05, ** = p< 0.01, *** =p< 0.001.). G, Endogenous ERα and Kdm2a or Nedd4 were knocked-down using siRNAs targeting in the ERα P-box mutated stable cell line. The enrichment of Pol II at selected enhancers was measured by RT-qPCR. Data are presented as mean ± s.d. (N = 3; unpaired t test; * = p< 0.05, ** = p< 0.01, *** =p< 0.001.). H, Working model: E2 treatment causing an increased enrichment of coactivators, such as P300, CBP, SRC3 at BAER enhancers. KDM2A interacts with the DBD of ERα, recruits NEDD4 to the enhancers, and dismisses Pol II to impede enhancer activation to eliminate target gene transcription.
NEDD4 is recruited to BAER enhancers by KDM2A and involved in dismissing Pol II from BAER enhancers
Because the KDM2A complex exhibits ubiquitin E3 ligase activity, we wondered which protein exerted this function in mediating Pol II ubiquitylation. NEDD4, is the major E3 ligase responsible for Pol II ubiquitylation in mammalian cells (Anindya et al., 2007), and has already been shown to be involved in human progesterone and glucocorticoid receptor-induced transcription programs (Imhof and McDonnell, 1996). Accordingly, we also observed that KDM2A not only interacts with ERα, but also with NEDD4 by co-immunoprecipitation (co-IP) assays (Figure 6A). This was further confirmed by analysis of Pol II co-IP protein complexes by mass spectrometry (Figure S7G), revealing the presence of KDM2A, ERα and NEDD4. ChIP-seq analysis also revealed a global increase of NEDD4 at BAER enhancers, but not at E2 activated enhancers, in MCF7 cells upon E2 treatment (Figure 7C). These interaction data and the coincident binding at BAER enhancers suggested that KDM2A might be required for NEDD4 recruitment. To ask whether KDM2A was indeed required for NEDD4 recruitment to BAER enhancers, we performed NEDD4 ChIP-seq in MCF7 cells. We found that the occupancy of NEDD4 at BAER enhancers was highly reduced upon Kdm2a knockdown (Figure 7D), indicating the KDM2A-dependent recruitment of NEDD4 at these enhancers. This was also further confirmed by ChIP-qPCR at the Plekhf2 and Sytl2 enhancers (Figure S7H).
In support of the role of NEDD4 in enhancer decommissioning, Pol II ChIP-qPCR was performed revealing that ERα-dependent Pol II dismissal on BAER enhancers, exemplified by Plekhf2e and Sytl2e, was rescued upon knockdown of either Kdm2a or Nedd4 (Figure S7I). This finding was also confirmed by genome-wide ChIP-seq analysis of Pol II enrichment at BAER enhancers, finding that the E2-dependent decrease in Pol II occupancy was rescued by knockdown of Nedd4, while knockdown of Nedd4 had essentially no effect on E2 activated enhancers (Figure 7E). To formally prove that KDM2A/NEDD4 complex could inhibit the transcription of enhancers, CRISPR-dCas9 tethered to KDM2A or to truncated NEDD4, encompassing the homologous to E6-AP carboxyl terminus (HECT) domain containing the ubiquitylase activity, was targeted by sgRNAs to select BAER enhancers in MCF7 cells. We found that expression of Sytl2 or Plekhf2 eRNAs was inhibited by the tethered KDM2A or truncated NEDD4 fusion proteins, and their target gene expression was also inhibited (Figure 7F); in contrast, there was no effect on transcription units not targeted by the sgRNAs used (Figure S7J). To further prove that KDM2A/NEDD4 complex is responsible for the indirect-bound ERα mediated Pol II dismissal at BAER enhancers, Kdm2a or Nedd4 was knocked-down by siRNAs in a HA-tagged P-box mutant ERα cell line. We found the either Kdm2a or Nedd4 knocked-down could reverse the dismissal of Pol II at selected BAER enhancers, which further confirmed the critical roles of KDM2A/NEDD4 in the decommissioning of BAER enhancers (Figure 7G).
To investigate whether KDM2A-mediated enhancer decommissioning might be broadly utilized in other systems, we analyzed published KDM2A ChIP-Seq in mouse embryonic stem cells (mESCs) (Blackledge et al., 2010). This revealed that 1,840 mESC enhancers marked by H3K4me1 (GSM723016) and H3K27Ac (GSM594578) are occupied by KDM2A (Figure S7K). In light of a previous study that assigned the enhancers and their target genes (Shen et al., 2012), it could be determined that knockdown of Kdm2a also caused an induction of the cognate target gene expression in mESCs (Figure S7L). Because KDM2A knock out (KO) mice exhibit embryonic lethality at E10.5–12.5 (Kawakami et al., 2015), further analysis of the functions of KDM2A on enhancer driven gene expression in mature tissues will require conditional KO strategies and would clearly be an interesting topic for future investigation.
Collectively, these data demonstrate that NEDD4 is recruited to the BAER enhancers by KDM2A, in turn dismissing Pol II from enhancers to which ERα is indirectly recruited, serving as the mechanism underlying their ERα-mediated enhancer decommissioning in MCF7 cells. Our data also suggest that the KDM2A-mediated repression strategy may be widely utilized in many regulatory contexts.
DISCUSSION
Estradiol-17β (E2) causes both the activation and repression of large transcriptional programs. Here, we have discovered that the repression program is largely mediated by the indirect recruitment of ERα to enhancers that are highly activated in the basal state (i.e., prior to exposure to E2). This is in contrast to the well-established transcriptional activation program, which is largely mediated by enhancers binding ERα in cis. Surprisingly, the indirectly-bound ERα causes the recruitment of KDM2A, which interacts with the DBD of ERα, decommissioning these active enhancers and causing the decrease of their cognate coding target gene transcription. Significantly, this does not require the demethylase activity of KDM2A, but rather its recruitment of NEDD4, which results in covalent modification and loss of Pol II at these enhancers. Knocking-down of either Kdm2a or Nedd4 rescues the enhancer decommissioning mediated by indirectly-bound ERα (Figure 7H).
Indirectly-bound ERα on BAER enhancers regulates transcriptional repression programs
Although active recruitment of corepressors to single gene promoters has been reported by several groups, with the application of genome wide ChIP-seq data, it became clear that ERα primarily functions as an enhancer-bound transcription factor. Indeed, only ~15% of ERα peaks were located in the broad promoter regions (+/− 1,000bp). Here, taking advantage of GRO-seq, we detected 1,205 E2 repressed genes, noting that for these genes, only 30 of their promoters bound ERα, and exhibited no enrichment in response to E2 treatment, suggesting that a promoter-based repression strategy could not be responsible for the majority of ERα-mediated target gene transcriptional repression events. The functional significance of BAER enhancers is validated by employing the CRSIPR-Cas9 gene editing strategy to knock out these enhancers, confirming that these enhancers indeed are required to activate their target gene expression under basal conditions, with the E2-dependent recruitment of ERα to these enhancers therefore repressing their cognate target genes.
RNA Polymerase II dismissal is a new enhancer decommissioning strategy
The E2-mediated repression program depends on the availability of the DBD of the indirectly-bound ERα, which permits recruitment of a specific complex causing dismissal of the Pol II at these initially eRNA-producing active enhancers, and represents a previously unappreciated type of repressive strategy. It occurs on basally-active enhancers, and these enhancers, while recruiting further level of coactivator machinery upon E2 treatment, also recruit a complex that ubiquitylates and dismisses Pol II, resulting in loss of enhancer activation function. An alternative outcome of indirect binding of a nuclear receptor is suggested by a recent paper that has reported that GR recruitment to non-GRE regions could cluster with directly bound GR to synergistically modulate the activity at direct GR binding sites (Vockley et al., 2016).
Meanwhile, trans-repression by liganded nuclear receptors based on actions at promoters has been observed for several members of this super-family (Glass and Saijo, 2010). In the presence of ligand, PPARγ and LXR become tethered and prevent dismissal of corepressor complexes. This is usually associated with sumoylation on the nuclear receptors and also with recruitment of corepressor complexes, including NCoR/SMRT/HDACs, executing their repression function by preventing coactivator exchange. In contrast, coactivators are actually further recruited to our BAER enhancers in response to ligand. Our results also reveal that expression levels of eRNAs, rather than enrichment of coactivators, severs as a more predictive indicator of enhancer function in the target gene activation.
Therefore, the mechanism we describe is distinct from previously-reported strategies, and is a mechanism that is likely to be widely utilized, exemplified by finding similar phenomena in ES cells. The broad utilization of repressive mechanisms by estrogen is also suggested by repressive actions of ERα in the hippocampus during memory formation (Cho et al., 2015), indicating the potentially widespread utilization of this repressive mechanism, deserving future study.
Pol II ubiquitylation is involved in the BAER enhancer decommissioning
Interestingly, in human breast cancer MCF7 cells, we do not observe extensive enrichment of KDM2A to the polycomb repressive complex 1 (PRC1)-depleted, non-methylated CpG islands, as had been reported in mouse embryonic stem cells (Blackledge et al., 2014). To exert its transcriptional repression function in MCF-7 cells, KDM2A recruits a different E3 ubiquitin-protein ligase, NEDD4, to the basally active enhancers, decommissioning them based on Pol II ubiquitylation. The fact that we observe that the effects on Pol II dismissal was prevented by treatment with MG132 is consistent with a previous report that transcriptional arrest at DNA lesions triggers NEDD4 recruitment and Pol II ubiquitylation (Anindya et al., 2007). Therefore, it appears that degradation of the ubiquitylated Pol II is an important mechanism underlying BAER enhancer decommissioning. While stalled Pol II found at many rapidly activated promoters exhibits rapid signal-dependent conversion to an elongating form (Hargreaves et al., 2009), ubiquitylation and degradation of Pol II can serve as a “last resort” to clear stalled Pol II from lesions when transcription-coupled nucleotide excision repair fails. Our findings here that ubiquitylated Pol II is associated with decreased eRNA transcription suggest the possibility that deubiquitylating enzymes might serve as available candidates for human cancer therapy, as has been suggested by several groups (Singhal et al., 2008).
STAR METHODS
Contact for Reagent and Resource Sharing
Please direct any requests for further information or reagents to the lead contact, Professor Michael. G. Rosenfeld (mrosenfeld@ucsd.edu), School of Medicine, University of California, San Diego, La Jolla, CA 92093, USA.
Experimental Model and Subject Details
MCF7 cells were maintained at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM GIBCO/Invitrogen) with phenol red, supplemented with 10% fetal bovine serum (FBS, GIBCO/Invitrogen). For hormone treatments, cells were incubated at 37°C and 5% CO2 for at least 3 days in phenol red-free DMEM (GIBCO/Invitrogen) supplemented with 5% charcoal dextran-stripped FBS (GIBCO/Invitrogen). 17-β-Estradiol (E2; Steraloids, Inc.) was added to a final concentration of 100 nM. The ethanol (EtOH) vehicle control was 0.1% in all samples. 46C murine ESCs were kindly gifted by Austin Smith. mESCs were maintained in 2i medium were grown in N2/B27 media with 50% Neurobasal (Gibco 21103–049) and 50% DMEM/F12 (Invitrogen 21331–020), 2 mM nonessential amino acids, glutamax, penicillin/streptomycin, 2-mercaptoethanol, N2 supplement (Invitrogen, 175020–01), B27 (Invitrogen, 17504–001), 1000 U/ml LIF, and 2i (ESGRO, ESG1121).
Method Details
Transfection of siRNA
For small interfering RNA (siRNAs), cells were transfected in 10-cm plates in regular DMEM without antibiotics. 20 l of 20 M siRNAs and Lipofectamine® 2000 reagent (Invitrogen Cat# 11668–019) were diluted in Opti-MEM® I Reduced Serum Medium (Invitrogen Cat# 11058–021), and incubated for 6 h, and then changed to phenol red-free medium. For siRNAs screen, cells were transfected in 24 well plates in regular DMEM without antibiotics. 1 l of 20 M siRNAs (Supplementary Table 2) was transfected with Lipofectamine® 2000 reagent (Invitrogen Cat# 11668–019). In both condition, two days later, cells were transfected with siRNA again in phenol red-free medium. Three days following transfection, cells were treated with EtOH or E2 for 4 h and harvested for RNA isolation, or treated for 1 h for GRO-seq assay.
shRNA Lentivirus Package and Infection
pLKO lentiviral shRNA constructs and control shRNA constructs were purchased from Sigma. Knockdown experiments with lentivirus shRNAs were conducted according to the standard lentivirus package and transduction protocols from Addgene. pLKO-based lentiviral shRNA plasmids were co-transfected with packaging plasmids (psPAX2 and pMD2.G) into 293T cells. Lentiviruses were harvested, concentrated, and used for MCF7 cell infection. Stable knockdown MCF7 cells were selected with 1 μ/ml puromycin and collected for experiments within 5 days. Before collection, the cells were grown for 3–4 days in stripping media containing 1 μ/ml puromycin for continued selection to achieve better knockdown.
RNA isolation and qRT-PCR
RNA was isolated using Trizol (Invitrogen), and total RNA was reversed transcribed using SuperScript III Reverse Transcriptase (Invitrogen). Quantitative PCRs were performed mostly with StepOne Plus (Applied Biosystem). The sequences of the primers used for the different gene targets are shown in Supplementary Table 2. β-actin mRNA or GAPDH was used as the internal control. Relative mRNA levels were calculated by theΔΔCt method with the vehicle (EtOH) used as the calibrator.
CRISPR/Cas9 Knockout Assay
We use two sgRNAs targeting the upstream and downstream of putative repressive enhancer respectively with relative high specificity predicted by the online software (http://crispr.mit.edu/). Then these two sgRNAs were cloned to lentiCRISPR vector (Addgene, Cat#49535). The strategy for cloning two sgRNA into lentiCRISPR vector was developed by Prof. Xingxu Huang lab (unpublished). Next, we transfected the lentiCRIPSR vector expressing Cas9 and two sgRNAs into HEK293T cells with the packaging plasmids pVSVg (Addgene, Cat#8454) and psPAX2 (Addgene Cat#12260). 48 hours later, viruses were harvested and infected MCF-7 cells. Then MCF7 cells were screened under puromycin selection and homozygous clones were selected using PCR and Sanger sequencing for genotyping deletion clones, and confirmed by droplet digital polymerase chain reaction (ddPCR). The sgRNA sequences and primers used for the different gene targets are shown in Supplementary Table 2.
Droplet Digital polymerase chain reaction
Droplet Digital PCR (ddPCR) was performed and data analyzed according to previous studies(Abyzov et al., 2012) and standard BioRad protocol (http://www.bioradiations.com/a-new-paradigm-for-precise-quantitation-of-rna/). Briefly, first design the primers and fluorescence-labeled probes for enhancer deletion region and outside control region. Extract genome DNA using QuickExtract DNA Extraction Solution (Epicentre). Mix DNA templates from wild type cell and Plekhf2 enhancer knock out cells, primers, and fluorescence-labeled probes with Bio-Rad’s ddPCR supermix. In the beginning, we used a gradient PCR to optimize the amplifying condition. Eventually, we used the program: 94°C 5 min (94°C 20 sec, 58°C 25 sec, 72°C 30 sec) for 40 cycles, 72°C, 3 min. Load the ddPCR reaction mix into the wells of a droplet generator cartridge to generate about 8 × 20,000 droplets from each run in the QX200 droplet generator, which made control DNA and deletion region DNA are randomly distributed in droplets. Next, transfer the droplets to a 96-well PCR plate and seal the plate and run the PCR using the optimized condition mentioned above. After PCR, load the 96-well PCR plate into the QX200 droplet reader. Positive and negative droplets in each sample are read. Finally, analyze concentrations with QuantaSoft™ software. All Primers for ddPCR are listed in Supplementary Table 2.
CRISPRi mediated enhancer inhibition
The dCas9-KRAB stable MCF-7 cell line was constructed by lenti-virus infection. The dCas9-KRAB lenti-virus was prepared by co-transfection of pVSVg (Addgene, Cat#8454) and psPAX2 (Addgene Cat#12260), and pHAGE-TRE-dCas9-KRAB vector (Addgene, Plasmid #50917) into HEK-293T cell using Lipofectamine2000 and harvest the lenti-virus in 48 h and 72 h after transfection. Then MCF-7 cells were infected with dCas9-KRAB lenti-virus. Two days after infection, G418 was used to select positive infected cells. Then, the sgRNAs expression vectors were constructed by cloning multiple sgRNAs to pLKO.1-U6–2sgRNA-ccdB-EF1a-Puromycin vector (from Prof. Xingxu Huang lab) following the above-mentioned strategy. 2 days later after infection, cells were cultured under phenol red-free DMEM (GIBCO/Invitrogen) supplemented with 5% charcoal dextran-stripped FBS (GIBCO/Invitrogen). Meanwhile, Puromycin (working concentration 1ug/ml) was added to select positive infected cells and doxycycline was also added to induce dCas9-KRAB expression. After three days selection and induction, the cells were harvested and RNA was extracted to analyze eRNAs and target gene expression. Two groups of sgRNAs were employed to target Plekhf2 enhancer. Group-1 (Plekhf2-sgRNA1 in Figs. 7F and S7J) with combination of four sgRNAs as following: ATTATTCTGTCCGCCATAGTTGG; CTGCAACGTGGTAGGCAAACAGG; TTTCCTCACCCGTAGGACTGTGG; ATTGTATCCAGGTTCCGGGAGG. Group-2 (Plekhf2-sgRNA2 in Figs. 7F and S7J) with combination of four sgRNAs as following: TCCGCCATAGTTGGAAGCATGGG; TTAGCAACCAGGATGCCCCATGGG; CCTCACCCGTAGGACTGTGGTGG; GAGCTGCAGGTATAAGCGCTGGG. Similarly, there were another two groups of sgRNAs were employed to target Sytl2 enhancer. Group-1 (Sytl2-sgRNA1 in Figs. 7F and S7J) with combination of four sgRNAs as following: TCACTCCCAGAAGACTTATCTGG; CATGGTTGTCCTGCTAGTAGGGG; ACTTTCAGTTCTACGCCCTGGGG; ATCCTTCATAGATCTGATACTGG. Group-2 (Sytl2-sgRNA2 in Figs. 7F and S7J) with combination of four sgRNAs as following: AGTGACTCATCATAGTAAACAGG; ACAGACTATTGAGTCATGTGTGG; GACAAATCATGTAGTCTCCGTGG; CCAAGTTGAGTGTATTCAGTGGG. Control sgRNA, ACGGAGGCTAAGCGTCGCAA containing no target in human genome. The PAM sequence was underlined.
dCas9 fused with KDM2A or truncated NEDD4 mediated enhancer inhibition
Amplification of KDM2A and truncated NEDD4 (with HECT domain) coding sequence by using primers: KDM2A-F: AGGgctagcgccaccATGGAACCCGAAGAAGAAAGG, KDM2A-R: AGGgctagcGGCATAATCGGGCACGTCATAGGGATAGCTGATCTTCTGTATCAGCTTCT C; NEDD4c-F: AGGgctagcgccaccATGAGAGCAGACTTCCTGAAGGCTCGA, NEDD4c-R: AGGgctagcggcgtagtcaggcacgtcgtaaggataATCAACTCCATCAAAGCCCT. The digested the CDS fragments and pST1374-Cas9-ZF-N-Blasticidin vector (a gift of Xingxu Huang lab, containing dCas9) by Nhe I, then ligated coding fragments with pST1374-Cas9-ZF-N-Blasticidin vector to expression dCas9-KDM2A or dCas9-trunc-NEDD4 fusion protein. After transfection of the plasmids into MCF-7 cells, two days later added blasticidin (working concentration 30 μg/ml) was added 2 days later to select positive expressed cells. After another three-days selection, cells were infected with sgRNA expression lenti-virus as mentioned in CRISPRi experiment, and selected with Puromycin for another three days in phenol red-free DMEM (GIBCO/Invitrogen) supplemented with 5% charcoal dextran-stripped FBS (GIBCO/Invitrogen), then cells were harvested and extracted RNA to detect mRNA and enhancer RNA expression.
Luciferase Reporter Assay
~1kb indicated enhancer region was cloned from MCF7 cells into pGL4.23 (Promega) vector encoding Firefly luciferase, then each of these constructs and Renilla TK vector (Promega) were co-transfected into MCF7 cells in a ratio of 100:3. The luciferase value was detected 3 days post-transfection by Dual-Luciferase® Reporter Assay System (Promega). For the terminator insertion assay, the terminator sequence was cloned from MCF7 genomic DNA, according to reported sequence, using primer CCNB1termF CCATCTAGATTTGTGTGAGTA and CCNB1_R3 TATGTCATCTGGTTCTATCAA that reported in the literature (Nojima et al., 2013). The PCR product was inserted into the luciferase construct within Plekhf2 or Sytl2 enhancer by homologous recombination. A random sequence was cloned from CCNB1 gene locus other than terminal region. Primers for cloning Plekhf2 enhancers were tgcGGTACCTGCAAAGGCCTCTGCTTTTGTA and tgcCTCGAG GTACATGTGCATATTGTGCAG. Primers for cloning Sytl2 enhancers were tgcCTCGAGCTGTGTTGATTCCCACTCAGA and tgcGGTACCCTGGTGACCCAATCATTTGT.
Immunoblotting, Coimmunoprecipitations and Mass Spectrometry Sample preparation
Cells were washed with ice-cold phosphate-buffered saline (PBS) and harvested in cold PBS. For the preparation of whole cell extracts, pellets were resuspended in lysis buffer containing 20 mM Tris-Cl (pH 8.0), 137 mM NaCl, 10% glycerol, 1% Nonidet P-40 (NP-40) and a mixture of protease inhibitors. Samples were sonicated by using a Bioruptor (Diagenode) for 16 min at medium power, with an interval of 30 s between pulses. Following sonication, samples were centrifuged for 2×5 min at 12,000×g. After preclearing, immunoprecipitation was performed overnight at 4°C by using the indicated antibodies and protein G-Sepharose. Following immunoprecipitation, five times of washes in 1 ml lysis buffer were performed at 4°C, and protein complexes were denatured in Laemmli sample buffer (2% SDS, 10% glycerol, 60 mM Tris-Cl [pH 6.8], 0.01% bromophenol blue, 100 mM dithiothreitol [DTT]) for 5 min at 95°C and resolved by NuPAGE® Novex® 4–12% Bis-Tris Protein Gels (Invitrogen Cat# NP0336PK2). After electronic transfer, the PVDF membrane was blocked by incubation at room temperature for 1 h in Blocker Casein in TBS (Thermo Scientific, Cat# 37532). Complexes were revealed by Clarity™ Western ECL substrate (Bio-Rad Cat# 170–5061), as recommended by the manufacturer. Mass-Spectrometry was performed in UCSD Mass-Spectrometry facility. 40 μg of anti-Pol II antibody was employed to pull down the 5×108 pre-cleared cell nuclei. Proteins were separated by SDS-PAGE gels and cut the gels with indicated molecular weight for mass spectrometry analysis in UCSD molecular mass spectrometry facility (MMSF). In brief, protein samples were diluted in TNE (50 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA) buffer. RapiGest SF reagent (Waters Corp.) was added to the mix to a final concentration of 0.1% and samples were boiled for 5 min. TCEP (Tris (2-carboxyethyl) phosphine) was added to 1 mM (final concentration) and the samples were incubated at 37°C for 30 min. Subsequently, the samples were carboxymethylated with 0.5 mg/ml of iodoacetamide for 30 min at 37°C followed by neutralization with 2 mM TCEP (final concentration). Proteins samples prepared as above were digested with trypsin (trypsin:protein ratio - 1:50) overnight at 37°C. RapiGest was degraded and removed by treating the samples with 250 mM HCl at 37°C for 1 h followed by centrifugation at 14000 rpm for 30 min at 4°C. The soluble fraction was then added to a new tube and the peptides were extracted and desalted using C18 desalting columns (Thermo Scientific, PI-87782).
LC-MS/MS
Trypsin-digested peptides were analyzed by ultra high-pressure liquid chromatography (UPLC) coupled with tandem mass spectroscopy (LC-MS/MS) using nano-spray ionization. The nano-spray ionization experiments were performed using a TripleTof 5600 hybrid mass spectrometer (ABSCIEX) interfaced with nano-scale reversed-phase UPLC (Waters corporation nano ACQUITY) using a 20 cm-75 micron ID glass capillary packed with 2.5-μm C18 (130) CSHTM beads (Waters corporation). Peptides were eluted from the C18 column into the mass spectrometer using a linear gradient (5–80%) of ACN (Acetonitrile) at a flow rate of 250 l/min for 1h. The buffers used to create the ACN gradient were: Buffer A (98% H2O, 2% ACN, 0.1% formic acid, and 0.005% TFA) and Buffer B (100% ACN, 0.1% formic acid, and 0.005% TFA). MS/MS data were acquired in a data-dependent manner in which the MS1 data was acquired for 250 ms at m/z of 400~1250 Da and the MS/MS data was acquired from m/z of 50~2,000 Da. The Independent data acquisition (IDA) parameters were as follows; MS1-TOF acquisition time of 250 milliseconds, followed by 50 MS2 events of 48 milliseconds acquisition time for each event. The threshold to trigger MS2 event was set to 150 counts when the ion had the charge state +2, +3 and +4. The ion exclusion time was set to 4 seconds. Finally, the collected data were analyzed using Protein Pilot 4.5 (ABSCIEX) for peptide identifications. MS data analysis is performed by the ProteomicS Performance Evaluation Pipeline Software (PSPEP) belonged ProteinPilot Software. The general approach to estimate false discovery rates in ProteinPilot Software is called “decoy database searching.” The principle of decoy searching is to search a collection of answers that are known to be wrong (“decoy” proteins), in addition to the database of interest (“target” proteins). The relative rate of reporting of identifications from the database of interest versus the known incorrect answers indicates the likelihood that wrong answers are reported from the database of interest. The FDR report is saved to the same location as the .group file and is named “Group Result File Name_FDR.xlsx”, where “Group Result File Name” is the name of the .group file. It is an Excel file which contains the raw data and false discovery rate analysis results for three levels: protein, peptide and spectral.
MCF7 Tet-On stable cell line construction
For KDM2A-HA cell line, MCF7 Tet-On® Advanced Cell Line (Cat #631153) was bought from Takara Clontech. KDM2A was cloned into the pREV-TRE vector with 2XHA tag at its C-term and Flag tag at its N-term. pRev-TRE-Flag-KDM2A-2XHA was transfected into MCF7 Tet-On cells with Tet-free serum supplied. Two days after transfections, cells were selected with Hygromycin for 2 weeks. Colonies were picked and verified by Western blot after Doxycycline induction. After titration, 1ug/ml Doxycycline was chosen to achieve similar level of expression of HA-tagged KDM2A compared with endogenous KDM2A. The biotinylated KDM2A with Flag tag cell line system were generated according to our previous paper (Liu et al., 2014).
Protein Preparation and Purification
Recombinant His-ERα DNA binding domain (amino acids 180–263) were expressed in E.coli strain BL21-CodonPlus® (DE3)-RIPL (Agilent Technologies). Bacteria pellet (about 10 g) were re-suspended in 50 ml lysis buffer (50 mM potassium phosphate, pH 7.2, 5% glycerol, 200 mM NaCl, 1x proteinase inhibitor, 1mg/ml lysozyme). The suspension was sonicated 15” for 12 times on ice and cell debris was removed by centrifuging at 9000 rpm × 15’ twice at 4 °C. The supernatant was incubated for 1 hour at 4 °C with 1 ml of a 50% slurry Ni-NTA (Qiagen) equilibrated in lysis buffer. The resin was poured onto a column, washed with 50 mM potassium phosphate, pH 7.2, 5% glycerol, 200 mM NaCl, 10 mM imidazole and the protein was eluted with 50 mM potassium phosphate, pH 7.2, 5% glycerol, 200 mM NaCl, 250 mM imidazole. Every 500 l elution was assigned to one fraction. The purified proteins were then subjected to PAGE gel separation and stained Coomassie brilliant blue to check the size and purity. Recombinant KDM2A protein was purchased from Signal Chem.
ERα and KDM2A interaction assay
For HA-tagged ERα pull-down, constructs expressing HA-tagged ERα protein truncations and Flag-tagged KDM2A protein were transfected into 293T cells for 2 days with hormone-deficient medium. Upon harvesting, the 293T cells were treated with 100 nM E2 for 1h then were collected with cold PBS. Cell pellet was lysed with lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 3 mM MgCl2, 1 mM CaCl2, 0.2% Triton X-100, 0.2% Nonidet NP-40, 10% glycerol, PIC freshly added) supplied with Benzonase to solubilize chromatin. After centrifugation, the supernatant was incubated with anti-HA antibody (Roche) overnight at 4°C. The next day, protein G beads were added to the lysate and incubated at 4°C for 2 hours, after which the beads were washed 5 times with lysis buffer. In the end, the beads were boiled with SDS sample buffer for Western blot analyses. For most IPs and co-IPs, about 3~5% of input was loading for western blots.
In vitro competitive binding assay of KDM2A and ERE to ERα DNA binding domain
30 μmole 2xERE oligonucleotides (Sense CGTCAGGTCACAGTGACCTGATGTCAGGTCACAGTGACCTGATC, Antisense TCGAGATCAGGTCACTGTGACCTGACATCAGGTCACTGTGACCTGACGGTAC) or control 60-mer oligonucleotides (Sense AAAAGCATAATAACCAGATAGAATTGGATCCAATTCTATCTGGTTATTATGC, Antisense GCATAATAACCAGATAGAATTGGATCCAATTCTATCTGGTTATTATGCTTTTTT) were annealed by 95°C 5 min and progressive cool down. ~2ug his-ERE DBD with no DNA, ERE, or random sequence were mixed in 500 l PBS buffer for 2hrs, and then loaded 2 g KDM2A protein to each sample for 2hrs before adding 0.5 g KDM2A antibody and incubated overnight at 4°C. 10 l Protein G magnetic beads were added for another 2 hrs and washed 4 times by PBS. Then the beads were boiled and subjected to Western blot for detecting bound his-ERα DBD.
In vitro ubiquitination assay of KDM2A-containing complex
Dox-induced -KDM2A MCF7 cells were inducted by 500 ng/ml Dox (Tet-on) or vehicle (Tetoff) for 24hrs and then lysed in lysis buffer (50 mM Tris-HCl, pH7.5, 150 mM NaCl, 0.5% NP-40, 2mM EDTA) for 45min. Biotinylated KDM2A protein-containing complex was immunoprecipitated by streptavidin magnetic beads (Sigma) and washed extensively followed by in vitro ubiquitination assay with Ubiquitin, Ubiquitin Activating Enzyme Solution (E1), UbcH5a (E2), Mg-ATP Solution and Ub E3 Ligase Buffer supplied by E3 Ligase Auto-Ubiquitination Assay Kit (Abcam, ab139469).
ChIP and ChIP-seq
Briefly, cells were cross-linked with 1% formaldehyde at room temperature for 10 min. For KDM2A ChIP, cells were double cross-linked with 2mM DSG (ProteoChem Cat# C1104) for 45 min and then for another 15 min with 1% formaldehyde (Sigma, F8775). In both situations, the cross-linking was quenched with 0.125M glycine for 5 min. Chromatin was fragmented using a Bioruptor (Diagenode) for 30 min at high power, with an interval of 30 s between pulses to get around 200bp fragments and precleared using 20 l Protein G Dynabeads (Life Technologies, Cat# 10009D). Subsequently, the soluble chromatin was incubated with 2–5 g antibodies at 4°C overnight. Immunoprecipitated complexes were collected using 30 l Protein G Dynabeads per reaction. Cloning, mutagenesis, and generation of ERα wild type and P-box mutation biotin and HA-tagged inducible MCF7 stable cell lines were described in a previous paper (Liu et al., 2014). We performed biotin ChIP or HA ChIP experiments for BLRP and HA-tagged ERα, and ERα P-box mutation stable cell lines following an earlier protocol(Heinz et al., 2010; Liu et al., 2014). Briefly, cross-linked protein-DNA complexes were pulled down by M-280 Streptavidin Magnetic beads (Life Technologies, Cat# 11205D) and the washing was performed under much more stringent conditions that included 2 washes with 1% SDS in TE (20 min each) and two washes with 1% Triton X-100 in TE. The washed streptavidin beads were then subjected to AcTEV protease (Life Technologies, Cat# 12575–015) digestion twice for tagged protein and DNA complex elution before de-crosslinking at 65°C overnight. For all ChIPs, after de-crosslinking overnight at 65°C, final ChIP DNA was extracted and purified using QIAquick spin columns (QIAGEN). The ChIP-seq libraries were constructed following Illumina’s ChIP-seq Sample prep kit. The library was amplified by 14 cycles of PCR.
PRO-seq
PRO-seq experiments were performed as previously reported (Kwak et al., 2013) with a few modifications. Briefly, 10 millions of MCF7 cells treated with E2 for 1 hr were washed 3 times with cold PBS and then sequentially swelled in swelling buffer (10mM Tris-HCl pH7.5, 2mM MgCl2, 3mM CaCl2) for 10 min on ice, harvested, and lysed in lysis buffer (swelling buffer plus 0.5% NP-40, 20 units of SUPERase-In, and 10% glycerol). The resultant nuclei were washed two more times with 10ml lysis buffer and finally resuspended in 100 μl of freezing buffer (50mM Tris-HCl pH8.3, 40% glycerol, 5mM MgCl2, 0.1mM EDTA). For the run-on assay, resuspended nuclei were mixed with an equal volume of reaction buffer (10 mM Tris-HCl pH 8.0, 5 mM MgCl2, 1 mM DTT, 300 mM KCl, 20 units of SUPERase-In, 1% sarkosyl, 100 M A/GTP, 100 μM biotin-11-C/UTP (Perkin-Elmer) and incubated for 5 min at 30°C. The resultant nuclear-runon RNA (NRO-RNA) was then extracted with TRIzol® LS reagent (Life Technologies, Cat# 10296–028) following manufacturer’s instructions. NRO-RNA was fragmented to 200–500nt by alkaline base hydrolysis on ice for 30 min and neutralized by adding 1× volume of 1 M Tris-HCl pH 6.8, Excessive salt and residual NTPs were removed by using P-30 column (Bio-Rad, Cat# 732–6250), followed by treatment with DNase I (Promega Cat# M6101) and antarctic phosphatase (NEB Cat# M0289L). Fragmented nascent RNA was bound to 10 μl of MyOne Streptavidin C1 dynabeads (Invitrogen, Cat# 65001) following the manufacturer’s instructions. The beads were washed twice in high salt (2 M NaCl, 50 mM Tris-HCl pH 7.5, 0.5% Triton X-100, 0.5 mM EDTA), once in medium salt (1M NaCl, 5 mM Tris-HCl pH 7.5, 0.1% Triton X-100, 0.5 mM EDTA), and once in low salt (5 mM Tris-HCl pH 7.5, 0.1% Triton X-100). Bound RNA was extracted from the bead using Trizol (Invitrogen, Cat# 15596–018) in two consecutive extractions, and the RNA fractions were pooled, followed by ethanol precipitation. The RNA fragments were then subjected to poly-A tailing reaction by poly-A polymerase (NEB, Cat# M0276L) for 30 min at 37°C. Subsequently, reverse transcription was performed using oNTI223 primer and superscript III RT kit (Life Technologies, Cat# 18080–044). The cDNA products were separated on a 10% polyacrylamide TBE-urea gel and only those fragments migrating between 100–400bp were excised and recovered by gel extraction. Next, the first-strand cDNA was circularized by CircLigase (Epicenter, Cat# CL4115K) and relinearized by APE1 (NEB, Cat# M0282L). Finally, cDNA template was amplified by PCR using the Phusion High-Fidelity enzyme (NEB, Cat# M0530L) according to the manufacturer’s instructions. The oligonucleotide primers oNTI200 and oNTI201 were used to generate DNA library for deep sequencing and the primer sequences were described in previous paper (Li et al., 2013).
In situ Hi-C
In situ Hi-C was essentially performed as described (Rao et al., 2014). Briefly, for each experiment, 2 × 106 cells, fixed for 10 minutes with 1% formaldehyde/PBS and washed twice with PBS, permeabilized for 7 minutes at 62°C in a PCR cycler with 200 μl lysis buffer (0.5% SDS, 50 mM Tris-HCl pH7.5, 10 mM NaCl, 1mM EDTA). Supernatant was removed and nuclei were resuspended in 25 μl 10% Triton X-100, 25 μl NEB 2 buffer, 195 μl water, and rotated for 15’ at 37°C. Chromatin was digested overnight at 37°C after adding 0.5 μl 1 M DTT and 4 μl 25 U/μl Mbo I and rotated at 8 RPM. MboI was inactivated by incubation at 62°C for 20 minutes. Overhangs were filled in by adding 35nM of dATP, dTTP, dGTP and 30 nM Biotin-14-dCTP (Invitrogen), with 25 U Klenow enzyme (Enzymatics) rotating for 40 minutes at room temperature. The reaction was stopped by adding 2.5 μl 0.5 M EDTA. DNA was ligated under rotation overnight at 16°C with 1200 U T4 DNA ligase (Enzymatics). Samples were digested for 15 minutes at 42°C with 1 μl 10 μg/μl RNase A. 33 μl of 5 M sodium chloride and 55 μl of 10% SDS were added and reverse crosslinked for 4 h at 65°C. Protein was digested with 10 μl of 20 mg/ml proteinase K (Life Technologies), incubated at 55°C for 120 minutes, shaking at 800 RPM, then 65°C for 90 minutes. DNA was extracted once with phenol/chloroform/isoamylalcohol (25:24:1) and once with CHCl3, and precipitated overnight at −20°C with 1.5 μl 20 mg/ml glycogen and 1412 μl 100% ethanol overnight. Pellets were dissolved in 131 μl TT (0.05% Tween 20/10 mM Tris pH=8) each. DNA was sheared with 300 bp Covaris protocol in snap cap tube in a Covaris E220 at 10 % duty cycle, intensity 140 W, 200 cycles/burst for 80” total time. Large DNA fragments (>400 bp) were depleted with speedbeeds and 6.45% PEG8000/2.5 M NaCl. Small DNA fragments were collected with 9.5% PEG8000/2.5 M NaCl and DNA was eluted in 50 μl TT for 5 minutes. DNA was captured with T1 Dynabeads (Invitrogen). DNA sequencing libraries were generated on beads using KAPA Library Preparation kit from Illumina. Libraries were PCR-amplified for 10 cycles using KAPA HiFi, size-selected to 225–425 bp insert size using speedbeads PEG8000/2.5 M NaCl solutions, and paired-end sequenced on an Illumina HiSeq 2500.
Chromatin Conformation Capture (3C) assay
MCF7 Cells were stripped for 3 days before treated with 100 nM E2 for 1hr. 3C procedures follow the reported protocol (Hagege et al., 2007) by BamHI enzyme. For test, 3-day stripped MCF7 cells were used, with or without T4 ligase ligation process. qPCR primers were designed at Sytl2 promoter (P) or enhancer (E) region as indicated, with a pair of primers at enhancer itself as genomic control. Since F1R3 PCR showed some background amplification whereas F1R5 was quite specific, qPCR was only performed by F1R5. Primers are listed in Supplementary Table 2.
Chromosome Conformation Capture Combined with high-throughput sequencing (4C-seq)
The protocol of 4C-seq largely followed a published protocol (Stadhouders et al., 2013) with modification. Briefly, 10 million cells were cross-linked with 1% formaldehyde for 10 min and nuclei were extracted. Nuclei were resuspended in restriction enzyme buffer and incubated with 0.3% SDS for 1h at 37°C and further incubated with 2% Triton X-100 for 1 h. 400U of DpnII restriction enzyme was added and incubated overnight. Restriction enzyme was heat inactivated at 65°C for 20 min. Ligation of DNA regions in close physical proximity was performed using 1000 U of T4 DNA ligase (NEB) for overnight. After de-crosslinking, the second digestion and ligation was performed using restriction enzyme NlaIII and T4 DNA ligase. 4C-seq libraries were amplified using PCR with the first primer designed on each viewpoint and the second primer designed beside the NlaIII site. Both primers contained illumina sequencing adaptors and barcode. 4C libraries were sequenced on the Illumina Hi-Seq 2500 using single-read 100-cycle runs. The 4C analysis was performed following the pipeline generated by previous report (van de Werken et al., 2012). 200 kb window of genome locus was used to visualize near-cis contact profile around the viewpoint. The linear mean method with 3 Kb resolution was chosen as statistic method to present normalized contact frequency on the top curve, and contact intensity was colored in bottom heatmap, which resolution was ranked from 2 Kb on the top to 50 Kb on the bottom. For Sytl2 locus, the view point of 4C-seq is chr11:85,523,062–85,523,104; for Plekhf2 locus, the view point of 4C-seq is chr8:96,125,141–96,125,203.
Deep Sequencing
For all ChIP-seqs and GRO-seqs, the extracted DNA libraries were sequenced with Illumina’s HiSeq 2500 system according to the manufacturer’s instructions. And DNA sequences generated by the Illumina Pipeline were aligned to the human genome (hg19) assembly using Bowtie2 (Langmead and Salzberg, 2012). The uniquely mapped tags were selected for further ChIP-seq data analysis and a maximum of 3 tags per genomic position were collected for further GRO-seq data analysis. The data were visualized by preparing custom tracks on the University of California, Santa Cruz (UCSC) genome browser using HOMER software package (Heinz et al., 2010). The total number of mappable reads was normalized to 107 for each experiment presented in this study.
Quantification and Statistical Analysis
Bioinformatic characterization of E2 Activated and BAER Enhancers
We followed our previously published method to define E2 activated and BAER enhancers (Li et al., 2013). Briefly, to determine ERα binding active enhancers, putative enhancers sites were first defined based on ChIP-seq enrichment of H3K27Ac (GSM1115992) flanking ± 500 bp from the center of the ERα peaks. Putative enhancers were defined by the following criteria: (1) regions were at least 1 kb away from annotated TSSs; (2) regions had at least 8 tags from H3K27Ac ChIP-seq normalized to 10 million tags; (3) regions had at least 10 tags from GRO-seq (GSE45822 & our GRO-seq data) normalized to 10 million tags when MCF7 cells were treated with E2; and (4) and fold changes (FC) of eRNA expression between E2 and EtOH condition are more than 1.5 (for E2 activated enhancers) or less than 0.67 (for BAER enhancers) and FDR<0.01.
Identification of ChIP-seq Peaks, Heatmap, and Tag Density Analyses
ChIP-seq peak identification, quality control, and motif analysis we performed using HOMER (Heinz et al., 2010) as described in our previously published methods. Briefly, Genome binding peaks of KDM2A were identified using the ‘findPeaks’ command in HOMER with setting of ‘– style factor’: 200 bp peaks with 3-fold enrichment and 0.01 FDR significance over local tags. To apply IDR to HOMER-identified ChIP-Seq peaks, we used the method described by Karmel (https://github.com/karmel/homer-idr). In summary, we created tag directories for each individual sample, allowing only one tag per base pair to remove any remaining duplicate reads (-tbp 1) and the combined replicates each treatment. We next made peak calls with a very low threshold as required for IDR (findPeaks LL1 -P 0.1 -LP 0.1 -poisson 0.1 -fragLength 150 -style factor) on the individual samples, combined replicates, individual pseudoreplicates, and combined pseudoreplicates. We then applied the HOMER-IDR program to format the data for the IDR R package to determine the IDR threshold and identify the top peaks above that threshold. Peaks from separate experiments were considered co-bound if their peak centers were located within 1kb region of each other. To generate histograms for the average distribution of tag densities, position-corrected, normalized tags in 100 bp windows were tabulated within the indicated distance from specific sites in the genome. Clustering plots for normalized tag densities at each genomic region were generated using HOMER and then clustered using Gene Cluster 3.0 (de Hoon et al., 2004) and visualized using Java TreeView (Saldanha, 2004).
Motif Analysis
For de novo motif analysis, transcription factor motif finding was performed on ± 200 bp relative to the peak center defined from ChIP-seq using HOMER (Heinz et al., 2010). Peak sequences were compared to random genomic fragments of the same size and normalized G/C content to identify motifs enriched in the ChIP-seq targeted sequence. Sequence logos were generated using WebLOGO (Crooks et al., 2004). Student’s two-tailed t test was used to assess the significance of the ERE motifs between the E2 activated and BAER enhancers.
GRO-seq and PRO-seq Analysis
GRO-seq and PRO-seq data analyses were performed as previously reported (Basnet et al., 2014). The sequencing reads were aligned to hg19 using Bowtie2 using very sensitive parameters. The common artifacts derived from clonal amplification were circumvented by considering maximal three tags from each unique genomic position as determined from the mapping data. To determine E2-dependent changes in gene body, the sequencing reads for RefSeq genes were counted over the first 13 kb of the entire gene body, excluding the 500 bp promoter-proximal region on the sense strand with respect to the gene orientation by using HOMER. EdgeR (Robinson et al., 2010) was used to compute the significance of the differential gene expression (FC ≥ 1.5, FDR ≤ 0.01). Additionally, a read density threshold (that is, normalized total read counts per kb) was used to exclude lowly expressed genes. GRO-seqs were normalized to 10 million tags, and HOMER was used to quantify eRNA expression by tabulating normalized tag numbers surrounding ± 1,000 bp from the center of the ERα peaks. eRNAs with a >1.5-fold change in GRO-seq or PRO-seq signals were considered to be differentially expressed.
In situ Hi-C analysis and visualization
In situ Hi-C data was analyzed using HiC-Pro version 2.7.8 (Servant et al., 2015) with default settings, mapped to human hg19. For data visualization, data was converted to .hic format using the hicpro2juicebox.sh script and juicebox_tools.7.0.jar, and visualized with Juicebox (Durand et al., 2016). Loops and contact domains from GM12878 cells (Rao et al., 2014) were overlaid over MCF-7 normalized matrices.
ChIP-seq and GRO-seq data visualization
Visualization of the data for ChIP-seq and GRO-seq was performed by organizing custom tracks onto the University of California, Santa Cruz, (UCSC) genome browser using HOMER software package. Experiments were normalized to 107 bp tags for the comparison between different tracks.
Statistical Analysis
For all qPCRs, the experiments were repeated at least three times, and one representative plot is shown in figures; the results were shown as mean ± SD and most P values were obtained using a two-tailed Student’s t-test.
Data and Software Availability
Software
See Key Resources Table.
Data Resources
The accession number for all the GRO-seq, PRO-seq and ChIP-seq data reported in this paper is GEO: GSE73958. The Raw image data for all the figures have been deposited at Mendeley with the https://data.mendeley.com/datasets/dt2ncczrms/draft?a=caf707f6–678d-43a0–8335-3c8b019ec6c3. We also employed some published ChIP-seq data from the Gene Expression Omnibus (GEO) database for FoxA1 under accession number GSE23852 (Tan et al., 2011), GATA3 under accession number GSE40129 (Theodorou et al., 2013), P300/CBP/SRC1/2/3 under accession number E-MTAB-785 (Zwart et al., 2011), ERα under accession number EMTAB-828(Schmidt et al., 2010), ERα ChIP-seq with siCTL and siFoxA1 treatment under accession number E-MTAB-233 (Hurtado et al., 2011), H3K27Ac and H3K4me1 under accession number GSE45822 (Li et al., 2013), and GRO-seq data for MCF7 with 1h E2 treatment under accession number GSE45822 (Li et al., 2013).
Supplementary Material
Supplementary Table 1 (Related to Figure 1). MCF7 cells were treated by E2 for 1h. All E2 regulated genes (FDR<0.01) detected by GRO-seq and calculated by EdgeR are listed.
Supplementary Table 2 (Related to STAR Methods). sgRNAs, PCR Primer Sets and siRNAs used in This Study.
Highlights.
Basally Active Estrogen Repressed (BAER) enhancers bind ERα indirectly.
ERα causes selective loss of Pol II on BAER enhancers.
The indirectly-bound ERα recruits KDM2A for enhancer decommissioning.
KDM2A brings NEDD4 to BAER enhancers, ubiquitylating/dismissing Pol II.
ACKNOWLEDGEMENTS
The authors are grateful to Janet Hightower for assistance with figure preparation, to Dr. Zhijie Liu for his KDM2A, ERα wild type and P-box mutant Tet-On BLRP-tagged stable cell lines, to Dr. Min Gyu Lee (MD Anderson Cancer Center) for providing KDM2A WT and H212A constructs, to Dr. Alana Welm (U of Utah), to Dr. Xingxu Huang (Shanghaitech University) for providing vectors for CRISPR-Cas9 and the method of multiple sgRNA cloning, to Dr. Barbara Parker (UCSD) for useful discussions, to Dr. Majid Ghassemian (UCSD) for assistance in mass spectrometry, and to members of the Rosenfeld laboratory for generous help during this work. We are also grateful to Drs. Christopher K. Glass, Ivan Garcia-Bassets and Francesca Telese for criticisms of the manuscript. C.J. is a recipient of Cancer Research Institute Irvington Postdoctoral Fellowship. Q.M. is a recipient of American Cancer Society Postdoctoral Fellowship (PF-16–211-01-TBE). M.G.R. is an investigator with the Howard Hughes Medical Institute. This work was supported by grants from NIH to M.G.R. (DK018477, DK039949, NS034934, and CA173903).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no conflict of interests.
REFERENCES
- Abyzov A, Mariani J, Palejev D, Zhang Y, Haney MS, Tomasini L, Ferrandino AF, Rosenberg Belmaker LA, Szekely A, Wilson M, et al. (2012). Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 492, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anindya R, Aygun O, and Svejstrup JQ (2007). Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Molecular cell 28, 386–397. [DOI] [PubMed] [Google Scholar]
- Arner E, Daub CO, Vitting-Seerup K, Andersson R, Lilje B, Drablos F, Lennartsson A, Ronnerblad M, Hrydziuszko O, Vitezic M, et al. (2015). Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji J, Rusconi S, and Schaffner W (1981). Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 299–308. [DOI] [PubMed] [Google Scholar]
- Basnet H, Su XB, Tan Y, Meisenhelder J, Merkurjev D, Ohgi KA, Hunter T, Pillus L, and Rosenfeld MG (2014). Tyrosine phosphorylation of histone H2A by CK2 regulates transcriptional elongation. Nature 516, 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackledge NP, Farcas AM, Kondo T, King HW, McGouran JF, Hanssen LL, Ito S, Cooper S, Kondo K, Koseki Y, et al. (2014). Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157, 1445–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackledge NP, Zhou JC, Tolstorukov MY, Farcas AM, Park PJ, and Klose RJ (2010). CpG islands recruit a histone H3 lysine 36 demethylase. Molecular cell 38, 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes E, and Pombo A (2009). Modifications of RNA polymerase II are pivotal in regulating gene expression states. EMBO reports 10, 1213–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Yu NK, Choi JH, Sim SE, Kang SJ, Kwak C, Lee SW, Kim JI, Choi DI, Kim VN, et al. (2015). Multiple repressive mechanisms in the hippocampus during memory formation. Science 350, 82–87. [DOI] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, and Lis JT (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome research 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, and Miyano S (2004). Open source clustering software. Bioinformatics 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- Drouin J, Sun YL, Chamberland M, Gauthier Y, De Lean A, Nemer M, and Schmidt TJ (1993). Novel glucocorticoid receptor complex with DNA element of the hormone-repressed POMC gene. The EMBO journal 12, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell systems 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukaya T, Lim B, and Levine M (2016). Enhancer Control of Transcriptional Bursting. Cell 166, 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, and Furlong EE (2014). Enhancer loops appear stable during development and are associated with paused polymerase. Nature 512, 96–100. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, and Saijo K (2010). Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nature reviews Immunology 10, 365–376. [DOI] [PubMed] [Google Scholar]
- Hagege H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W, and Forne T (2007). Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nature protocols 2, 1722–1733. [DOI] [PubMed] [Google Scholar]
- Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, and Kraus WL (2011). A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Horng T, and Medzhitov R (2009). Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138, 129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojfeldt JW, Agger K, and Helin K (2013). Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov 12, 917–930. [DOI] [PubMed] [Google Scholar]
- Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, and Carroll JS (2011). FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nature genetics 43, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imhof MO, and McDonnell DP (1996). Yeast RSP5 and its human homolog hRPF1 potentiate hormone-dependent activation of transcription by human progesterone and glucocorticoid receptors. Molecular and cellular biology 16, 2594–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami E, Tokunaga A, Ozawa M, Sakamoto R, and Yoshida N (2015). The histone demethylase Fbxl11/Kdm2a plays an essential role in embryonic development by repressing cell-cycle regulators. Mechanisms of development 135, 31–42. [DOI] [PubMed] [Google Scholar]
- Khan SA, Rogers MA, Khurana KK, Meguid MM, and Numann PJ (1998). Estrogen receptor expression in benign breast epithelium and breast cancer risk. Journal of the National Cancer Institute 90, 37–42. [DOI] [PubMed] [Google Scholar]
- Kim YW, Lee S, Yun J, and Kim A (2015). Chromatin looping and eRNA transcription precede the transcriptional activation of gene in the beta-globin locus. Bioscience reports 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk MS, Hughes JR, Garrick D, Lynch MD, Sharpe JA, Sloane-Stanley JA, McGowan SJ, De Gobbi M, Hosseini M, Vernimmen D, et al. (2012). Intragenic enhancers act as alternative promoters. Molecular cell 45, 447–458. [DOI] [PubMed] [Google Scholar]
- Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, et al. (2011). NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Molecular cell 44, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, and Lis JT (2013). Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, et al. (2012). Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, et al. (2013). Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498, 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, and Rosenfeld MG (2016). Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nature reviews Genetics 17, 207–223. [DOI] [PubMed] [Google Scholar]
- Liu Z, Merkurjev D, Yang F, Li W, Oh S, Friedman MJ, Song X, Zhang F, Ma Q, Ohgi KA, et al. (2014). Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell 159, 358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Jiang S, Garee JP, Verdin E, Lee AV, O’Malley BW, Zhang M, Belaguli NS, and Oesterreich S (2010). Histone deacetylase 7 and FoxA1 in estrogen-mediated repression of RPRM. Molecular and cellular biology 30, 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Dienstbier M, Murphy S, Proudfoot NJ, and Dye MJ (2013). Definition of RNA polymerase II CoTC terminator elements in the human genome. Cell reports 3, 1080–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview--extensible visualization of microarray data. Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Schwalie PC, Ross-Innes CS, Hurtado A, Brown GD, Carroll JS, Flicek P, and Odom DT (2010). A CTCF-independent role for cohesin in tissue-specific transcription. Genome research 20, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe JW, Chapman L, Finch JT, and Rhodes D (1993). The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell 75, 567–578. [DOI] [PubMed] [Google Scholar]
- Schwabe JW, Neuhaus D, and Rhodes D (1990). Solution structure of the DNA-binding domain of the oestrogen receptor. Nature 348, 458–461. [DOI] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, and Barillot E (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome biology 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Xu W, Guo R, Rong B, Gu L, Wang Z, He C, Zheng L, Hu X, Hu Z, et al. (2016). Suppression of Enhancer Overactivation by a RACK7-Histone Demethylase Complex. Cell 165, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. (2012). A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal S, Taylor MC, and Baker RT (2008). Deubiquitylating enzymes and disease. BMC biochemistry 9 Suppl 1, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somesh BP, Reid J, Liu WF, Sogaard TM, Erdjument-Bromage H, Tempst P, and Svejstrup JQ (2005). Multiple mechanisms confining RNA polymerase II ubiquitylation to polymerases undergoing transcriptional arrest. Cell 121, 913–923. [DOI] [PubMed] [Google Scholar]
- Stadhouders R, Kolovos P, Brouwer R, Zuin J, van den Heuvel A, Kockx C, Palstra RJ, Wendt KS, Grosveld F, van Ijcken W, et al. (2013). Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nature protocols 8, 509–524. [DOI] [PubMed] [Google Scholar]
- Stender JD, Kim K, Charn TH, Komm B, Chang KC, Kraus WL, Benner C, Glass CK, and Katzenellenbogen BS (2010). Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Molecular and cellular biology 30, 3943–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Keim CD, Wang J, Kazadi D, Oliver PM, Rabadan R, and Basu U (2013). E3-ubiquitin ligase Nedd4 determines the fate of AID-associated RNA polymerase II in B cells. Genes & development 27, 1821–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SK, Lin ZH, Chang CW, Varang V, Chng KR, Pan YF, Yong EL, Sung WK, and Cheung E (2011). AP-2gamma regulates oestrogen receptor-mediated long-range chromatin interaction and gene transcription. The EMBO journal 30, 2569–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Okamoto K, Teye K, Umata T, Yamagiwa N, Suto Y, Zhang Y, and Tsuneoka M (2010). JmjC enzyme KDM2A is a regulator of rRNA transcription in response to starvation. The EMBO journal 29, 1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodorou V, Stark R, Menon S, and Carroll JS (2013). GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome research 23, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, and Zhang Y (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816. [DOI] [PubMed] [Google Scholar]
- van de Werken HJ, Landan G, Holwerda SJ, Hoichman M, Klous P, Chachik R, Splinter E, Valdes-Quezada C, Oz Y, Bouwman BA, et al. (2012). Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nature methods 9, 969–972. [DOI] [PubMed] [Google Scholar]
- Vockley CM, D’Ippolito AM, McDowell IC, Majoros WH, Safi A, Song L, Crawford GE, and Reddy TE (2016). Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell 166, 1269–1281 e1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart W, Theodorou V, Kok M, Canisius S, Linn S, and Carroll JS (2011). Oestrogen receptor-co-factor-chromatin specificity in the transcriptional regulation of breast cancer. The EMBO journal 30, 4764–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 (Related to Figure 1). MCF7 cells were treated by E2 for 1h. All E2 regulated genes (FDR<0.01) detected by GRO-seq and calculated by EdgeR are listed.
Supplementary Table 2 (Related to STAR Methods). sgRNAs, PCR Primer Sets and siRNAs used in This Study.