

ABSTRACT
Metastatic reprogramming toward malignant tumor progression relies on the activation of oncogenic regulators, yet the cellular determinants remain elucidated. Through identification of aberrant prognostic cancer genes, we identified paraspeckle component 1 (PSPC1) functions as a master activator of metastatic reprogramming by activating epithelial-to-mesenchymal transition (EMT), stemness and TGF-β1 pro-metastatic switch.
Keywords: PSPC1, EMT, CSCs and TGF-β1 pro-metastatic reprogramming
Metastasis is an inefficient but lethal process that requires cancer cells of versatile self-reprogramming capabilities to achieve transition of epithelial-to- mesenchymal (EMT) and stemness for promoting tumor dissemination and metastasis. Cancer cells with EMT phenotype involved polarized epithelial cells into motile mesenchymal feature1 are commonly associated with increasing populations of cancer stem cells (CSCs), migration, invasion and dissemination to distant organs led to poor outcomes of cancer patients.
To uncover prognostic cancer genes with aberrant expression and residing in copy number alteration loci of cancer genomes,2 we identified paraspeckle component 1 (PSPC1) located at chromosome 13q12.11 with altered chromosome copy number and gene expression in multiple cancers.3 Higher PSPC1 expression in compared to the corresponding normal tissues is correlated to the late tumor stages and poor patient survival in breast, lung and liver cancers.
Since upregulation of PSPC1 is associated with poor prognosis of cancer patients, we took advantages of knowing altered genomic alterations in cancer cell lines and demonstrated PSPC1 pro-metastatic properties by gain-of-function and lose-of- function assays in PSPC1-overexpressing SK-Hep1 cells (liver) and PSPC1- knockdown A549 (lung), MDA-MB-231 (breast) and PC3 (prostate) cells. In addition to showing PSPC1-potentiated expression of EMT factors, self-renewal related genes, and stemness markers to promote cancer cell migration, invasion, spheroid formation, tumor formation and metastasis, we also validated the oncogenic roles of Pspc1 in spontaneous mammary (MMTV-PyMT) and lung (K-rasLSL-G12D/+p53fl/fl) (TP53, best known as p53) cancer mouse models. We concluded that expression of human or mouse PSPC1 stimulated the EMT and stemness of cancer cells and promoted their tumorigenesis and metastasis.
It is increasingly recognized that cancer cell unleash certain transcriptional reprograming to promote tumor progression such as activation of core transcription factors (Core-TFs) Snail, Twist or Nanog to control EMT and CSCs status during tumor metastasis. However, the “upstream modulator” regulated these core-TFs for activating pro-metastatic reprogramming is poorly elucidated. After transcriptome and gene set enrichment analysis, we found PSPC1 is the master modulator to activate signature gene sets of EMT, CSCs, TGF-β signaling and cell proliferation. With confirmations of PSPC1 increased core EMT-TFs (Snail, Slug, and Twist) and CSC-TFs (Nanog, Oct4, and Sox2) expression through transcriptional regulation, PSPC1 is the critical master transcriptional activator to potentiate the pro-metastatic reprogramming genes expression in tumor progression.4
To succeed the metastatic cascade, the sheltered niche enriched with cytokines and growth factors including TGF-β1 provides the soil for metastasis.5 PSPC1 specifically interacted with phosphorylated-Smad2/3 in the nucleus in a TGF-β1 signaling dependent manner to activate TGF-β1 autocrine signaling. Targeting TGF-β1 pathway by TGFBR1 inhibitor treatment (SB431542) or specific Smad2/3 knockdown abolished PSPC1-mediated core EMT-TFs/CSC-TFs expression, cellular migration, and CSC populations. Interestingly, PSPC1-enhanced TGF-β1 autocrine signaling regardless of somatic mutations on members of TGF-β1/Smads signaling pathway in cancer cells especially in gastrointestinal cancers provides unprecedented mechanistic insights to sustain a TGF-β-enriched tumor microenvironment.
TGF-β1 plays opposite and dichotomous functions in tumor progression and the differential TGF-β1 responses might be due to the difference of cellular context. In normal or early transforming cells, TGF-β1 functions as tumor suppressor by inhibiting proliferation and stimulating apoptosis, whereas, in advanced tumors, cells confer non-cytostatic TGF-β1 responses to increase pro-metastatic migration, EMT and stemness.6 Previous efforts for identifying Smad2/3 partners as contextual determinants for TGF-β1 pro-metastatic switch from cytostatic to metastatic roles was unsatisfactory. For instance, FoxO and C/EBP-β are two well-known Smad cofactors that activated p15 and p21 gene expression and suppressed c-Myc transcription.7,8 14-3-3ζ reduced p53-Smads complex and diminish TGF-β suppressor effect but stabilized GLI2-Smads for promoting bone metastasis.9
Our in silico analysis revealed that PSPC1-Smads-upregulated and PSPC1- Smads-downregulated groups involved in cell growth and movement as well as in cell death and apoptosis, respectively. Knockdown PSPC1 decreased TGF-β1-induced migration and sphere formation, whereas, increased TGF-β1-induced growth inhibition in cancer cells. Besides, non-cancerous cells (Beas2B, MCF10A, HaCaT cells) with high PSPC1 expression showed resistance to TGF-β1-mediated cytostasis. Moreover, our CHIP (Chromatin immunoprecipitation)-reCHIP and expression experiments on known Smad2/3 target genes revealed that Smad2/3 preferentially bind to tumor suppressor gene promoter (p21, p57 and DAPK1) in PSPC1 deficient cancer cells, whereas the transcription complex bind at key pro-metastatic gene promoter (Snail, Slug, Twist, Nanog, Oct4, and Sox2) in PSPC1 highly expressed cancer cells (Figure 1). Our results suggested that PSPC1 upregulation is the cellular determinant of Smad2/3 targets to reprogram the TGF-β signaling from cytostatic response to highly aggressive one.
Figure 1.
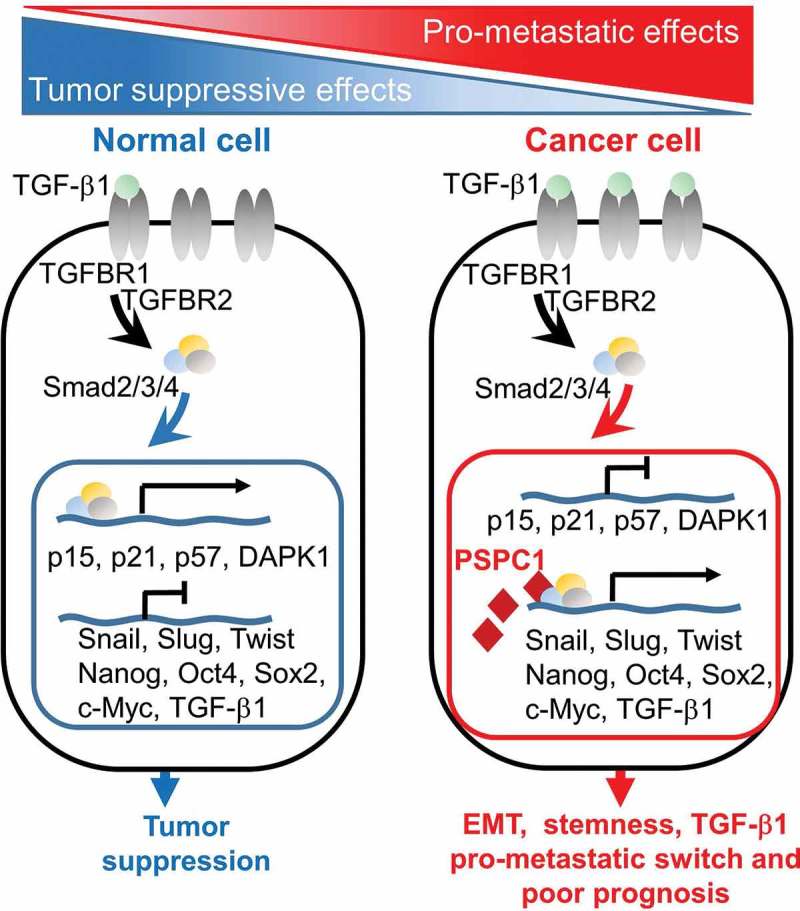
Paraspeckle component 1 (PSPC1) switches TGF-β1 signaling from cytostasis towards epithelial-to-mesenchymal transition (EMT), stemness and metastasis. In normal or pre-malignant tissues with low PSPC1 expression, Smad2/3/4 complex is translocated into nucleus and targets cytostasis-related genes upon TGF-β1 signaling activation. However, in advanced tumor with high PSPC1 expression, Smad2/3/4 is recruited by PSPC1 for activating promoters of EMT and stemness and promoting metastatic programming. PSPC1/TGF-β1-dependent pro-metastatic switch is sustained by increasing TGF-β1 cytokine expression and autocrine activation.
Another interesting feature for PSPC1 activated gene set is the signature of c-Myc target genes in related to increase of cell proliferation. C-Myc suppression has been known as the indicator of TGF-β1/Smad2/3-mediated cell cycle arrest. Activation of c-Myc by PSPC1 could suggest that PSPC1 selectively de-repressed its expression to induce cancer cell proliferation and accompanied with PSPC1-mediated EMT and stemness to facilitate tumor metastasis.
Notably, abundant expression of TGF-β1 is known to be critical for cancer malignancy through paracrine signaling such as secreted from stromal cancer- associated fibroblasts (CAFs) and tumor associated macrophages (TAMs) in tumor microenvironment.10 Future studies of PSPC1-meditaed cytokine reprogramming in the tumor microenvironment will be critical for further understanding the underlining mechanisms of metastatic reprogramming.
With validation of IHC analysis for co-existence of PSPC1/TGF-β1/p-Smad2/p-Smad3 axis in lung and breast patient-derived tumor tissue samples and the emerging anticancer strategies on countering tumor metastasis, targeting PSPC1 might halt tumor metastasis via suppression of EMT, stemness and metastatic TGF-β1 signaling for maintaining cancer cells in dormancy. In summary, identification of PSPC1 not only improves our understanding of pro-metastatic reprogramming in tumor progression but also serves as a theranostic target for anti-metastasis.
Funding Statement
This research was supported by Academia Sinica and Ministry of Science and Technology (MOST 107-0210-01-19-01) and by the MOST, Taiwan: (MOST 106-2321-B-001-051 and MOST104-2320-B-001-009MY3).
Disclosure of Potential Conflicts of Interest
The authors declare no potential conflicts of interest.
References
- 1.Thiery JP, Acloque H, Huang RY, Nieto MA.. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Shiau CK, Gu DL, Chen CF, Lin CH, Jou YS.. IGRhCellID: integrated genomic resources of human cell lines for identification. Nucleic Acids Res. 2011;39(Database issue):D520–D524. doi: 10.1093/nar/gkq1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeh HW, Hsu EC, Lee SS, Lang YD, Lin YC, Chang CY, Lee SY, Gu DL, Shih JH. Ho CM and others. PSPC1 mediates TGF-beta1 autocrine signalling and Smad2/3 target switching to promote EMT, stemness and metastasis. Nat Cell Biol. 2018;20(4):479–491. doi: 10.1038/s41556-018-0062-y. [DOI] [PubMed] [Google Scholar]
- 4.Knott GJ, Bond CS, Fox AH. The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res. 2016;44(9):3989–4004. doi: 10.1093/nar/gkw271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. https://www.ncbi.nlm.nih.gov/pubmed/12809600. [DOI] [PubMed] [Google Scholar]
- 6.Seoane J, Gomis RR. TGF-beta family signaling in tumor suppression and cancer progression. Cold Spring Harb Perspect Biol. 2017;9(12):a022277. doi: 10.1101/cshperspect.a022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–868. https://www.ncbi.nlm.nih.gov/pubmed/10102273. [DOI] [PubMed] [Google Scholar]
- 8.Gomis RR, Alarcon C, He W, Wang Q, Seoane J, Lash A, Massague J. A FoxO-Smad synexpression group in human keratinocytes. Proc Natl Acad Sci U S A. 2006;103(34):12747–12752. doi: 10.1073/pnas.0605333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, Acharya S, Sahin O, Zhang Q, Saito Y, Yao J, Wang H, Li P, Zhang L. Lowery FJ and others. 14-3-3zeta turns TGF-beta’s function from tumor suppressor to metastasis promoter in breast cancer by contextual changes of Smad partners from p53 to Gli2. Cancer Cell. 2015;27(2):177–192. doi: 10.1016/j.ccell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer. 2013;13(11):788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]