

Abstract
Synthetic derivatives of spiro[pyrrolidinyl-3,3′-oxindole] alkaloids (coerulescine analogues) were investigated as new ligands for aminergic G-protein coupled receptors (GPCRs). The chemical starting point 2′-phenylspiro[indoline-3,3′-pyrrolidin]-2-one scaffold was identified by virtual fragment screening utilizing ligand- and structure based methods. As a part of the hit-to-lead optimization a structure-activity relationship analysis was performed to explore the differently substituted 2′-phenyl-derivatives, introducing the phenylsulphonyl pharmacophore and examining the corresponding reduced spiro[pyrrolidine-3,3′-indoline] scaffold. The optimization process led to ligands with submicromolar affinities towards the 5-HT6 receptor that might serve as viable leads for further optimization.
Keywords: oxindole, indoline, coerulescine, 5-HT6R, G-protein coupled receptor
1. Introduction
The recent isolation of naturally occurring and biologically active spiropyrrolidinyl-oxindole alkaloids initiated a significant research on synthetic derivatives [1], particularly compounds with the spiro[indoline-3,3′-pyrrolidine]-2-one ring system. Spiropyrrolidinyl-oxindole alkaloids were isolated from different species including Gelsemium sempervirens, Aspidosperma, Mitragyna, Ourouparia, Rauwolfia, Vinca species [2]. The most prominent examples are rynchophylline (1), formosanine (2), coerulescine (3), horsfiline (4) and elacomine (5) that show diverse biological activity (Figure 1). Although these compounds have a common tryptamine derived motif, a ChEMBL-analysis [3] revealed that no representatives have been ever tested against serotonergic targets. Developing ligand-(FrAGs) [4] and structure-based (FrACS) [5] methods for the identification of aminergic receptor ligands we identified 6 as a micromolar 5-hydroxytryptamine receptor 6 (5-HT6R) ligand.
Figure 1.

Representative natural spiropyrrolidinyl-oxindole alkaloids. The tryptamine scaffold is shown as red skeleton.
The 5-HT6R is a member of the Class A G-protein coupled receptors (aminergic family) considered to be a current and promising drug target for the treatment of several central nervous system related indications, such as: cognitive, learning and memory deficits related to Alzheimer’s disease [6], Parkinson’s disease [7] and schizophrenia [8]. Chemical similarity to the endogenous agonist serotonin explains the most frequent heteroaromatic ring systems routinely used in 5-HT6R ligands that include indoles, indolines, indazoles, pyrrolo[2,3-b]pyridines, pyrazolo[1,5-a]pyridines, [1,2,3]-triazolo[1,5-a]pyrimidines and further 5 + 6 condensed N-heterocycles. Pharmacophore based approaches on 5-HT6R antagonists are typically focused to the canonical pharmacophore model [9] (Figure 2), which is defined as having two hydrophobic rings/ring systems connected by a hydrogen bond acceptor (e.g., sulfonyl, sulfonamide linker). Optionally, a positively ionizable residue is included in one of the hydrophobic sites of the ligands, typically offering an interacting moiety with the D3.32 aspartate residue of aminergic GPCR’s as a key factor of aminergic 7TM-receptor activation [10]. A further pharmacophore feature might contain an additional intramolecular hydrogen bond donor moiety further stabilizing the binding conformation of the ligands [11] (see Figure 2). Selectivity among other aminergic GPCR’s was shown [12] to be accessible through omitting the positively ionizable group in the 5-HT6R antagonists. Bis(hetero)arylsulphonyl- and sulfonamide substituents also contribute to 5-HT6R affinity and selectivity.
Figure 2.

General pharmacophore model of 5-HT6R antagonists. PI: positive ionisable portion; HYD1,2: hydrophobic sites; iHBD: intramolecular hydrogen bond donor site; HBA: hydrogen bond acceptor feature; HSi,j: substituents on the hydrophobic sites.
The tryptamine derived scaffold of 6 and its alignment with the published 5-HT6R pharmacophore [9] prompted us to explore the structure-activity relationship around the spiropyrrolidinyl-oxindole core. Here we report the results of our hit-to-lead program, which led to an identification of promising 5-HT6R ligands of this chemotype.
2. Results and Discussion
2.1. Identification and Early Structure-Activity Data on Spiro[pyrrolidine-3,3′-oxindole] Derivatives
Fragment libraries containing a couple hundreds or even thousands of small polar molecules are routinely used for hit identification at the early stage of drug discovery. Fragment screening provides diverse chemotypes and significant operational freedom for the further optimization of promising hits. Inspired by these advantages in a recent study we developed a strategy for aminergic focused fragment libraries using a sequential filtering methodology applying ligand- and structure-based scoring functions [4,5]. The prospective validation was performed on our in-house library of 1183 fragments. A physicochemical-property based scoring, followed by docking the fragments into an ensemble of carefully selected aminergic GPCR X-ray structures (PDB ID: 3PBL, 3RZE, 4IB4, 4IAQ, 3UON, 4MQT, 4LDE, 2RH1, 3NY9). This resulted in a set of 36 top ranked hit molecules which were measured on an aminergic target not being included in the original docking ensemble, namely 5-HT6R. We demonstrate the usefulness of the method for comprehensive aminergic focused screenings. Out of the four hits with low micromolar inhibitory results, the structurally novel 2′-(3-fluorophenyl)spiro[indoline-3,3′-pyrrolidin]-2-one (6) possessing the spiro[pyrrolidine-3,3′-oxindole] scaffold (Table 1), was selected for further optimization.
Table 1.
Serotonergic G-protein coupled receptor (GPCR) panel of 6 as measured in binding assays of four serotonin receptors (Ki values are in µM).
| ID | Structure | 5-HT1A | 5-HT2A | 5-HT6 | 5-HT7 |
|---|---|---|---|---|---|
| 6 | 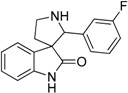 |
16.91 | 5.05 | 6.75 | 8.48 |
As the next step of exploring the structure-activity relationship substructure search in the MCULE purchasable database [13] of 5 million compounds resulted in further 887 spiro[pyrrolidine-3,3′-oxindole] derivatives. The molecules were prepared by Schrödinger’s LigPrep [14] creating possible conformers, tautomers and protonation states by default settings. The ligands were then projected to single precision molecular docking analysis on a nine membered ensemble of molecular dynamics frames of an 5-HT6R homology model [15] (built using 5-HT2BR X-ray crystal structure [16] in complex with ergotamine as template (PDB ID: 4IB4)). The docking poses were filtered in a post-processing step keeping only binding modes forming hydrogen bonds towards Asp1063.32, Asn2886.55 and optionally Ser1935.43, occupying a primary hydrophobic cleft defined by Trp2816.48, Phe2846.51 and Phe2856.52. A secondary hydrophobic subpocket was defined by the pose filtering criteria as following: Val1073.33, Ala1574.56, Leu1604.59, Pro1614.60, Leu1644.63. The satisfactory poses of the molecules were scored to have the best ranking in possibly all of the 9 frames using consensus ranking [5]. Altogether ten compounds (7–16) were purchased and tested in our serotonergic panel (Table 2).
Table 2.
Serotonergic GPCR panel of derivatives substituted in the 2′-phenyl moiety as measured in binding assays of four serotonin receptors (Ki values are in µM).
| ID | Structure | 5-HT1A | 5-HT2A | 5-HT6 | 5-HT7 |
|---|---|---|---|---|---|
| 7 | 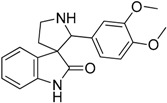 |
7.07 | 5.45 | 2.26 | 2.69 |
| 8 | 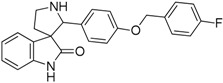 |
1.74 | 2.28 | 3.18 | 1.09 |
| 9 | 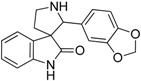 |
9.84 | 2.69 | 3.20 | 8.82 |
| 10 | 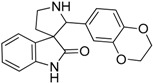 |
12.08 | 4.33 | 4.11 | 12.74 |
| 11 | 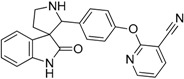 |
49.11 | 8.68 | 6.20 | 58.96 |
| 12 | 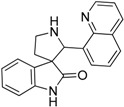 |
45.67 | 1.12 | 6.63 | 10.45 |
| 13 | 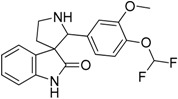 |
7.14 | 5.77 | 7.73 | 7.49 |
| 14 | 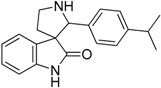 |
7.76 | 4.54 | 8.62 | 8.98 |
| 15 | 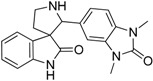 |
40.50 | 23.70 | 9.49 | 46.72 |
| 16 | 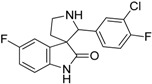 |
not active | 53.89 | 16.54 | not active |
In spite of the structural diversity, all virtual screening hits were lacking substitution at the oxindole scaffold. However, compounds 7, 9 and 10 having oxygen-containing substituents in the 3,4-positions of the 2′-phenyl ring showed somewhat improved affinity compared to the initial compound 6. The moderate improvement in affinity and the lack of selectivity amongst the closely related serotonergic targets have driven us to interpret the results in the context of the known 5-HT6R pharmacophore patterns [17] (Figure 3). This analysis suggests that introducing a phenylsulfonyl moiety either to the 1-nitrogen at the oxindole ring or to the 1′-nitrogen at the pyrrolidine ring would be beneficial for both the affinity and selectivity of this chemotype.
Figure 3.

(a) 1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidin]-2-one (17) superimposed onto the classical pharmacophore; (b) 1′-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidin]-2-one (18) fitting to the pharmacophore pattern lacking a positive ionisable moiety.
2.2. Hit-to-Lead Optimization of Spiro[pyrrolidine-3,3′-oxindoles]
Detailed elaboration of the spiro[pyrrolidine-3,3′-oxindole] scaffold required a viable synthesis strategy for the preparation of designed analogues. The first, conventional approach is based on an intramolecular Mannich-reaction used in case of several alkaloids including (±)-horsfiline (4) [18] and Spirotryprostatin B [19]. An alternative approach is the oxidative reaction of tryptolines induced by tert-butyl hypochlorite, N-bromosuccinimide, N-chlorosuccinimide, sodium tungstate, lead tetraacetate, or osmium tetroxide [20,21]. The reaction is completed by the subsequent elimination of water that finally results in the reorganization of the ring system to spiro[pyrrolidine-3,3′-oxindoles] (Scheme 1).
Scheme 1.

Mechanism of the succinimide assisted oxidative spiro-rearrangement of tryptolines.
Further, sophisticated approaches such as [1,3]-dipolar cycloaddition reactions of azomethine ylides [22], radical cyclization by AIBN [23], intramolecular Heck reaction [24] and asymmetric nitroolefination of oxindoles [25] are also available, however, used less frequently.
For the effective exploration of the structure-activity relationship we need a feasible and universal approach with acceptable functional group tolerance and high variability around the spiro-oxindole core. Considering the requirements of the early stage optimization program we aimed a synthesis strategy that
uses readily accessible, simple starting materials
applies well-documented, readily available reagents
has key intermediates to access a wide variety of derivatives
is not necessarily stereoselective at this stage of the optimization
The oxidative spiro-rearrangement reactions offer a wide variety of oxidative reagents [20,21,26,27,28,29,30,31,32,33] and the corresponding starting materials are readily accessible through Pictet-Spengler reaction of tryptamines [34] (Scheme 2).
Scheme 2.

Synthesis of tryptoline by Pictet-Spengler condensation.
Following this synthesis strategy, tryptamine (23) was used as a starting material for the synthesis of the common intermediate tryptoline (25). The Pictet-Spengler condensation reaction was performed by glyoxylic acid-monohydrate in aqueous medium [35]. The crude acid intermediate 24 was decarboxylated in refluxing concentrated hydrochloric acid affording the tetrahydro-β-carboline. The first synthetic route depicted in Scheme 3 starts with the N-acylation of the tryptoline (25). The application of Cbz (carboxybenzyl) [36] protecting group was necessary for two reasons: 1. achieve the sulfonylation at the oxindol-nitrogen, 2. the spiro-rearrangement reaction is not occurring in case of unsubstituted pyrido-nitrogen. N-chlorosuccinimide [37] was used in the first experiments as halogenating reagent, providing the Cbz-protected spiro[pyrrolidine-3,3′-oxindole]. The phenylsulfonylation was performed either by using lithium-hexamethyl disilazane [38] and sodium hydride [39] as deprotonating agents and acid scavengers. Finally, the deprotection of the Cbz-group by hydrogenation [40] resulted in the desired 1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidin]-2-one (17) compound. Following an alternative way, the more basic pyrido-nitrogen of tryptoline 29 might be first sulfonylated [41] followed by the N-bromosuccinimide assisted spiro-cyclization to 18 [42].
Scheme 3.

Synthesis of 1- and 1′-phenylsulfonyl derivatives.
Compounds 17 and 18 showed improved selectivity towards the 5-HT6R (Table 3) as compared to the non-sulfonylated 2′-phenyl derivatives, investigated in the first batch (compounds 15–24), however, no improvement in the binding affinity was detected.
Table 3.
Serotonergic GPCR panel of the N-phenylsulfonylated derivatives at N′ and N-positions as measured in binding assays of four serotonin receptors (Ki values are in µM).
| ID | Structure | 5-HT1A | 5-HT2A | 5-HT6 | 5-HT7 |
|---|---|---|---|---|---|
| 17 | 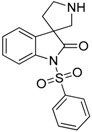 |
not active | not active | 12.30 | 58.31 |
| 18 | 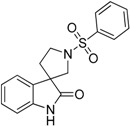 |
not measured | 87.35 | 6.86 | not active |
Facilitating the potency optimization, we followed the classical 5-HT6R pharmacophore pattern and tried to remove the polar interacting hydrogen bond acceptor feature in the oxindole ring of 17. For better understanding of the indole, indoline and oxindole-related chemical space of known 5-HT6R ligands we collected all compounds, with at least 0.1 μM activity towards h5-HT6R from the ChEMBL database.
Substructure filtering by the query of 30 (Figure 4) revealed that altogether 3 examples were found for oxindoles, as 5-HT6R ligands. All hits belong to the 3′-phenylspiro[indoline-3,2′-thiazolidine]-2,4′-dione scaffold 31 that was identified by Hostetler et al. [43]. Searching for the 1-(phenylsulfonyl) indoline scaffold 32, 27 actives were found with the structure 33. Out of the 27 active molecules 4 possess a positive ionizable moiety built upon the phenyl side of the sulfonyl and all of the remaining examples are equipped by the classical tryptamine-type amine feature. Searching by the 1-(phenylsulfonyl)indole 34 query resulted in 397 examples with the structure 35. The structures were analyzed based on the chain-length distance between the 3-carbon of the indole ring and the positive ionizable basic nitrogen atom, if any. We found 20 examples representing indoles with attached basic nitrogen (all tertiary amines). Only five compounds were identified with a single methylene linker out of which 2 were primary amines and 3 were tertiary amines. The largest part of the with structure 34 contains ethylene linker (201 examples out of 397) of which 123 were tertiary amines, 51 were secondary amines and 19 were primary amines. Much less compounds have 3 atom long linkage, 54 examples were identified, with 21 tertiary amines and 33 secondary amines and no primary amines. No ligands were found with basic nitrogen’s being further than 3 atoms from the indole core.
Figure 4.

Substructure analysis of ChEMBL 5-HT6R active compounds.
To conclude, we have aimed to synthesize different 1-(phenylsulfonyl)indolines (scaffold 32), being substituted in the 1′-pyrrolidine nitrogen with either basic-nitrogen containing, or lipophilic groups, presented in Scheme 4—compounds 39a–b. Based on these considerations, first we benzylated [44] the tryptoline (25) to afford 36a, further converting it to the corresponding spiro-derivative [45] 37a. The reduction of the oxindole oxo-group was performed by applying borane-tetrahydrofuran complex in refluxing absolutized tetrahydrofuran [46]. It was an important observation, that the reduction step has to precede the sulfonylation, to avoid the formation of side products when treating the sulfonamide with the boronic reagent. The phenyl-sulfonylated product 39a was afforded either by applying LiHMDS [38] or trietylamine/dimethylaminopyridine [47] reagents. The pyridine-4-ylmethyl derivative (39b) was synthesized based on the same route, however, as a first step, the tryptoline (25) was treated with 4-chloromethylpyridine [48,49].
Scheme 4.

Synthesis of N′-substituted and N-phenylsulfonylated indolines.
We also intended to examine the effect of only transforming our initial test compound 17 to the corresponding reduced indoline, however the debenzylation step of 39a afforded unexpected products (Scheme 5, 40a and 40b). In case, when methanol was applied as solvent for the debenzylation by catalytic hydrogenation, a methylated product 40a has formed and respectively, the use of ethanol afforded an ethyl-alkylated derivative 40b.
Scheme 5.

Synthesis of N′-methyl and ethyl derivatives.
However, both the alkylated derivatives showed some improvement of binding affinity to reach the low micromolar range, the compound possessing the biggest lipophilic, bulky group 43a has produced the best, submicromolar binding affinity, with both selectivity against the other closely related serotonergic targets (Table 4).
Table 4.
Serotonergic GPCR panel of N-phenylsulfonyl indolines with different N′-substituents as measured in binding assays of four serotonin receptors (Ki values are in µM).
| ID | Structure | 5-HT1A | 5-HT2A | 5-HT6 | 5-HT7 |
|---|---|---|---|---|---|
| 39a | 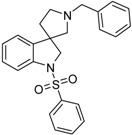 |
not measured | 4.76 | 0.76 | 55.76 |
| 39b | 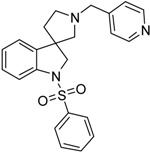 |
not active | 26.85 | 6.60 | not active |
| 40a | 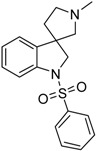 |
not measured | 4.52 | 1.85 | 40.17 |
| 40b | 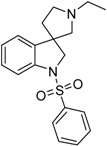 |
not measured | 3.82 | 2.42 | not active |
The submicromolar affinity of 39a prompted us investigating the substituent vectors at both the benzylic and the phenylsulfonyl rings by walking fluorine substituents around. This methodology, often referred as fluoro-scan has been used effectively for the identification of sites tolerant to functionalization. In particular, fluoro-scan explores the impact of enhanced lipophilicity, H-bonding and/or filling a small pocket [50]. In case of the N′-benzylic substitution [13] pattern, 2-, 3- and 4-fluorobenzylchlorides were used as alkylating agents in order to synthesize the corresponding fluorinated indolines (Scheme 6, 44a–c).
Scheme 6.

Fluoro-scan of the N′-benzyl direction.
The substitution of benzylic ring with fluorine caused a minor decrease in affinity, thus has shown, that growing towards this direction is not beneficial (Table 5). On the other hand, however, some improvement in the selectivity was observed.
Table 5.
Serotonergic GPCR panel of the fluorobenzyl derivatives as measured in binding assays of three serotonin receptors (Ki values are in µM).
| ID | Structure | 5-HT1A | 5-HT6 | 5-HT7 |
|---|---|---|---|---|
| 44a | 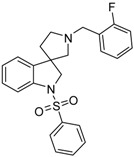 |
not active | 4.53 | 78.93 |
| 44b | 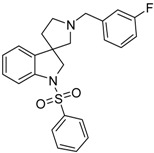 |
not active | 1.97 | 31.49 |
| 44c | 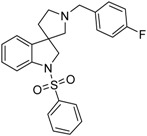 |
not active | 1.77 | 15.74 |
The corresponding fluorophenylsulfonyl derivatives 46a–c were synthesized from compound 38a intermediate, previously synthesized (Scheme 7).
Scheme 7.

Fluoro-scan of the N-phenylsulfonyl direction.
The alternative growing direction towards the phenylsulfonyl ring (compounds 46a–c) showed improvement in the affinities (see Table 6), compared to the results of 39a, also retaining the good selectivity against the 5-HT7 subtype. These data suggest, that position 2 of the phenylsulfonyl group of 46a is beneficial and therefore it is worth to explore this vector during the forthcoming lead optimization program.
Table 6.
Serotonergic GPCR panel of the fluorophenylsulfonyl derivatives as measured in binding assays of three serotonin receptors (Ki values are in µM).
| ID | Structure | 5-HT1A | 5-HT6 | 5-HT7 |
|---|---|---|---|---|
| 46a | 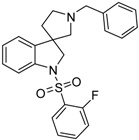 |
not active | 0.19 | 42.57 |
| 46b | 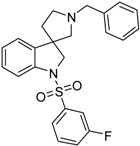 |
not active | 0.71 | 76.00 |
| 46c | 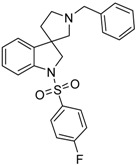 |
not active | 2.45 | 56.05 |
2.3. Binding Mode Analysis of the Optimized 5-HT6R Ligand
We have performed a docking analysis of the 1′-benzyl-1-((2-fluorophenyl)sulfonyl)-spiro[indoline-3,3′-pyrrolidine] (46a) 5-HT6R antagonist using the receptor model reported earlier [15]. Docking of 46a by Schrödinger—Glide either with, or without applying any constraints (requiring hydrogen bonding with both Asn2886.55 and/or Ser1935.43) resulted in consistent docking poses inside the orthosteric binding pocket (Figure 5).
Figure 5.

Binding mode of compound 46a in the orthosteric pocket of 5-HT6R.
The indoline core occupies the primary hydrophobic cavity of the binding pocket (formed by Trp2816.48, Phe2846.51, Phe2856.52), while the protonated quaternary pyrrolidine-nitrogen is oriented towards the conserved Asp1063.32, forming a well-established hydrogen bond. The phenyl-sulfonyl moiety of 46a is sitting at a secondary hydrophobic cleft defined by Val1073.33, Ala1574.56, Leu1604.59, Pro1604.60 and Leu1644.63, positioning the sulfonyl-linker as hydrogen-bond acceptor against Ser1935.43 and Asn2886.55. Interestingly, the fluorine atom as hydrogen bond acceptor is offering a possible polar interaction with the Asn2886.55 residue, underlining the preference of the ortho-substitution at the phenyl-sulfonyl ring.
3. Conclusions
The structure-activity relationship around the original spiropyrrolidinyl-oxindole hit (6) was first investigated using the “SAR-by-catalog” approach that resulted in some moderate improvement in affinity and a low level of selectivity. Therefore, we decided to explore the novel 5-HT6R chemotype by a more conventional medicinal chemistry strategy. A synthetic tree (summarized in Scheme 8) of the synthesized oxindoles and indoles was elaborated starting from the core tryptoline intermediate (25).
Scheme 8.

Spiro[pyrrolidine-3,3′-oxindoles] and spiro[pyrrolidine-3,3′-indolines] as 5HT6 ligands.
The removal of the bulky lipophilic site at the 2′-position of the pyrrolidine ring and insertion of the classical phenylsulfonyl part resulted in notable selectivity across the serotoninergic GPCR panel of 4 receptors. The next optimization step was analyzing the chemistry of known indole, indoline and oxindole-based 5-HT6R inhibitors in ChEMBL. We have synthesized different 1-(phenylsulfonyl) indolines, being substituted in the 1′-pyrrolidine nitrogen with either basic-nitrogen containing, or lipophilic groups. As a result, 1′-benzyl-1-(phenylsulfonyl) spiro[indoline-3,3′-pyrrolidine] (39a) and its corresponding 2-, 3-substituted analogues 46a–b were identified as 5-HT6R starting points. This opened a new chemical space for the under-represented spiro[pyrrolidine-3,3′-indoline] chemotype.
4. Materials and Methods
All chemical reagents used were purchased from commercial chemical suppliers. The NMR experiments were performed at 500 MHz (1H) on a Varian VNMR SYSTEM spectrometer. Chemical shifts are referenced to the residual solvent signals, 2.50 ppm for 1H in DMSO-d6 and 7.28 ppm for 1H in CDCl3. The MS measurements were performed on Shimadzu LCMS2020 LC/MS system. Flash chromatography was performed using Teledyne ISCO CombiFlash Lumen+ Rf. Purifications by preparative-HPLC were performed with Hanbon NS4205 Binary high pressure semi-preparative HPLC. Thin-layer chromatography was performed on TLC Silica Gel 60 F254. High resolution mass spectrometric measurements were performed using a Q-TOF Premier mass spectrometer (Waters Corporation, Milford, MA, USA) in positive electrospray ionization mode. Compounds 7–16 were purchased from Mcule Inc.
4.1. General Procedures for the Synthesis of Spiro[pyrrolidine-3,3′-oxindole] and Spiro[pyrrolidine-3,3′-indoline] Derivatives
4.1.1. Procedures to Afford 1-(Phenylsulfonyl)spiro[indoline-3,3′-pyrrolidin]-2-one (17)
Synthesis of Benzyl 2-Oxospiro[indoline-3,3′-pyrrolidine]-1′-carboxylate (27)
To the solution of benzyl 3,4-dihydro-1H-pyrido[3,4-b]indole-2(9H)-carboxylate (26) (918 mg, 3 mmol) in 10 mL tetrahydrofuran was added 10 mL distilled water and triethylamine (0.436 mL, 3.15 mmol, 1.05 equiv.) at room temperature. Afterwards N-chlorosuccinimide (431 mg, 3.24 mmol, 1.08 equiv.) was added at 0 °C in portions. The reaction mixture was stirred for 1.5 h at room temperature. The reaction mixture was diluted with 10 mL brine and was extracted using 2 × 10 mL ethyl acetate. The combined organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The crude material was purified using flash chromatography (gradient: dichloromethane: methanol 0% to 5% methanol) to give 27 as a yellow solid. Yield: 399 mg (41%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 7.42–7.28 (m, 8H, ArH-benzyl, ArH-4, ArH-5, ArH-6), 7.14 (d, J = 7.8 Hz, 1H, ArH-7), 5.11 (s, 2H, benzyl CH2), 3.70–3.53 (m, 4H, pyrrolidine H-2′, pyrrolidine H-5′), 2.19–2.05 (m, 2H, H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 176.9 (C-2), 154.8 (C=O), 142.8 (C-3a), 141.5 (C-7a), 135.9 (benzyl C-1), 128.5 (benzyl C-3, benzyl C-5), 127.9 (benzyl C-4, benzyl C-2, benzyl C-6), 123.8 (C-4), 122.1 (C-5), 109.3 (C-7), 67.9 (benzyl CH2), 57.7 (C-3), 45.1 (pyrrolidine C-2′), 43.3 (pyrrolidine C-5′), 34.2 (pyrrolidine C-4′). MS (DUIS+) m/z 323 [M + H]+.
Synthesis of Benzyl 2-oxo-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine]-1′-carboxylate (28)
(1) Procedure Using Sodium-Hydride
To the solution of benzyl 2-oxospiro[indoline-3,3′-pyrrolidine]-1′-carboxylate (27) (175 mg, 0.542 mmol) in 5 mL anhydrous tetrahydrofuran was added sodium-hydride (60%, 28 mg, 0.705 mmol; 1.3 equiv.) at 0 °C. After stirring the reaction mixture at room temperature for 0.5 h, benzenesulfonyl chloride (0.076 mL, 0.596 mmol, 1.1 equiv.) was added dropwise at 0 °C. The reaction mixture was stirred overnight at room temperature, then it was quenched by 10 mL brine and was extracted using ethyl acetate (2 × 10 mL). The collected organic phases were dried using sodium sulfate, filtered and evaporated under reduced pressure. The crude product was purified using flash chromatography (gradient: hexane: ethyl acetate 0% to 20%) to afford 28 as a colorless solid. Yield: 94 mg (38%).
(2) Procedure Using Lithium-Hexamethyl-Disilazane
To the solution of benzyl 2-oxospiro[indoline-3,3′-pyrrolidine]-1′-carboxylate (27) (161 mg, 0.5 mmol) and benzenesulfonyl chloride (0.076 mL, 0.6 mmol, 1.2 equiv.) in 5 mL anhydrous tetrahydrofuran was added lithiumhexamethyl-disilazane (1M in THF, 0.85 mL, 0.85 mmol, 1.7 equiv.) at 0 °C and the reaction mixture was stirred at this temperature for 0.5 h. After quenching the reaction by 10 mL brine, the reaction mixture was extracted using ethyl acetate (2 × 10 mL) and the collected organic phases were dried over sodium sulfate, filtered and evaporated under reduced pressure. The crude product was purified using flash chromatography (gradient: hexane: ethyl acetate 0% to 20%) to give 28 as a white solid. Yield: 230 mg (99%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 8.02 (d, J = 7.7 Hz, 2H, ArH-3″, ArH-5″), 7.77 (m, 2H, ArH-4″, ArH-7), 7.67 (m, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.43–7.30 (m, 7H, ArH-4, ArH-6, benzyl ArH-2,3,4,5,6), 7.23 (t, J = 7.4 Hz, 1H, ArH-5), 5.09 (m, 2H, benzyl CH2), 3.65–3.50 (m, 4H, pyrrolidine H-2′, pyrrolidine H-5′), 2.25–2.09 (m, 2H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 170.1 (C-2), 154.3 (C=O), 136.2 (benzyl C-1), 135.5 (C-7a), 134.8 (phenyl-sulfonyl C-1″), 133.9 (C-3a), 133.1 (phenyl-sulfonyl C-4″), 129.2 (C-3″, C-5″), 128.5 (benzyl C-3, benzyl C-5), 128.0 (C-6, benzyl C-4), 127.5 (benzyl C-2, benzyl C-6), 127.4 (C-2″, C-6″), 123.3 (C-4), 121.8 (C-5), 113.8 (C-7), 67.5 (benzyl CH2), 66.1 (C-3), 46.4 (C-2′), 46.1 (C-5′), 30.3 (C-4′). MS (DUIS+) m/z 463 [M + H]+.
Synthesis of 1-(Phenylsulfonyl)spiro[indoline-3,3′-pyrrolidin]-2-one (17)
To the solution of benzyl 2-oxo-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine]-1′-carboxylate (28) (180 mg, 0.389 mmol) in methanol was added palladium (10%) on charcoal (90 mg, 5 w/w %) in an autoclave. The reactor was filled with 5 atm hydrogen gas and was stirred for 1 h at room temperature. The reaction mixture was then filtered on Celite and was evaporated under reduced pressure. The crude material was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 5% methanol) to give 17 as an orange solid. Yield: 108 mg (85%). 1H-NMR (500 MHz, CDCl3) δ ppm 8.10 (d, J = 8.0 Hz, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.91 (d, J = 8.4 Hz, 1H, phenyl-sulfonyl ArH-4″), 7.65 (t, J = 7.2 Hz, 1H, ArH-7), 7.53 (t, J = 7.2 Hz, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.47 (d, J = 7.2 Hz, 1H, ArH-4), 7.32 (t, J = 8.0 Hz, 1H, ArH-6), 7.19 (t, J = 8.0 Hz, 1H, ArH-5), 3.08 (m, 1H, H-2′), 2.93 (m, 1H, pyrrolidine H-2′), 2.74–2.69 (m, 2H, pyrrolidine H-5′), 2.28–2.23 (m, 1H, pyrrolidine H-4′), 2.11–2.04 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 175.2 (C-2), 166.8 (C-7a), 140.4 (phenyl-sulfonyl C-1″), 135.2 (C-3a), 133.0 (phenyl-sulfonyl C-4″), 129.5 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 128.4 (C-6), 127.1 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 122.5 (C-4), 121.7 (C-5), 109.6 (C-7), 55.8 (C-3), 48.3 (pyrrolidine C-2′), 47.2 (pyrrolidine C-5′), 33.0 (pyrrolidine C-4′). MS (DUIS+) m/z 329 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C17H17N2O3S: 329.0960; found: 329.0949.
4.1.2. Synthesis of 1′-(Phenylsulfonyl)spiro[indoline-3,3′-pyrrolidin]-2-one (18)
To the solution of 2-(phenylsulfonyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (29) (312 mg, 1 mmol) in tetrahydrofuran: water (10:10 mL) was added 10 mL glacial acetic acid at 0 °C. N-bromosuccinimide (178 mg, 1 mmol, 1 equiv.) was added slowly in portions at 0 °C. The reaction mixture was stirred for 1.5 h at 0 °C. Afterwards the reaction was quenched at 0 °C by 10 mL concentrated sodium-carbonate solution and was stirred for 0.5 h, allowing to warm up to room temperature. The mixture was extracted using ethyl acetate (2 × 10 mL). The combined organic phases were washed with concentrated sodium-hydrogen carbonate (2 × 10 mL) and by brine (2 × 10 mL). The organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The crude product was purified using flash chromatography (gradient: hexane: ethyl acetate 0% to 20% ethyl acetate) to afford 18 as a colorless solid. Yield: 180 mg (55%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 10.47 (s, 1H, NH), 7.87 (d, J = 7.3 Hz, 2H, phenyl-sulfonyl ArH-3″, ArH-5″), 7.76 (t, J = 7.3 Hz, 1H, phenyl-sulfonyl ArH-4″), 7.67 (t, J = 7.3 Hz, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.15 (t, J = 7.3 Hz, 1H, ArH-4), 6.82 (t, J = 8.5 Hz, 2H, ArH-5, ArH-6), 6.69 (d, J = 7.3 Hz, 1H, ArH-7), 3.56 (t, J = 6.7 Hz, 1H, pyrrolidine H-2′), 3.35 (t, J = 7.3 Hz, 1H, pyrrolidine H-2′), 3.35 (d, J = 3.5 Hz, 2H, pyrrolidine H-5′), 2.10–2.05 (m, 1H, pyrrolidine H-4′), 1.94–1.88 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 181.4 (C-2), 167.0 (C-3a), 141.3 (C-7a), 135.8 (phenyl-sulfonyl C-1″), 133.3 (phenyl-sulfonyl C-4″), 129.5 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 128.3 (C-6), 127.4 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 122.4 (C-4), 121.8 (C-5), 109.6 (C-7), 55.6 (C-3), 47.4 (pyrrolidine C-2′, pyrrolidine C-5′), 35.8 (pyrrolidine C-4′). MS (DUIS+) m/z 329 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C17H17N2O3S: 329.0960; found: 329.0957.
4.1.3. Procedures to Afford 1′-Benzyl-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (39a)
To the solution of 1′-benzylspiro[indoline-3,3′-pyrrolidin]-2-one (37a) (500 mg, 1.7985 mmol) in 15 mL absolutized tetrahydrofuran was added borane/tetrahydrofuran (1M) complex solution (4.495 mL, 4.495 mmol, 2.5 equiv.) and the reaction mixture was refluxed overnight. The reaction was allowed to cool down to room temperature and was quenched using 30 mL concentrated ammonium-chloride solution and was stirred for 20 min. Afterwards, 20 mL ethyl acetate was used for extraction. The combined organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure to afford 400 mg 38a, used as a crude product in the further steps without purification. To the solution of crude 1′-benzylspiro[indoline-3,3′-pyrrolidine] (38a) (400 mg 1.513 mmol) (in anhydrous dichloromethane was added triethylamine (0.419 mL, 3.03 mmol, 2 equiv.) and N,N-dimethylpyridin-4-amine (10 mg, 0.076 mmol, 0.5 equiv.) at room temperature. Afterwards benzenesulfonyl chloride (0.203 mL, 1.59 mmol, 1.05 equiv.) was added to the solution at 5 °C. The reaction mixture was stirred at room temperature overnight. The reaction was quenched using 10 mL 10% HCl solution and was extracted by 10 mL dichloromethane. The dichloromethane phase was collected and dried over sodium-sulfate. The drying agent was filtered-off and the solution was evaporated under reduced pressure. The crude product was purified by flash chromatography to afford 39a as yellow oil. Yield: 621 mg (85% calculated to 37a). 1H-NMR (500 MHz, DMSO-d6) δ ppm 7.73 (d, J = 7.6 Hz, 2H, phenyl-sulfonyl H-2″, phenyl-sulfonyl H-6″), 7.58 (t, J = 7.6 Hz, 1H, H-6), 7.46 (t, J = 7.9 Hz, 3H, phenyl-sulfonyl H-3″, phenyl-sulfonyl H-4″, phenyl-sulfonyl H-5″), 7.31 (t, J = 7.6 Hz, 2H, phenyl-sulfonyl H-3″, phenyl-sulfonyl H-5″), 7.27–7.21 (m, 5H, benzyl H-1-5), 7.05 (t, J = 7.6 Hz, 1H, H-5), 3.89 (d, J = 10.9 Hz, 1H, H-2), 3.78 (d, J = 10.9 Hz, 1H, H-2), 3.52 (s, 2H, benzyl CH2), 2.69 (m, 1H, pyrrolidine H-2′), 2.57 (m, 1H, pyrrolidine H-2′), 2.24 (d, J = 9.1 Hz, 1H, pyrrolidine H-5′), 2.15 (d, J = 9.2 Hz, 1H, pyrrolidine H-5′), 1.91–1.85 (m, 1H, pyrrolidine H-4′), 1.78–1.73 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 141.0 (C-3a), 139.0 (C-7a), 134.2 (benzyl C-1), 129.8 (phenyl-sulfonyl C-1″), 129.8 (phenyl-sulfonyl C-4″), 128.7 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 128.7 (benzyl C-3, benzyl C-5), 127.4 (benzyl C-2, benzyl C-6), 127.4 (C-6), 124.9 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″, benzyl C-4), 124.2 (C-4), 121.8 (C-5), 114.6 (C-7), 66.7 (pyrrolidine C-2′), 63.5 (benzyl CH2), 59.3 (C-3), 53.3 (pyrrolidine C-5′), 49.8 (C-4′). MS (DUIS+) m/z 405 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H25N2O2S: 405.1637; found: 405.1620.
4.1.4. Procedures to Afford 1-(Phenylsulfonyl)-1′-(pyridin-4-ylmethyl)spiro[indoline-3,3′-pyrrolidine] (39b)
Synthesis of 1′-(Pyridin-4-ylmethyl)spiro[indoline-3,3′-pyrrolidin]-2-one (37b)
To the solution of the crude 2-(pyridin-4-ylmethyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (36b) (812 mg) in tetrahydrofuran:water (10:10 mL) was added 10 mL glacial acetic acid at 0 °C. N-bromosuccinimide (549 mg, 3.0874 mmol) was added slowly in portions at 0 °C. The reaction mixture was stirred for 1.5 h at 0 °C. Afterwards the reaction was quenched at 0 °C by 20 mL concentrated sodium-carbonate solution and was stirred for 0.5 h, allowing to warm up to room temperature. The mixture was extracted using ethyl-acetateethyl acetate (2 × 10 mL). The combined organic phases were washed with concentrated sodium-hydrogen carbonate (2 × 10 mL) and by brine (2 × 10 mL). The organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The crude product was purified using flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) to afford 37b as a yellow solid. Yield: 293 mg (35% calculated to 25). 1H-NMR (500 MHz, DMSO-d6) δ ppm 11.63 (s, 1H, NH), 8.52 (d, J = 5.6 Hz, 2H, pyridine ArH-2, pyridine ArH-6), 7.59 (d, J = 7.2 Hz, 1H, ArH-4), 7.40 (d, J = 8.0 Hz, 1H, ArH-7), 7.32 (d, J = 5.5 Hz, 2H, pyridine ArH-3, pyridine ArH-5), 7.23 (t, J = 7.6 Hz, 1H, ArH-6), 7.05 (t, J = 7.3 Hz, 1H, ArH-5), 4.72 (s, 2H, pyridyl-methyl CH2), 3.65 (t, J = 6.7 Hz, 2H, pyrrolidine H-2′), 3.31 (m, 2H, pyrrolidine H-5′), 3.02 (t, J = 6.7 Hz, 2H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 179.3 (C=O), 148.5 (pyridine C-2, pyridine C-6), 147.2 (pyridine C-1), 141.6 (C-7a), 134.7 (C-3a), 128.1 (C-6), 123.5 (C-4), 122.4 (pyridine C-3, pyridine C-5), 109.7 (C-7), 61.2 (pyridyl-methylene CH2), 60.1 (C-3), 54.4 (pyrrolidine C-2′), 53.6 (pyrrolidine C-5′), 34.7 (pyrrolidine C-4′). MS (DUIS+) m/z 280 [M + H]+.
Synthesis of 1-(Phenylsulfonyl)-1′-(pyridin-4-ylmethyl)spiro[indoline-3,3′-pyrrolidine] (39b)
To the solution of 1′-(pyridin-4-ylmethyl)spiro[indoline-3,3′-pyrrolidin]-2-one (37b) (289 mg, 1.05 mmol) in 10 mL anhydrousdry tetrahydrofuran was added borane/tetrahydrofuran (1 M) complex solution (2.63 mL, 2.63 mmol, 2.5 equiv.) and the reaction mixture was refluxed overnight. The reaction was allowed to cool down to room temperature and was quenched using 15 mL concentrated ammonium-chloride solution and was stirred for 20 min. Afterwards, 10 mL ethyl acetate was used for extraction. The combined organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure to afford 292 mg 38b, used as a crude product in the further steps without purification. To the solution of crude 1′-(pyridin-4-ylmethyl)spiro[indoline-3,3′-pyrrolidine] (38b) (0.292 mg) in 5 mL anhydrous dichloromethane was added triethylamine (0.307 mL, 2.22 mmol) and N,N-dimethylpyridin-4-amine (6.8 mg, 0.0555 mmol) at room temperature. Afterwards benzenesulfonyl chloride (204 mg, 1.161 mmol) was added to the solution at 5 °C. The reaction mixture was stirred at room temperature overnight. The reaction was quenched using 10 mL 10% HCl solution and was extracted by 5 mL dichloromethane. The dichloromethane phase was collected and dried over sodium-sulfate. The drying agent was filtered-off and the solution was evaporated under reduced pressure. The crude product was purified by flash chromatography to afford 39b as yellow solid. Yield: 0.003 mg. 1H-NMR (500 MHz, DMSO-d6) δ ppm 8.50 (d, J = 6.2 Hz, 2H, pyridine ArH-2, pyridine ArH-6), 7.72 (d, J = 7.9 Hz, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.58–7.53 (m, 3H, pyridine ArH-3, pyridine ArH-5, phenyl-sulfonyl ArH-4″), 7.49 (d, J = 7.8 Hz, 1H, ArH-7), 7.43 (t, J = 8.0 Hz, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.28–7.23 (m, 2H, ArH-4, ArH-6), 7.07 (t, J = 7.1 Hz, 1H, ArH-5), 3.90 (d, J = 11.1 Hz, 1H, pyridyl methyl CH2), 3.83 (d, J = 11.1 Hz, 1H, pyridyl methyl CH2), 3.74 (d, J = 15.2 Hz, 1H, H-2), 3.67 (d, J = 15.2 Hz, 1H, H-2), 2.77 (m, 1H, pyrrolidine H-2′), 2.62 (m, 1H, pyrrolidine H-2′), 2.17 (m, 2H, pyrrolidine H-5′), 1.95–1.89 (m, 1H, pyrrolidine H-4′), 1.82–1.78 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 148.6 (pyridine C-2, pyridine C-6), 141.0 (pyridine C-1), 143.9 (C-3a), 143.1 (C-7a), 139.5 (phenyl-sulfonyl C-1″), 134.1 (phenyl-sulfonyl C-4″), 130.0 (phenyl-sulfonyl C-3,” phenyl-sulfonyl C-5″), 128.1 (C-6), 127.3 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6”), 127.6 (C-4), 124.2 (pyridine C-3, pyridine C-5), 124.8 (C-5), 114.1 (C-7), 65.6 (pyrrolidine C-2′), 61.5 (benzyl CH2), 60.1 (C-2), 53.0 (C-3), 51.3 (pyrrolidine C-5′), 32.9 (pyrrolidine C-4′). MS (DUIS+) m/z 406 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C23H24N3O2S: 406.1589; found: 406.1586.
4.1.5. Synthesis of 1′-Methyl-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (40a)
The solution of 1′-benzyl-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (39a) (2 mg, 0.495 mmol) in 5 mL methanol was filled into an autoclave. Palladium (10%) on charcoal (100 mg, 5 w/w %) was added to the solution and reaction was stirred overnight under 5 bar hydrogen atmosphere at 50 °C. The reaction mixture was filtered through a pad of Celite and was evaporated under reduced pressure. The crude residual was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) to afford 40a as a colorless solid. Yield: 37 mg (23%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 7.79 (d, J = 7.5 Hz, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.66 (t, J = 7.2 Hz, 1H, phenyl-sulfonyl ArH-4″), 7.56 (t, J = 7.9 Hz, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.48 (d, J = 7.9 Hz, 1H, ArH-7), 7.22 (m, 2H, ArH-4, H-6), 7.03 (t, J = 7.9 Hz, 1H, ArH-5), 3.87 (d, J = 11.8 Hz, 1H, H-2), 3.75 (d, J = 11.8 Hz, 1H, H-2), 2.62 (m, 1H, pyrrolidine H-2′), 2.44 (m, 1H, pyrrolidine H-2′), 2.24 (d, J = 9.1 Hz, 1H, pyrrolidine H-5′), 2.17 (s, 3H, methyl CH3), 2.15 (d, J = 9.1 Hz, 1H, pyrrolidine H-5′), 1.85–1.80 (m, 1H, pyrrolidine H-4′), 1.74–1.69 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 143.1 (C-3a, C-7a), 133.1 (phenyl-sulfonyl C-1″, phenyl-sulfonyl C-4″), 129.2 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 127.5 (C-6, phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 124.1 (C-4), 114.5 (C-5), 110.0 (C-7), 69.1 (pyrrolidine C-2′), 60.2 (pyrrolidine C-5′), 55.5 (C-3), 52.8 (C-2), 42.3 (pyrrolidine N(1′)-methyl), 32.5 (pyrrolidine C-4′). MS (DUIS+) m/z 329 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C18H21N2O2S: 343.1324; found: 343.1320.
4.1.6. Synthesis of 1′-Ethyl-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (40b)
The solution of 1′-benzyl-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (39a) (100 mg, 0.2475 mmol) in 5 mL methanol was filled into an autoclave. Palladium (10%) on charcoal (500 mg, 5 w/w %) was added to the solution and reaction was stirred overnight under 5 bar hydrogen atmosphere at 50 °C. The reaction mixture was filtered through a pad of Celite and was evaporated under reduced pressure. The crude residual was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) to afford 40b as a colorless solid. Yield: 19 mg (22%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 7.78 (m, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.66 (m, 1H, phenyl-sulfonyl ArH-4″), 7.56 (m, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.50–7.47 (m, 1H, ArH-7), 7.24–7.21 (m, 2H, ArH-4, ArH-6), 7.04 (t, J = 7.6 Hz, 1H, ArH-5), 3.86 (d, J = 10.8 Hz, 1H, H-2), 3.75 (d, J = 10.8 Hz, 1H, H-2), 2.68 (m, 1H, pyrrolidine H-2′), 2.44 (m, 1H, pyrrolidine H-2′), 2.34 (q, J = 6.4 Hz, 2H, ethyl CH2), 2.24 (m, 1H, pyrrolidine H-5′), 2.11 (q, J = 8.9 Hz, 1H, pyrrolidine H-5′), 1.86–180 (m, 1H, pyrrolidine H-4′), 1.73–167 (m, 1H, pyrrolidine H-4′), 0.93 (t, J = 7.6 Hz, 3H, ethyl CH3). 13C-NMR (125 MHz, DMSO-d6) δ ppm 143.3 (C-3a, C-7a), 133.1 (phenyl-sulfonyl C-1″, phenyl-sulfonyl C-4″), 129.5 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 127.7 (C-6, phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 124.1 (C-4), 114.1 (C-5), 110.1 (C-7), 69.1 (pyrrolidine C-2′), 60.2 (pyrrolidine C-5′), 55.3 (C-3), 52.1 (C-2), 50.0 (pyrrolidine N(1′)-ethyl CH2), 32.5 (pyrrolidine C-4′), 13.6 (pyrrolidine N(1′)-ethyl CH3). MS (DUIS+) m/z 343 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C19H23N2O2S: 343.1480; found: 343.1496.
4.1.7. Procedures to Afford 1′-(2-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44a)
Synthesis of 1′-(2-Fluorobenzyl)spiro[indoline-3,3′-pyrrolidin]-2-one (42a)
To the solution of 41a (1.54 g, 5.5 mmol) in tetrahydrofuran: water (30:30 mL) was added 30 mL glacial acetic acid at 0 °C. N-bromosuccinimide (979 mg, 5.5 mmol, 1 equiv.) was added slowly in portions at 0 °C. The reaction mixture was stirred for 1.5 h at 0 °C. Afterwards the reaction was quenched at 0 °C by 40 mL concentrated sodium-carbonate solution and was stirred for 0.5 h, allowing to warm up to room temperature. The mixture was extracted using ethyl acetate (2 × 20 mL). The combined organic phases were washed with concentrated sodium-hydrogen carbonate (2 × 20 mL) and by brine (2 × 20 mL). The organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The product 42a was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 970 mg (56%). 1H-NMR (500 MHz, DMSO-d6) δ ppm 10.31 (s, 1H, NH), 8.01 (d, J = 8.8 Hz, 1H, benzyl ArH-6), 7.77 (d, J = 7.9 Hz, 1H, ArH-4), 7.65 (t, J = 7.9 Hz, 1H, benzyl ArH-4), 7.47 (t, J = 6.2 Hz, 1H, benzyl ArH-5), 7.24 (t, J = 8.4 Hz, 1H, ArH-6), 7.16 (m, 1H, benzyl ArH-3), 6.94 (t, J = 7.5 Hz, 1H, ArH-5), 6.80 (d, J = 7.5 Hz, 1H, ArH-7), 3.75 (s, 2H, benzyl CH2), 3.06 (m, 1H, pyrrolidine H-2′), 2.76 (d, J = 8.9 Hz, 1H, pyrrolidine H-5′), 2.69 (d, J = 8.9 Hz, 1H, pyrrolidine H-5′), 2.61 (q, J = 7.9 Hz, 1H, pyrrolidine H-2’), 2.20–2.15 (m, 1H, pyrrolidine H-4′), 1.92–1.87 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 181.5 (C=O), 178.0 (benzyl C-2), 141.6 (C-7a), 137.0 (C-3a), 135.5 (benzyl C-6), 130.2 (benzyl C-4), 127.6 (C-6), 124.7 (benzyl C-1), 123.9 (benzyl C-5), 123.3 (C-4), 122.3 (C-5), 115.7 (benzyl C-3), 109.6 (C-7), 64.0 (C-3), 53.86 (benzyl CH2), 52.9 (pyrrolidine C-2′), 51.7 (pyrrolidine C-5′), 37.1 (pyrrolidine C-4′). MS (DUIS+) m/z 297 [M + H]+.
Synthesis of 1′-(2-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44a)
Compound 43a was prepared according to the same procedure as 38a, starting from 42a (100 mg, 0.3378 mmol), using borane-tetrahydrofuran (0.6757 mL, 0.6757 mmol, 2.5 equiv.), to afford 10 mg crude product used in the next step without further purification. Compound 44a was prepared according to the same procedure as 39a, starting from crude 43a (10 mg, 0.03545 mmol), using benzylsulfonyl chloride (50 mg, 0.07445 mmol, 1.05 equiv.), triethylamine (0.010 mL, 0.0709 mmol, 2 equiv.) and N,N-dimethylpyridin-4-amine (0.2 mg, 0.0017 mmol, 0.5 equiv.). The product was purified by preparative-HPLC (gradient: water: acetonitrile 0% to 100% acetonitrile) Yield: 2 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 8.22 (t, J = 6.7 Hz, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.88 (m, 1H, phenyl-sulfonyl ArH-4″), 7.69 (m, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.53 (t, J = 6.2 Hz, 1H, benzyl ArH-4), 7.47 (m, 1H, ArH-7), 7.33 (t, J = 7.8 Hz, 1H, benzyl ArH-5), 7.23 (t, J = 7.8 Hz, 1H, benzyl ArH-6), 7.17–7.08 (m, 2H, ArH-5, ArH-6), 6.73 (d, J = 7.0 Hz, 2H, benzyl ArH-3), 4.22 (m, 2H, H-2), 3.98 (m, 2H, benzyl CH2), 3.75 (m, 1H, pyrrolidine H-2′), 3.40 (m, 1H, pyrrolidine H-2′), 3.14 (m, 1H, pyrrolidine H-5′), 2.98 (m, 1H, pyrrolidine H-5′), 2.28 (m, 1H, pyrrolidine H-4′), 1.51 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, CDCl3) δ ppm 159.3 (benzyl C-2), 144.1 (C-3a), 142.5 (C-7a), 137.3 (phenyl-sulfonyl C-1″), 134.2 (phenyl-sulfonyl C-4″), 130.3 (benzyl C-6), 129.5 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 129.2 (benzyl C-4), 128.2 (C-6), 127.6 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 125.8 (benzyl C-1), 125.3 (benzyl C-5), 125.0 (C-4), 121.6 (C-5), 115.7 (benzyl C-3), 113.0 (C-7), 62.3 (C-2′), 59.6 (benzyl CH2), 55.8 (C-3), 54.9 (C-5′), 53.6 (C-2), 33.1 (C-4′). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2FS: 423.1543; found: 423.1531.
4.1.8. Procedures to Afford 1′-(3-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44b)
Synthesis of 1′-(3-Fluorobenzyl)spiro[indoline-3,3′-pyrrolidin]-2-one (42b)
To the solution of 41b (1.681 g, 6 mmol) in tetrahydrofuran: water (30:30 mL) was added 30 mL glacial acetic acid at 0 °C. N-bromosuccinimide (1.069 g, 6 mmol, 1 equiv.) was added slowly in portions at 0 °C. The reaction mixture was stirred for 1.5 h at 0 °C. Afterwards the reaction was quenched at 0 °C by 40 mL concentrated sodium-carbonate solution and was stirred for 0.5 h, allowing to warm up to room temperature. The mixture was extracted using ethyl acetate (2 × 20 mL). The combined organic phases were washed with concentrated sodium-hydrogen carbonate (2 × 20 mL) and by brine (2 × 20 mL). The organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The product 42b was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 1.31 g (76% calculated to 29). 1H-NMR (500 MHz, DMSO-d6) δ ppm 10.31 (s, 1H, NH), 7.38–7.33 (m, 2H, benzyl H-5, benzyl ArH-6), 7.21–7.16 (m, 3H, ArH-4, benzyl H-2, benzyl ArH-4), 7.04 (m, 1H, ArH-6), 6.98 (t, J = 7.0 Hz, 1H, ArH-5), 6.81 (d, J = 8.0 Hz, 1H, ArH-7), 3.72 (s, 2H, benzyl CH2), 3.07 (m, 1H, pyrrolidine H-2′), 2.74 (d, J = 8.8 Hz, 1H, pyrrolidine H-5′), 2.66 (d, J = 8.8 Hz, 1H, pyrrolidine H-2′), 2.56 (m, 1H, pyrrolidine H-5′), 2.20 (m, 1H, pyrrolidine H-4′), 1.90 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, CDCl3) δ ppm 181.96 (C=O), 161.2 (benzyl C-3), 142.3 (C-7a), 141.4 (benzyl C-1), 130.4 (benzyl C-5), 127.8 (benzyl-C6, C-6), 122.1 (benzyl C-2, benzyl C-4), 109.4 (C-7), 64.7 (benzyl CH2), 63.9 (C-3), 53.9 (pyrrolidine C-2′), 52.7 (C-5′), 36.8 (pyrrolidine C-4′). MS (DUIS+) m/z 297 [M + H]+.
Synthesis of 1′-(3-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44b)
Compound 43b was prepared according to the same procedure as 38a, starting from 42b (385 mg, 1.3 mmol), using borane-tetrahydrofuran (3.250 mL, 3.25 mmolm 2.5 equiv.), to afford 338 mg crude product used in the next time without further purification. Compound 44b was prepared according to the same procedure as 39a, starting from crude 43b (338 mg, 1.197 mmol), using benzylsulfonyl chloride (221 mg, 1.2579 mmol, 1.05 equiv.), triethylamine (0.331 mL, 2.396 mmol, 2 equiv.) and N,N-dimethylpyridin-4-amine (7 mg, 0.0599 mmol, 0.5 equiv.). The product was purified by preparative HPLC (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 11 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 7.83 (d, J = 8.2 Hz, 1H, ArH-7), 7.77 (d, J = 7.6 Hz, 1H, benzyl ArH-6), 7.73 (d, J = 7.6 Hz, 1H, ArH-4), 7.64 (d, J = 8.2 Hz, 1H, benzyl ArH-4), 7.58–7.53 (m, 1H, benzyl ArH-5), 7.50–7.39 (m, 3H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-4″, phenyl-sulfonyl ArH-5″), 7.37–7.27 (m, 2H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″), 7.16–7.12 (m, 1H, phenyl-sulfonyl ArH-6), 7.09–7.02 (m, 1H, phenyl-sulfonyl ArH-5), 6.99 (m, 1H, benzyl H-2), 4.23 (m, 1H, H-2), 4.11 (m, 1H, H-2), 3.93–3.71 (m, 2H, benzyl CH2), 3.23 (m, 1H, pyrrolidine H-2′), 2.94 (m, 1H, pyrrolidine H-2′), 2.83 (m, 1H, pyrrolidine H-5′), 2.69 (m, 1H, pyrrolidine H-5′), 2.39–2.25 (m, 1H, pyrrolidine H-4′), 2.18 (m, 1H, pyrrolidine H-5′). 13C-NMR (125 MHz, CDCl3) δ ppm 163.0 (benzyl C-3), 143.6 (C-3a), 143.5 (C-7a), 137.6 (phenyl-sulfonyl C-1″), 134.2 (phenyl-sulfonyl C-4″), 130.4 (benzyl C-5), 129.5 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 129.1 (C-6), 128.0 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 127.7 (benzyl C-6), 125.6 (benzyl C-1), 125.3 (C-4), 124.1 (C-5), 121.8, 115.8 (benzyl C-2), 113.4 (C-7), 113.3 (benzyl C-4), 61.2 (benzyl CH2), 59.9 (pyrrolidine C-2′), 55.3 (C-3), 54.8 (pyrrolidine C-5′), 53.6 (C-2), 33.1 (pyrrolidine C-4’). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2SF: 423.1543; found: 423.1558.
4.1.9. Procedures to Afford 1′-(4-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44c)
Synthesis of 1′-(4-Fluorobenzyl)spiro[indoline-3,3′-pyrrolidin]-2-one (42c)
To the solution of 41c (2.361 g, 8.4321 mmol) in tetrahydrofuran: water (30:30 mL) was added 30 mL glacial acetic acid at 0 °C. N-bromosuccinimide (1.501 g, 8.4321 mmol, 1 equiv.) was added slowly in portions at 0 °C. The reaction mixture was stirred for 1.5 h at 0 °C. Afterwards the reaction was quenched at 0 °C by 40 mL concentrated sodium-carbonate solution and was stirred for 0.5 h, allowing to warm up to room temperature. The mixture was extracted using ethyl acetate (2 × 20 mL). The combined organic phases were washed with concentrated sodium-hydrogen carbonate (2 × 20 mL) and by brine (2 × 20 mL). The organic phases were dried over sodium-sulfate, filtered and evaporated under reduced pressure. The product 42c was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 320 mg (19% calculated to 25). 1H-NMR (500 MHz, DMSO-d6) δ ppm 10.30 (s, 1H, NH), 7.41–7.35 (m, 3H, benzyl H-2, benzyl ArH-6, ArH-4), 7.16 (m, 3H, benzyl ArH-3, benzyl ArH-5, ArH-6), 6.96 (t, J = 7.2 Hz, 1H, ArH-5), 6.81 (d, J = 7.4 Hz, 1H, ArH-7), 5.73 (s, 2H, benzyl CH2), 3.03 (m, 1H, pyrrolidine H-5′), 2.73 (d, J = 9.3 Hz, 1H, pyrrolidine H-2′), 2.64 (d, J = 9.3 Hz, 1H, pyrrolidine H-2′), 2.58–2.53 (m, 1H, pyrrolidine H-5′), 2.21–2.16 (m, 1H, pyrrolidine H-4′), 1.95–1.87 (m, 1H, pyrrolidine H-4′) 13C-NMR (125 MHz, DMSO-d6) δ ppm 179.1 (C=O), 161.9 (benzyl C-4), 141.6 (C-7a), 138.2 (benzyl C-1), 134.6 (C-3a), 130.7 (benzyl C-2, benzyl C-6), 128.0 (C-6), 123.3 (C-4), 122.3 (C-2), 115.4 (benzyl C-3, benzyl C-5), 109.6 (C-7), 64.0 (benzyl CH2), 58.3 (C-3), 54.0 (pyrrolidine C-2′), 52.9 (pyrrolidine C-5′), 37.0 (pyrrolidine C-4′). MS (DUIS+) m/z 297 [M + H]+.
Synthesis of 1′-(4-Fluorobenzyl)-1-(phenylsulfonyl)spiro[indoline-3,3′-pyrrolidine] (44c)
Compound 43c was prepared according to the same procedure as 38a, starting from 42c (310 mg, 1.0473 mmol), using borane-tetrahydrofuran (2.620 mL, 2.620 mmol, 2.5 equiv.), to afford 461 mg crude product used in the next step without further purification. Compound 44c was prepared according to the same procedure as 39a, starting from crude 43c (100 mg, 0.354 mmol), using benzylsulfonyl chloride (131 mg, 0.745 mmol, 1.05 equiv.), triethylamine (0.099 mL, 0.7092 mmol) and N,N-dimethylpyridin-4-amine (2 mg, 0.01773 mmol). The product 44c was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 10 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 7.84–7.77 (m, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.74–7.70 (m, 1H, phenyl-sulfonyl ArH-4″), 7.67–7.57 (m, 3H, phenyl-sulfonyl ArH-2″, phenyl-sulfonyl ArH-6″, ArH-7), 7.50–7.45 (m, 2H, benzyl ArH-2, benzyl ArH-6), 7.43–7.40 (m, 1H, ArH-4), 7.24–7.20 (m, 1H, ArH-6), 7.16–7.12 (m, 2H, benzyl ArH-3, benzyl ArH-5), 7.09 (m, 1H, ArH-5), 4.39 (m, 1H, H-2), 4.29 (m, 1H, H-2), 4.18 (m, 1H, benzyl CH2), 413.06 (m, 1H, benzyl CH2), 3.90 (m, 1H, pyrrolidine H-2′), 3.70 (m, 1H, pyrrolidine H-2′), 3.06 (m, 1H), 2.68 (m, 1H, pyrrolidine H5′), 2.33 (m, 1H, pyrrolidine H-5′), 2.33 (m, 1H, pyrrolidine H-4′), 1.75 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, CDCl3) δ ppm 159.8 (benzyl C-4), 144.1 (C-3a), 142.6 (C-7a), 137.5 (benzyl C-1), 134.2 (phenyl-sulfonyl C-1″, phenyl-sulfonyl C-4″), 130.7 (benzyl C-2, benzyl C-6), 129.2 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 128.3 (C-6), 127.7 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 125.8 (C-4), 125.2 (C-5), 115.5 (benzyl C-3, benzyl C-5), 113.1 (C-7), 62.4 (pyrrolidine C-2′), 59.8 (benzyl CH2), 55.5 (C-3), 55.0 (pyrrolidine C-5′), 53.7 (C-2), 33.0 (pyrrolidine C-4′). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2SF: 423.1543; found: 423.1542.
4.1.10. Synthesis of 1′-Benzyl-1-((2-fluorophenyl)sulfonyl)spiro[indoline-3,3′-pyrrolidine] (46a)
Compound 46a was prepared according to the same procedure as 39a, starting from crude 38a (15 mg, 0.0567 mmol), using 2-fluorobenzylsulfonyl chloride (23 mg, 0.11819 mmol), triethylamine (0.015 mL, 0.1188 mmol, 2.1 equiv.) and N,N-dimethylpyridin-4-amine (0.7 mg, 0.006 mmol, 0.1 equiv.). The product was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 6.5 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 8.00 (t, J = 6.7 Hz, 1H, phenyl-sulfonyl ArH-4″), 7.53 (m, 3H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-6″, ArH-7), 7.41–7.34 (m, 5H, benzyl ArH-2, benzyl ArH-3, benzyl ArH-5, benzyl ArH-6, ArH-4), 7.27 (t, J = 8.1 Hz, 1H, phenyl-sulfonyl ArH-5″), 7.17 (t, J = 8.1 Hz, 1H, benzyl ArH-4), 7.10 (t, J = 9.4 Hz, 1H, ArH-6), 7.04 (t, J = 7.6 Hz, 1H, ArH-5), 4.16–4.07 (m, 5H, benzyl CH2, H-2, pyrrolidine H-2′), 3.34 (m, 1H, pyrrolidine H-2′), 3.17 (m, 1H, pyrrolidine H-5′), 3.05 (m, 1H, pyrrolidine H-5′), 2.36 (m, 1H, pyrrolidine H-4′), 2.14 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, DMSO-d6) δ ppm 155.6 (phenyl-sulfonyl C-2″), 149.7 (C-3a), 142.8 (C-7a), 133.2 (benzyl C-1), 131.8 (phenyl-sulfonyl C-4″), 131.0 (phenyl-sulfonyl C-6″), 129.6 (benzyl C-3, benzyl C-5), 128.4 (benzyl C-2, benzyl C-6), 124.2 (phenyl-sulfonyl C-5″), 122.9 (benzyl C-4), 122.5 (C-4), 121.0 (C-5), 120.4 (phenyl-sulfonyl C-1″), 116.8 (phenyl-sulfonyl C-3″), 111.3 (C-7), 63.2 (pyrrolidine C-2′), 62.6 (benzyl CH2), 55.1 (C-3), 52.5 (pyrrolidine C-5′), 49.2 (C-2), 37.1 (pyrrolidine C-4′). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2SF: 423.1543; found: 423.1550.
4.1.11. Synthesis of 1′-Benzyl-1-((3-fluorophenyl)sulfonyl)spiro[indoline-3,3′-pyrrolidine] (46b)
Compound 45b was prepared according to the same procedure as 39a, starting from crude 38a (15 mg, 0.0567 mmol), using 3-fluorobenzylsulfonyl chloride (23 mg, 0.1188 mmol, 2.1 equiv.), triethylamine (0.015 mL, 0.1188 mmol, 2.1 equiv.) and N,N-dimethylpyridin-4-amine (0.7 mg, 0.006 mmol, 0.1 equiv.). The product was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 5.5 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 8.25 (m, 1H, phenyl-sulfonyl ArH-2″), 7.65–7.58 (m, 2H, phenyl-sulfonyl ArH-5″, phenyl-sulfonyl ArH-6″), 7.48–7.38 (m, 6H, ArH-7, benzyl ArH-2–6), 7.29–7.25 (m, 2H, ArH-4, ArH-6), 7.21 (m, 1H, phenyl-sulfonyl ArH-4″), 7.08 (t, J = 7.6 Hz, 1H, ArH-5), 4.16 (d, J = 12.7 Hz, 1H, benzyl CH2), 4.09 (d, J = 12.7 Hz, 1H, benzyl CH2), 3.99 (d, J = 10.9 Hz, 1H, H-2), 3.85 (d, J = 10.9 Hz, 1H, H-2), 3.33–3.24 (m, 2H, pyrrolidine H-2′), 3.12 (d, J = 11.6 Hz, 1H, pyrrolidine H-5′), 3.00 (d, J = 11.6 Hz, 1H, pyrrolidine H-5′), 2.26–2.20 (m, 1H, pyrrolidine H-4′), 2.05–2.00 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, CDCl3) δ ppm 158.6 (phenyl-sulfonyl C-3″), 146.8 (C-3a), 145.4 (C-7a), 137.5 (benzyl C-1), 130.5 (phenyl-sulfonyl C-1″), 129.7 (phenyl-sulfonyl C-5″), 129.0 (benzyl C-3, benzyl C-5), 128.7 (benzyl C-2, benzyl C-6), 124.5 (C-6), 122.8 (benzyl C-4), 122.7 (phenyl-sulfonyl C-6″), 121.7 (C-4), 120.2 (C-5), 117.7 (phenyl-sulfonyl C-4″), 114.3 (phenyl-sulfonyl C-2″), 109.5 (C-7), 63.0 (pyrrolidine C-2′), 62.6 (benzyl CH2), 55.4 (C-3), 52.7 (pyrrolidine C-5′), 49.2 (C-2), 37.9 (pyrrolidine C-4′). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2SF: 423.1543; found: 423.1547.
4.1.12. Synthesis of 1′-Benzyl-1-((4-fluorophenyl)sulfonyl)spiro[indoline-3,3′-pyrrolidine] (46c)
Compound 46c was prepared according to the same procedure as 39a, starting from crude 38a (15 mg, 0.0567 mmol), using 4-fluorobenzylsulfonyl chloride (23 mg, 0.1182 mmol, 2.1 equiv.), triethylamine (0.015 mL, 0.1188 mmol, 2.1 equiv.) and N,N-dimethylpyridin-4-amine (0.7 mg, 0.006 mmol, 0.1 equiv.). The product was purified by flash chromatography (gradient: dichloromethane: methanol 0% to 10% methanol) Yield: 7.7 mg. 1H-NMR (500 MHz, CDCl3) δ ppm 7.80 (m, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.63 (d, J = 7.9 Hz, 1H, ArH-7), 7.47 (m, 2H, benzyl H-3, benzyl ArH-5), 7.40 (m, 3H, benzyl ArH-2, benzyl ArH-4, benzyl ArH-6), 7.28 (m, 2H, phenyl-sulfonyl ArH-3″, phenyl-sulfonyl ArH-5″), 7.09–7.05 (m, 3H, ArH-4, ArH-5, ArH-6), 4.10 (d, J = 12.3 Hz, 1H, H-2), 4.02–3.96 (m, 2H, benzyl CH2), 3.84 (d, J = 11.3 Hz, 1H, H-2), 3.25 (m, 1H, pyrrolidine H-2′), 3.16 (m, 1H, pyrrolidine H-2′), 2.94 (m, 1H, pyrrolidine H-5′), 2.89 (m, 1H, pyrrolidine H-5′), 2.24–2.18 (m, 1H, pyrrolidine H-4′), 2.03–1.99 (m, 1H, pyrrolidine H-4′). 13C-NMR (125 MHz, CDCl3) δ ppm 163.7 (phenyl-sulfonyl C-4″), 152.7 (C-3a), 150.6 (C-7a), 147.0 (benzyl C-1), 129.6 (phenyl-sulfonyl C-1″), 129.5 (phenyl-sulfonyl C-2″, phenyl-sulfonyl C-6″), 129.2 (benzyl C-3, benzyl C-5), 129.1 (benzyl C-2, benzyl C-6), 128.8 (C-6), 128.6 (benzyl C-4), 125.6 (C-4), 124.4 (C-5), 122.8 (phenyl-sulfonyl C-3″, phenyl-sulfonyl C-5″), 116.1 (C-7), 63.1 (pyrrolidine C-2′), 62.4 (benzyl CH2), 54.1 (C-3), 52.8 (pyrrolidine C-5′), 49.1 (pyrrolidine C-2′), 36.6 (pyrrolidine C-4′). MS (DUIS+) m/z 423 [M + H]+. HRMS (ESI+): m/z [M + H]+ calcd. for C24H24N2O2SF: 423.1543; found: 423.1563.
4.2. General Procedure for the Serotonergic Screening Assays
4.2.1. Cell Culture and Preparation of Cell Membranes
HEK293 cells with stable expression of human serotonin 5-HT1A, 5-HT6 or 5-HT7b receptors (all prepared with the use of Lipofectamine 2000) or CHO-K1 cells with plasmid containing the sequence coding for the human serotonin 5-HT2A receptor (PerkinElmer) were maintained at 37 °C in a humidified atmosphere with 5% CO2 and were grown in Dulbeco’s Modifier Eagle Medium containing 10% dialysed fetal bovine serum and 500 μg/mL G418 sulphate. For membranes preparations, cells were subcultured in 150 cm2 flasks, grown to 90% confluence, washed twice with prewarmed to 37 °C phosphate buffered saline (PBS) and were pelleted by centrifugation (200 g) in PBS containing 0.1 mM EDTA and 1 mM dithiothreitol. Prior to membrane preparations pellets were stored at −80 °C.
4.2.2. Radioligand Binding Assays
Cell pellets were thawed and homogenized in 10 volumes of assay buffer using an Ultra Turrax tissue homogenizer and centrifuged twice at 35,000 g for 20 min at 4 °C, with incubation for 15 min at 37 °C in between. The composition of the assay buffers was as follows: for 5-HT1AR: 50 mM Tris–HCl, 0.1 mM EDTA, 4 mM MgCl2, 10 µM pargyline and 0.1% ascorbate; for 5-HT2AR: 50 mM Tris–HCl, 0.1 mM EDTA, 4 mM MgCl2 and 0.1% ascorbate; for 5-HT6R: 50 mM Tris–HCl, 0.5 mM EDTA and 4 mM MgCl2, for 5-HT7bR: 50 mM Tris–HCl, 4 mM MgCl2, 10 µM pargyline and 0.1% ascorbate. All assays were incubated in a total volume of 200 μL in 96-well microtiter plates for 1 h at 37 °C, except for 5-HT1AR and 5-HT2AR which were incubated at room temperature for 1 h and 1.5 h respectively. The process of equilibration is terminated by rapid filtration through Unifilter plates with a 96-well cell harvester and radioactivity retained on the filters was quantified on a Microbeta plate reader (PerkinElmer).
For displacement studies the assay samples contained as radioligands: 2.5 nM [3H]-8-OH-DPAT (5002.4 GBq/mmol) for 5-HT1AR; 1 nM [3H]-Ketanserin (1975.8 GBq/mmol) for 5-HT2AR; 2 nM [3H]-LSD (3093.2 GBq/mmol) for 5-HT6R or 0.8 nM [3H]-5-CT (1450.4 GBq/mmol) for 5-HT7R. Non-specific binding is defined with 10 µM of 5-HT in 5-HT1AR and 5-HT7R binding experiments, whereas 10 µM of chlorpromazine or 10 µM of methiothepine were used in 5-HT2AR and 5-HT6R assays, respectively. Each compound was tested in triplicate at 7–8 concentrations (10−11–10−4 M). The inhibition constants (Ki) were calculated from the Cheng-Prusoff equation [47]. Results were expressed as means of at least three separate experiments (SD ≤ 24%).
4.2.3. Docking Procedure
The homology model of h-5HT6R [15] was prepared for docking experiment my Schrödinger’s Protein Preparation Wizard by default settings for protomer-state optimization and restrained minimization by OPLS_2005 force field. Single precision docking was performed with Schrödinger’s Glide by default settings. The ligand (46a) was prepared for docking by Schrödinger’s LigPrep, generating possible ionization states (at pH range of 7.0 ± 2.0), tautomers and stereoisomers. Constrained docking was set to two required interactions (at least 1 match): hydrogen bond formed with Asn2886.55 and/or hydrogen bond formed with Ser1935.43.
Acknowledgments
The authors are grateful to László Drahos and Ágnes Gömöry (RCNS, Hungary) for the exact mass (high-resolution MS) measurements and we are thankful for Aaron Keeley (RCNS, Hungary) for carefully reading the manuscript and for Stefan Mordalski (Institute of Pharmacology, Poland) for helpful discussions. The support of the National Brain Research Program (KTIA_NAP_13-1-2013-0001) is acknowledged.
Author Contributions
G.M.K. and A.J.B. conceived the study; Á.A.K. performed the syntheses, G.S. performed the in vitro measurements.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors.
References
- 1.Russel J.S. Oxindoles and spirocyclic variations: Strategies for C3 functionalization. Top. Heterocycl. Chem. 2011;26:397–432. doi: 10.1007/7081. [DOI] [Google Scholar]
- 2.Bindra J.S. Oxindole alkaloids. Alkaloids Chem. Physiol. 1973;14:83–121. doi: 10.1016/S1876-081360219-5. [DOI] [Google Scholar]
- 3.ChEMBL. [(accessed on 12 October 2017)]; Available online: Https://www.ebi.ac.uk/chembl/
- 4.Kelemen Á.A., Ferenczy G.G., Keseru G.M. A desirability function-based scoring scheme for selecting fragment-like class A aminergic GPCR ligands. J. Comput. Aided Mol. Des. 2015;29:59–66. doi: 10.1007/s10822-014-9804-5. [DOI] [PubMed] [Google Scholar]
- 5.Kelemen Á.A., Kiss R., Ferenczy G.G., Kovács L., Flachner B., Lorincz Z., Keseru G.M. Structure-based consensus scoring scheme for selecting class A aminergic GPCR fragments. J. Chem. Inf. Model. 2016;56:412–422. doi: 10.1021/acs.jcim.5b00598. [DOI] [PubMed] [Google Scholar]
- 6.Upton N., Chuang T.T., Hunter A.J., Virley D.J. 5-HT6 receptor antagonists as novel cognitive enhancing agents for Alzheimer’s disease. Neurotherapeutics. 2008;5:458–469. doi: 10.1016/j.nurt.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Messina D., Annesi G., Serra P., Nicoletti G., Pasqua A., Annesi F., Tomaino C., Cirò-Candiano I.C., Carrideo S., Caracciolo M., et al. Association of the 5-HT6 receptor gene polymorphism C267T with Parkinson’s disease. Neurology. 2002;58:828–829. doi: 10.1212/WNL.58.5.828. [DOI] [PubMed] [Google Scholar]
- 8.Codony X., Vela J.M., Ramírez M.J. 5-HT6 receptor and cognition. Curr. Opin. Pharmacol. 2011;11:94–100. doi: 10.1016/j.coph.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Holenz J., Mercè R., Díaz J.L., Guitart X., Codony X., Dordal A., Romero G., Torrens A., Mas J., Andaluz B., et al. Medicinal chemistry driven approaches toward novel and selective serotonin 5-HT6 receptor ligands. J. Med. Chem. 2005;48:1781–1795. doi: 10.1021/jm049615n. [DOI] [PubMed] [Google Scholar]
- 10.Kobilka B.K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta Biomembr. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivachtchenko A.V. Sulfonyl-containing modulators of serotonin 5-HT6 receptors and their pharmacophore models. Russ. Chem. Rev. 2014;83:439–473. doi: 10.1070/RC2014v083n05ABEH004371. [DOI] [Google Scholar]
- 12.Staroń J., Warszycki D., Kalinowska-Tłuścik J., Satała G., Bojarski A.J. Rational design of 5-HT6R ligands using a bioisosteric strategy: Synthesis, biological evaluation and molecular modelling. RSC Adv. 2015;5:25806–25815. doi: 10.1039/C5RA00054H. [DOI] [Google Scholar]
- 13.Mcule. [(accessed on 12 October 2017)]; Available online: https://mcule.com/
- 14.Small-Molecule Drug Discovery Suite 2017-3. Schrödinger, LLC; New York, NY, USA: 2017. [Google Scholar]
- 15.Vass M., Jójárt B., Bogár F., Paragi G., Keseru G.M., Tarcsay Á. Dynamics and structural determinants of ligand recognition of the 5-HT6 receptor. J. Comput. Aided Mol. Des. 2015;29:1137–1149. doi: 10.1007/s10822-015-9883-y. [DOI] [PubMed] [Google Scholar]
- 16.Wacker D., Wang C., Katritch V., Han G.W., Huang X.-P., Vardy E., McCorvy J.D., Jiang Y., Chu M., Siu F.Y., et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelemen Á.A., Mordalski S., Bojarski A.J., Keserű G.M. Computational Modeling of Drugs against Alzheimer’s Disease. Vol. 132. Humana Press; New York, NY, USA: 2017. Computational modeling of drugs for Alzheimer’s disease: Design of serotonin 5-HT6 antagonists; pp. 419–461. [Google Scholar]
- 18.Laronze J.-Y., Bascop S.I., Sápi J., Levy J. On the synthesis of the oxindole alkaloid: (±)-Horsfiline. Heterocycles. 1994;38 doi: 10.3987/COM-93-6639. [DOI] [Google Scholar]
- 19.Overman L.E., Rosen M.D. Total synthesis of (−)-spirotryprostatin B and three stereoisomers. Angew. Chem. Int. Ed. 2000;39:4596–4599. doi: 10.1002/1521-3773(20001215)39:24<4596::AID-ANIE4596>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 20.Finch N., Taylor W.I. The conversion of tetrahydro-β-carboline alkaloids into oxindoles. The structures and partial syntheses of mitraphylline and rhyn cophylline. J. Am. Chem. Soc. 1962;84:1318–1320. doi: 10.1021/ja00866a062. [DOI] [Google Scholar]
- 21.Wang H., Ganesan A. A biomimetic total synthesis of (−)-spirotryprostatin B and related studies. J. Org. Chem. 2000;65:4685–4693. doi: 10.1021/jo000306o. [DOI] [PubMed] [Google Scholar]
- 22.Sebahar P.R., Williams R.M. The asymmetric total synthesis of (+)- and (−)-spirotryprostatin B. J. Am. Chem. Soc. 2000;122:5666–5667. doi: 10.1021/ja001133n. [DOI] [Google Scholar]
- 23.Jones K., Wilkinson J. A total synthesis of horsfiline via aryl radical cyclisation. J. Chem. Soc. Chem. Commun. 1992;5:1767–1769. doi: 10.1039/c39920001767. [DOI] [Google Scholar]
- 24.Abelman M.M., Oh T., Overman L.E. Intramolecular alkene arylations for rapid assembly of polycyclic systems containing quaternary centers. A new synthesis of spirooxindoles and other fused and bridged ring systems. J. Org. Chem. 1987;52:4130–4133. doi: 10.1021/jo00227a038. [DOI] [Google Scholar]
- 25.Fuji K., Node M., Nagasawa H., Naniwa Y., Terada S. Asymmetric Induction via addition-elimination process: Nitroolefination of α-substituted lactones. J. Am. Chem. Soc. 1986;108:3855–3856. doi: 10.1021/ja00273a065. [DOI] [Google Scholar]
- 26.Stahl R., Borschberg H.-J. A Reinvestigation of the oxidative rearrangement of yohimbane-type alkaloids. Part A. Formation of pseudoindoxyl (=1,2-dihydro-3H-indol-3-one) derivatives. Helv. Chim. Acta. 1994;77:1331–1345. doi: 10.1002/hlca.19940770514. [DOI] [Google Scholar]
- 27.Shavel J., Zinnes H., Oxindole Alkaloids I. Oxidative-rearrangement of indole alkaloids to their oxindole analogs. J. Am. Chem. Soc. 1962;84:1320–1321. doi: 10.1021/ja00866a063. [DOI] [Google Scholar]
- 28.Martin S.F., Mortimore M. New methods for the synthesis of oxindole alkaloids. Total syntheses of isopteropodine and pteropodine. Tetrahedron Lett. 1990;31:4557–4560. doi: 10.1016/S0040-4039(00)97675-5. [DOI] [Google Scholar]
- 29.Takayama H., Seki N., Kitajima M., Aimi N., Seki H., Sakai S.-I. Indoloquinolizidine and yohimbine were converted to the corresponding Na-methoxyindoles and Na-methoxyoxindoles by the oxidation of the dihydroindole derivatives with H2O2 and sodium tungstate. Heterocycles. 1992;33:121–125. doi: 10.3987/COM-91-S41. [DOI] [Google Scholar]
- 30.Yu P., Cook J.M. Diastereospecific synthesis of ketooxindoles. Potential intermediates for the synthesis of alstonisine as well as for voachalotine related oxindole alkaloids. Tetrahedron Lett. 1997;38:8799–8802. doi: 10.1016/S0040-4039(97)10420-8. [DOI] [Google Scholar]
- 31.Pellegrini C., Strässler C., Weber M., Borschberg H.J. Synthesis of the oxindole alkaloid (−)-horsfiline. Tetrahedron Asymmetry. 1994;5:1979–1992. doi: 10.1016/S0957-4166(00)86273-4. [DOI] [Google Scholar]
- 32.Edmondson S.D., Danishefsky S.J. The total synthesis of spirotryprostatin A. Angew. Chem. Int. Ed. 1998;37:1138–1140. doi: 10.1002/(SICI)1521-3773(19980504)37:8<1138::AID-ANIE1138>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 33.Edmondson S., Danishefsky S.J., Sepp-Lorenzino L., Rosen N. Total synthesis of spirotryprostatin A, leading to the discovery of some biologically promising analogues. J. Am. Chem. Soc. 1999;121:2147–2155. doi: 10.1021/ja983788i. [DOI] [Google Scholar]
- 34.Pictet A., Spengler T. Über die Bildung von Isochinolin Derivaten durch Einwirkung von Methylal auf Phenylethylamin, Phenylalanin und Tyrosin. Eur. J. Inorg. Chem. 1911;44:2030–2036. doi: 10.1002/cber.19110440309. [DOI] [Google Scholar]
- 35.Zhao Y., Yu S., Sun W., Liu L., Lu J., McEachern D., Shargary S., Bernard D., Li X., Zhao T., et al. A potent small-molecule inhibitor of the MDM2-p53 interaction (MI-888) achieved complete and durable tumor regression in mice. J. Med. Chem. 2013;56:5553–5561. doi: 10.1021/jm4005708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kath J.C., Tom N.J., Cox E.D., Bhattacharya S.K. Heteroaromatic Bicyclic Derivatives Useful as Anticancer Agents. 6,867,201 B2. U.S. Patent. 2002 Aug 22;
- 37.Powell N.A., Kohrt J.T., Filipski K.J., Kaufman M., Sheehan D., Edmunds J.E., Delaney A., Wang Y., Bourbonais F., Lee D.Y., et al. Novel and selective spiroindoline-based inhibitors of sky kinase. Bioorg. Med. Chem. Lett. 2012;22:190–193. doi: 10.1016/j.bmcl.2011.11.036. [DOI] [PubMed] [Google Scholar]
- 38.Li C., Chan C., Heimann A.C., Danishefsky S.J. On the rearrangement of an azaspiroindolenine to a precursor to phalarine: Mechanistic insights. Angew. Chem. Int. Ed. 2007;46:1444–1447. doi: 10.1002/anie.200604071. [DOI] [PubMed] [Google Scholar]
- 39.Ottoni O., Cruz R., Krammer N.H. Regioselective nitration of 3-acetylindole and its N-acyl and N-sulfonyl derivatives. Tetrahedron Lett. 1999;40:1117–1120. doi: 10.1016/S0040-4039(98)02636-7. [DOI] [Google Scholar]
- 40.Lakshmaiah G., Kawabata T., Shang M., Fuji K. Total synthesis of (−)-horsfiline via asymmetric nitroolefination. J. Org. Chem. 1999;64:1699–1704. doi: 10.1021/jo981577q. [DOI] [PubMed] [Google Scholar]
- 41.Aurora Fine Chemicals Inc. [(accessed on 22 April 2017)]; Available online: http://www.aurorafinechemicals.com/
- 42.Mitchell I.S., Blake J.F., Xu R., Kallan N.C., Xiao D., Spencer K.L., Bencsik J.R., Liang J., Safina B., Zhang B., et al. Hydroxylated and Methoxylated Cyclopenta [D] Pyrimidines as Akt Protein Kinase Inhibitors. No. CA26,56,622 A1. Patent. 2007 Jul 5;
- 43.Hostetler G., Dunn D., McKenna B.A., Kopec K., Chatterjee S. 1-Thia-4,7-diaza-spiro[4.4]nonane-3,6-dione: A structural motif for 5-hydroxytryptamine 6 receptor antagonism. Chem. Biol. Drug Des. 2014;83:149–153. doi: 10.1111/cbdd.12240. [DOI] [PubMed] [Google Scholar]
- 44.Protiva M., Vejdělek Z.J., Jílek J.O., Macek K. Synthetische modelle hypotensiv wirksamer alkaloide V. Einige weitere derivate des tryptamins und 1,2,3,4-tetrahydronorharmans. Collect. Czechoslov. Chem. Commun. 1959;24:3978–3987. doi: 10.1135/cccc19593978. [DOI] [Google Scholar]
- 45.Bell S.E.V., Brown R.F.C., Eastwood F.W., Horvath J.M. An approach to some spiro oxindole alkaloids through cycloaddition reactions of 3-methylideneindolin-2-one. Aust. J. Chem. 2000;53 doi: 10.1071/CH00016. [DOI] [Google Scholar]
- 46.Lim Y.-H., Guo Z., Ali A., Edmondson S.D., Liu W., Gallo-Ettienne G.V., Wu H., Gao Y.-D., Stamford A.M., Yu Y., et al. Factor XIa Inhibitors. No. WO201,5164,308. Patent. 2015 Oct 29;
- 47.Allen J., Brasseur D.M., De Bruin B., Denoux M., Pérard S., Philippe N., Roy S.N. The use of biocatalysis in the synthesis of labelled compounds. J. Label. Compd. Radiopharm. 2007;50:342–346. doi: 10.1002/jlcr.1388. [DOI] [Google Scholar]
- 48.Manda S., Khan S.I., Jain S.K., Mohammed S., Tekwani B.L., Khan I.A., Vishwakarma R.A., Bharate S.B. Synthesis, antileishmanial and antitrypanosomal activities of N-substituted tetrahydro-β-carbolines. Bioorg. Med. Chem. Lett. 2014;24:3247–3250. doi: 10.1016/j.bmcl.2014.06.030. [DOI] [PubMed] [Google Scholar]
- 49.Sevrin M., Morel C., Menin J., George P. 2-(Methyl(4-piperidinyl))-1,2,3,4-tetrahydro-9H-pyrido(3,4-b Indole Derivatives, Their Preparation and Therapeutical Use. No. EP0,302,788 B1. Patent. 1992 Feb 5;
- 50.Shah P., Westwell A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007;22:527–540. doi: 10.1080/14756360701425014. [DOI] [PubMed] [Google Scholar]