

ABSTRACT
The crosstalk between cellular energy status and epigenetic modifications remains largely elusive. We recently uncovered that upon energy restriction, AMP-activated protein kinase (AMPK) phosphorylates enhancer of zeste homolog 2 (EZH2), which disrupts the integrity of the polycomb repressive complex 2 (PRC2), thus inhibiting PRC2 oncogenic functions in ovarian and breast cancer cells.
KEYWORDS: AMPK, PRC2, EZH2, energy restriction
Polycomb repressive complex 2 (PRC2) governs gene expression by promoting the trimethylation of histone H3 lysine 27 (H3K27) at target gene promoters, a program leads to epigenetic silencing. It has been well documented that PRC2 maintains pluripotency in embryonic stem (ES) cells by repressing the expression of developmental regulators such as the Hox family genes. The PRC2 complex consists of three core subunits, the catalytic subunit EZH2 or EZH1, which catalyzes H3K27 methylation, as well as the scaffolding components SUZ12 and EED, which participate in PRC2-nucleosome interaction.1 As the primary catalytic component for PRC2 in most cell types, EZH2 gain-of-function mutations have been shown to associate with the development of B-cell lymphoma.2 A large body of clinical studies suggest an oncogenic role for EZH2 in lymphoma and many types of solid tumors including prostate, breast, and ovarian cancers. Different from the hot-spot oncogenic mutations found in B-cell lymphoma, EZH2 is rather overexpressed in most solid tumors. Numerous compounds that target EZH2 have been developed and some of them have achieved promising outcomes in clinical trials. However, recent reports unexpectedly reveal a tumor suppressor role of EZH2 in leukemia and melanoma, thus indicating a more complex and context-dependent role of EZH2 in tumorigenesis.
The functions of PRC2 are tightly controlled by various mechanisms.1 Long non-coding RNAs (lncRNAs) XIST and HOTAIR have been shown to promote the recruitment of PRC2 to target gene loci. In addition, the EZH2 protein is subjected to phosphorylation by multiple kinases at different sites. Akt has been shown to promote EZH2 Ser21 phosphorylation, which attenuates EZH2's binding with histone H3.3 CDK1 catalyzes EZH2 Thr345 and Thr487 phosphorylation during G2/M phases to regulate EZH2-lncRNA and EZH2-PRC2 interactions.4 In our recent work published in Molecular Cell,5 we identified the AMP-activated protein kinase (AMPK) as an upstream factor that phosphorylates EZH2 at Thr311. Anti-H3K27me3 chromatin immunoprecipitation coupled with deep sequencing (ChIP-seq) revealed a global elevation of H3K27 trimethylation (H3K27me3) in Ampkα1−/−;Ampkα2−/− mouse embryonic fibroblasts (MEFs), in which the two catalytic subunit isoforms of the AMPK heterotrimeric complex are deleted. These results suggest a negative regulation of PRC2/EZH2 function by the AMPK kinase.
AMPK senses cellular ATP levels through an allosteric binding to AMP, which activates the kinase.6 When treating cells with AMPK agonists, such as A769662, metformin, or 2-Deoxy-D-glucose, accompanied with the upregulation of phospho-Thr311 (pT311), global H3K27me3 levels were reduced. Further in-depth examination of the expression of known PRC2 target genes revealed a dramatic increse of these genes upon AMPK activation. Mechanistically, we demonstrated that phosphorylation of EZH2 at Thr311 disrupts its association with the PRC2 core subunit SUZ12, leading compromised PRC2 integrity and enzymatic activity (Fig. 1). Recent structural studies unveiled that Thr311 resides in a region connecting the SANT1 and SANT2 domains of EZH2, and has been shown to interact with the VEFS domain of SUZ12.7 The crucial role of SUZ12-EZH2 interaction to escort EZH2 to the PRC2 genomic target thus underscores the importance of AMPK-mediated EZH2 phosphorylation in fine tuning PRC2 function.
Figure 1.
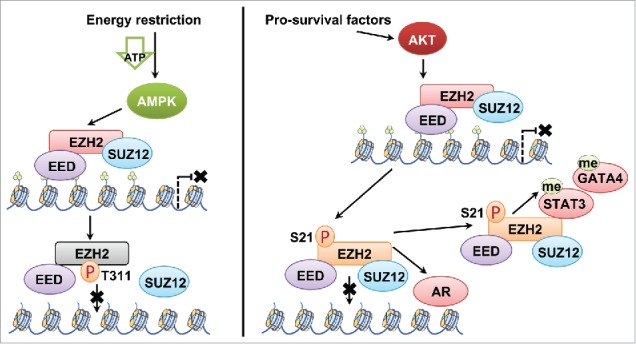
Distinct mechanisms of AMPK and Akt in controlling PRC2 activity. AMPK phosphorylates Thr311-EZH2, which disrupts the interaction between SUZ12 and EZH2 to inhibit the PRC2 holoenzyme activity. Like pS21-EZH2, pT311-EZH2 results in reduced H3K27 tri-methylation. However, unlike pS21-EZH2, pT311-EZH2 disrupts the integrity of the PRC2 holoenzyme, thereby mainly serves to inhibit the oncogenic role of PRC2. On the other hand, Akt phosphorylates Ser21-EZH2 to disrupt the interaction between PRC2 and the H3 tail, leading to reduced H3K27 tri-methylation. However, pS21-EZH2 could still exert its oncogenic function via PRC2-mediated methylation of non-histone proteins such as STAT3, or through associating with AR to function as a transcriptional cofactor, both of which contribute to the context-dependent oncogenic role of pS21-EZH2.
Given the opposing roles of Akt and AMPK in various cellular processes, it is mysterious that both Akt and AMPK attenuate PRC2/EZH2 function as the H3K27me3 writer. Recent discoveries on the non-histone methyltransferase activity of PRC2/EZH2 may shed light on the difference between Akt-mediated Ser21 and AMPK-mediated Thr311 phosphorylation of EZH2. Although PRC2/EZH2 is detached from the canonical PRC2 target promoters after Ser21 phosphorylation,3 EZH2 is detoured to associate with an AR-containing transcription complex to facilitate castration-resistant prostate cancer development,8 or to catalyze GATA4 and STAT3 methylation.9 These findings coherently support the notion that although Akt suppresses canonical EZH2 function, due to the preserved PRC2 integrity upon Ser21 phosphorylation, EZH2 still demonstrates its versatility in driving tumorigenesis. Our findings, on the other hand, elucidate a suppressive role of AMPK in modulating PRC2 oncogenic function through a direct phosphorylation of EZH2 at Thr311, a site located in the center of the PRC2 core complex, thus may have a more profound effect in inhibiting the catalytic activity of PRC2 (Fig. 1).
The identification of AMPK as a negative regulator for EZH2 suggests the use of AMPK agonists to sensitize anti-EZH2 inhibitors in EZH2-amplified or overexpressed cancers. Among currently available AMPK agonists, metformin, a first-line drug for treating patients with type 2 diabetes mellitus (T2DM), displays anti-tumor functions in various types of cancers including ovarian and breast cancers.10 It is noteworthy that metformin activates AMPK indirectly through inhibiting the mitochondria function to produce ATP, thus it is tempting to further evaluate if a direct AMPK agonist like A769662 could play a more specific and potent role to cooperate with anti-EZH2 agent in harnessing the tumorigenicity of EZH2-hyperactive cancer cells.
Funding Statement
This work was supported by the NIH under grant CA183914 and the Florida Breast Cancer Foundation.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. doi: 10.1038/nature09784. PMID:21248841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamagishi M, Uchimaru K. Targeting EZH2 in cancer therapy. Curr Opin Oncol. 2017;29:375–81. doi: 10.1097/CCO.0000000000000390. PMID:28665819. [DOI] [PubMed] [Google Scholar]
- 3.Cha T-L, Zhou BP, Xia W, Wu Y, Yang C-C, Chen C-T, Ping B, Otte AP, Hung M-C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310:306–10. doi: 10.1126/science.1118947. PMID:16224021. [DOI] [PubMed] [Google Scholar]
- 4.Caretti G, Palacios D, Sartorelli V, Puri PL. Phosphoryl-EZH-ion. Cell Stem Cell. 2011;8:262–5. doi: 10.1016/j.stem.2011.02.012. PMID:21362566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wan L, Xu K, Wei Y, Zhang J, Han T, Fry C, Zhang Z, Wang YV, Huang L, Yuan M, et al.. Phosphorylation of EZH2 by AMPK Suppresses PRC2 Methyltransferase Activity and Oncogenic Function. Mol Cell. 2018;69:279–291.e5. doi: 10.1016/j.molcel.2017.12.024. PMID:29351847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121–35. doi: 10.1038/nrm.2017.95. PMID:28974774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chittock EC, Latwiel S, Miller TCR, Müller CW. Molecular architecture of polycomb repressive complexes. Biochem Soc Trans. 2017;45:193–205. doi: 10.1042/BST20160173. PMID:28202673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, et al.. EZH2 Oncogenic Activity in Castration Resistant Prostate Cancer Cells is Polycomb-Independent. Science. 2012;338:1465–9. doi: 10.1126/science.1227604. PMID:23239736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Comet I, Riising EM, Leblanc B, Helin K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat Rev Cancer. 2016;16:803–10. doi: 10.1038/nrc.2016.83. PMID:27658528. [DOI] [PubMed] [Google Scholar]
- 10.Pernicova I, Korbonits M. Metformin-Mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–56. doi: 10.1038/nrendo.2013.256. PMID:24393785. [DOI] [PubMed] [Google Scholar]