

Abstract
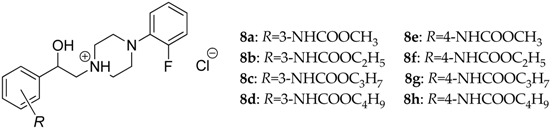
Novel 1-(2-{3-/4-[(alkoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)-piperazin-1-ium chlorides (alkoxy = methoxy to butoxy; 8a–h) have been designed and synthesized through multistep reactions as a part of on-going research programme focused on finding new antimycobacterials. Lipophilic properties of these compounds were estimated by RP-HPLC using methanol/water mobile phases with a various volume fraction of the organic modifier. The log kw values, which were extrapolated from intercepts of a linear relationship between the logarithm of a retention factor k (log k) and volume fraction of a mobile phase modifier (ϕM), varied from 2.113 (8e) to 2.930 (8h) and indicated relatively high lipophilicity of these salts. Electronic properties of the molecules 8a–h were investigated by evaluation of their UV/Vis spectra. In a next phase of the research, the compounds 8a–h were in vitro screened against M. tuberculosis CNCTC My 331/88 (identical with H37Rv and ATCC 2794), M. kansasii CNCTC My 235/80 (identical with ATCC 12478), a M. kansasii 6 509/96 clinical isolate, M. avium CNCTC My 330/80 (identical with ATCC 25291) and M. avium intracellulare ATCC 13950, respectively, as well as against M. kansasii CIT11/06, M. avium subsp. paratuberculosis CIT03 and M. avium hominissuis CIT10/08 clinical isolates using isoniazid, ethambutol, ofloxacin, ciprofloxacin or pyrazinamide as reference drugs. The tested compounds 8a–h were found to be the most promising against M. tuberculosis; a MIC = 8 μM was observed for the most effective 1-(2-{4-[(butoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8h). In addition, all of them showed low (insignificant) in vitro toxicity against a human monocytic leukemia THP-1 cell line, as observed LD50 values > 30 μM indicated. The structure–antimycobacterial activity relationships of the analyzed 8a–h series are also discussed.
Keywords: N-arylpiperazines, arylaminoethanols, lipophilicity, electronic properties, Mycobacterium tuberculosis H37Rv
1. Introduction
An N-arylpiperazine privileged scaffold [1] has been found in the chemical structure of many effective antimycobacterial agents [2,3,4,5,6,7,8]. Some of these compounds have been characterized by a very typical structural arrangement [5,6,7,8], i.e., the N-aryl- or variously substituted N-phenylpiperazine moiety, connecting hydrocarbon chain and terminal heterocyclic fragment. Efforts to combine the N-arylpiperazine and “ethambutol-like“ structural frameworks were supported by comprehensive structure–activity relationships (SAR) studies of homopiperazin-1,4-diyl-containing derivatives (e.g., a molecule SQ775; Figure 1a), ethambutol (EMB; Figure 1b) and the diamines structurally based on EMB [9,10,11,12]. Among the synthesized compounds, N-geranyl-N′-(2-adamanthan-1-yl)ethane-1,2-diamine (Figure 1c) showed promising efficiency [12]. The outlined systematic research led to N-ArPA molecules (Figure 1d), in which structure the distance between two nitrogens, presence of β-aminoalcohol motifs and short connecting chains were crucial for their in vitro antimycobacterial activity [13]. The derivatives N-ArPA were very effective against Mycobacterium tuberculosis CNCTC My 331/88 (identical with M. tuberculosis H37Rv) and multi-drug resistant (MDR) M. tuberculosis 43 strain, which showed resistance to rifampicin (RIF) and isoniazid (INH).
Figure 1.

Chemical structures of: (a) compound SQ775; (b) ethambutol (EMB); (c) N-geranyl-N´-(2-adamanthan-1-yl)ethane-1,2-diamine; and (d) chiral N-arylpiperazine-based aminoalcohols (N-ArPA), which showed a notable in vitro efficiency against M. tuberculosis CNCTC My 331/88 [9,10,11,12,13].
It was also concluded that removal or significant alteration of basicity of either amino group led to loss of potency [9,13]. In addition, the presence of the R = 2-/4-F substituent and OH group with the (R)-configuration at the carbon of a connecting chain (Figure 1d) resulted in higher in vitro antimycobacterial efficiency than in a case of EMB. On the other hand, the remaining R substituents (H, Cl; Figure 1d) caused decrease in activity [13].
Regarding the design of original antimycobacterials, a carbamate (NHCOO) functionality is structurally related to hybrid amide-ester features and, in general, displays very good chemical and proteolytic stabilities [14]. The carbamate functionality imposes a degree of conformational restriction due to the delocalization of non-bonded electrons on nitrogen into the carboxyl moiety. In addition, this functionality participates in hydrogen bonding through the carboxyl group and the backbone NH [15]. Therefore, a substitution on the O- and N-termini of the carbamate offers opportunities to modulate biological properties and improve stability and pharmacokinetic features [14,15].
The idea to introduce a lipophilic 3-/4-alkoxy or 3-/4-alkoxycarbonylamino moiety (alkoxy = methoxy to butoxy) into a chemical structure of newly designed molecules was based on previous studies focused on the synthesis and in vitro biological evaluation of the N-arylpiperazines and phenylcarbamic acid derivatives [16,17,18,19,20,21]. Their antimycobacterial activity increased with elongation of this chain until a maximum in efficiency was reached [19,20,21]. Further increase in its length led to the decrease in potency. The observed dependence was approximated by a parabolic function and described as a cut-off effect. That phenomenon was comprehensively reviewed and rationalized in number of mechanistic ways by Hansch and Clayton [22] as well as Balgavý and Devínsky [23] more than two decades later.
Regarding the conclusions published in papers [19,20,21], it might be expected that eventual incorporation of the alkoxycarbonylamino group into a 2-position of a phenyl ring would cause the decrease in antimycobacterial activity.
It was also believed that the presence of the linear alkoxy side chain would be very favorable in terms of interactions with target structures located in biomembranes of mycobacterial strains, especially M. tuberculosis H37Rv. Branching or substitution of the alkoxy with the alkyl group is reported to have caused decrease in efficiency [19,20,21,24].
It was found that a suitable modification of the aromatic ring attached to a piperazin-1,4-diyl framework might not result in loss of activity. The derivatives containing a pyrimidin-2-yl fragment were slightly more efficient than the ones with a pyridin-2-yl moiety (a series MM; Figure 2). The molecules MM [18] were able to effectively in vitro fight M. tuberculosis My 331/88, M. kansasii 6 509/96, M. tuberculosis 7375/1998, a strain resistant to INH, RIF, rifabutine (RFB) and streptomycin (STM), respectively, as well as M. tuberculosis Prague 1, an extremely-resistant strain to INH, RIF, RFB, STM, EMB, ofloxacin (OFLX), gentamicin (GTM) and amikacin (AK), respectively [18].
Figure 2.

N-Arylpiperazine derivatives containing a 2-hydroxyethane-1,2-diyl connecting chain (a series MM), which were in vitro screened against some mycobacterial strains [18].
Inspired and encouraged by given importance of the 3-/4-alkoxycarbonylamino, β-aminoalcohol and 4-(2-fluorophenyl)piperazin-1-yl fragments in a chemical structure of effective antimycobacterials, the present study was focused on the synthesis of original racemic compounds in order to find out if they would be efficient against some of following mycobacterial strains in vitro, namely, M. tuberculosis CNCTC My 331/88 (identical with H37Rv and ATCC 2794), M. kansasii CNCTC My 235/80 (identical with ATCC 12478), a M. kansasii 6 509/96 clinical isolate, M. avium CNCTC My 330/80 (identical with ATCC 25291) as well as M. avium intracellulare ATCC 13950, M. kansasii CIT11/06, M. avium subsp. paratuberculosis CIT03 and M. avium hominissuis CIT10/08 clinical isolates, respectively.
Despite a fact that currently designed molecules contained a stereogenic centre (Table 1), the synthesis of racemates has been regarded as a reasonable strategy in conceptual development of original antimycobacterial agents to verify the relevancy of proposed structural frameworks [16,18,25,26,27].
Table 1.
Chemical structure of the compounds 8a–h, their lipophilicity indices RM (RP-TLC) and log k (RP-HPLC) as well as retention times tr (RP-HPLC) estimated in the mobile phases consisted of a various volume ratio (v/v) of a methanol (MeOH) organic modifier and water.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Comp. | 1 RM | Mobile phase MeOH/water (v/v) | |||||||
| 60:40 | 70:30 | 80:20 | 85:15 | ||||||
| tr (min) | log k | tr (min) | log k | tr (min) | log k | tr (min) | log k | ||
| 8a | −0.55 | 6.593 | 0.612 | 3.587 | 0.248 | 2.493 | −0.034 | 2.200 | −0.156 |
| 8b | −0.35 | 7.707 | 0.695 | 4.893 | 0.444 | 2.907 | 0.095 | 2.433 | −0.056 |
| 8c | −0.16 | 8.933 | 0.771 | 5.907 | 0.552 | 3.193 | 0.166 | 2.620 | 0.010 |
| 8d | 0.01 | 10.021 | 0.829 | 7.820 | 0.702 | 3.586 | 0.245 | 2.830 | 0.074 |
| 8e | −0.02 | 5.307 | 0.491 | 3.180 | 0.163 | 2.360 | −0.085 | 2.120 | −0.196 |
| 8f | 0.19 | 7.153 | 0.656 | 3.953 | 0.312 | 2.620 | 0.010 | 2.275 | −0.121 |
| 8g | 0.39 | 8.280 | 0.732 | 5.320 | 0.492 | 3.033 | 0.128 | 2.533 | −0.020 |
| 8h | 0.63 | 11.035 | 0.881 | 7.287 | 0.665 | 3.543 | 0.240 | 2.814 | 0.069 |
1 RM, Lipophilicity index (RP-TLC). Silica gel plates (stationary phases) were impregnated by 1% silicone oil in heptane.
2. Results and Discussion
2.1. Chemistry
2.1.1. Synthesis and Spectral Characteristics
Designer N-arylpiperazines were synthesized via multistep reactions, exploring the impact of their lipohydrophilic and electronic properties on the in vitro activity against selected mycobacterial strains. In addition, the in vitro toxicity profile of the final molecules against a human monocytic leukemia THP-1 cell line was inspected.
Following the outlined objectives, the compounds were synthesized according to Scheme 1 and Scheme 2 as follows. Initially, 3-aminoacetophenone (1a) and 4-aminoacetophenone (1b) were employed as convenient starting compounds. They were treated with alkyl chloroformates 2a–d (alkyl = methyl to butyl) at presence of pyridine in acetone to afford colourless alkyl (3-/4-acetylphenyl)-carbamates 3a–h [28] in the yields that varied from 89% to 99% (Scheme 1).
Scheme 1.

Synthesis of the alkyl [3-/4-(bromoacetyl)phenyl]carbamates 4a–h (alkyl = methyl to butyl), Reagents and conditions: (i) ClCOOR′ (R′ = methyl to butyl; 2a–d), pyridine; (ii) Br2, chloroform.
Scheme 2.

Synthesis of the final 1-(2-{3-/4-[(alkoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)piperazin-1-ium chlorides 8a–h (alkoxy = methoxy to butoxy), Reagents and conditions: (i) TEA, THF; (ii) NaBH4, methanol; (iii) a saturated solution of hydrogen chloride in diethyl ether.
When designing the chemical structure of target molecules, 2-aminoacetophenone was not considered a suitable starting structure due to a possible undesired cyclization of resulting intermediates [29,30]. In addition, a weak in vitro antimycobacterial activity of final products would be probably observed [19,20,21].
The molecules 3a–h underwent α-bromination of an acetyl group because of the dropwise addition bromine in chloroform. This reaction procedure took place at a sufficiently high rate in chloroform at room temperature with constant stirring [31]. Resulting alkyl [3-/4-(bromoacetyl)phenyl]carbamates 4a–h (alkyl = methyl to butyl; Scheme 1) were achieved with 75% to 93% yields.
A substitution of bromine by 1-(2-fluorophenyl)piperazine 5 [32] in the presence of triethylamine (TEA) in anhydrous tetrahydrofuran (THF) led to colourless alkyl {3-/4-[(4-(2-fluorophenyl)piperazin-1-yl)acetyl]phenyl}carbamates 6a–h (Scheme 2). The intermediates 6a–h were prepared in moderate to good yields that ranged from 75% to 96%. Next, they were transformed into alkyl {3-/4-[2-(4-(2-fluorophenyl)piperazin-1-yl)-1-hydroxy-ethyl]phenyl}carbamates 7a–h by nucleophilic addition of hydride anions (Scheme 2) using simple and convenient reduction with sodium borohydride [32]. The molecules 7a–h were synthesized in 80% to 94% yields.
Detailed spectral characteristics (1H-NMR, 13C-NMR, HR-MS or ESI-MS) of the thirty-two prepared intermediates 3a–h, 4–h, 6a–h and 7a–h, are given in the Materials and Methods section of a current paper.
Addition of a saturated solution of hydrogen chloride in diethyl ether into a particular solution of the compounds 7a–h in chloroform led to the desired 1-(2-{3-/4-[(alkoxycarbonyl)amino]phen-yl}-2-hydroxyethyl)-4-(2-fluorophenyl)piperazin-1-ium chlorides 8a–h. The crude products 8a–h were purified by recrystallization from acetone providing 84% to 96% yields (Table 1, Scheme 2).
Purity of the final salts 8a–h was verified by thin-layer chromatography (TLC) using petroleum ether/diethyl amine eluant (10:3, v/v) as a mobile phase. Spots were observed under iodine vapours/UV light at a wavelength (λ) of 254 nm. The position and length of an 3-/4-alkoxycarbonylamino fragment influenced values of a retardation factor (Rf), as expected. The 3-positional isomers 8a–d showed slightly higher Rf values (Rf = 0.40–0.73) compared to those of the 4-substituted ones 8e–h (0.31–0.56). The Rf values have been provided in detail in the Materials and Methods section of a current paper.
The newly synthesized target substances 8a–h were fully characterized by their IR, 1H-NMR, 13C-NMR and ESI-MS spectral values, which were in full accordance with proposed structures. Analyzing the IR spectra of 8a–h, bands typical for stretching vibrations υ (C = O) were observed in the region from 1730 cm−1 to 1718 cm−1. Identity of aromatic rings was confirmed by presence of υ (C = C) at around 1600 cm−1. The recorded IR spectra also afforded vibrations in the range from 1552 cm−1 to 1543 cm−1 due to δ(N–H). The bands between 1234 cm−1 and 1221 cm−1 were related to asymmetric stretching of a C–O–C fragment. The in-plane deformation vibrations (δip) at around 1020 cm−1 and out-of-plane deformation vibrations (δoop) at around 850 cm−1 of a =C–H group were also observed.
In the 1H-NMR, signals of particular protons were verified on basis of their chemical shift (δ), multiplicities and coupling constants in DMSO-d6. Regarding the 3-alkoxycarbonylamino substituent-containing molecules 8a–d, a proton signal of a carbamoyloxy group was detected in the δ interval from 9.60 ppm to 9.57 ppm. A shift of this chain to a 4-position (compounds 8e–h) led to slightly higher δ values recognized from 9.71 ppm to 9.67 ppm. The δ chemical shift between 154.90 ppm and 153.25 ppm (doublet) was assigned to the carbon atom of a C–F bond in the 13C-NMR spectra of prepared salts 8a–h. The carbon of a carbamoyloxy group was identified in the δ range from 153.91 ppm to 152.91 ppm.
Elemental analyses of the synthesized derivatives 8a–h indicated that addition of a saturated solution of hydrogen chloride in diethyl ether caused a protonation of only one nitrogen of a piperazin-1,4-diyl fragment. This was due to a positive mesomeric effect of the nitrogen atom towards an aromatic ring. This conclusion was also evidenced by mass spectral values of these compounds, for which particular [M + H]+ molecular peaks were observed. Current elemental analyses results (% C, H, N) were within ±0.40% of theoretical values for the proposed monohydrochlorides.
2.1.2. Lipohydrophilic Properties
Lipophilicity has been the physicochemical parameter continually attracting prime interest in QSAR and SAR studies as a predominant descriptor of pharmacodynamic, pharmacokinetic and toxic aspects of the antimycobacterial drugs [33,34,35,36].
A partition coefficient P (or its logarithm) between water or a phosphate buffer and octan-1-ol has been used as a preferential experimental expression of lipophilic properties of a compound. However, the log P parameter is losing that role as the method of a choice due to some methodological drawbacks and limitations, which were extensively described and explained in [37]. Chromatographic methods have been therefore developed and used successfully to estimate the lipophilicity of organic compounds [37].
Lipophilicity indices RM and k (log k) of the compounds 8a–h were estimated by the reversed-phase thin-layer chromatography (RP-TLC) and reversed-phase high-performance liquid chromatography (RP-HPLC). The reason for more detailed chromatographic characterization of the salts 8a–h was their better solubility in polar media (mobile phases) compared to free bases 7a–h.
In addition, previous in vitro antimycobacterial assays [18] employed structurally very similar compounds MM as hydrochlorides (Figure 2). This approach was very beneficial due to improvement in their solubility in tested media compared to free bases.
Based on previous experience, it would be more precise to evaluate the lipohydrophilic properties and antimycobacterial activities of 8a–h and compare the observed values to those related to the MM series.
Calculations of the RM and log k parameters were detailed in the Materials and Methods section of a current paper.
In the RP-TLC, silica gel plates impregnated by a hand with a variously concentrated silicone oil in heptane (a strong hydrophobic agent) were used as a non-polar stationary phase [37]. Optimal differences in RM values within both homological groups 8a–d and 8e–h were observed if 1% silicone oil in heptane was chosen (Tables 1 and S1 in Supplementary Materials).
The calculated RM values for the 8a–d series varied from −0.55 to 0.01, the molecules 8e–h showed higher RMs from −0.02 to 0.63 (Table 1). These RM parameters were considered at least useful as a “quick and rough” estimation of lipophilicity.
Octadecyl-functionalized silica gel was used as a stationary phase in the RP-HPLC evaluation of 8a–h. A gradient of two solvents at different volume ratios modulated retention properties of a stationary phase [38]. Liquid binary mixtures of methanol (MeOH) with water were employed as mobile phases in a present isocratic RP-HPLC method. The MeOH organic modifier was preferred because of making a reversed-phase chromatographic system closer to the octan-1-ol/water partitioning one in terms of sensitivity to H-bond donor properties of investigated compounds [39,40]. The modifier was applied in different volume concentrations that varied from 60% to 85% (v/v).
The isocratic separation was possible and in addition, reasonable retention of the analyzed compounds 8a–h was observed in all mobile phases. The estimated k parameters were found in an acceptable interval from 0.5 to 20 [39] and were listed in Table S2 (Supplementary Materials).
Increase in a volume concentration of MeOH led to shortening of tr and log k values for all molecules 8a–h (Table 1).
The 3-alkoxycarbonylamino substituent-containing derivatives 8a–d showed higher tr and log k parameters than their 4-positional isomers 8e–h (Table 1), albeit excluding compounds 8d and 8h. Elongation of an R substituent led to the increase in tr and log k values within both groups 8a–d and 8e–h (Table 1). Lower log k parameters of a molecule 8e compared to those of a derivative 8a (Table S2 in Supplementary Materials) would be a result of “linearity“ of the 4-substituted molecule as well as interactions between the mobile phase and methoxy moiety of 8e. Hydrogen atoms of this group were more acidic due to movement of electrons as a consequence of a different electronegativity of carbon and oxygen so the ability of the compound 8e to form hydrogen bonds with a particular MeOH-containing mobile phase would be enhanced.
The highest log k values were observed for 1-(2-{3-[(butoxycarbonyl)amino]phenyl}-2-hydr-oxyethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8d) and its positional isomer, 1-(2-{4-[(butoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8h; Table 1).
Extrapolation of the estimated log k parameters to elution with 100% water, i.e., the calculation of log kw values, has become a widely accepted approach. The log kw descriptor has been considered more efficient predictor of a biological activity than the log k itself because it reduced influence of an organic mobile phase modifier what was necessary to obtain measurable elution [41,42,43]. The log kw values were extrapolated from intercepts of a linear relationship between the log k and volume fraction of a mobile phase modifier (ϕM) using a Snyder-Soczewiński relationship [42,43,44]. The linear relationship was justified by a correlation coefficient (R) > 0.9900 and adjusted coefficient of determination (Adj. R2) > 0.9700 for all fitting models, excluding the one connected with the compound 8d (Table 2). Anyway, values of the calculated statistical descriptors related to 8d were also satisfactory (R = 0.9730, Adj. R2 = 0.9201; Table 2).
Table 2.
Extrapolated log kw values (RP-HPLC) of the analyzed molecules 8a–h and statistical descriptors (RSS, R, Adj. R2, RMSE, NoR, F and Prob > F), which characterized a linear relationship between the log k and ϕM values for a particular compound. The ϕM parameter was a volume fraction of MeOH in the isocratic elution RP-HPLC.
| Comp. | log kw | 1 S | 2 RSS | 3 R | 4 Adj. R2 | 5 RMSE | 6 NoR | 7 F | 8 Prob > F |
|---|---|---|---|---|---|---|---|---|---|
| 8a | 2.430 | 3.0678 | 0.0023 | 0.9967 | 0.9902 | 0.0337 | 0.0477 | 305.70 | 0.0033 ** |
| 8b | 2.546 | 3.0529 | 0.0017 | 0.9975 | 0.9925 | 0.0294 | 0.0415 | 398.96 | 0.0025 ** |
| 8c | 2.679 | 3.1244 | 0.0051 | 0.9930 | 0.9791 | 0.0504 | 0.0713 | 141.72 | 0.0070 ** |
| 8d | 2.796 | 3.1641 | 0.0208 | 0.9730 | 0.9201 | 0.1019 | 0.1441 | 35.58 | 0.0270 * |
| 8e | 2.113 | 2.7386 | 0.0019 | 0.9965 | 0.9896 | 0.0311 | 0.0440 | 285.13 | 0.0035 ** |
| 8f | 2.512 | 3.1156 | 0.0009 | 0.9988 | 0.9964 | 0.0208 | 0.0294 | 826.63 | 0.0012 ** |
| 8g | 2.600 | 3.0739 | 0.0027 | 0.9962 | 0.9885 | 0.0367 | 0.0519 | 259.09 | 0.0038 ** |
| 8h | 2.930 | 3.3441 | 0.0081 | 0.9903 | 0.9710 | 0.0637 | 0.0901 | 101.50 | 0.0097 ** |
1 S, Slope; 2 RSS, residual sum of squares; 3 R, correlation coefficient; 4 Adj. R2, adjusted coefficient of determination; 5 RMSE, root mean squared error (standard deviation); 6 NoR, norm of residuals; 7 F, Fisher´s significance ratio (Fisher´s F-test); 8 Prob > F, probability of obtaining the F Ratio (significance of a whole model). Indication of a significance level of the F Ratio was as follows: * (one star), significant; ** (two stars), very significant.
The extrapolated log kw values of the analyzed compounds 8a–h (Table 2) were in accordance with their elution order and hydrophobicity and ranged from 2.113 (8e) to 2.930 (8h). Higher log kw values were observed for the derivatives 8a–c compared to 8e–g. Butoxycarbonylamino substituent-containing compounds 8d and 8h were found to be the most lipophilic, as proven by their log kw of 2.796 (8d) and 2.930 (8h), respectively (Table 2).
The slope S of a regression line used to obtain log kw encoded notable information regarding a specific hydrophobic surface area and could serve as indicative measure of uniformity of a retention mechanism. If uniformity was observed, a convenient model between the slope(s) and intercept(s) was anticipated [45]. The currently calculated S parameters varied from 2.7386 (8e) to 3.3441 (8h; Table 2). The slope S was related to a specific hydrophobic surface of a compound and could be used as alternative measure of its lipophilicity [46].
Statistically extremely significant relationship between the log kw and S values was described by Equation (1). The model was characterized by the Prob > F parameter, which was in the range from 0 to <0.0010:
| S = 0.6457 ( ± 0.0882) × log kw + 1.4219 ( ± 0.2208) | (1) |
| RSS = 0.0199, R = 0.9483, Adj. R2 = 0.8826, RMSE = 0.0575, NoR = 0.1409, F = 53.61, Prob > F = 0.0003, n = 8 |
Based on the calculated statistical descriptors provided above, the uniformity of a retention mechanism of the studied derivatives 8a–h was proven and suitability of selected mobile phases was confirmed for lipophilicity evaluation.
2.1.3. Electronic Properties
Electronic properties of the inspected compounds 8a–h (Table 3) were characterized by logarithms of molar absorption coefficients (log ε) of their methanolic solutions (c = 3.0 × 10−5 M) investigated in the UV/Vis region of the spectrum.
Table 3.
Wavelengths of the observed absorption maxima (λ1, λ2(Ch-T) and λ3) and logarithms of the molar absorption coefficients (log ε) of compounds´ methanolic solutions (c = 3.0 × 10−5 M), which were investigated in the UV/Vis region of a spectrum.
| Comp. | λ1 (nm) | log ε1 | λ2(Ch-T) (nm) | 1 log ε2(Ch-T) | λ3 (nm) | log ε3 |
|---|---|---|---|---|---|---|
| 8a | 210 | 4.30 | 238 | 4.30 | 276 | 3.45 |
| 8b | 210 | 4.31 | 238 | 4.33 | 276 | 3.40 |
| 8c | 210 | 4.30 | 238 | 4.37 | 276 | 3.42 |
| 8d | 210 | 4.31 | 238 | 4.32 | 276 | 3.49 |
| 8e | 210 | 4.61 | 240 | 4.67 | 274 | 3.67 |
| 8f | 208 | 4.59 | 240 | 4.60 | 274 | 3.60 |
| 8g | 210 | 4.47 | 240 | 4.54 | 274 | 3.52 |
| 8h | 208 | 4.34 | 240 | 4.42 | 274 | 3.42 |
1 log ε2(Ch-T), Logarithms of molar absorption coefficients observed at the charge-transfer absorption maximum λ2(Ch-T) = 238–240 nm.
The solutions showed three absorption maxima in a near ultraviolet (quarz) region of the electromagnetic spectrum between 200 and 400 nm [47], e.g., λ1 = 208–210 nm, λ2(Ch-T) = 238–240 nm and λ3 = 274–276 nm, respectively (Table 3).
The log ε2(Ch-T) parameters of the compounds 8a–d observed at a charge-transfer absorption maximum λ2(Ch-T) were found in a narrow interval from 4.30 (8a) to 4.37 (8c). The methanolic solutions of 8e–h were characterized by higher log ε2(Ch-T) values than the ones of 8a–d and varied from 4.42 (8h) to 4.67 (8e; Table 3). In addition, elongation of the 4-side chain led to lower log ε values related to all observed absorption maxima (Table 3).
2.2. Biological Assays
2.2.1. In Vitro Antimycobacterial Activity and Structure–Activity Relationships
The compounds 8a–h were initially tested in vitro against M. tuberculosis CNCTC My 331/88 (identical with H37Rv and ATCC 2794), M. avium CNCTC My 330/80 (identical with ATCC 25291), M. avium intracellulare ATCC 13950 and M. kansasii CNCTC My 235/80 (identical with ATCC 12478), respectively, as well as against M. kansasii 6 509/96, M. kansasii CIT11/06, M. avium subsp. paratuberculosis CIT03 and M. avium hominissuis CIT10/08 clinical isolates by methods described earlier [17,48,49,50].
The MIC was defined as the lowest concentration of a particular compound, which (i) inhibited growth of M. tuberculosis CNCTC My 331/88, M. avium CNCTC My 330/80, M. kansasii CNCTC My 235/80 or M. kansasii 6 509/96 [48]; (ii) prevented a visual colour change from blue to pink when testing susceptibility of the M. kansasii CIT11/06, M. avium subsp. paratuberculosis CIT03, M. avium hominissuis CIT10/08 or M. avium intracellulare ATCC 13950 strain. The MIC for given mycobacteria was defined as 90% or greater reduction of their growth (IC90) compared to a control [17,49,50].
The efficiency of newly synthesized molecules 8a–h was compared to the activity of reference drugs, i.e., isonicotinic acid hydrazide (isoniazid, INH), ethambutol (EMB), ofloxacin (OFLX), ciprofloxacin (CPX) or pyrazinamide (PZA) under same experimental conditions.
Next sections of the current research were focused specifically on the most susceptible strains, i.e., M. tuberculosis CNCTC My 331/88, M. kansasii CNCTC My 235/80 and M. kansasii 6 509/96, respectively. The in vitro activities (MIC values) of the most promising N-arylpiperazines were highlighted by a bold font style in gray (Table 4).
Table 4.
The in vitro activity (the MIC values were expressed in the μM units) of currently screened compounds 8a–h and reference drugs isoniazid (INH), ethambutol (EMB) and ofloxacin (OFLX) against M. tuberculosis My 331/88 (M. tuberculosis H37Rv; MT My 331/88), M. kansasii My 235/80 (MK My 235/80), M. kansasii 6 509/96 (MK 6 509/96) and M. avium My 330/88 (MA My 330/88), respectively.
| Comp. | MIC (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MT My 331/88 | MK My 235/80 | MK 6 509/96 | MA My 330/88 | |||||||
| 1 14 d | 2 21 d | 3 7 d | 14 d | 21 d | 7 d | 14 d | 21 d | 14 d | 21 d | |
| 8a | 250 | 250 | 125 | 500 | 1000 | 125 | 500 | 500 | 500 | 500 |
| 8b | 125 | 125 | 62.5 | 250 | 250 | 62.5 | 250 | 250 | 250 | 250 |
| 8c | 62.5 | 62.5 | 62.5 | 125 | 125 | 32 | 125 | 125 | 125 | 250 |
| 8d | 32 | 32 | 32 | 62.5 | 62.5 | 16 | 32 | 62.5 | 62.5 | 62.5 |
| 8e | 125 | 125 | 125 | 500 | 500 | 125 | 500 | 500 | 250 | 500 |
| 8f | 32 | 62.5 | 125 | >250 | >250 | 62.5 | >125 | >125 | >250 | >250 |
| 8g | 16 | 16 | 125 | >250 | >250 | 125 | >250 | >250 | >250 | >250 |
| 8h | 8 | 8 | 62.5 | >125 | >125 | 125 | >250 | >250 | >250 | 250 |
| INH | 0.5 | 0.5 | >250 | >250 | >250 | 4 | 8 | 8 | >250 | >250 |
| EMB | 1 | 2 | 1 | 2 | 2 | 1 | 2 | 2 | 16 | 16 |
| OFLX | 1 | 2 | 0.5 | 1 | 1 | 0.5 | 0.5 | 1 | 32 | 62.5 |
1 14 d, 14-Day cultivation; 2 21 d, 21-day cultivation; 3 7d, 7-day cultivation. The in vitro activities (MIC values) of the most promising N-arylpiperazines were highlighted by a bold font style in gray.
The position of a side chain notably affected the activity of tested derivatives 8a–h against M. tuberculosis CNCTC My 331/88 (Table 4). After 14- and 21-day cultivation (14-d/21-d), the 4-positional isomers were more active, with the MIC values ranging from 8 μM (8h) to 125 μM (8e), than the 3-positional ones, which possessed the MICs from 32 μM (8d) to 250 μM (8a).
Among all the in vitro screened molecules, INH standard was found to be the most active with the MIC = 0.5 μM (14-d/21-d).
Introduction of a 4-(pyrimidin-2-yl)piperazin-1-yl fragment instead of the 4-(2-fluorophen-yl)piperazin-1-yl one led to the derivatives MM, which showed a comparable efficiency [18] to the compounds 8h and 8d, especially if they contained R = C3H7/C4H9 (Figure 2). Similarly, presence of an 3-alkoxyphenylcarbamoyloxy moiety (alkoxy = methoxy to butoxy) and elongation of a connecting chain resulted in the molecules IM (Figure 3) with a comparable in vitro activity [51] to 8a–h.
Figure 3.

N-Arylpiperazines containing a 2-hydroxypropane-1,3-diyl connecting chain (a series IM), which were in vitro screened against M. tuberculosis H37Rv [51].
Isosteric replacement of the carbamoyloxy with a carboxy group in a structure of the compounds IM and introduction of an 4-alkoxycarbonylamino side chain (alkoxy = methoxy to butoxy) at the aromatic ring resulted in decreased in vitro efficiency of such modified racemic derivatives against M. tuberculosis H37Rv [16].
The compounds 8a–d were more efficient against M. kansasii My 235/80 and M. kansasii 6 509/96 than the substances 8e–h. The most active compound against both mycobacteria was 8d with the MIC = 16 μM and 62.5 μM, respectively, depending on a particular strain and also on the number of days of incubation. Increase in length of the side chain resulted in lower MIC values of 8a–d against both tested M. kansasii strains. The observed MIC values were, however, higher compared to the ones related to EMB with the MIC = 1 μM and 2 μM (14-d/21-d), or OFLX, which showed the MIC = 0.5 μM and 1 μM, respectively (14-d/21-d; Table 4).
The in vitro activity of screened compounds 8a–h against a non-tuberculous INH-resistant M. avium CNCTC My 330/80 was apparently dependent on the position of the alkoxycarbonylamino chain R. Its presence in the 3-position (8a–d) led to the MIC values varying from 62.5 μM (8d) to 500 μM (8a; 14-d/21-d). However, a potential of 4-substituent-containing derivatives (8e–h) to fight given mycobacterium was insufficient (MIC > 250 μM; Table 4).
The activity of the most active substance 8d (MIC = 62.5 μM; 14-d/21-d) against M. avium CNCTC My 330/80 was comparable to the effectiveness of OFLX (MIC = 32 μM and 62.5 μM, respectively; 14-d/21-d). The EMB reference drug was slightly more active (MIC = 16 μM; 14-d/21-d) than 8d. Elongation of an 3-R side chain led to more potent compounds (Table 4).
The molecules 8a–h were in vitro practically inactive against M. kansasii CIT11/06, M. avium subsp. paratuberculosis CIT03, M. avium intracellulare ATCC 13950 and M. avium hominissuis CIT10/08, respectively (MIC ≥ 295 μM; Table 5). The CPX standard was the most effective among all investigated compounds (MIC > 91 μM and 181 μM, respectively; Table 5).
Table 5.
The in vitro activity (the MIC values were expressed in the μM units) of the inspected compounds 8a–h and reference drugs isoniazid (INH), ciprofloxacin (CPX) and pyrazinamide (PZA) against M. kansasii CIT11/06 (MK CIT11/06), M. avium subsp. paratuberculosis CIT03 (MAP CIT03), M. avium intracellulare ATCC 13950 (MAI ATCC 13950) and M. avium hominissuis CIT10/08 (MAH CIT10/08), respectively.
| Comp. | MIC (μM) | |||
|---|---|---|---|---|
| MK | MAP | MAI | MAH | |
| CIT11/06 | CIT03 | ATCC 13950 | CIT10/08 | |
| 8a | >610 | >610 | >610 | >610 |
| 8b | 295 | >590 | >590 | >590 |
| 8c | >571 | >571 | >571 | >571 |
| 8d | >553 | > 53 | >553 | >553 |
| 8e | >610 | >610 | >610 | >610 |
| 8f | 295 | >590 | >590 | >590 |
| 8g | >571 | >571 | >71 | >571 |
| 8h | >553 | >553 | >553 | >553 |
| INH | >1823 | >1823 | >1823 | >1823 |
| CPX | >91 | 181 | 181 | 181 |
| PZA | >2031 | >2031 | >2031 | 487 |
The lipophilicity has been considered one of the most important factors, which critically influenced a compound´s activity in penetrating mycobacterial cell walls [48,52,53]. To explore this statement in detail, relationships between the log kw (independent variable) and activity values (dependent variable) of the compounds 8a–h were inspected. For purposes of the current SAR study, observed MIC values were transformed into log (1/MIC [M]) units.
The INH, EMB and OFLX standard drugs were not included in the investigated models because of being structurally different and, in addition, different modes of their action have been proposed [54,55,56,57,58,59,60].
Linear regression analyses were carried out using the Origin Pro 9.0.0 software (OriginLab Corporation, Northampton, MA, USA). More details about particular statistical parameters and significance levels were provided in the Materials and Methods section of a current paper.
Regarding the 8a–h set, analyses related to M. tuberculosis My 331/88 (M. tuberculosis H37Rv; 14-d/21-d) were expressed by Equations (2) and (3):
MT My 331/88 (14-d), 8a–h:
| log (1/MIC [M]) = 1.4383 ( ± 0.5893) × log kw + 0.6071 ( ± 1.5239) | (2) |
| RSS = 0.8867, R = 0.7059, Adj. R2 = 0.4146, RMSE = 0.3844, NoR = 0.9417, F = 5.95, Prob > F = 0.0504, n = 8 |
MT My 331/88 (21-d), 8a–h:
| log (1/MIC [M]) = 1.4819 ( ± 0.5598) × log kw + 0.4586 ( ± 1.4476) | (3) |
| RSS = 0.8002, R = 0.7340, Adj. R2 = 0.4619, RMSE = 0.3652, NoR = 0.8945, F = 7.01, Prob > F = 0.0382, n = 8 |
Based on given statistical parameters, only Equation (3) described a statistically significant model, for which the Prob > F value was found the interval from 0.0100 to <0.0500.
If the analysis was separately applied to the groups 8a–d and 8e–h, much more convenient values of statistical descriptors were calculated for 8a–d (14-d/21-d) as follows: RSS = 0.0003, R = 0.9997, Adj. R2 = 0.9991, RMSE = 0.0119, NoR = 0.0168, F = 3143.04, Prob > F = 0.0003, n = 4. The statistically extremely significant models (Table S3 in Supplementary Materials) were characterized by Equations (S1) and (S3).
On the other hand, the relationships connected with the 8e–h series were statistically insignificant, as proven by Equation (S2) and (S4), respectively (Table S3).
Focusing on the M. kansasii My 235/80 strain after 7-day cultivation (7-d), only the derivatives 8b–d showed interesting MIC values of 32 μM or 62.5 μM (Table 4). However, the linear regression model involving the log kw and log (1/MIC [M]) values related to the 8a–d set was statistically insignificant, as expected (Equation (S5) in Table S3).
Differing effectacy of the compounds 8a–d against M. tuberculosis My 331/88 (M. tuberculosis H37Rv) and M. kansasii My 235/80 (Table 4) was probably a consequence of diverse composition of the bacterial membrane of those strains [61,62].
Lipophilicity could play a crucial role in a mechanism of action of the compounds 8a–d against the M. kansasii 6 509/96 clinical isolate (7-d; Table 4), as provided by Equation (4).
MK 6 509/96 (7-d), 8a–d:
| log (1/MIC [M]) = 2.4098 (± 0.0576) × log kw − 1.9466 ( ± 0.1507) | (4) |
| RSS = 0.0005, R = 0.9994, Adj. R2 = 0.9982, RMSE = 0.0158, NoR = 0.0224, F = 1750.57, Prob > F = 0.0005, n = 4 |
Despite of very convenient values of the statistical descriptors, a main limitation of the approach was a low number of the compounds (n = 4) involved in this analysis.
The log ε2(Ch-T) values (independent variable), which were observed at the charge-transfer absorption maximum λ2(Ch-T), were taken into a special consideration (Table 3), because they could be the most sensitive to the differences in position and electronic properties of a particular alkoxycarbonylamino substituent [47].
The relationship between the log ε2(Ch-T) parameters and log (1/MIC [M]) values connected with the in vitro screening of 8a–h against M. tuberculosis My 331/88 (M. tuberculosis H37Rv; 14-d) provided a bilinear course. Based on this fitting, maximal efficiency of the tested compounds could be observed if their log ε2(Ch-T) values were approximately 4.43 (Figure 4).
Figure 4.

Bilinear relationship between the log ε2(Ch-T) and log (1/MIC [M]) parameters of the investigated compounds 8a–h. The dependent variable values were taken from the in vitro screening of these derivatives against M. tuberculosis CNCTC My 331/88 (M. tuberculosis H37Rv; 14-d).
Regarding the M. kansasii My 235/80 and M. kansasii 6 509/96 strains, no significant relationships between the in vitro activity of the compounds 8a–h and their electronic features were observed.
2.2.2. In Vitro Cytotoxicity Screening
Cytotoxicity of the compounds 8a–h was inspected as the LD50 value, i.e., a lethal dose to 50% of a cell population, which was derived from survival rate curves. The highest dose of all tested derivatives in a medium (30 μM) did not lead to a significant lethal effect on a human monocytic leukemia THP-1 cell line.
The tested molecules showed low (insignificant) toxicity of LD50 > 30 μM against given cell line. Moreover, the compounds 8a and 8e increased proliferation of the THP-1 cells in 24 h when compared to a control. Relative survival rate (in percentages) of the THP-1 cells for all tested derivatives was found to be over 80%, excluding the molecule 8d, where it was 79% at the highest tested concentration of 30 μM (Figure S1 in Supplementary Materials). Only the compounds with the IC50 < 10 μM could be considered antiproliferative (cytotoxic) agents [63], and the highest tested concentration used for the current toxicity assay was 3-fold this value.
As the LD50 values of oxaliplatin and camptothecin standard drugs assessed in this cell line were considerably lower (1.7 ± 0.64 μM and 0.16 ± 0.07 μM, respectively), the discussed compounds 8a–h were deemed non-toxic agents suitable for further design and development of novel antimycobacterials.
3. Materials and Methods
3.1. General Information
All reagents used for syntheses were commercially available from Alpha Aesar (Lancashire, UK), Fluka Chemie (Buchs, Switzerland), Lachema (Brno, Czech Republic), LachNer (Neratovice, Czech Republic), Lancaster (Ward Hill, MA, USA), Merck (Darmstadt, Germany) or Sigma-Aldrich (Dorset, UK) in sufficient quality and were used without additional purification. Solvents were dried and freshly distilled before use.
Thin-layer chromatography (TLC) Kieselgel 60 F254 plates (Merck) visualized by UV irradiation (λ = 254 nm) were used to monitor reactions and purity of synthesized compounds.
Melting point (Mp or mp) values of prepared intermediates and final compounds, respectively, were determined on the Kofler hot plate apparatus HMK (Franz Kustner Nacht GK, Dresden, Germany) and left uncorrected. The mps of some intermediates were already published, i.e., 3a: 102–104 °C [64]; 3b: 111–112 °C [64] and 113–114 °C [65], respectively; 3d: 53–55 °C [64]; 3e: 168 °C [64] and 160–162 °C [66], respectively; 3f: 158 °C [67], 157–158 °C [68,69] and 160–161 °C [64], respectively; 3h: 87–88.5 °C [64]; 4a: 99–103 °C [70]; 4b: 108–110 °C [71]; 4d: 80–86 °C [70]; 4e: 200–201 °C [72].
The Rf values of prepared salts 8a–h were obtained by the TLC on 10-cm aluminium sheets pre-coated with silica gel 60 F254 (0.25 mm thickness; Merck) in glass developing chambers using petroleum ether/diethylamine (10:3, v/v) eluant as a mobile phase. Spots were located under iodine vapours/UV light at λ = 254 nm.
The 1H- and 13C-NMR spectral analyses were carried out on the FT-NMR spectrometer (Varian Co., Palo Alto, CA, USA) operating at 300 MHz (1H-NMR) and 75 MHz (13C-NMR), respectively, in dried DMSO-d6 using tetramethylsilane (TMS; Sigma-Aldrich, Darmstadt, Germany) as an internal standard.
Chemical shifts were reported in a δ scale in parts per million (ppm), coupling constants J were given in Hertz (Hz) and spin multiplicities were expressed as follows: s (singlet), d (doublet), dd (double doublet), t (triplet), q (quartet), m (multiplet). A complete assignment of 1H- and 13C-NMR resonances was based on an interpretation of standard NMR values.
The FT-IR (IR) spectra were obtained by the ATR technique on the FT-IR spectrophotometer Impact 410 (Thermo Fisher Scientific, West Palm Beach, FL, USA). The absorption frequencies ῦmax were reported in reciprocal centimeters (cm−1) in a recorded range from 4000 cm−1 to 400 cm−1.
The mass spectra (HR-MS) of prepared intermediates 3a–h, 4a–h and 6a–h (Scheme 1 and Scheme 2) were measured using the high-performance liquid chromatograph Dionex UltiMate® 3000 (Thermo Fischer Scientific) coupled with the LTQ Orbitrap XL™ Hybrid Ion Trap-Orbitrap Fourier Transform Mass Spectrometer (Thermo Fischer Scientific) with injection into HESI-II in a positive mode.
The liquid chromatography mass spectra of the compounds 7a–h and 8a–h (Scheme 2, Table 1) were carried out on the Agilent 1100 LC/MSD Trap (Agilent Technologies, Santa Clara, CA, USA) in a positive mode using electrospray ionization at atmospheric pressure. The particular compound was dissolved in methanol (c = 1 mg/mL) and the solution was passed through the XDB SOX 2.1 mm column (Agilent Technologies) with a 1.8 μm particle size at the pressure of 400 bar. The UV detection was performed at λ = 254 nm. Nebulization gas (N2) flow was 8 L/min, pressure was 40 psi. The MS electrospray operated at capillary voltage of 3.5 kV and temperature was set to 350 °C (ESI-MS). Fragments were described as a relationship between atomic mass units and charge (m/z), a recorded interval was from 50 m/z to 1000 m/z.
The elemental analysis (% C, H, N) of the compounds 8a–h was carried out by the Perkin-Elmer 2400 Series-II Elemental Analyzer (Perkin-Elmer, Waltham, MA, USA) and all the derivatives were within ±0.40% of calculation.
The chromatographic HPLC-Diode Array Detection apparatus for the determination of capacity (retention) factor k (log k) values was the LC Agilent Infinity System (Agilent Technologies, Santa Clara, CA, USA) equipped with a Infinity 1260 gradient pump with a degasser, a 1260 HiPals automatic injector, a column thermostat 1290, a photo-diode array detector Infinity 1290 (all the equipments were obtained by Agilent Technologies) and personal computer with the Agilent ChemStation software (Agilent Technologies) for the registration of values and calibration procedures. The chromatographic column Eclipse plus RP C18, 150 × 4.6 mm i.d., a 5 μm particle size (Agilent Technologies), was used and thermostated at 35 °C.
The analyses were performed at pressure ranged from 7 MPa to 15 MPa. The detection wavelength was set to 260 nm, injection sample volume was 5 μL with a flow rate of 1.0 mL/min in all RP-HPLC analyses.
LC-MS Methanol (J. T. Baker Chemicals Co., Phillipsburg, NJ, USA) and HPLC-quality water (Sigma-Aldrich, Darmstadt, Germany) were used for a preparation of mobile phases. Water was firstly deionized and purified by the Millipore Simplicity 185 Ultrapure water purification system (Millipore, Billerica, MA, USA).
The UV/Vis spectra of methanolic solutions of the analyzed compounds 8a–h (c = 3.0 × 10−5 M) were estimated on the 8452A Diode Array spectrophotometer HP-8452A (Hewlett Packard, Palo Alto, CA, USA). Methanol for UV-spectroscopy (Merck) was used for the preparation of these solutions.
Results of the UV/Vis analyses were collected and stored digitally using the ChemStation controller software (Agilent Technologies, Waldbronn, Germany). The HP-8452A apparatus measured a complete range of a spectrum from 190 nm to 820 nm.
3.2. Synthesis of Compounds
3.2.1. General Procedure For the Preparation of Alkyl (3-/4-Acetylphenyl)carbamates (3a–h)
Into a stirred solution of 3-aminoacetophenone 1a (CAS Registry Number 99-03-6; 5.00 g, 37 mmol) or 4-aminoacetophenone 1b (CAS Registry Number 99-92-3; 5.00 g, 37 mmol) and pyridine (3.0 mL, 37 mmol) in 20 mL of acetone, a solution of methyl chloroformate 2a (CAS Registry Number 79-22-1; 3.5 mL, 37 mmol), ethyl chloroformate 2b (CAS Registry Number 541-41-3; 4.0 mL, 37 mmol), propyl chloroformate 2c (CAS Registry Number 109-61-5; 4.5 mL, 37 mmol) or butyl chloroformate 2d (CAS Registry Number 592-34-7; 5.0 mL, 37 mmol) in 5 mL of acetone, was added dropwise. The particular mixture was heated to reflux for 3 h [28]. When the reaction was completed (TLC control), the solvents were removed in vacuo, crude solid products 3a–h were washed with distilled water and recrystallized from absolute ethanol. Full characterization data for the compounds 3a–h (Scheme 1), isolated as colourless solids, are given below.
Methyl (3-acetylphenyl)carbamate (3a); CAS Registry Number 87743-55-3). Yield 6.80 g (95%); Mr 193.19; Mp 103–104 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.87 (s, 1H, NHCOO), 8.06 (s, 1H, Ar–H), 7.71 (d, 1H, Ar–H, J = 7.7 Hz), 7.61 (d, 1H, Ar–H, J = 7.3 Hz), 7.44 (t, 1H, Ar–H, J = 8.2 Hz), 3.69 (s, 3H, COOCH3), 2.55 (s, 3H, COCH3); 13C-NMR (DMSO-d6) δC (ppm): 197.47, 153.92, 139.49, 137.37, 129.01, 122.56, 122.36, 117.26, 51.53, 26.51. HR-MS: for C10H11O3N [M – H]+ calculated 192.06552 m/z, found 192.06728 m/z.
Ethyl (3-acetylphenyl)carbamate (3b; CAS Registry Number 39569-24-9). Yield 7.50 g (98%); Mr 207.19; Mp 112–113 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.83 (s, 1H, NHCOO), 8.07 (s, 1H, Ar–H), 7.68 (d, 1H, Ar–H, J = 8.1 Hz), 7.60 (d, 1H, Ar–H, J = 7.7 Hz), 7.42 (t, 1H, Ar–H, J = 7.8 Hz), 4.14 (q, 2H, CH2CH3, J = 7.1 Hz), 2.53 (s, 3H, COCH3), 1.24 (t, 3H, CH2CH3, J = 7.1 Hz ); 13C-NMR (DMSO-d6) δC (ppm): 197.53, 153.50, 139.60, 137.35, 129.01, 122.60, 122.33, 117.25, 60.21, 26.57, 14.37. HR-MS: for C11H13O3N [M – H]+ calculated 206.08117 m/z, found 206.08297 m/z.
Propyl (3-acetylphenyl)carbamate (3c). Yield 7.30 g (89%); Mr 221.19; Mp 101–103 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.85 (s, 1H, NHCOO), 8.09 (s, 1H, Ar–H), 7.70 (d, 1H, Ar–H, J = 8.1 Hz), 7.61 (d, 1H, Ar–H, J = 8.1 Hz), 7.44 (t, 1H, Ar–H, J = 7.9 Hz), 4.06 (t, 2H, CH2CH2CH3, J = 6.6 Hz), 2.53 (s, 3H, COCH3), 1.72–1.56 (m, 2H, CH2CH2CH3), 0.94 (t, 3H, CH2CH2CH3, J = 7.5 Hz); 13C-NMR (DMSO-d6) δC (ppm): 197.45, 153.59, 139.60, 137.36, 128.96, 122.57, 122.27, 117.28, 65.73, 26.51, 21.76, 10.06. HR-MS: for C12H15O3N [M – H]+ calculated 220.09682 m/z, found 220.09854 m/z.
Butyl (3-acetylphenyl)carbamate (3d; CAS Registry Number 72531-03-4). Yield 7.90 g (91%); Mr 235.19; Mp 58–59 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.83 (s, 1H, NHCOO), 8.07 (s, 1H, Ar–H), 7.68 (d, 1H, Ar–H, J = 7.7 Hz), 7.60 (d, 1H, Ar–H, J = 7.7 Hz), 7.42 (t, 1H, Ar–H, J = 7.9 Hz), 4.09 (t, 2H, CH2CH2CH2CH3, J = 6.6 Hz), 2.54 (s, 3H, COCH3), 1.64–1.54 (m, 2H, CH2CH2CH2CH3), 1.46–1.33 (m, 2H, CH2CH2CH2CH3), 0.92 (t, 3H, CH2CH2CH2CH3, J = 6.2 Hz); 13C-NMR (DMSO-d6) δC (ppm): 197.53, 153.60, 139.60, 137.35, 129.01, 122.59, 122.30, 117.28, 63.94, 30.46, 26.56, 18.49, 13.45. HR-MS: for C13H17O3N [M – H]+ calculated 234.11247 m/z, found 234.11425 m/z.
Methyl (4-acetylphenyl)carbamate (3e; CAS Registry Number 60677-43-2). Yield 7.05 g (99%); Mr 193.19; Mp 168–170 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.09 (s, 1H, NHCOO), 7.91 (d, 2H, Ar–H, J = 8.8 Hz), 7.59 (d, 2H, Ar–H, J = 8.8 Hz), 3.70 (s, 3H, COOCH3), 2.52 (s, 3H, COCH3); 13C-NMR (DMSO-d6) δC (ppm): 196.28, 153.72, 143.60, 130.99, 129.43, 117.20, 51.79, 26.24. HR-MS: for C10H11O3N [M – H]+ calculated 192.06552 m/z, found 192.06728 m/z.
Ethyl (4-acetylphenyl)carbamate (3f; CAS Registry Number 5520-79-6). Yield 7.60 g (99%); Mr 207.19; Mp 161–163 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.08 (s, 1H, NHCOO), 7.91 (d, 2H, Ar–H, J = 8.8 Hz), 7.59 (d, 2H, Ar–H, J = 8.8 Hz), 4.13 (q, 2H, CH2CH3, J = 7.1 Hz), 2.52 (s, 3H, COCH3), 1.24 (t, 3H, CH2CH3, J = 7.1 Hz); 13C-NMR (DMSO-d6) δC (ppm): 196.28, 153.38, 143.73, 130.94, 129.42, 117.19, 60.44, 26.23, 14.39. HR-MS: for C11H13O3N [M – H]+ calculated 206.08117 m/z, found 206.08295 m/z.
Propyl (4-acetylphenyl)carbamate (3g). Yield 8.05 g (98%); Mr 221.19; Mp 125–126 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.07 (s, 1H, NHCOO), 7.91 (d, 2H, Ar–H, J = 8.8 Hz), 7.60 (d, 2H, Ar–H, J = 8.8 Hz), 4.07 (t, 2H, CH2CH2CH3, J = 6.6 Hz), 2.51 (s, 3H, COCH3), 1.75–1.57 (m, 2H, CH2CH2CH3), 0.94 (t, 3H, CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 196.28, 153.37, 143.71, 130.93, 129.42, 117.20, 65.96, 26.22, 21.73, 10.11. HR-MS: for C12H15O3N [M – H]+ calculated 220.09682 m/z, found 220.09852 m/z.
Butyl (4-acetylphenyl)carbamate (3h; CAS Registry Number 72531-04-5). Yield 8.23 g (95%); Mr 235.19; Mp 89–91 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.05 (s, 1H, NHCOO), 7.91 (d, 2H, Ar–H, J = 8.8 Hz), 7.60 (d, 2H, Ar–H, J = 8.8 Hz), 4.11 (t, 2H, CH2CH2CH2CH3, J = 6.6 Hz), 2.51 (s, 3H, COCH3), 1.69–1.55 (m, 2H, CH2CH2CH2CH3), 1.48–1.30 (m, 2H, CH2CH2CH2CH3), 0.92 (t, 3H, CH2CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 196.27, 153.36, 143.69, 130.92, 129.40, 117.17, 64.14, 30.40, 26.22, 18.47, 13.45. HR-MS: for C13H17O3N [M – H]+ calculated 234.11247 m/z, found 234.11425 m/z.
3.2.2. General Procedure For the Preparation of Alkyl [3-/4-(Bromoacetyl)phenyl]carbamates (4a–h)
Into a stirred solution of a particular alkyl (3-/4-acetylphenyl)carbamate, i.e., 3a, 3e (6.96 g, 36 mmol), 3b, 3f (7.46 g, 36 mmol), 3c, 3g (7.97 g, 36 mmol), 3d or 3h (8.47 g, 36 mmol), in 80 mL of chloroform, a solution of bromine (1.9 mL, 36 mmol) in 10 mL of chloroform was added dropwise and stirred for 3 h at laboratory temperature. When the reaction was completed (TLC control), the solvent was removed in vacuo giving a crude solid product [31]. Intermediates 4a–h (Scheme 1) were recrystallized from propan-2-ol. Full characterization parameters for the compounds 4a–h, isolated as colourless solids, are given below.
Methyl [3-(bromoacetyl)phenyl]carbamate (4a). Yield 7.40 g (75%); Mr 272.09; Mp 99–103 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.92 (s, 1H, NHCO), 8.08 (s, 1H, Ar–H), 7.78–7.66 (m, 2H, Ar–H), 7.47 (t, 1H, Ar–H, J = 8.1 Hz), 4.89 (s, 2H, COCH2Br), 3.68 (s, 3H, COOCH3); 13C-NMR (DMSO-d6) δC (ppm): 191.39, 153.92, 139.70, 134.51, 129.17, 123.28, 122.98, 117.62, 51.64, 33.61. HR-MS: for C10H10O3BrN [M – H]+ calculated 269.97603 m/z, found 269.97781 m/z.
Ethyl [3-(bromoacetyl)phenyl]carbamate (4b; CAS Registry Number 88541-97-3).Yield 9.30 g (90%); Mr 286.09; Mp 105–108 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.89 (s, 1H, NHCO), 8.10 (s, 1H, Ar–H), 7.78–7.66 (m, 2H, Ar–H), 7.46 (t, 1H, Ar–H, J = 7.7 Hz), 4.88 (s, 2H, COCH2Br), 4.14 (q, 2H, CH2CH3, J = 7.3 Hz), 1.25 (t, 3H, CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 191.43, 153.48, 139.67, 134.51, 129.17, 123.29, 122.95, 117.60, 60.27, 33.72, 14.36. HR-MS: for C11H12O3BrN [M – H]+ calculated 283.99168 m/z, found 283.99340 m/z.
Propyl [3-(bromoacetyl)phenyl]carbamate (4c). Yield 9.60 g (89%); Mr 300.09; Mp 104–107 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.90 (s, 1H, NHCO), 8.10 (s, 1H, Ar–H), 7.78–7.65 (m, 2H, Ar–H), 7.46 (t, 1H, Ar–H, J = 7.9 Hz), 4.88 (s, 2H, COCH2Br), 4.05 (t, 2H, CH2CH2CH3, J = 6.6 Hz), 1.73–1.55 (m, 2H, CH2CH2CH3), 0.93 (t, 3H, CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 191.43, 153.59, 139.81, 134.51, 129.17, 123.30, 122.93, 117.61, 65.81, 33.70, 21.76, 10.13. HR-MS: for C12H14O3BrN [M – H]+ calculated 298.00733 m/z, found 298.00913 m/z.
Butyl [3-(bromoacetyl)phenyl]carbamate (4d). Yield 9.60 g (85%); Mr 314.19; Mp 82–85 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.89 (s, 1H, NHCO), 8.10 (s, 1H, Ar–H), 7.77–7.65 (m, 2H, Ar–H), 7.46 (t, 1H, Ar–H, J = 7.9 Hz), 4.88 (s, 2H, COCH2Br), 4.10 (t, 2H, CH2CH2CH2CH3, J = 6.6 Hz), 1.64–1.54 (m, 2H, CH2CH2CH2CH3), 1.47–1.34 (m, 2H, CH2CH2CH2CH3), 0.92 (t, 3H, CH2CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 191.41, 153.57, 139.79, 134.50, 129.16, 123.27, 122.91, 117.61, 63.99, 33.64, 30.43, 18.46, 13.43. HR-MS: for C13H16O3BrN [M – H]+ calculated 312.02298 m/z, found 312.02474 m/z.
Methyl [4-(bromoacetyl)phenyl]carbamate (4e; CAS Registry Number 942316-98-5). Yield 7.71 g (79%); Mr 272.09; Mp 200–202 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.17 (s, 1H, NHCOO), 7.97 (d, 2H, Ar–H, J = 8.8 Hz), 7.61 (d, 2H, Ar–H, J = 8.8 Hz), 4.85 (s, 2H, COCH2Br), 3.71 (s, 3H, COOCH3); 13C-NMR (DMSO-d6) δC (ppm): 190.15, 153.71, 144.30, 130.17, 127.90, 117.32, 51.90, 33.46. HR-MS: for C10H10O3BrN [M – H]+ calculated 269.97603 m/z, found 269.97781 m/z.
Ethyl [4-(bromoacetyl)phenyl]carbamate (4f). Yield 9.63 g (93%); Mr 286.09; Mp 174–176 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.14 (s, 1H, NHCOO), 7.96 (d, 2H, Ar–H, J = 8.8 Hz), 7.62 (d, 2H, Ar–H, J = 8.8 Hz), 4.85 (s, 2H, COCH2Br), 4.17 (q, 2H, CH2CH3, J = 6.7 Hz), 1.27 (t, 3H, CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 190.12, 153.22, 144.38, 130.14, 127.81, 117.29, 60.56, 33.46, 14.31. HR-MS: for C11H12O3BrN [M – H]+ calculated 283.99168 m/z, found 283.9938 m/z.
Propyl [4-(bromoacetyl)phenyl]carbamate (4g). Yield 9.26 g (86%); Mr 300.09; Mp 154–155 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.14 (s, 1H, NHCOO), 7.96 (d, 2H, Ar–H, J = 8.8 Hz), 7.62 (d, 2H, Ar–H, J = 8.8 Hz), 4.84 (s, 2H, COCH2Br), 4.08 (t, 2H, CH2CH2CH3, J = 6.6 Hz), 1.75–1.57 (m, 2H, CH2CH2CH3), 0.94 (t, 3H, CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 190.15, 153.36, 144.41, 130.16, 127.82, 117.32, 66.07, 33.48, 21.73, 10.14. HR-MS: for C12H14O3BrN [M – H]+ calculated 298.00733 m/z, found 298.00911 m/z.
Butyl [4-(bromoacetyl)phenyl]carbamate (4h). Yield 9.64 g (85%); Mr 314.19; Mp 152–154 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.13 (s, 1H, NHCOO), 7.96 (d, 2H, Ar–H, J = 8.8 Hz), 7.62 (d, 2H, Ar–H, J = 8.8 Hz), 4.84 (s, 2H, COCH2Br), 4.12 (t, 2H, CH2CH2CH2CH3, J = 6.6 Hz), 1.70–1.56 (m, 2H, CH2CH2CH2CH3), 1.48–1.30 (m, 2H, CH2CH2CH2CH3), 0.92 (t, 3H, CH2CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 190.12, 153.31, 144.38, 130.13, 127.79, 117.28, 64.23, 33.43, 30.38, 18.47, 13.45. HR-MS: for C13H16O3BrN [M – H]+ 312.02298 m/z, found 312.02474 m/z.
3.2.3. General Procedure For the Preparation of Alkyl {3-/4-[(4-(2-Fluorophenyl)piperazin-1-yl)-acetyl]phenyl}carbamates (6a–h)
A solution of a particular alkyl [3-/4-(bromoacetyl)phenyl]carbamate, i.e., 4a, 4e (1.50 g, 5.5 mmol), 4b, 4f (1.57 g, 5.5 mmol), 4c, 4g (1.65 g, 5.5 mmol), 4d or 4h (1.73 g, 5.5 mmol) in 30 mL of anhydrous tetrahydrofuran (THF) was added dropwise into a stirred solution of 1-(2-fluorophenyl)piperazine 5 (CAS Registry Number 111-15-0; 1.00 g, 5.5 mmol) and triethylamine (TEA; 0.8 mL, 5.5 mmol) in 20 mL of anhydrous THF [32]. The particular mixture was stirred for 3 h at laboratory temperature. When the reaction was completed (TLC control), the solvents were removed in vacuo and remaining solid was treated with 100 mL of distilled water and 100 mL of chloroform. The organic layer was washed with distilled water, dried over anhydrous sodium sulphate and evaporated in vacuo giving crude solid products. Prepared intermediates 6a–h (Scheme 2) were recrystallized from acetone. Full characterization parameters for the compounds 6a–h, isolated as colourless solids, are provided below.
Methyl {3-[(4-(2-fluorophenyl)piperazin-1-yl)acetyl]phenyl}carbamate (6a). Yield 1.80 g (86%); Mr 371.39; Mp 103–105 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.86 (s, 1H, NHCO), 8.09 (s, 1H, Ar–H), 7.75–7.60 (m, 2H, Ar–H), 7.40 (t, 1H, Ar–H, J = 7.9 Hz), 7.07–6.98 (m, 4H, Ar–H), 3.87 (s, 2H, COCH2N), 3.68 (s, 3H, COOCH3), 3.10–3.00 (m, 4H, 3,5-piperazine), 2.75–2.60 (m, 4H, 2,6-piperazine); 13C-NMR (DMSO-d6) δC (ppm): 196.47, 154.86 (d, J = 242.8 Hz), 153.92, 139.76 (d, J = 8.4 Hz), 139.45, 136.44, 128.89, 124.66 (d, J = 3.0 Hz), 122.63, 122.19, 122.09 (d, J = 7.6 Hz), 119.15 (d, J = 2.3 Hz), 117.35, 115.37 (d, J = 20.5 Hz), 63.55, 52.56, 51.58, 50.00 (d, J = 3.1 Hz). HR-MS: for C20H22O3FN3 [M – H]+ calculated 370.15615 m/z, found 370.15791 m/z.
Ethyl {3-[(4-(2-fluorophenyl)piperazin-1-yl)acetyl]phenyl}carbamate (6b). Yield 1.64 g (75%); Mr 385.44; Mp 127–129 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.84 (s, 1H, NHCO), 8.11 (s, 1H, Ar–H), 7.75–7.60 (m, 2H, Ar–H), 7.42 (t, 1H, Ar–H, J = 7.9 Hz), 7.10–6.94 (m, 4H, Ar–H), 4.13 (q, 2H, CH2CH3, J = 7.1 Hz), 3.87 (s, 2H, COCH2N), 3.10–3.00 (m, 4H, 3,5-piperazine), 2.75–2.60 (m, 4H, 2,6-piperazine), 1.24 (t, 3H, CH2CH3, J = 7.0 Hz); 13C-NMR (DMSO-d6) δC (ppm): 196.47, 154.89 (d, J = 242.8 Hz), 153.50, 139.77 (d, J = 8.4 Hz), 139.55, 136.41, 128.92, 124.68 (d, J = 3.0 Hz), 122.66, 122.15, 122.15 (d, J = 7.6 Hz), 119.17 (d, J = 2.3 Hz), 117.32, 115.78 (d, J = 20.5 Hz), 63.55, 60.21, 52.58, 49.99 (d, J = 3.1 Hz), 14.37. HR-MS: for C21H24O3FN3 [M – H]+ calculated 384.17180 m/z, found 384.17360 m/z..
Propyl {3-[(4-(2-fluorophenyl)piperazin-1-yl)acetyl]phenyl}carbamate (6c). Yield 1.78 g (80%); Mr 399.47; Mp 124–126 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.84 (s, 1H, NHCO), 8.11 (s, 1H, Ar–H), 7.75–7.60 (m, 2H, Ar–H), 7.42 (t, 1H, Ar–H, J = 7.9 Hz), 7.10–6.94 (m, 4H, Ar–H), 4.06 (t, 2H, CH2CH2CH3, J = 6.6 Hz), 3.87 (s, 2H, COCH2N), 3.10–3.00 (m, 4H, 3,5-piperazine), 2.70–2.55 (m, 4H, 2,6-piperazine), 1.74–1.56 (m, 2H, CH2CH2CH3), 0.94 (t, 3H, CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 196.47, 154.89 (d, J = 242.8 Hz), 153.62, 139.79 (d, J = 8.4 Hz), 139.58, 136.41, 128.95, 124.74 (d, J = 3.0 Hz), 122.66, 122.15 (d, J = 7.6 Hz), 122.16, 119.17 (d, J = 2.3 Hz), 117.32, 115.80 (d, J = 20.5 Hz), 65.77, 63.56, 52.61, 49.98 (d, J = 3.1 Hz), 21.79, 10.14. HR-MS: for C22H26O3FN3 [M – H]+ calculated 398.18745 m/z, found 398.18917 m/z.
Butyl {3-[(4-(2-fluorophenyl)piperazin-1-yl)acetyl]phenyl}carbamate (6d). Yield 1.85 g (81%); Mr 413.50; Mp 112–114 °C; 1H-NMR (DMSO-d6) δH (ppm): 9.84 (s, 1H, NHCO), 8.11 (s, 1H, Ar–H), 7.75–7.60 (m, 2H, Ar–H), 7.42 (t, 1H, Ar–H, J = 7.9 Hz), 7.11–6.94 (m, 4H, Ar–H), 4.10 (t, 2H, CH2CH2CH2CH3, J = 6.2 Hz), 3.87 (s, 2H, COCH2N), 3.15–3.00 (m, 4H, 3,5-piperazine), 2.80–2.60 (m, 4H, 2,6-piperazine), 1.68–1.55 (m, 2H, CH2CH2CH2CH3), 1.48–1.33 (m, 2H, CH2CH2CH2CH3), 0.91 (t, 3H, CH2CH2CH2CH3, J = 6.8 Hz); 13C-NMR (DMSO-d6) δC (ppm): 196.47, 154.89 (d, J = 242.8 Hz), 153.59, 139.77 (d, J = 8.4 Hz), 139.55, 136.43, 128.87, 124.65 (d, J = 3.0 Hz), 122.63, 122.10, 122.10 (d, J = 7.6 Hz), 119.13 (d, J = 2.3 Hz), 117.35, 115.75 (d, J = 20.5 Hz), 63.93, 63.56, 52.58, 49.98 (d, J = 3.1 Hz), 30.46, 18.47, 13.42. HR-MS: for C23H28O3FN3 [M – H]+ calculated 412.20310 m/z, found 412.20488 m/z.
Methyl {4-[(4-(2-fluorophenyl)piperazin-1-yl)acetyl]phenyl}carbamate (6e). Yield 1.71 g (81%); Mr 371.39; Mp 178–180 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.09 (s, 1H, NHCO), 7.97 (d, 2H, Ar–H, J = 8.4 Hz), 7.59 (d, 2H, Ar–H, J = 8.4 Hz), 7.17–6.91 (m, 4H, Ar–H), 3.84 (s, 2H, COCH2N), 3.70 (s, 3H, COOCH3), 3.05–2.95 (m, 4H, 3,5-piperazine), 2.75–2.60 (m, 4H, 2,6-piperazine); 13C-NMR (DMSO-d6) δC (ppm): 195.13, 154.85 (d, J = 242.8 Hz), 153.66, 143.63, 139.70 (d, J = 8.4 Hz), 129.92, 129.39, 124.62 (d, J = 3.0 Hz), 122.10 (d, J = 7.6 Hz), 119.14 (d, J = 2.3 Hz), 117.14, 115.74 (d, J = 20.5 Hz), 63.32, 52.56, 51.73, 49.89 (d, J = 3.1 Hz). HR-MS: for C20H22O3FN3 [M – H]+ calculated 370.15615 m/z, found 370.15787 m/z.
Ethyl {4-[(4-(2-fluorophenyl)piperazin-1-yl)acetyl]phenyl}carbamate (6f). Yield 1.82 g (86%); Mr 385.44; Mp 172–174 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.05 (s, 1H, NHCO), 7.96 (d, 2H, Ar–H, J = 8.4 Hz), 7.59 (d, 2H, Ar–H, J = 8.4 Hz), 7.17–6.91 (m, 4H, Ar–H), 4.13 (q, 2H, CH2CH3, J = 7.0 Hz), 3.82 (s, 2H, COCH2N), 3.05–2.95 (m, 4H, 3,5-piperazine), 2.75–2.60 (m, 4H, 2,6-piperazine), 1.25 (t, 3H, CH2CH3, J = 7.0 Hz); 13C-NMR (DMSO-d6) δC (ppm): 195.27, 154.85 (d, J = 242.8 Hz), 153.21, 143.69, 139.73 (d, J = 8.4 Hz), 129.92, 129.36, 124.62 (d, J = 3.0 Hz), 122.13 (d, J = 7.6 Hz), 119.14 (d, J = 2.3 Hz), 117.13, 115.76 (d, J = 20.5 Hz), 63.38, 60.39, 52.58, 49.94 (d, J = 3.1 Hz), 14.25. HR-MS: for C21H24O3FN3 [M – H]+ calculated 384.17180 m/z, found 384.17360 m/z.
Propyl {4-[(4-(2-fluorophenyl)piperazin-1-yl)acetyl]phenyl}carbamate (6g). Yield 1.71 g (75%); Mr 399.47; Mp 140–142 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.06 (s, 1H, NHCO), 7.96 (d, 2H, Ar–H, J = 8.4 Hz), 7.59 (d, 2H, Ar–H, J = 8.4 Hz), 7.17–6.91 (m, 4H, Ar–H), 4.06 (t, 2H, CH2CH2CH3, J = 6.6 Hz), 3.83 (s, 2H, COCH2N), 3.05–2.95 (m, 4H, 3,5-piperazine), 2.75–2.60 (m, 4H, 2,6-piperazine), 1.75–1.57 (m, 2H, CH2CH2CH3), 0.93 (t, 3H, CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 195.28, 154.87 (d, J = 242.8 Hz), 153.33, 143.72, 139.75 (d, J = 8.4 Hz), 129.92, 129.37, 124.64 (d, J = 3.0 Hz), 122.08 (d, J = 7.6 Hz), 119.16 (d, J = 2.3 Hz), 117.14, 115.74 (d, J = 20.5 Hz), 65.93, 63.40, 52.59, 49.98 (d, J = 3.1 Hz), 21.69, 10.05. HR-MS: for C22H26O3FN3 [M – H]+ calculated 398.18745 m/z, found 398.18917 m/z.
Butyl {4-[(4-(2-fluorophenyl)piperazin-1-yl)acetyl]phenyl}carbamate (6h). Yield 2.23 g (96%); Mr 413.50; Mp 154–156 °C; 1H-NMR (DMSO-d6) δH (ppm): 10.06 (s, 1H, NHCO), 7.96 (d, 2H, Ar–H, J = 8.4 Hz), 7.59 (d, 2H, Ar–H, J = 8.4 Hz), 7.16–6.91 (m, 4H, Ar–H), 4.10 (t, 2H, CH2CH2CH2CH3, J = 6.6 Hz), 3.82 (s, 2H, COCH2N), 3.05–2.95 (m, 4H, 3,5-piperazine), 2.75–2.60 (m, 4H, 2,6-piperazine), 1.69–1.55 (m, 2H, CH2CH2CH2CH3), 1.46–1.30 (m, 2H, CH2CH2CH2CH3), 0.91 (t, 3H, CH2CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 195.33, 154.90 (d, J = 242.8 Hz), 153.36, 143.74, 139.78 (d, J = 8.4 Hz), 129.95, 129.40, 124.67 (d, J = 3.0 Hz), 122.13 (d, J = 7.6 Hz), 119.17 (d, J = 2.3 Hz), 117.17, 115.77 (d, J = 20.5 Hz), 64.16, 63.42, 52.61, 50.00 (d, J = 3.1 Hz), 30.39, 18.46, 13.40. HR-MS: for C23H28O3FN3 [M – H]+ calculated 412.20310 m/z, found 412.20490 m/z.
3.2.4. General Procedure For the Preparation of Alkyl {3-/4-[2-(4-(2-Fluorophenyl)piperazin-1-yl)-1-hydroxyethyl]phenyl}carbamates (7a–h)
The synthesized alkyl {3-/4-[(4-(2-fluorophenyl)piperazin-1-yl)acetyl]phenyl}carbamates, i.e., 6a, 6e (1.49 g, 4.0 mmol), 6b, 6f (1.54 g, 4.0 mmol), 6c, 6g (1.60 g, 4.0 mmol), 6d or 6h (1.45 g, 4.0 mmol), were dissolved in hot methanol (50 mL) and solid NaBH4 (0.30 g, 8.0 mmol) was added in small portions [32]. The mixtures were refluxed for 1 h. When the reaction was completed (TLC control), the solvent was evaporated in vacuo, residua were treated with 100 mL of distilled water and 100 mL of chloroform. The organic layer was washed with distilled water, dried over anhydrous sodium sulphate and evaporated in vacuo to give crude solid products 7a–h, which were crystallized from acetone. Full characterization parameters for the compounds 7a–h (Scheme 2), isolated as colourless solids, are given below.
Methyl {3-[2-(4-(2-fluorophenyl)piperazin-1-yl)-1-hydroxyethyl]phenyl}carbamate (7a). Yield 1.20 g (83%); Mr 373.41; Mp 103–105 °C; IR (ATR, cm−1): 3382 (υ NH), 2951 (υas CH2), 2816 (υs CH2), 1726 (υ C = O), 1553 (δ NH), 1497 (υ CN), 1229 (υas COC), 1079 (υs CO), 1020 (δip = C–H), 850 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 9.61 (s, 1H, NHCO), 7.48 (s, 1H, Ar–H), 7.35 (d, 1H, Ar–H, J = 8.1 Hz), 7.22 (t, 1H, Ar–H, J = 8.1 Hz), 7.16–6.90 (m, 5H, Ar–H), 5.06 (d, 1H, CHOH, J = 3.7 Hz), 4.73–4.65 (m, 1H, OH), 3.66 (s, 3H, COOCH3), 3.10–2.95 (m, 4H, 3,5-piperazine), 2.75–2.60 (m, 4H, 2,6-piperazine), 2.55–2.38 (m, 2H, CH(OH)CH2N); 13C-NMR (DMSO-d6) δC (ppm): 154.83 (d, J = 242.8 Hz), 153.88, 145.20, 139.80 (d, J = 8.4 Hz), 138.73, 128.05, 124.61 (d, J = 3.0 Hz), 121.97 (d, J = 7.6 Hz), 120.08, 119.02 (d, J = 2.3 Hz), 116.76, 116.00, 115.72 (d, J = 20.5 Hz), 69.76, 66.10, 53.02, 51.34, 50.00 (d, J = 3.1 Hz); ESI-MS: for C20H24O3FN3 [M + H]+ calculated 373.42138 m/z, found 373.42095 m/z.
Ethyl {3-[2-(4-(2-fluorophenyl)piperazin-1-yl)-1-hydroxyethyl]phenyl}carbamate (7b). Yield 1.30 g (82%); Mr 387.46; Mp 118–120 °C; IR (ATR, cm−1): 3463 (υ NH), 2937 (υas CH2), 2833 (υs CH2), 1703 (υ C = O), 1548 (δ NH), 1498 (υ CN), 1240 (υas COC), 1082 (υs CO), 1018 (δip = C–H), 852 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 9.57 (s, 1H, NHCO), 7.50 (s, 1H, Ar–H), 7.34 (d, 1H, Ar–H, J = 8.1 Hz), 7.21 (t, 1H, Ar–H, J = 8.1 Hz), 7.13–6.91 (m, 5H, Ar–H), 5.05 (d, 1H, CHOH, J = 3.7 Hz), 4.73–4.65 (m, 1H, OH), 4.12 (q, 2H, CH2CH3, J = 7.1 Hz), 3.10–2.95 (m, 4H, 3,5-piperazine), 2.75–2.60 (m, 4H, 2,6-piperazine), 2.52–2.37 (m, 2H, CH(OH)CH2N), 1.24 (t, 3H, CH2CH3, J = 7.0 Hz ); 13C-NMR (DMSO-d6) δC (ppm): 154.83 (d, J = 242.8 Hz), 153.44, 145.18, 139.82 (d, J = 8.4 Hz), 138.82, 128.04, 124.64 (d, J = 3.0 Hz), 121.98 (d, J = 7.6 Hz), 120.01, 119.02 (d, J = 2.3 Hz), 116.78, 115.99, 115.76 (d, J = 20.5 Hz), 69.79, 66.11, 59.86, 53.03, 50.01 (d, J = 3.1 Hz), 14.39; ESI-MS: for C21H26O3FN3 [M + H]+ calculated 387.44796 m/z, found 387.44805 m/z.
Propyl {3-[2-(4-(2-fluorophenyl)piperazin-1-yl)-1-hydroxyethyl]phenyl}carbamate (7c). Yield 1.30 g (80%); Mr 401.49; Mp 103–105 °C; IR (ATR, cm−1): 3262 (υ NH), 2942 (υas CH2), 2829 (υs CH2), 1726 (υ C = O), 1545 (δ NH), 1497 (υ CN), 1226 (υas COC), 1081 (υs CO), 1025 (δip = C–H), 848 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 9.58 (s, 1H, NHCO), 7.51 (s, 1H, Ar–H), 7.34 (d, 1H, Ar–H, J = 8.1 Hz), 7.21 (t, 1H, Ar–H, J = 8.1 Hz), 7.13–6.91 (m, 5H, Ar–H), 5.05 (d, 1H, CHOH, J = 3.7 Hz), 4.73–4.65 (m, 1H, OH), 4.03 (t, 2H, CH2CH2CH3, J = 6.6 Hz), 3.07–2.93 (m, 4H, 3,5-piperazine), 2.73–2.59 (m, 4H, 2,6-piperazine), 2.55–2.38 (m, 2H, CH(OH)CH2N), 1.73–1.55 (m, 2H, CH2CH2CH3), 0.93 (t, 3H, CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 154.83 (d, J = 242.8 Hz), 153.57, 145.23, 139.85 (d, J = 8.4 Hz), 138.87, 128.08, 124.67 (d, J = 3.0 Hz), 122.03 (d, J = 7.6 Hz), 120.04, 119.06 (d, J = 2.3 Hz), 116.81, 116.02, 115.77 (d, J = 20.5 Hz), 69.80, 66.16, 65.46, 53.07, 50.05 (d, J = 3.1 Hz), 21.82, 10.13; ESI-MS: for C22H28O3FN3 [M + H]+ calculated 401.47454 m/z, found 401.47421 m/z.
Butyl {3-[2-(4-(2-fluorophenyl)piperazin-1-yl)-1-hydroxyethyl]phenyl}carbamate (7d). Yield 1.50 g (90%); Mr 415.52; Mp 107–110 °C; IR (ATR, cm−1): 3294 (υ NH), 2940 (υas CH2), 2830 (υs CH2), 1706 (υ C = O), 1541 (δ NH), 1497 (υ CN), 1228 (υas COC), 1082 (υs CO), 1018 (δip = C–H), 845 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 9.57 (s, 1H, NHCO), 7.51 (s, 1H, Ar–H), 7.33 (d, 1H, Ar–H, J = 8.1 Hz), 7.20 (t, 1H, Ar–H, J = 8.1 Hz), 7.13–6.91 (m, 5H, Ar–H), 5.04 (d, 1H, CHOH, J = 3.7 Hz), 4.73–4.65 (m, 1H, OH), 4.07 (t, 2H, CH2CH2CH2CH3, J = 6.6 Hz), 3.10–2.95 (m, 4H, 3,5-piperazine), 2.75–2.60 (m, 4H, 2,6-piperazine), 2.53–2.37 (m, 2H, CH(OH)CH2N), 1.67–1.53 (m, 2H, CH2CH2CH2CH3), 1.47–1.29 (m, 2H, CH2CH2CH2CH3), 0.92 (t, 3H, CH2CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 154.85 (d, J = 242.8 Hz), 153.56, 145.21, 139.83 (d, J = 8.4 Hz), 138.85, 128.07, 124.65 (d, J = 3.0 Hz), 122.01 (d, J = 7.6 Hz), 120.02, 119.03 (d, J = 2.3 Hz), 116.78, 115.97, 115.77 (d, J = 20.5 Hz), 69.80, 66.14, 63.64, 53.07, 50.04 (d, J = 3.1 Hz), 30.52, 18.49, 13.45; ESI-MS: for C23H30O3FN3 [M + H]+ calculated 415.50112 m/z, found 415.50207 m/z.
Methyl {4-[2-(4-(2-fluorophenyl)piperazin-1-yl)-1-hydroxyethyl]phenyl}carbamate (7e). Yield 1.42 g (94%); Mr 373.41; Mp 143–146 °C; IR (ATR, cm−1): 3340 (υ NH), 2975 (υas CH2), 2808 (υs CH2), 1702 (υ C = O), 1552 (δ NH), 1502 (υ CN), 1234 (υas COC), 1063 (υs CO), 1021 (δip = C–H), 855 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 9.59 (s, 1H, NHCO), 7.38 (d, 2H, Ar–H, J = 8.4 Hz), 7.25 (d, 2H, Ar–H, J = 8.4 Hz), 7.16–6.90 (m, 4H, Ar–H), 4.96 (d, 1H, CHOH, J = 3.7 Hz), 4.75–4.63 (m, 1H, OH), 3.64 (s, 3H, COOCH3), 3.12–2.90 (m, 4H, 3,5-piperazine), 2.75–2.55 (m, 4H, 2,6-piperazine), 2.55–2.35 (m, 2H, CH(OH)CH2N); 13C-NMR (DMSO-d6) δC (ppm): 154.85 (d, J = 242.8 Hz), 153.92, 139.82 (d, J = 8.4 Hz), 138.55, 137.69, 126.26, 124.64 (d, J = 3.0 Hz), 121.98 (d, J = 7.6 Hz), 119.05 (d, J = 2.3 Hz), 117.87, 115.74 (d, J = 20.5 Hz), 69.36, 66.04, 53.03, 51.37, 50.05 (d, J = 3.1 Hz); ESI-MS: for C20H24O3FN3 [M + H]+ calculated 373.42138 m/z, found 373.42142 m/z.
Ethyl {4-[2-(4-(2-fluorophenyl)piperazin-1-yl)-1-hydroxyethyl]phenyl}carbamate (7f). Yield 1.40 g (88%); Mr 387.46; Mp 142–145 °C; IR (ATR, cm−1): 3340 (υ NH), 2977 (υas CH2), 2808 (υs CH2), 1711 (υ C = O), 1549 (δ NH), 1503 (υ CN), 1234 (υas COC), 1060 (υs CO), 1018 (δip = C–H), 860 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 9.54 (s, 1H, NHCO), 7.38 (d, 2H, Ar–H, J = 8.4 Hz), 7.22 (d, 2H, Ar–H, J = 8.4 Hz), 7.15–6.91 (m, 4H, Ar–H), 4.96 (d, 1H, CHOH, J = 3.7 Hz), 4.71–4.61 (m, 1H, OH), 4.08 (q, 2H, CH2CH3, J = 7.0 Hz), 3.06–2.91 (m, 4H, 3,5-piperazine), 2.71–2.54 (m, 4H, 2,6-piperazine), 2.52–2.34 (m, 2H, CH(OH)CH2N), 1.21 (t, 3H, CH2CH3, J = 7.0 Hz); 13C-NMR (DMSO-d6) δC (ppm): 154.85 (d, J = 242.8 Hz), 153.47, 139.82 (d, J = 8.4 Hz), 138.46, 137.76, 126.23, 124.63 (d, J = 3.0 Hz), 121.97 (d, J = 7.6 Hz), 119.03 (d, J = 2.3 Hz), 117.82, 115.73 (d, J = 20.5 Hz), 69.36, 66.05, 59.88, 53.03, 50.07 (d, J = 3.1 Hz), 14.39; ESI-MS: for C21H26O3FN3 [M + H]+ calculated 387.44796 m/z, found 387.44822 m/z.
Propyl {4-[2-(4-(2-fluorophenyl)piperazin-1-yl)-1-hydroxyethyl]phenyl}carbamate (7g). Yield 1.53 g (92%); Mr 401.49; Mp 149–152 °C; IR (ATR, cm−1): 3339 (υ NH), 2966 (υas CH2), 2808 (υs CH2), 1698 (υ C = O), 1550 (δ NH), 1505 (υ CN), 1235 (υas COC), 1073 (υs CO), 1015 (δip = C–H), 857 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 9.56 (s, 1H, NHCO), 7.39 (d, 2H, Ar–H, J = 8.4 Hz), 7.24 (d, 2H, Ar–H, J = 8.4 Hz), 7.16–6.89 (m, 4H, Ar–H), 4.98 (d, 1H, CHOH, J = 3.7 Hz), 4.73–4.61 (m, 1H, OH), 4.01 (t, 2H, CH2CH2CH3, J = 6.6 Hz), 3.08–2.91 (m, 4H, 3,5-piperazine), 2.68–2.54 (m, 4H, 2,6-piperazine), 2.54–2.33 (m, 2H, CH(OH)CH2N), 1.71–1.52 (m, 2H, CH2CH2CH3), 0.92 (t, 3H, CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 154.86 (d, J = 242.8 Hz), 153.60, 139.82 (d, J = 8.4 Hz), 138.46, 137.79, 126.24, 124.64 (d, J = 3.0 Hz), 122.00 (d, J = 7.6 Hz), 119.05 (d, J = 2.3 Hz), 117.84, 115.75 (d, J = 20.5 Hz), 69.38, 66.05, 65.48, 53.05, 50.06 (d, J = 3.1 Hz), 21.79, 10.11; ESI-MS: for C22H28O3FN3 [M + H]+ calculated 401.47454 m/z, found 401.47510 m/z.
Butyl {4-[2-(4-(2-fluorophenyl)piperazin-1-yl)-1-hydroxyethyl]phenyl}carbamate (7h). Yield 1.62 g (94%); Mr 415.52; Mp 140–143 °C; IR (ATR, cm−1): 3336 (υ NH), 2954 (υas CH2), 2808 (υs CH2), 1698 (υ C = O), 1549 (δ NH), 1527 (υ CN), 1228 (υas COC), 1076 (υs CO), 1018 (δip = C–H), 852 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 9.56 (s, 1H, NHCO), 7.40 (d, 2H, Ar–H, J = 8.4 Hz), 7.25 (d, 2H, Ar–H, J = 8.4 Hz), 7.16–6.89 (m, 4H, Ar–H), 4.99 (d, 1H, CHOH, J = 3.7 Hz), 4.72–4.59 (m, 1H, OH), 4.06 (t, 2H, CH2CH2CH2CH3, J = 6.6 Hz), 3.09–2.94 (m, 4H, 3,5-piperazine), 2.64–2.55 (m, 4H, 2,6-piperazine), 2.55–2.36 (m, 2H, CH(OH)CH2N), 1.65–1.52 (m, 2H, CH2CH2CH2CH3), 1.46–1.28 (m, 2H, CH2CH2CH2CH3), 0.91 (t, 3H, CH2CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 154.87 (d, J = 242.8 Hz), 153.62, 139.84 (d, J = 8.4 Hz), 138.47, 137.81, 126.26, 124.66 (d, J = 3.0 Hz), 122.02 (d, J = 7.6 Hz), 119.07 (d, J = 2.3 Hz), 117.86, 115.76 (d, J = 20.5 Hz), 69.38, 66.07, 63.67, 53.05, 50.06 (d, J = 3.1 Hz), 30.52, 18.49, 13.43; ESI-MS: for C23H30O3FN3 [M + H]+ calculated 415.50112 m/z, found 415.50154 m/z.
3.2.5. General Procedure For the Preparation of 1-(2-{3-/4-[(Alkoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)piperazin-1-ium Chlorides (8a–h)
The solution of a particular alkyl {3-/4-[2-(4-(2-fluorophenyl)piperazin-1-yl)-1-hydroxyeth-yl]phenyl}carbamate, i.e, 7a, 7e (0.71 g, 1.9 mmol), 7b, 7f (0.74 g, 1.9 mmol), 7c, 7g (0.76 g, 1.9 mmol), 7d or 7h (0.79 g, 1.9 mmol), in 40 mL of chloroform was treated with a saturated solution of hydrogen chloride in diethyl ether and stirred for 5 h at laboratory temperature. The solvents were removed in vacuo and solid crude products 8a–h were crystallized from acetone. Full characterization parameters for the target compounds 8a–h (Scheme 2), isolated as colourless solids, are provided below.
1-(2-Hydroxy-2-{3-[(methoxycarbonyl)amino]phenyl}ethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8a). Yield 0.75 g (88%); Mr 409.89; Mp 139–141 °C; Rf 0.40; IR (ATR, cm−1): 3266 (υ NH), 2952 (υas CH2), 2815 (υs CH2), 1718 (υ C = O), 1612 (υ C = C), 1552 (δ NH), 1491 (υ CN), 1233 (υas COC), 1081 (υs CO), 1020 (δip = C–H), 848 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 10.71 (m, 1H, NH+), 9.60 (s, 1H, NHCO), 7.51 (s, 1H, Ar–H), 7.44 (t, 1H, J = 8.0 Hz), 7.35 (d, 1H, Ar–H, J = 8.0 Hz), 7.25 (t, 1H, Ar–H, J = 8.0 Hz), 7.30–7.15 (m, 5H, Ar–H), 5.25 (dd, 1H, CHOH, J = 4.1 Hz, J = 9.0 Hz), 4.70–4.65 (m, 1H, OH), 3.65 (s, 3H, COOCH3), 3.65–3.55 (m, 4H, 3,5-piperazine), 4.10–3.25 (m, 6H, CH(OH)CH2N + 2,6-piperazine); 13C-NMR (DMSO-d6) δC (ppm): 153.25 (d, J = 249.1 Hz), 152.91, 144.68, 139.37 (d, J = 8.3 Hz), 137.21, 128.16, 124.35 (d, J = 3.1 Hz), 121.42 (d, J = 7.6 Hz), 120.15, 118.46 (d, J = 2.2 Hz), 116.24, 116.07, 115.11 (d, J = 20.4 Hz), 69.55, 66.14, 53.34, 51.26, 50.07 (d, J = 3.0 Hz); ESI-MS: for C20H25O3FN3 [M + H]+ calculated 374.42932 m/z, found 374.42873 m/z. Anal. Calcd. for C20H25O3ClFN3 (409.89): C, 58.61%; H, 6.15%; N, 10.25%. Found: C, 58.65%; H, 6.18%; N, 10.15%.
1-(2-{3-[(Ethoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8b). Yield 0.80 g (91%); Mr 423.91; Mp 117–119 °C; Rf 0.53; IR (ATR, cm−1): 3425 (υ NH), 2983 (υas CH2), 2852 (υs CH2), 1718 (υ C = O), 1610 (υ C = C), 1547 (δ NH), 1496 (υ CN), 1229 (υas COC), 1068 (υs CO), 1018 (δip = C–H), 852 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 10.73 (m, 1H, NH+), 9.57 (s, 1H, NHCO), 7.50 (s, 1H, Ar–H), 7.45 (t, 1H, Ar–H, J = 8.0 Hz), 7.35 (d, 1H, Ar–H, J = 8.1 Hz), 7.27–7.15 (m, 5H, Ar–H), 5.24 (dd, 1H, CHOH, J = 4.1 Hz, J = 9.0 Hz), 4.73–4.65 (m, 1H, OH), 4.20 (q, 2H, CH2CH3, J = 7.0 Hz), 3.60–3.50 (m, 4H, 3,5-piperazine), 4.00–3.25 (m, 6H, CH(OH)CH2N + 2,6-piperazine), 1.30 (t, 3H, CH2CH3, J = 7.0 Hz); 13C-NMR (DMSO-d6) δC (ppm): 154.81 (d, J = 241.4 Hz), 153.47, 146.78, 139.94 (d, J = 8.4 Hz), 138.57, 128.16, 124.02 (d, J = 3.0 Hz), 121.64 (d, J = 7.5 Hz), 119.78, 119.07 (d, J = 2.3 Hz), 116.35, 116.28, 115.01 (d, J = 20.4 Hz), 69.74, 65.89, 59.34, 53.07, 50.12 (d, J = 3.1 Hz), 14.21; ESI-MS: for C21H27O3FN3 [M + H]+ calculated 388.45590 m/z, found 388.45615 m/z. Anal. Calcd. for C21H27O3ClFN3 (423.91): C, 59.50%; H, 6.42%; N, 9.91%. Found: C, 59.33%; H, 6.31%; N, 10.06%.
1-(2-Hydroxy-2-{3-[(propoxycarbonyl)amino]phenyl}ethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8c). Yield 0.82 g (91%); Mr 437.94; Mp 99–101 °C; Rf 0.66; IR (ATR, cm−1): 3402 (υ NH), 2964 (υas CH2), 2837 (υs CH2), 1724 (υ C = O), 1612 (υ C = C), 1546 (δ NH), 1498 (υ CN), 1224 (υas COC), 1072 (υs CO), 1022 (δip = C–H), 850 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 10.78 (m, 1H, NH+), 9.58 (s, 1H, NHCO), 7.50 (s, 1H, Ar–H), 7.44 (t, 1H, Ar–H, J = 8.0 Hz), 7.35 (d, 1H, Ar–H, J = 8.0 Hz), 7.20 (t, 1H, Ar–H, J = 8.1 Hz), 7.30–7.18 (m, 5H, Ar–H), 5.24 (dd, 1H, CHOH, J = 4.0 Hz, J = 9.0 Hz), 4.73–4.65 (m, 1H, OH), 4.12 (t, 2H, CH2CH2CH3, J = 7.0 Hz), 3.60–3.45 (m, 4H, 3,5-piperazine), 4.00–3.42 (m, 6H, CH(OH)CH2N + 2,6-piperazine), 1.69–1.57 (m, 2H, CH2CH2CH3), 0.95 (t, 3H, CH2CH2CH3, J = 7.0 Hz); 13C-NMR (DMSO-d6) δC (ppm): 154.80 (d, J = 242.7 Hz), 153.78, 145.15, 138.19 (d, J = 8.4 Hz), 137.45, 128.49, 124.02 (d, J = 3.0 Hz), 122.09 (d, J = 7.7 Hz), 119.97, 119.03 (d, J = 2.3 Hz), 116.76, 116.08, 115.29 (d, J = 20.5 Hz), 69.54, 66.17, 65.76, 53.27, 50.03 (d, J = 3.1 Hz), 21.81, 10.15; ESI-MS: for C22H29O3FN3 [M + H]+ calculated 402.48248 m/z, found 402.48197 m/z. Anal. Calcd. for C22H29O3ClFN3 (437.94): C, 60.34%; H, 6.67%; N, 9.59%. Found: C, 60.12%; H, 6.81%; N, 9.37%.
1-(2-{3-[(Butoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8d). Yield 0.88 g (95%); Mr 451.97; Mp 98–100 °C; Rf 0.73; IR (ATR, cm−1): 3416 (υ NH), 2959 (υas CH2), 2837 (υs CH2), 1730 (υ C = O), 1604 (υ C = C), 1543 (δ NH), 1495 (υ CN), 1221 (υas COC), 1074 (υs CO), 1016 (δip = C–H), 840 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 10.78 (m, 1H, NH+), 9.57 (s, 1H, NHCO), 7.50 (s, 1H, Ar–H), 7.44 (t, 1H, Ar–H, J = 8.1 Hz), 7.34 (d, 1H, Ar–H, J = 8.1 Hz), 7.25–7.18 (m, 5H, Ar–H), 5.25 (dd, 1H, CHOH, J = 4.0 Hz, J = 9.0 Hz), 4.73–4.65 (m, 1H, OH), 4.18 (t, 2H, CH2CH2CH2CH3, J = 7.0 Hz), 3.60–3.45 (m, 4H, 3,5-piperazine), 4.00–3.40 (m, 6H, CH(OH)CH2N + 2,6-piperazine), 1.67–1.58 (m, 2H, CH2CH2CH2CH3, J = 7.0), 1.40–1.30 (m, 2H, CH2CH2CH2CH3), 0.93 (t, 3H, CH2CH2CH2CH3, J = 7.0 Hz); 13C-NMR (DMSO-d6) δC (ppm): 154.90 (d, J = 241.4 Hz), 153.13, 145.61, 139.70 (d, J = 8.5 Hz), 138.62, 128.17, 124.34 (d, J = 3.1 Hz), 122.14 (d, J = 7.6 Hz), 120.07, 119.26 (d, J = 2.3 Hz), 116.35, 115.82, 115.61 (d, J = 20.4 Hz), 69.81, 66.38, 63.94, 53.23, 50.17 (d, J = 3.1 Hz), 30.60, 18.26, 13.33; ESI-MS: for C23H31O3FN3 [M + H]+ calculated 416.50906 m/z, found 416.50915 m/z. Anal. Calcd. for C23H31O3ClFN3 (451.97): C, 61.12%; H, 6.91%; N, 9.30%. Found: C, 61.02%; H, 6.85%; N, 9.42%.
1-(2-Hydroxy-2-{4-[(methoxycarbonyl)amino]phenyl}ethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8e). Yield 0.78 g (84%); Mr 409.89; Mp 218–220 °C; Rf 0.31; IR (ATR, cm−1): 3377 (υ NH), 2963 (υas CH2), 2831 (υs CH2), 1719 (υ C = O), 1610 (υ C = C), 1550 (δ NH), 1487 (υ CN), 1234 (υas COC), 1073 (υs CO), 1020 (δip = C–H), 854 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 10.42 (s, 1H, NH+), 9.71 (s, 1H, NHCO), 7.47 (d, 2H, Ar–H, J = 8.5 Hz), 7.32 (d, 2H, Ar–H, J = 8.5 Hz), 7.23–7.00 (m, 4H, Ar–H), 6.20 (s, 1H, OH), 5.14 (dd, 1H, CHOH, J = 4.8 Hz, J = 7.8 Hz), 3.66 (s, 3H, COOCH3), 3.75–3.11 (m, 10H, CH(OH)CH2N + piperazine); 13C-NMR (DMSO-d6) δC (ppm): 154.73 (d, J = 244.6 Hz), 153.91, 138.62, 138.25 (d, J = 8.6 Hz), 135.32, 126.41, 124.94 (d, J = 3.1 Hz), 123.27 (d, J = 7.8 Hz), 119.51 (d, J = 2.2 Hz), 117.93, 116.10 (d, J = 20.3 Hz), 66.15, 61.59, 52.32, 51.54, 50.30, 46.82, 46.51; ESI-MS: for C20H25O3FN3 [M + H]+ calculated 374.42932 m/z, found 374.42951 m/z. Anal. Calcd. for C20H25O3ClFN3 (409.89): C, 58.61%; H, 6.15%; N, 10.25%. Found: C, 58.62%; H, 6.31%; N, 10.06%.
1-(2-{4-[(Ethoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8f). Yield 0.79 g (91%); Mr 423.91; Mp 208–210 °C; Rf 0.45; IR (ATR, cm−1): 3354 (υ NH), 2980 (υas CH2), 2851 (υs CH2), 1719 (υ C = O), 1600 (υ C = C), 1552 (δ NH), 1519 (υ CN), 1232 (υas COC), 1079 (υs CO), 1016 (δip = C–H), 858 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 10.46 (m, 1H, NH+), 9.67 (s, 1H, NHCO), 7.47 (d, 2H, Ar–H, J = 8.5 Hz), 7.32 (d, 2H, Ar–H, J = 8.5 Hz), 7.25–7.00 (m, 4H, Ar–H), 6.21 (s, 1H, OH), 5.14 (dd, 1H, CHOH, OH, J = 4.8 Hz, J = 7.8 Hz), 4.12 (q, 2H, CH2CH3, J = 7.0 Hz), 3.78–3.14 (m, 10H, CH(OH)CH2N + piperazine), 1.24 (t, 3H, CH2CH3, J = 7.0 Hz); 13C-NMR (DMSO-d6) δC (ppm): 154.74 (d, J = 244.4 Hz), 153.36, 138.78, 138.21 (d, J = 8.7 Hz), 135.20, 126.34, 124.89 (d, J = 3.3 Hz), 123.31 (d, J = 8.0 Hz), 119.45 (d, J = 2.2 Hz), 117.91, 116.11 (d, J = 20.5 Hz), 66.33, 61.74, 60.56, 52.34, 50.30, 46.76, 46.64, 14.38; ESI-MS: for C21H27O3FN3 [M + H]+ calculated 388.45590 m/z, found 388.45541 m/z. Anal. Calcd. for C21H27O3ClFN3 (423.91): C, 59.50%; H, 6.42%; N, 9.91%. Found: C, 59.45%; H, 6.39%; N, 10.02%.
1-(2-Hydroxy-2-{4-[(propoxycarbonyl)amino]phenyl}ethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8g). Yield 0.77 g (86%); Mr 437.94; Mp 212–214 °C; Rf 0.50; IR (ATR, cm−1): 3392 (υ NH), 2973 (υas CH2), 2847 (υs CH2), 1720 (υ C = O), 1605 (υ C = C), 1543 (δ NH), 1501 (υ CN), 1231 (υas COC), 1079 (υs CO), 1012 (δip = C–H), 855 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 10.56 (m, 1H, NH+), 9.68 (s, 1H, NHCO), 7.48 (d, 2H, Ar–H, J = 8.5 Hz), 7.33 (d, 2H, Ar–H, J = 8.5 Hz), 7.23–7.00 (m, 4H, Ar–H), 6.20 (s, 1H, OH), 5.15 (dd, 1H, CHOH, J = 4.8 Hz, J = 7.9 Hz), 4.03 (t, 2H, CH2CH2CH3, J = 6.8 Hz), 3.79–3.16 (m, 10H, CH(OH)CH2N + piperazine), 1.63 (m, 2H, CH2CH2CH3, J = 7.1 Hz), 0.93 (t, 3H, CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 154.74 (d, J = 244.7 Hz), 153.45, 138.78, 138.21 (d, J = 8.7 Hz), 135.32, 126.42, 124.89 (d, J = 3.3 Hz), 123.31 (d, J = 7.9 Hz), 119.45 (d, J = 2.2 Hz), 117.91, 116.10 (d, J = 20.2 Hz), 66.29, 65.56, 61.77, 52.23, 50.42, 46.80, 46.64, 21.78, 10.21; ESI-MS: for C22H29O3FN3 [M + H]+ calculated 402.48248 m/z, found 402.48217 m/z. Anal. Calcd. for C22H29O3ClFN3 (437.94): C, 60.34%; H, 6.67%; N, 9.59%. Found: C, 60.21%; H, 6.75%; N, 9.43%.
1-(2-{4-[(Butoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8h). Yield 0.89 g (96%); Mr 451.97; Mp 219–220 °C; Rf 0.56; IR (ATR, cm−1): 3361 (υ NH), 2957 (υas CH2), 2825 (υs CH2), 1722 (υ C = O), 1610 (υ C = C), 1546 (δ NH), 1499 (υ CN), 1232 (υas COC), 1075 (υs CO), 1020 (δip = C–H), 852 (δoop = C–H); 1H-NMR (DMSO-d6) δH (ppm): 10.46 (m, 1H, NH+), 9.67 (s, 1H, NHCO), 7.48 (d, 2H, Ar–H, J = 8.5 Hz), 7.33 (d, 2H, Ar–H, J = 8.5 Hz), 7.24–7.01 (m, 4H, Ar–H), 6.22 (s, 1H, OH), 5.14 (dd, 1H, CHOH, J = 4.8 Hz, J = 7.8 Hz), 4.07 (t, 2H, CH2CH2CH2CH3, J = 6.6 Hz), 3.78–3.16 (m, 10H, CH(OH)CH2N + piperazine), 1.62 (q, 2H, CH2CH2CH2CH3, J = 7.0 Hz), 1.38 (m, 2H, CH2CH2CH2CH3, J = 7.4 Hz), 0.92 (t, 3H, CH2CH2CH2CH3, J = 7.3 Hz); 13C-NMR (DMSO-d6) δC (ppm): 154.74 (d, J = 244.4 Hz), 153.52, 138.81, 138.23 (d, J = 8.6 Hz), 135.35, 126.41, 124.89 (d, J = 3.1 Hz), 123.33 (d, J = 7.3 Hz), 119.50 (d, J = 2.2 Hz), 117.92, 116.10 (d, J = 20.2 Hz), 66.29, 63.78, 61.74, 52.34, 50.32, 46.82, 46.61, 30.52, 18.48, 13.47; ESI-MS: for C23H31O3FN3 [M + H]+ calculated 416.50906 m/z, found 416.50934 m/z. Anal. Calcd. for C23H31O3ClFN3 (451.97): C, 61.12%; H, 6.91%; N, 9.30%. Found: C, 61.05%; H, 6.90%; N, 9.37%.
3.3. Lipophilicity Parameter Determination
Lipohydrophilic properties of the final compounds 8a–h were characterized by the RM (RP-TLC), log k and log kw (RP-HPLC) parameters, respectively.
3.3.1. Reversed-Phase Thin-Layer Chromatography (RP-TLC)
The RM values were calculated according to Equation (5) from the Rf parameters observed in a SM mobile phase: hydrochloric acid (c = 1.0 M)/acetone (4:1, v/v):
| RM = log (1/Rf – 1) | (5) |
For the experiments, aluminium sheets pre-coated with silica gel 60 F254 (0.25 mm thickness; Merck) were impregnated by a variously concentrated silicone oil in heptane, which ranged from 1% to 5%. The plates were separately spotted with 2 µL of methanolic solutions of each compound (c = 1 mg/mL), starting points were 1 cm from a bottom edge of these plates. The chromatographic plates were developed in glass developing chambers saturated for 30 min by the SM mobile phase. Development was carried out upon 15 cm from a starting line by an ascending technique [73,74]. After being developed, the plates were dried at room temperature. Detection of zones was performed under iodine vapours/UV light at λ = 254 nm. Each experiment was run in triplicate at 21 °C, the RM values of analytes were calculated separately for each run.
Optimal differences in RM values within both homological groups 8a–d and 8e–h were observed if 1% silicone oil in heptane was chosen. The Rfs were found in a range from 0.19 (8h) to 0.78 (8a; Table 1). All the calculated average RM values were summarized in Table S1 (Supplementary Materials).
3.3.2. Reversed-Phase High-Performance Liquid Chromatography (RP-HPLC)
Methanol (MeOH)/water mobile phases with a various volume ratio of the organic modifier and water (60:40, 70:30, 80:20 and 85:15, respectively; v/v) were chosen. A concentration of all analyzed compounds 8a–h was c = 0.25 mg/mL. An queous solution of potassium nitrate (c = 0.1 M) was used for the determination of dead time t0 = 1.295 min. The capacity (retention) factor k values (Table 1) were calculated by Equation (6) as follows:
| k = (tr – t0)/t0 | (6) |
where the tr and t0 parameters were the retention times of a solute (tr) and unretained compound (potassium nitrate; t0), respectively. The observed retention (tr) and dead (t0) times were means of three independent determinations [73,74].
The log kw values, i.e., the logarithms of extrapolated capacity (retention) factors for 100% water in the isocratic RP-HPLC, were determined from intercepts of linear plots between the log k and ϕM (a volume fraction of an organic modifier in the isocratic elution RP-HPLC) according to Equation (7):
| log k = log kw – S × ϕM | (7) |
where the S parameter represented the slope of a regression curve, which was related to the solvent strength of a pure organic solvent [46,75].
3.4. Electronic Properties Determination
The log ε values characterizing methanolic solutions of the analyzed compounds 8a–h (c = 3.0 × 10−5 M) were estimated at λ1 = 208–210 nm, λ2(Ch-T) = 238–240 nm and λ3 = 274–276 nm (Table 3), respectively, in a near ultraviolet (quarz) region of the electromagnetic spectrum between 200 and 400 nm [47]. The log ε values for the observed absorption maxima were calculated according to the Lambert-Beer´s law discussed in [47], for example, and expressed by Equation (8):
| A = ε × c × l | (8) |
where the A parameter represented the absorbance of a solution, the descriptor ε was a molar absorption coefficient (in the L/mol/cm units) and l was the path length (in the cm units).
3.5. Biological Assays
3.5.1. In Vitro Antimycobaterial Evaluation
Mycobacterial strains. The in vitro activity of compounds 8a–h was inspected against Mycobacterium tuberculosis CNCTC My 331/88 (identical with H37Rv and ATCC 2794, respectively; dilution of the strain was 10−3 M), M. kansasii CNCTC My 235/80 (identical with ATCC 12478; 10−4 M), the M. kansasii 6 509/96 clinical isolate (10−4 M) and M. avium CNCTC My 330/80 (identical with ATCC 25291; 10−5 M), respectively, in the Laboratory for Mycobacterial Diagnosis and Tuberculosis (Institute of Public Health in Ostrava, Czech Republic). These strains were purchased from the National Reference Laboratory – Czech National Collection of Type Cultures (CNCTC; The National Institute of Public Health, Prague, Czech Republic), excluding M. kansasii 6 509/96, which was clinically isolated because the INH-resistant M. kansasii strains have not been found in Czech Republic or Slovak Republic yet.
M. avium intracellulare ATCC 13950 as well as the M. kansasii CIT11/06, M. avium subsp. paratuberculosis CIT03 and M. avium hominissuis CIT10/08 clinical isolates, respectively, were obtained from the Department of Biological Sciences (Cork Institute of Technology, Bishopstown, Cork, Ireland).
Standard drugs. The isoniazid (INH), ethambutol (EMB), ofloxacin (OFLX), ciprofloxacin (CPX) and pyrazinamide (PZA) reference drugs were purchased from Sigma-Aldrich (Darmstadt, Germany), showing the purity of analytical grade.
Determination of a minimum inhibitory concentration (MIC) against M. tuberculosis CNCTC My 331/88, M. kansasii CNCTC My 235/80, M. kansasii 6 509/96 and M. avium CNCTC My 330/80. Efficiency of the compounds 8a–h and standard drugs against given mycobacteria were determined in the Šula semisynthetic medium (Sevac, Prague, Czech Republic) by a dilution-micromethod [48,76].
In a brief, each mycobacterial strain was simultaneously inoculated into Petri plates containing a Löwenstein-Jensen medium for sterility control and growth of inoculum. All inspected compounds were added to the medium as solutions in dimethyl sulfoxide (DMSO; Sigma-Aldrich, Irvine, UK). In the assays, following concentrations of the solutions were used: 1000, 500, 250, 125, 62.5, 32, 16, 8, 4, 2, 1, 0.5, 0.25 and 0.125 μM, respectively. The inoculated plates kept in microtone bags were incubated at 37 °C. Particular reading was carried out on a stand with a bottom magnifying mirror, macroscopically, with the use of a magnifying glass. The growth in the plates was evaluated after 7, 14 and 21 days (M. kansasii CNCTC My 235/80, M. kansasii 6 509/96) and after 14 and 21 days (M. tuberculosis CNCTC My 331/88, M. avium CNCTC My 330/80), respectively [48,76].
The value of a minimum inhibitory concentration (MIC) was the lowest concentration (on the above concentration scale) of a tested compound, which inhibited growth of the mycobacteria [48,76]. The evaluation was repeated three times and the MIC values, reported in Table 4 in the μM units, were the same.
Determination of a minimum inhibitory concentration (MIC) against M. avium intracellulare ATCC 13950, M. kansasii CIT11/06, M. avium subsp. paratuberculosis CIT03 and M. avium hominissuis CIT10/08. Those mycobacterial strains were grown in a Middlebrook broth (MB), supplemented with Oleic-Albumin-Dextrose-Catalase Supplement (OADC; Becton Dickinson, Oxford, UK). Identification of isolates was performed using biochemical and molecular protocols.
At log phase growth, a culture sample (10 mL) was centrifuged at 15,000 rpm/20 min using a bench top centrifuge Model CR 4-12 (Jouan Inc., London, UK). Following removal of a supernatant, the pellet was washed in a fresh Middlebrook 7H9GC broth (Difco, Detroit, MI, USA), and re-suspended in a fresh supplemented MB (10 mL). Turbidity was adjusted to match the McFarland standard No. 1 containing 3 × 108 Colony Forming Units (CFU) with the MB broth. Further 1:20 dilution of a culture was performed in the broth.
Susceptibility of given mycobacterial strains was investigated in a 96-well plate format. In the experiments, sterile deionised water (300 μL) was added to all outer-perimeter wells of plates to minimize evaporation of a medium in the test wells during the incubation process. Each tested compound (100 μL) was incubated with each of mycobacterial species (100 μL). The dilutions of tested compounds were prepared in triplicate, their final concentrations ranged from 500 μg/mL to 15 μg/mL. The screened derivatives were prepared in DMSO (Sigma-Aldrich, London, UK) and dilutions were made in the supplemented MB broth. The plates were sealed with a parafilm and incubated at 37 °C for 7 days. Following the incubation, 10% addition of a water-soluble dye, the alamarBlue reagent (AbD Serotec, Kidlington, UK) was mixed into each well. Absorbance readings at λ = 570 nm and 600 nm were taken, initially for a background subtraction and after 24 h re-incubation. The subtraction is necessary for strongly coloured compounds, where the colour may interfere with interpretation of any colour change. For non-interfering compounds, a blue colour in a well was interpreted as absence of growth and pink colour was scored as growth [17,49,50].
The MIC value was defined as the lowest concentration of a compound, at which no visible bacterial growth was observed. In other words, the MIC was the lowest concentration that prevented a visual colour change from blue to pink. The MIC for mentioned mycobacteria was defined as 90% or greater growth reduction (IC90) compared to a control. The MIC (IC90) parameter has been routinely and widely used in bacterial assays as a standard detection limit according to the Clinical and Laboratory Standards Institute [49,50].
Clinically used antimycobacterial drugs INH, CPX and PZA, respectively, were applied as standards. The observed MIC values were listed in Table 5 in the μM units.
3.5.2. In Vitro Antiproliferative (Cytotoxicity) Screening
Human monocytic leukemia THP-1 cells were obtained from the European Collection of Cell Cultures (ECACC; Salisbury, UK; Methods of characterization: DNA Fingerprinting (Multilocus probes) and isoenzyme analysis). The cells were routinely cultured in the RPMI 1640 medium (Lonza, Verviers, Belgium) supplemented with 10% fetal bovine serum (Sigma-Aldrich, Darmstadt, Germany), 2% L-glutamine, 1% penicillin and streptomycin (Lonza, Verviers, Belgium) at 37 °C with 5% CO2. The cells were passaged at approximately 1-week intervals and were routinely tested for absence of mycoplasma by a Hoechst 33258 staining method.
The tested compounds 8a–h were dissolved in DMSO (Sigma-Aldrich, Darmstadt, Germany) and added in five increasing concentrations to the cell suspension in the culture medium. The maximum concentration of DMSO in the assays never exceeded 0.1%. Subsequently, the cells were incubated for 24 h at 37 °C with 5% CO2 at various compounds´ concentrations varying from 0.37 μM to 30 μM in the RPMI 1640 medium.
Cell toxicity was determined using the Cytotoxicity Detection KitPLUS Lactate Dehydrogenase (LDH) assay kit (Roche Diagnostics, Mannheim, Germany) and used according to manufacturer’s instructions. For the LDH assays, the cells were seeded into 96-well plates (5 × 104 cells/well in 100 μL culture medium) in triplicate in the serum-free RPMI 1640 medium, and measurements at λ = 492 nm (Synergy 2 Multi-Mode Microplate Reader; BioTek, Winooski, VT, USA) were taken 24 h after treatment with the tested compounds [77,78].
Median lethal dose values, LD50, were deduced through the construction of a dose-response curve. All values were evaluated using the GraphPad Prism 5.00 software (GraphPad Software, San Diego, CA, USA).
3.6. Calculations and Statistical Analyses
Regression equations and statistical characteristics were calculated and visualized by the Origin Pro 9.0.0 software (OriginLab Corporation, Northampton, MA, USA).
In a current research, those statistical parameters were calculated: Residual sum of squares (RSS), correlation coefficient (R), adjusted coefficient of determination (Adj. R2), root mean squared error (standard deviation; RMSE) and norm of residuals (NoR), respectively. The study provided Analysis of Variance (ANOVA) outputs as well, i.e., Fisher´s F-test (Fisher´s significance ratio; F) and probability of obtaining the F Ratio (Prob > F).
The RSS descriptor was used to measure amount of variance in a data set that was not explained by the regression model. The RSS parameter was a measure of amount of error remaining between the regression function and data set. Relatively smaller RSS explained greater amount of data [79,80].
The R parameter was based on a method of covariance. The coefficient was used to measure the strength of a relationship between two (continuous) variables. The Adj. R2 value penalized the R2 data for addition of regressors, which did not contribute to explanatory power of a model. Indeed, the Adj. R2 value was never larger than the R2 one; it would be decreased by the adding of other regressors, and might be even negative for poorly fitting models [81,82,83].
The RMSE was an unambiguous indicator of error for numerical predictions. The parameter provided standard deviation of a model prediction error. Relatively smaller value indicated better model performance [84]. The NoR parameter was the square root of RMSE and was used as measure for goodness of fit when comparing different fits [85].
The F value, as a parameter obtained by the ANOVA, could determine whether the means of three or more groups were different. The ANOVA approach tested an effect of a categorical predictor variable on a continuous dependent variable and used the F-test to statistically test equality of means [79].
The Prob > F data provided a p-value for the test and measured probability of obtaining the F Ratio as large as what was observed, given that all parameters except the intercept were zero [81,82].
Indication of significance level of the F Ratio by stars was as follows: one star, i.e., statistically significant relationship defined by the Prob > F value in the range from 0.0100 to <0.0500; two stars, i.e., statistically very significant model defined by the Prob > F parameter in the interval from 0.0010 to <0.0100; and three stars, i.e., statistically extremely significant relationship with the Prob > F value in the interval from 0 to <0.0010. Finally, a statistically insignificant model was connected with the Prob > F parameter ≥ 0.0500 [82].
5. Conclusions
In summary, original N-arylpiperazines 8a–h were prepared by multistep procedures and characterized by spectral values (1H-NMR, 13C-NMR, IR and ESI-MS) and elemental analyses (% C, H, N). The compounds contained a flexible 3-/4-alkoxycarbonylamino group (alkoxy = methoxy to butoxy), 2-hydroxyethane-1,2-diyl connecting chain and 4-(2-fluorophenyl)piperazin-1-yl moiety (Table 1). These fragments have been separately found in a chemical structure of various compounds with a notable in vitro efficiency against some tuberculous strains of mycobacteria.
Lipohydrophilic properties of the molecules 8a–h were preliminary evaluated by the RP-TLC using silica gel plates impregnated with 1% silicone oil in heptane. Calculated RM values of 3-alkoxycarbonylamino substituent-containing compounds (8a–d) ranged from −0.55 (8a) to 0.01 (8d), the 4-substituted ones (8e–h) showed higher RM parameters varying from −0.02 (8e) to 0.63 (8h).
Linearly extrapolated logarithms of retention factors corresponding to 100% water as a mobile phase, log kw values (RP-HPLC), characterized the lipohydrophilic properties of the molecules 8a–h (Table 2) more reliably and precisely than any arbitrary selected isocratic log k parameters. These log kw values were in accordance with elution order and hydrophobicity of 8a–h and ranged from 2.113 (8e) to 2.930 (8h). The derivatives 8a–c showed higher log kw values (2.430–2.796) than the molecules 8e–g (2.113–2.600). The compounds containing the longest side chain, i.e., 1-(2-{3-[(butoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)piperazin-1-ium chloride (8d) and 1-(2-{4-[(butoxycarbonyl)amino]phenyl}-2-hydroxyethyl)-4-(2-fluorophenyl)-piperazin-1-ium chloride (8h) were found to be the most lipophilic, as proven by their log kw of 2.796 (8d) and 2.930 (8h), respectively (Table 2).
Regarding the extrapolation calculations, the slopes S of regression lines varied from 2.7386 (8e) to 3.3441 (8h; Table 2). The S parameter was related to a specific hydrophobic surface of a particular compound and could be used as the alternative measure of its lipophilicity.
Electronic properties of the inspected compounds 8a–h were characterized by logarithms of molar absorption coefficients (log ε) of their methanolic solutions (c = 3.0 × 10−5 M) investigated in the UV/Vis region of a spectrum.
The solutions showed three absorption maxima in a near ultraviolet (quarz) region of the electromagnetic spectrum, e.g., λ1 = 208–210 nm, λ2(Ch-T) = 238–240 nm and λ3 = 274–276 nm, respectively (Table 3). The log ε2(Ch-T) parameters of the compounds 8a–d observed at a charge-transfer absorption maximum λ2(Ch-T) were found in a narrow interval from 4.30 (8a) to 4.37 (8c). The methanolic solutions of 8e–h were characterized by higher log ε2(Ch-T) values than the ones of 8a–d and these parameters ranged from 4.42 (8h) to 4.67 (8e; Table 3). In addition, elongation of a 4-side chain led to lower log ε values related to all observed absorption maxima (Table 3).
The racemic compounds 8a–h were in vitro screened against Mycobacterium tuberculosis CNCTC My 331/88 (identical with H37Rv and ATCC 2794), M. kansasii CNCTC My 235/80 (identical with ATCC 12478), a M. kansasii 6 509/96 clinical isolate, M. avium CNCTC My 330/80 (identical with ATCC 25291) and M. avium intracellulare ATCC 13950 as well as against M. kansasii CIT11/06, M. avium subsp. paratuberculosis CIT03 and M. avium hominissuis CIT10/08 clinical isolates, respectively (Table 4 and Table 5). The biological evaluation revealed the most promising potential of the 8a–h set against M. tuberculosis, M. kansasii My 235/80 and M. kansasii 6 509/96, respectively.
A position and length of a side chain notably affected the activity of presently investigated N-arylpiperazine derivatives against M. tuberculosis CNCTC My 331/88. The 4-positional isomers were more effective, with the MIC values ranging from 8 μM (8h) to 125 μM (8e), than the 3-positional ones, which possessed the MICs from 32 μM (8d) to 250 μM (8a). Among all in vitro screened molecules, the INH standard was found to be the most active with the MIC = 0.5 μM (14-d/21-d; Table 4).
The compounds 8a–d were more efficient against M. kansasii My 235/80 and M. kansasii 6 509/96 than the derivatives 8e–h. The most active molecule against given mycobacteria was 8d with the MIC = 16 μM and 62.5 μM, respectively, depending on a particular strain and also on the number of days of incubation. Increase in length of the side chain resulted in lower MIC values of 8a–d against these mycobacterial strains. The observed MIC parameters were, however, higher compared to the ones related to EMB with the MIC = 1 μM and 2 μM (14-d/21-d), or OFLX, which showed the MIC = 0.5 μM and 1 μM, respectively (14-d/21-d; Table 4).
The activity of 8a–h against a non-tuberculous INH-resistant M. avium CNCTC My 330/80 was apparently dependent on the position of an alkoxycarbonylamino chain. Its presence in the 3-position (8a–d) led to the MIC values varying from 62.5 μM (8d) to 500 μM (8a; 14-d/21-d). However, a potential of the 4-substituent-containing derivatives (8e–h) to fight given mycobacterium was insufficient (MIC > 250 μM; Table 4).
The efficiency of the most active substance 8d (MIC = 62.5 μM; 14-d/21-d) against M. avium CNCTC My 330/80 was comparable to the effectiveness of OFLX (MIC = 32 μM and 62.5 μM, respectively; 14-d/21-d); reference EMB drug was moderately more active (MIC = 16 μM; 14-d/21-d). Elongation of a 3-side chain led to more promising compounds (Table 4).
Regarding current SAR studies, lipophilic properties represented by extrapolated log kw values seemed to be considerably more important for the in vitro activity of the 8a–d set against M. tuberculosis and M. kansasii 6 509/96 compared to the lipophilic features of the compounds 8e–h.
The log ε2(Ch-T) values (Table 3) observed at the charge-transfer absorption maximum λ2(Ch-T) were also taken into a special consideration, because they could be the most sensitive to the differences in a position and electronic properties of the alkoxycarbonylamino substituent.
A relationship between the log ε2(Ch-T) parameters and log (1/MIC [M]) values connected with the in vitro screening of 8a–h against M. tuberculosis My 331/88 (14-d) provided a bilinear course. Maximal efficiency of these compounds could be observed if their log ε2(Ch-T) values were approximately 4.43 (Figure 4).
If attention was paid to the M. kansasii My 235/80 and M. kansasii 6 509/96 strains, no significant relationships between the in vitro activity of the compounds 8a–h and their electronic features were observed.
Favorable cytotoxicity profiles of the molecules 8a–h were proved by the LD50 values > 30 μM, which were estimated on a human monocytic leukemia THP-1 cell line. Moreover, the least lipophilic methoxy group-containing derivatives 8e (log kw = 2.113) and 8a (log kw = 2.430) increased proliferation of the THP-1 cells in 24 h when compared to a control.
Overall, the results of current in vitro biological evaluation and initial SAR investigations of the molecules 8a–h considered them very promising candidates for further structural optimization, which could lead to even more effective antimycobacterials. Regarding these findings, the authors of the study were inspired and encouraged to synthesize enantiomerically pure compounds and explore their in vitro efficiency, especially against M. tuberculosis CNCTC My 331/88, M. kansasii My 235/80 or M. kansasii 6 509/96, in further phases of the research programme
Acknowledgments
The authors very gratefully acknowledge a financial support received especially from the Faculty of Pharmacy, Comenius University in Bratislava (Slovak Republic). The research was also supported by the grant projects VEGA 1/0873/15, KEGA 022UK-4/2015 and Science Foundation Ireland Project Ref: 12/R1/2335. Part of the experiments was carried out in the Toxicological and Antidoping Center at the Faculty of Pharmacy, Comenius University in Bratislava (Slovak Republic) and this support was also very acknowledged. The research was also partially supported by Sanofi-Aventis Pharma Slovakia, s.r.o. (Slovak Republic).
Abbreviations
The following abbreviations are used in this manuscript:
| 7-d | 7-Day incubation |
| 14-d/21-d | 14-/21-Day incubation |
| ϕM | Volume fraction of a mobile phase modifier (RP-HPLC) |
| CPX | Ciprofloxacin |
| Adj. R2 | Adjusted coefficient of determination (statistical analysis) |
| DMSO | Dimethyl sulfoxide |
| EMB | Ethambutol |
| F | Fisher´s F-test (Fisher´s significance ratio; statistical analysis) |
| INH | Isoniazid |
| k | Capacity (retention) factor (RP-HPLC) |
| log kw | Lipophilicity index; values extrapolated from intercepts of a linear relationship between the logarithm of retention factor k (log k) and volume fraction of a mobile phase modifier (ϕM; RP-HPLC) |
| MDR | Multi-drug resistant |
| MeOH | Methanol |
| MA | Mycobacterium avium |
| MIC | Minimum inhibitory concentration (in the μΜ units) |
| MK | Mycobacterium kansasii |
| Mp/mp | Melting point |
| MT | Mycobacterium tuberculosis |
| NoR | Norm of residuals (statistical analysis) |
| OFLX | Ofloxacin |
| Prob > F | Probability of obtaining the F Ratio (statistical analysis) |
| PZA | Pyrazinamide |
| RM | Lipophilicity index (RP-TLC) |
| RMSE | Root mean squared error (statistical analysis) |
| RSS | Residual sum of squares (statistical analysis) |
| S | Slope (RP-HPLC) |
| SAR | Structure–activity relationship(s) |
| tr | Retention time of a compound (RP-HPLC) |
Supplementary Materials
The supplementary materials are available online at www.mdpi.com/link.
Author Contributions
T.G. synthesized and spectrally characterized the compounds; I.M. spectrally characterized the compounds, investigated their lipophilic and electronic properties, analyzed the data related to the in vitro antimycobacterial investigation, created the concept and designed the study, investigated, statistically characterized and interpreted the SAR results, wrote and revised the paper; J.Cs. designed the chemical structure of compounds under the study, contributed reagents/materials tools; J.J. and I.S. conceived the in vitro antimycobacterial screening of synthesized compounds, analyzed data related to the in vitro antimycobacterial evaluation of the molecules; J.S., J.O.M. and A.C. performed the in vitro antimycobacterial evaluation of the compounds and interpreted the results, contributed reagents/materials tools; A.C. revised the paper; P.M. investigated spectral and lipophilic properties of the synthesized compounds, contributed reagents/materials tools; S.K. and P.K. performed the in vitro antiproliferative (cytotoxic) activity of the synthesized molecules, contributed reagents/materials tools. The authors have approved the final version of manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 8a–h are available from the authors Tomáš Goněc and Ivan Malík.
References
- 1.Evans B.E., Rittle K.E., Bock M.G., DiPardo R.M., Freidinger R.M., Whitter W.L., Lundell G.F., Veber D.F., Anderson P.S., Chang R.S.L., et al. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 2.Shaquiquzzaman M., Verma G., Marella A., Akhter M., Akhtar W., Khan M.F., Tasneem S., Alam M.M. Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents. Eur. J. Med. Chem. 2015;102:487–529. doi: 10.1016/j.ejmech.2015.07.026. [DOI] [PubMed] [Google Scholar]
- 3.Bobesh K.A., Renuka J., Srilakshmi R.R., Yellanki S., Kulkarni P., Yogeeswari P., Sriram D. Replacement of cardiotoxic aminopiperidine linker with piperazine moiety reduces cardiotoxicity? Mycobacterium tuberculosis novel bacterial topoisomerase inhibitors. Bioorg. Med. Chem. 2016;24:42–52. doi: 10.1016/j.bmc.2015.11.039. [DOI] [PubMed] [Google Scholar]
- 4.Xu Z., Zhang S., Feng L.-S., Li X.-N., Huang G.-Ch., Chai Y., Lv Z.-S., Guo H.-Y., Liu M.-L. Synthesis and in vitro antimycobacterial and antibacterial activity of 8-OMe ciprofloxacin-hydrozone/azole hybrids. Molecules. 2017;22:1171. doi: 10.3390/molecules22071171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kayukova L.A., Orazbaeva M.A., Bismilda V.L., Chingisova L.T. Synthesis and antituberculosis activity of O-aroyl-β-(4-phenylpiperazin-1-yl)propioamidooximes. Pharm. Chem. J. 2010;44:17–20. doi: 10.1007/s11094-010-0467-9. [DOI] [Google Scholar]
- 6.Keng Yoon Y., Ashraf Ali M., Choon T.S., Ismail R., Chee Wei A., Suresh Kumar R., Osman H., Beevi F. Antituberculosis: Synthesis and antimycobacterial activity of novel benzimidazole derivatives. Biomed. Res. Int. 2013 doi: 10.1155/2013/926309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sriram D., Yogeeswari P., Senthilkumar P., Sangaraju D., Nelli R., Banerjee D., Bhat P., Manjashetty T.H. Synthesis and antimycobacterial evaluation of novel phthalazin-4-ylacetamides against log- and starved phase cultures. Chem. Biol. Drug. Des. 2010;75:381–391. doi: 10.1111/j.1747-0285.2010.00947.x. [DOI] [PubMed] [Google Scholar]
- 8.Malinka W., Świątek P., Śliwińska M., Szponar B., Gamian A., Karczmarzyk Z., Fruziński A. Synthesis of novel isothiazolopyridines and their in vitro evaluation against Mycobacterium and Propionibacterium acnes. Bioorg. Med. Chem. 2013;21:5282–5291. doi: 10.1016/j.bmc.2013.06.027. [DOI] [PubMed] [Google Scholar]
- 9.Bogatcheva E., Hanrahan C., Nikonenko B., Samala R., Chen P., Gearhart J., Barbosa F., Einck L., Nacy C.A., Protopopova M. Identification of new diamine scaffolds with activity against Mycobacterium tuberculosis. J. Med. Chem. 2006;49:3045–3048. doi: 10.1021/jm050948+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shepherd R.G., Baughn C., Cantrall M.L., Goodstein B., Thomas J.P., Wilkinson R.G. Structure–activity studies leading to ethambutol, a new type of antituberculous compound. Ann. N. Y. Acad. Sci. 1966;135:686–710. doi: 10.1111/j.1749-6632.1966.tb45516.x. [DOI] [PubMed] [Google Scholar]
- 11.Lee R.E., Protopopova M., Crooks E., Slayden R.A., Terrot M., Barry C.E., III Combinatorial lead optimization of [1,2]-diamines based on ethambutol as potential antituberculosis preclinical candidates. J. Comb. Chem. 2003;5:172–187. doi: 10.1021/cc020071p. [DOI] [PubMed] [Google Scholar]
- 12.Stavrakov G., Valcheva V., Philipova I., Doytchinova I. Novel camphane-based anti-tuberculosis agents with nanomolar activity. Eur. J. Med. Chem. 2013;70:372–379. doi: 10.1016/j.ejmech.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 13.Petkova Z., Valcheva V., Momekov G., Petrov P., Dimitrov V., Doytchinova I., Stavrakov G., Stoyanova M. Antimycobacterial activity of chiral aminoalcohols with camphane scaffold. Eur. J. Med. Chem. 2014;81:150–157. doi: 10.1016/j.ejmech.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh A.K., Brindisi M. Organic carbamates in drug design and medicinal chemistry. J. Med. Chem. 2015;58:2895–2940. doi: 10.1021/jm501371s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moraczewski A.L., Banaszynski L.A., From A.M., White C.E., Smith B.D. Using hydrogen bonding to control carbamate C−N rotamer equilibria. J. Org. Chem. 1998;63:7258–7262. doi: 10.1021/jo980644d. [DOI] [PubMed] [Google Scholar]
- 16.Kečkéšová S., Sedlárová E., Čižmárik J., Garaj V., Csöllei J., Mokrý P., Andriamainty F., Malík I., Kaustová J. Antimycobacterial activity of novel derivatives of arylcarbonyloxyaminopropanols. Čes. Slov. Farm. 2009;58:203–207. [Google Scholar]
- 17.Tengler J., Kapustíková I., Peško M., Govender R., Keltošová S., Mokrý P., Kollár P., O’Mahony J., Coffey A., Kráľová K., et al. Synthesis and biological evaluation of 2-hydroxy-3-[(2-aryloxyethyl)amino]propyl-4-[(alkoxycarbonyl)amino]benzoates. Sci. World J. 2013;2013 doi: 10.1155/2013/274570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maruniak M., Sedlárová E., Csöllei J., Kapustíková I., Mokrý P., Malík I., Havranová Sichrovská Ľ., Stanzel L. Study of physicochemical properties and antimycobacterial activity of phenylcarbamic acid derivatives. In: Sedlárová E., Malík I., Garaj V., Maruniak M., editors. Advances in Pharmaceutical Chemistry. 1st ed. KO and KA Company; Bratislava, Slovak Republic: 2016. pp. 68–76. [Google Scholar]
- 19.Waisser K., Dražková K., Čižmárik J., Kaustová J. Antimycobacterial activity of basic ethylesters of alkoxy-substituted phenylcarbamic acids. Folia Microbiol. 2003;48:45–50. doi: 10.1007/BF02931274. [DOI] [PubMed] [Google Scholar]
- 20.Waisser K., Dražková K., Čižmárik J., Kaustová J. Antimycobacterial activity of piperidinylpropyl esters of alkoxy-substituted phenylcarbamic acids. Folia Microbiol. 2003;48:585–587. doi: 10.1007/BF02993463. [DOI] [PubMed] [Google Scholar]
- 21.Waisser K., Dražková K., Čižmárik J., Kaustová J. A new group of potential antituberculotics: Hydrochlorides of piperidinylalkyl esters of alkoxy-substituted phenylcarbamic acids. Folia Microbiol. 2004;49:265–268. doi: 10.1007/BF02931041. [DOI] [PubMed] [Google Scholar]
- 22.Hansch C., Clayton J.M. Lipophilic character and biological activity of drugs II. The parabolic case. J. Pharm. Sci. 1973;62:1–21. doi: 10.1002/jps.2600620102. [DOI] [PubMed] [Google Scholar]
- 23.Balgavý P., Devínsky F. Cut-off effects in biological activities of surfactants. Adv. Colloid Interface Sci. 1996;12:23–63. doi: 10.1016/0001-8686(96)00295-3. [DOI] [PubMed] [Google Scholar]
- 24.Waisser K., Dražková K., Čižmárik J., Kaustová J. Influence of lipophilicity on the antimycobacterial activity of the hydrochlorides of piperidinylethyl esters of ortho-substituted phenylcarbamic acids. Sci. Pharm. 2004;72:43–49. doi: 10.3797/scipharm.aut-04-05. [DOI] [Google Scholar]
- 25.Upadhayaya R.S., Kulkarni G.M., Vasireddy N.R., Vandavasi J.K., Dixit S.S., Sharma V., Chattopadhyaya J. Design, synthesis and biological evaluation of novel triazole, urea and thiourea derivatives of quinoline against Mycobacterium tuberculosis. Bioorg. Med. Chem. 2009;13:4681–4692. doi: 10.1016/j.bmc.2009.04.069. [DOI] [PubMed] [Google Scholar]
- 26.Upadhayaya R.S., Vandavasi J.K., Kardile R.A., Lahore S.V., Dixit S.S., Deokar H.S., Shinde P.D., Sarmah M.P., Chattopadhyaya J. Novel quinoline and naphthalene derivatives as potent antimycobacterial agents. Eur. J. Med. Chem. 2010;45:1854–1867. doi: 10.1016/j.ejmech.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 27.Parai M.K., Panda G., Chaturvedi V., Manju Y.K., Sinha S. Thiophene containing triarylmethanes as antitubercular agents. Bioorg. Med. Chem. Lett. 2008;18:289–292. doi: 10.1016/j.bmcl.2007.10.083. [DOI] [PubMed] [Google Scholar]
- 28.Kettmann V., Csöllei J., Račanská E., Švec P. Synthesis and structure–activity relationships of new β-adrenoreceptor antagonists. Evidence for the electrostatic requirements for β-adrenoreceptor antagonists. Eur. J. Med. Chem. 1991;26:843–851. doi: 10.1016/0223-5234(91)90127-9. [DOI] [Google Scholar]
- 29.Kiss A., Potor A., Hell Z. Heterogeneous catalytic solvent-free synthesis of quinoline derivatives via the Friedländer Reaction. Catal. Lett. 2008;125:250–253. doi: 10.1007/s10562-008-9573-7. [DOI] [Google Scholar]
- 30.Broutin P.-E., Hilty P., Thomas A.W. An efficient synthesis of ortho-N-Boc-arylmethyl ketone derivatives. Tetrahedron Lett. 2003;44:6429–6432. doi: 10.1016/S0040-4039(03)01597-1. [DOI] [Google Scholar]
- 31.Kolosov M.A., Orlov V.D. 5-Thiazolyl derivatives of 4-aryl-3,4-dihydropyrimidin-2(1H)-ones. Chem. Heterocycl. Compd. 2008;44:1418–1420. doi: 10.1007/s10593-009-0204-z. [DOI] [Google Scholar]
- 32.Hu B., Ellingboe J., Han S., Largis E., Lim K., Malamas M., Mulvey R., Niu C., Oliphant A., Pelletier J., et al. Novel (4-piperidin-1-yl)-phenyl sulfonamides as potent and selective human β3 agonists. Bioorg. Med. Chem. 2001;9:2045–2059. doi: 10.1016/S0968-0896(01)00114-6. [DOI] [PubMed] [Google Scholar]
- 33.Pan Y., Li P., Xie S., Tao Y., Chen D., Dai M., Hao H., Huang L., Wang Y., Wang L., et al. Synthesis, 3D-QSAR analysis and biological evaluation of quinoxaline 1,4-di-N-oxide derivatives as antituberculosis agents. Bioorg. Med. Chem. Lett. 2016;26:4146–4153. doi: 10.1016/j.bmcl.2016.01.066. [DOI] [PubMed] [Google Scholar]
- 34.Pancholia S., Dhameliya T.M., Shah P., Jadhavar P.S., Sridevi J.P., Yogeshwari P., Sriram D., Chakraborti A.K. Benzo[d]thiazol-2-yl(piperazin-1-yl)methanones as new anti-mycobacterial chemotypes: Design, synthesis, biological evaluation and 3D-QSAR studies. Eur. J. Med. Chem. 2016;116:187–199. doi: 10.1016/j.ejmech.2016.03.060. [DOI] [PubMed] [Google Scholar]
- 35.Rajkhowa S., Deka R.C. DFT Based QSAR/QSPR models in the development of novel anti-tuberculosis drugs targeting Mycobacterium tuberculosis. Curr. Pharm. Des. 2014;20:4455–4473. doi: 10.2174/1381612819666131118165824. [DOI] [PubMed] [Google Scholar]
- 36.Joshi S.D., More U.A., Aminabhavi T.M., Badiger A.M. Two- and three-dimensional QSAR studies on a set of antimycobacterial pyrroles: CoMFA, topomer CoMFA, and HQSAR. Med. Chem. Res. 2014;23:107–126. doi: 10.1007/s00044-013-0607-3. [DOI] [Google Scholar]
- 37.Pliška V., Testa B., van de Waterbeemd H. Lipophilicity in drug action and toxicology. In: Mannhold R., Kubinyi H., Timmerman H., editors. Methods and Principles of Medicinal Chemistry. Volume 4. Wiley-VCh Publishers; Weinheim, Germany: 1996. pp. 1–6. [Google Scholar]
- 38.Ottaviani M.F., Leonardis I., Cappiello A., Cangiotti M., Mazzeo R., Trufelli H., Palma P. Structural modifications and adsorption capability of C18-silica/binary solvent interphases studied by EPR and RP-HPLC. J. Colloid Interface Sci. 2010;352:512–519. doi: 10.1016/j.jcis.2010.08.080. [DOI] [PubMed] [Google Scholar]
- 39.Snyder L.R., Dolan J.W. Initial experiments in high-performance liquid chromatographic method development I. Use of a starting gradient run. J. Chromatogr. A. 1996;721:3–14. doi: 10.1016/0021-9673(95)00770-9. [DOI] [Google Scholar]
- 40.Du Ch.M., Valko K., Bevan Ch., Reynolds D., Abraham M.H. Rapid method for estimating octanol–water partition coefficient (log Poct) from isocratic RP-HPLC and a hydrogen bond acidity term (A) J. Liqud Chromatogr. Relat. Technol. 2001;24:635–649. doi: 10.1081/JLC-100103400. [DOI] [Google Scholar]
- 41.Terada H. Determination of log Poct by high-performance liquid chromatography, and its application in the study of Quantitative Structure–Activity Relationships. Quant. Struct. Act. Relat. 1986;5:81–88. doi: 10.1002/qsar.19860050302. [DOI] [Google Scholar]
- 42.Snyder L.R., Dolan J.W., Grant J.R. Gradient elution in high-performance liquid chromatography: I. Theoretical basis for reversed-phase systems. J. Chromatogr. A. 1979;165:3–30. doi: 10.1016/S0021-9673(00)85726-X. [DOI] [Google Scholar]
- 43.Valkó K., Snyder L.R., Glajch J.L. Retention in reversed-phase liquid chromatography as a function of mobile-phase composition. J. Chromatogr. A. 1993;656:501–520. doi: 10.1016/0021-9673(93)80816-Q. [DOI] [Google Scholar]
- 44.Soczewiński E. Mechanistic molecular model of liquid–solid chromatography: Retention–eluent composition relationships. J. Chromatogr. A. 2002;965:109–116. doi: 10.1016/S0021-9673(01)01278-X. [DOI] [PubMed] [Google Scholar]
- 45.Vrakas D., Panderi I., Hadjipavlou-Litina D., Tsantili-Kakoulidou A. Investigation of the relationships between log P and various chromatographic indices for a series of substituted coumarins. Evaluation of their similarity/dissimilarity using multivariate statistics. QSAR Comb. Sci. 2005;24:254–260. doi: 10.1002/qsar.200430898. [DOI] [Google Scholar]
- 46.Sztanke K., Markowski W., Świeboda R., Polak B. Lipophilicity of novel antitumour and analgesic active 8-aryl-2,6,7,8-tetrahydroimidazo[2,1-c][1,2,4]triazine-3,4-dione derivatives determined by reversed-phase HPLC and computational methods. Eur. J. Med. Chem. 2010;45:2644–2649. doi: 10.1016/j.ejmech.2010.01.068. [DOI] [PubMed] [Google Scholar]
- 47.Yadav L.D.S. Ultraviolet and visible spectroscopy. In: Yadav L.D.S., editor. Organic Spectroscopy. Springer; Amsterdam, The Netherlands: 2005. pp. 7–51. [Google Scholar]
- 48.Férriz J.M., Vávrová K., Kunc F., Imramovský A., Stolaříková J., Vavříková E., Vinšová J. Salicylanilide carbamates: Antitubercular agents active against multidrug-resistant Mycobacterium tuberculosis strains. Bioorg. Med. Chem. 2010;18:1054–1061. doi: 10.1016/j.bmc.2009.12.055. [DOI] [PubMed] [Google Scholar]
- 49.Clinical and Laboratory Standards Institute (CLSI) Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard. 8th ed. CLSI; Wayne, NJ, USA: 2012. pp. 10–56. CLSI Document M11-A8. [PubMed] [Google Scholar]
- 50.Clinical and Laboratory Standards Institute (CLSI) Performance Standards for Antimicrobial Susceptibility Testing. 24th ed. CLSI; Wayne, NJ, USA: 2014. pp. 106–211. Informational Supplement M100-S24. [Google Scholar]
- 51.Waisser K., Doležal R., Čižmárik J., Malík I., Kaustová J. The potential antituberculotics of the series of 2-hydroxy-3-(4-phenylpiperazin-1-yl)-propylphenylcarbamates. Folia Pharm. Univ. Carol. 2007;35–36:45–48. [Google Scholar]
- 52.Doležal M., Zitko J., Kešetovičová D., Kuneš J., Svobodová M. Substituted N-phenylpyrazine-2-carboxamides: Synthesis and antimycobacterial evaluation. Molecules. 2009;14:4180–4189. doi: 10.3390/molecules14104180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Čižmárik J., Waisser K., Doležal R. QSAR Study of antimicrobial activity of esters of substituted phenylcarbamic acid. Acta Fac. Pharm. Univ. Comen. 2008;55:90–95. [Google Scholar]
- 54.Timmins G.S., Deretic V. Mechanisms of action of isoniazid. Mol. Microbiol. 2006;62:1220–1227. doi: 10.1111/j.1365-2958.2006.05467.x. [DOI] [PubMed] [Google Scholar]
- 55.Forbes M., Kuck N.A., Peets E.A. Mode of action of ethambutol. J. Bacteriol. 1962;84:1099–1103. doi: 10.1128/jb.84.5.1099-1103.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jena L., Waghmare P., Kashikar S., Kumar S., Harinath B.C. Computational approach to understanding the mechanism of action of isoniazid, an anti-TB drug. Int. J. Mycobacteriol. 2014;3:276–282. doi: 10.1016/j.ijmyco.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 57.Kuck N.A., Peets E.A., Forbes M. Mode of action of ethambutol on Mycobacterium tuberculosis, strain H37Rv. Am. Rev. Respir. Dis. 1963;87:905–906. doi: 10.1164/arrd.1963.87.6.905. [DOI] [PubMed] [Google Scholar]
- 58.Mikusová K., Slayden R.A., Besra G.S., Brennan P.J. Biogenesis of the mycobacterial cell wall and the site of action of ethambutol. Antimicrob. Agents Chemother. 1995;39:2484–2489. doi: 10.1128/AAC.39.11.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lata M., Sharma D., Kumar B., Deo N., Tiwari P.K., Bisht D., Venkatesan K. Proteome analysis of ofloxacin and moxifloxacin induced Mycobacterium tuberculosis isolates by proteomic approach. Protein Pept. Lett. 2015;22:362–371. doi: 10.2174/0929866522666150209113708. [DOI] [PubMed] [Google Scholar]
- 60.Aubry A., Pan X.S., Fisher L.M., Jarlier V., Cambau E. Mycobacterium tuberculosis DNA gyrase: Interaction with quinolones and correlation with antimycobacterial drug activity. Antimicrob. Agents Chemother. 2004;48:1281–1288. doi: 10.1128/AAC.48.4.1281-1288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brennan P.J. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis. 2003;83:91–97. doi: 10.1016/S1472-9792(02)00089-6. [DOI] [PubMed] [Google Scholar]
- 62.De Wijs H., Jollès P. Cell walls of three strains of mycobacteria (Mycobacterium phlei, Mycobacterium fortuitum and Mycobacterium kansasii): Preparation, analysis and digestion by lysozymes of different origins. Biochim. Biophys. Acta. 1964;83:326–332. doi: 10.1016/0926-6526(64)90010-2. [DOI] [PubMed] [Google Scholar]
- 63.Suffness M., Douros J. Current status of the NCI plant and animal product program. J. Nat. Prod. 1982;45:1–14. doi: 10.1021/np50019a001. [DOI] [PubMed] [Google Scholar]
- 64.Witek S., Bielawski J., Bielawska A. Synthesis of N-(formylphenyl)- and N-(acetophenyl) derivatives of urea and carbamic acid. J. Prakt. Chem. 1979;321:804–812. doi: 10.1002/prac.19793210512. [DOI] [Google Scholar]
- 65.Takeuchi H., Mastubara E. Electrophilic aromatic N-substitution by ethoxycarbonylnitrenium ion generated from ethyl azidoformate in the presence of trifluoroacetic acid. J. Chem. Soc. Perkin Trans. 1984;1:981–985. doi: 10.1039/p19840000981. [DOI] [Google Scholar]
- 66.Park Ch.-H., Givens R.S. New photoactivated protecting groups. 6. p-Hydroxyphenacyl: A phototrigger for chemical and biochemical probes. J. Am. Chem. Soc. 1997;119:2453–2463. doi: 10.1021/ja9635589. [DOI] [Google Scholar]
- 67.Basterfield S., Woods E.L., Wright H.N. Studies in urethans. III. The preparation of various substituted urethans. J. Am. Chem. Soc. 1926;48:2371–2375. doi: 10.1021/ja01420a018. [DOI] [Google Scholar]
- 68.Smith Broadbent H., Chu C.-Y. The carbethoxylation products of p-aminoacetophenone and p-dimethylaminoacetophenone. J. Am. Chem. Soc. 1953;75:226–227. doi: 10.1021/ja01097a502. [DOI] [Google Scholar]
- 69.Sigman E.M., Autrey T., Schuster G.B. Aroylnitrenes with singlet ground states: Photochemistry of acetyl-substituted aroyl and aryloxycarbonyl azides. J. Am. Chem. Soc. 1988;110:4297–4305. doi: 10.1021/ja00221a032. [DOI] [Google Scholar]
- 70.Vettorazzi M., Angelina E., Lima S., Gonec T., Otevrel J., Marvanova P., Padrtova T., Mokry P., Bobal P., Acosta L.M., et al. An integrative study to identify novel scaffolds for sphingosine kinase 1 inhibitors. Eur. J. Med. Chem. 2017;139:461–481. doi: 10.1016/j.ejmech.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bietti G., Cereda E., Donetti A., del Soldato P., Giachetti A., Micheletti R. Guanidino-heterocyclyl-phenyl-amidines and Salts Thereof. No. US4548944 A. [(accessed on 12 November 2017)];U.S. Patent. Available online: https://encrypted.google.com/patents/US4548944?cl=un.
- 72.Rather J.B., Reid E.E. The identification of acids. IV. Phenacyl esters. J. Am. Chem. Soc. 1919;41:75–83. doi: 10.1021/ja01458a009. [DOI] [Google Scholar]
- 73.Dross K., Rekker R.F., de Vries G., Mannhold R. The lipophilic behaviour of organic compounds: 3. The search for interconnections between reversed-phase chromatographic data and log Pfoct values. Quant. Struct. Act. Relat. 1999;18:549–557. doi: 10.1002/(SICI)1521-3838(199812)17:06<549::AID-QSAR549>3.3.CO;2-T. [DOI] [Google Scholar]
- 74.Kulig K., Malawska B. Estimation of the lipophilicity of antiarrhythmic and antihypertensive active 1-substituted pyrrolidin-2-one and pyrrolidine derivatives. Biomed. Chromatogr. 2003;17:318–324. doi: 10.1002/bmc.246. [DOI] [PubMed] [Google Scholar]
- 75.Özden S., Atabey D., Yıldız S., Göker H. Synthesis, potent anti-staphylococcal activity and QSARs of some novel 2-anilinobenzazoles. Eur. J. Med. Chem. 2008;43:1390–1402. doi: 10.1016/j.ejmech.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 76.Imramovsky A., Pesko M., Kralova K., Vejsova M., Stolarikova J., Vinsova J., Jampilek J. Investigating spectrum of biological activity of 4- and 5-chloro-2-hydroxy-N-[2-(arylamino)-1-alkyl-2-oxoethyl]benz-amides. Molecules. 2011;16:2414–2430. doi: 10.3390/molecules16032414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gonec T., Kos J., Zadrazilova I., Pesko M., Govender R., Chambel B., Pereira D., Kollar P., Imramovsky A., O’Mahony J., et al. Antibacterial and herbicidal activity of ring-substituted 2-hydroxynaphthalene-1-carboxanilides. Molecules. 2013;18:9397–9419. doi: 10.3390/molecules18089397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gonec T., Kos J., Zadrazilova I., Pesko M., Keltosova S., Tengler J., Bobal P., Kollar P., Cizek A., Kralova K., et al. Antimycobacterial and herbicidal activity of ring-substituted 1-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2013;21:6531–6541. doi: 10.1016/j.bmc.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 79.Morgan J.A., Tatar J.F. Calculation of the residual sum of squares for all possible regressions. Technometrics. 1972;14:317–325. doi: 10.1080/00401706.1972.10488918. [DOI] [Google Scholar]
- 80.Cheng B., Tong H. On residual sums of squares in non-parametric autoregression. Stoch. Process. Their Appl. 1983;48:157–174. doi: 10.1016/0304-4149(93)90112-H. [DOI] [Google Scholar]
- 81.Kubinyi H. QSAR: Hansch Analysis and Related Approaches. In: Mannhold R., Krogsgaard-Larsen P., Timmerman H., editors. Methods and Principles in Medicinal Chemistry. Volume 1. Wiley-VCh Verlag; Weinheim, Germany: 1993. pp. 22–56. [Google Scholar]
- 82.Weisberg S. Multiple Regression. In: Weisberg S., editor. Applied Linear Regression. 3rd ed. Wiley-Interscience (John Wiley and Sons); Hoboken, NJ, USA: 2005. pp. 47–68. [DOI] [Google Scholar]
- 83.Nakagawa S., Schielzeth H. General and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol. Evol. 2013;4:133–142. doi: 10.1111/j.2041-210x.2012.00261.x. [DOI] [Google Scholar]
- 84.Mevik B.H., Cederkvist H.R. Mean squared error of prediction (MSEP) estimates for principal component regression (PCR) and partial least squares regression (PLSR) J. Chemom. 2004;18:422–429. doi: 10.1002/cem.887. [DOI] [Google Scholar]
- 85.Ying X., Yang L., Zha H. A fast algorithm for multidimensional ellipsoid-specific fitting by minimizing a new defined vector norm of residuals using semidefinite programming. IEEE Trans. Pattern Anal. Mach. Intell. 2012;34:1856–1863. doi: 10.1109/TPAMI.2012.109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.