

ABSTRACT
We recently reported that expression of the costimulatory molecule and microbial sensor SLAMF1 (signaling lymphocytic activation molecule family 1, also known as CD150) is lost in chronic lymphocytic leukemia (CLL) patients characterized by a shorter overall survival. SLAMF1 modulates CLL responses to chemokines and regulates autophagy. Loss of SLAMF1 renders CLL cells relatively unresponsive to autophagy-inducing drugs, including B-cell CLL/lymphoma 2 (BCL2) inhibitors.
KEYWORDS: Autophagy, CLL, SLAMF1
Abbreviations
- BCL2
B-cell CLL/lymphoma 2
- JNK1/2
Jun N-terminal kinase 1/2
- LC3B
microtubule associated protein 1 light chain 3 beta
- NOX2
NADPH oxidase 2
- pS70
phospho-serine70
- ROS
reactive oxygen species
- SLAMF1
signaling lymphocytic activation molecule family 1
- VPS34
phosphatidylinositol 3-kinase catalytic subunit type 3
Chronic lymphocytic leukemia (CLL) is a highly heterogeneous disease. Recent studies have concentrated on the identification of molecular markers characterizing the more aggressive form of the disease, whereas little is known about the genetic structure or markers associated with CLL cases with a favorable prognosis.1,2
Our work, recently published in the Journal of Clinical Investigation,3 shows that signaling lymphocytic activation molecule family 1 (SLAMF1, also known as CD150) is expressed by the subset of CLL cases characterized by a relatively favorable prognosis. In contrast, SLAMF1 expression is lost in the subset of patients characterized by the presence of molecular markers of aggressive disease and a shorter time to first treatment and overall survival.
SLAMF1 is a self-ligand adhesion/co-stimulatory molecule that belongs to a family of 9 membrane receptors. In addition to its involvement in T-B lymphocyte interactions, SLAMF1 regulates Gram-negative bacterial phagosome functions through the ubiquitous cellular autophagy machinery and serves as a receptor for the measles virus.4,5 Our results, obtained by comparing gene expression in SLAMF1+ versus SLAMF1− cells, suggest that loss of SLAMF1 expression directly contributes to a worse clinical outcome of patients with leukemia. Cell movement and vesicle trafficking clearly emerged as the 2 main cellular processes affected. Consistent with these findings, chemotaxis toward CXCL12 was increased in SLAMF1− primary CLL cells, suggesting that this subset of cells may have an advantage by localizing within the lymph nodes. This finding is in line with the recent view of CLL as a compartmentalized disease with a proliferative component hosted in lymphoid organs,6 where an ideal cocktail of antigen and co-stimulatory signals drive leukemic cell proliferation.
The second set of genes modulated upon SLAMF1 silencing are involved in vesicle trafficking, an intriguing finding in the light of the reported involvement of SLAMF1 in organization of the autophagy macrocomplex, at least in mouse macrophages.5 Using primary CLL cells and a cell line model, we confirmed that SLAMF1 ligation using an agonistic monoclonal antibody activates a signaling pathway that culminates in the processing of microtubule associated protein 1 light chain 3 β (LC3B), a hallmark of autophagy. The intermediate steps in this cascade were identified using specific signaling inhibitors and co-immunoprecipitation experiments. In detail, we detected increased intracellular levels of reactive oxygen species (ROS) upon SLAMF1 activation, most likely due to activation of the membrane NAPDH oxidase 2 (NOX2) complex. These ROS can in turn lead to phosphorylation of Jun N-terminal kinase 1/2 (JNK1/2), a member of the MAP kinase family, which then phosphorylates B-cell CLL/lymphoma 2 (BCL2) on a critical serine residue. The resultant phosphorylation-induced structural changes in BCL2 are critical for the regulation of its molecular interactions. Specifically, we observed that phosphorylation at serine 70 caused rapid detachment of BCL2 from its partner molecule beclin 1. This is the first step in the organization of the autophagy macrocomplex: the free beclin 1 serves as scaffold for phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3 or VPS34), which provides energy for complex formation, and other key protein components. Our data indicate that SLAMF1 is also directly associated with beclin1 and VPS34 (Fig. 1). It remains to be determined what exactly triggers SLAMF1 activation. We observed that autophagy was induced upon ligation of SLAMF1 with an agonistic monoclonal antibody, suggesting that the antibody is standing in for a physiological ligand, possibly a second SLAMF1 molecule. Experimental confirmation of this would suggest that SLAMF1-SLAMF1 interactions, which occur regularly during normal T-B lymphocyte interactions, might promote autophagy, a mechanism that is increasingly becoming recognized as important in the regulation of lymphocyte proliferation, energy metabolism, and cell death.
Figure 1.
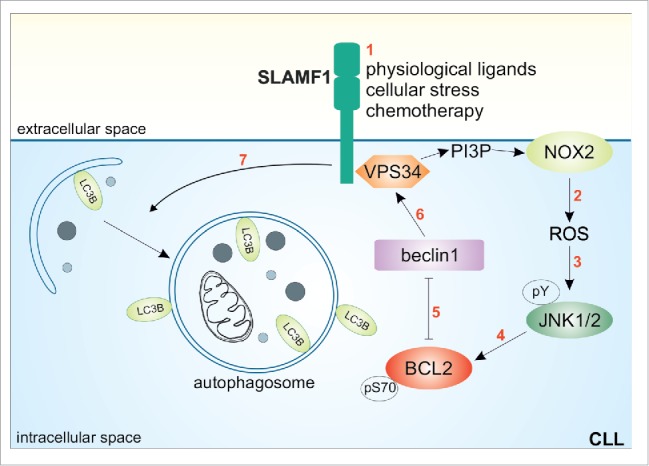
Molecular mechanism of SLAMF1-induced autophagy in CLL cells. Upon activation (1), SLAMF1 initiates a signaling pathway that increases NOX2-mediated reactive oxygen species (ROS) production (2), which is in turn responsible for JNK1/2 tyrosine phosphorylation (3). JNK1/2 then phosphorylates BCL2 at serine 70 (4), causing its dissociation from beclin 1 and leaving it free to assemble the autophagic complex including VPS34 and SLAMF1 itself. As a consequence, autophagosome formation is induced (7), leading to execution of autophagy.
The translational relevance of this study comes from the role of autophagy in cancer. Autophagy is a membrane-trafficking mechanism that delivers cytoplasmic constituents into the lysosome for bulk degradation. Besides maintaining cellular homeostasis under normal growth conditions, autophagy critically controls cellular responses to stressful conditions.7 In cancer, depending on the cellular context, autophagy may enable tumor cells to survive chemotherapy-mediated stress or it may maintain cellular homeostasis by removing damaged organelles and preventing the genomic damage that leads to cancer.8 In leukemia, several studies have shown that autophagy is activated upon treatment with different chemotherapeutic agents, thus inducing cell death. This is also true for CLL, in which many currently used drugs including fludarabine, dexamethasone, idelalisib, and the novel BCL2 antagonists are effective through activating autophagy.9,10 We observed that CLL cells lacking SLAMF1 were less responsive to fludarabine and the BH3 mimetic ABT-737. These results were supported by experiments comparing therapy responses in SLAMF1high and SLAMF1low subclones that were sorted from the same CLL patient and thus shared the same genetic background.
In conclusion, our work provides evidence that expression of the surface receptor SLAMF1 is necessary for CLL homeostasis by changing chemotactic responses of CLL cells and by modulating autophagy. These effects potentially affect recirculation of tumor cells to and from lymphoid organs as well as drug responses, providing a likely explanation for the unfavorable clinical outcome experienced by CLL patients who lack SLAMF1 expression. Thus, restoring SLAMF1 expression and functions in CLL cells would be of therapeutic value for patients with aggressive disease.
Funding Statement
Italian Association for Cancer Research (IG12754); Human Genetics Foundation (HGF001).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. New Engl J Med 2005; 352:804-15; PMID:15728813; https://doi.org/23023714 10.1056/NEJMra041720 [DOI] [PubMed] [Google Scholar]
- 2.Gaidano G, Foa R, Dalla-Favera R. Molecular pathogenesis of chronic lymphocytic leukemia. J Clin Invest 2012; 122:3432-8; PMID:23023714; https://doi.org/ 10.1172/JCI64101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bologna C, Buonincontri R, Serra S, Vaisitti T, Audrito V, Brusa D, Pagnani A, Coscia M, D'Arena G, Mereu E, et al.. SLAMF1 regulation of chemotaxis and autophagy determines CLL patient response. J Clin Invest 2016; 126:181-94; PMID:26619119; https://doi.org/ 10.1172/JCI83013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calpe S, Wang N, Romero X, Berger SB, Lanyi A, Engel P, Terhorst C. The SLAM and SAP gene families control innate and adaptive immune responses. Adv Immunol 2008; 97:177-250; PMID:18501771; https://doi.org/ 10.1016/S0065-2776(08)00004-7 [DOI] [PubMed] [Google Scholar]
- 5.Berger SB, Romero X, Ma C, Wang G, Faubion WA, Liao G, Compeer E, Keszei M, Rameh L, Wang N, et al.. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages. Nat Immunol 2010; 11:920-7; PMID:20818396; https://doi.org/ 10.1038/ni.1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaisitti T, Aydin S, Rossi D, Cottino F, Bergui L, D'Arena G, Bonello L, Horenstein AL, Brennan P, Pepper C, et al.. CD38 increases CXCL12-mediated signals and homing of chronic lymphocytic leukemia cells. Leukemia 2010; 24:958-69; PMID:20220774; https://doi.org/ 10.1038/leu.2010.36 [DOI] [PubMed] [Google Scholar]
- 7.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Molecular cell 2010; 40:280-93; PMID:20965422; https://doi.org/ 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, et al.. Autophagy in malignant transformation and cancer progression. EMBO J 2015; 34:856-80; PMID:25712477; https://doi.org/ 10.15252/embj.201490784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al.. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435:677-81; PMID:15902208; https://doi.org/ 10.1038/nature03579 [DOI] [PubMed] [Google Scholar]
- 10.Mahoney E, Lucas DM, Gupta SV, Wagner AJ, Herman SE, Smith LL, Yeh YY, Andritsos L, Jones JA, Flynn JM, et al.. ER stress and autophagy: new discoveries in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Blood 2012; 120:1262-73; PMID:22740450; https://doi.org/ 10.1182/blood-2011-12-400184 [DOI] [PMC free article] [PubMed] [Google Scholar]