

ABSTRACT
Oncogenic MLL-fusion proteins often hijack essential molecular mechanisms during leukemogenesis. The histone methyltransferase SETD2 was implicated in the regulation of transcription, DNA damage and other cellular processes. Recent studies identified a critical role for SETD2 in MLL-rearranged leukemia. These results may help to unravel important functions of SETD2 in hematopoiesis.
Keywords: AML, MLL-rearranged leukemia, fusion proteins, SETD2
Chromosomal translocations often result in the generation of oncogenic fusion proteins. Acute myeloid leukemia (AML), an aggressive cancer of white blood cells, features the highest number of gene fusions among all types of cancer.1 The gene encoding the histone methyltransferase MLL (Mixed Lineage Leukemia) is involved in translocations with over 135 fusion partner genes, resulting in the expression of over 70 known MLL-fusion proteins in leukemia.2 Moreover, it has been recognized that patients expressing MLL-fusion proteins have a particularly poor prognosis.3
MLL-fusion proteins are embedded in large multi-protein complexes whose architecture determines their molecular functions. A large number of interaction partners of MLL-fusion proteins have been characterized, and many of them have been shown to be necessary for the initiation and maintenance of MLL-rearranged leukemia.3 However, although critical effectors have been identified for some MLL-fusion proteins, it is not clear whether the large and molecularly diverse family of MLL-fusions share critical effectors that could represent attractive actionable targets.
We devised an experimental pipeline to unearth conserved molecular mechanisms across the large repertoire of known MLL-fusion proteins. We used functional proteomics to survey the molecular composition of distantly related MLL-fusion protein complexes in an isogenic background. This analysis resulted in a protein-protein interaction network of 960 proteins that shared 2047 interactions. This high number of protein interaction partners suggests that many proteins engage in indirect, rather than direct interactions with MLL-fusion proteins. Overall, each MLL-fusion protein interacted with a unique set of 70–110 proteins, highlighting a high degree of conservation in the protein interactome of MLL-fusions.
Reasoning that critical effectors of MLL-rearranged leukemia might be enriched among conserved physical interaction partners, we focused on 128 proteins, which reproducibly copurified with the majority of MLL-fusion proteins investigated by us. To systematically characterize the functional contribution of these conserved MLL interaction partners to leukemia development we employed a series of RNA interference (RNAi) screens in human leukemia cell lines. Any candidate whose RNAi-mediated knockdown selectively impaired proliferation of MLL-fusion expressing cells would qualify as a specific effector of MLL-fusion proteins. This strategy identified the histone methyltransferase SETD2 (SET domain containing 2) as a factor that is required for the proliferation of leukemia cells expressing MLL-fusions. RNAi- and CRISPR/Cas9-mediated perturbation of SETD2 expression led to proliferation arrest and induced terminal myeloid differentiation of cell lines and primary cells expressing MLL-fusions. Interestingly, the proliferation and differentiation status of leukemia cells without MLL-rearrangement were largely unaffected by SETD2 perturbation, indicating an MLL-fusion specific function of SETD2.
SETD2 is the only enzyme reported to catalyze tri-methylation of Lys-36 of histone H3 (H3K36me3).4 The H3K36me3 mark associates with actively transcribed genes and correlates with elongating RNA polymerase II. In addition, an interaction between H3K36me3 and other histone marks has been previously proposed.4 Chromatin immunoprecipitation with reference exogenous genome (ChIP-Rx) experiments in AML cells identified crosstalk between H3K36me3 and the H3K79me2 histone mark, which is catalyzed by DOT1L (Disruptor of Telomeric silencing 1-like). The methyltransferase DOT1L is a known effector of many MLL-fusion proteins, and the H3K79me2 mark is highly enriched on MLL-target genes. We found that H3K36me3 levels were highly correlated with the H3K79me2 mark across the genome, being particularly increased at MLL-target genes. The co-localization of these two histone marks on MLL-target genes strengthens the argument that MLL-leukemia requires SETD2. However, the functional implications of this dual signature remain to be elucidated.
Additionally, SETD2 has been implicated in the regulation of DNA damage repair pathways. The SETD2-dependent H3K36me3 histone mark can be recognized by PWWP domains, which are present in a variety of proteins associated with the DNA damage response (Figure1).4 RNAi-mediated loss of SETD2 in MLL-fusion-expressing cell lines caused increased DNA damage, activation of the TP53 (tumor protein p53) signaling pathway and apoptosis. Interestingly, this SETD2-dependent phenotype was strongly enhanced by treatment with the small molecule DOT1L inhibitor Pinometostat (EPZ5676), indicating that H3K79me2-H3K36me3 crosstalk is involved in maintaining genomic integrity in MLL-fusion expressing AML.
Figure 1.
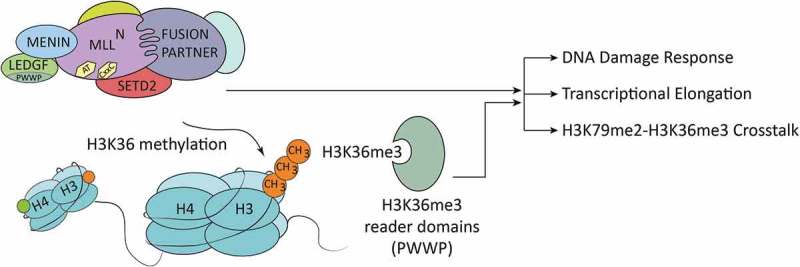
Potential functions of SETD2 in the context of MLL-rearranged leukemia. Schematic drawing of an MLL- (Mixed Lineage Leukemia) fusion protein complex. Among other partners, the complex contains MENIN (MEN1), PC4/SFRS1 interacting protein 1 (PSIP1, LEDGF), and the histone methyltransferase SET domain containing 2 (SETD2), which interact with the N-terminus of the MLL complex. Functional domains in the N-terminus of MLL are indicated: AT = AT-hooks, a DNA binding domain; CxxC = motif recognizing un-methylated CpG dinucleotides. The histone methyltransferase SETD2 (SET domain containing 2) interacts with the MLL complex. SETD2-dependent deposition of H3K36me3 is recognized and bound by PWWP domain-containing proteins that are involved in various cellular processes, such as DNA damage repair and transcriptional elongation.
A large body of evidence suggests that SETD2 acts as a tumor suppressor in various types of cancer, including clear renal cell carcinoma and leukemia.5,6 Loss of SETD2 was shown to cause a proliferative advantage of MLL-fusion-expressing cells, establishing a tumor suppressive role of SETD2 in AML.5 In contrast, findings from other laboratories indicate that MLL-fusion expressing AML cells are strongly dependent on SETD2 for survival and proliferation 7,8. Using a conditional Setd2 knockout in the context of an MLL-AF9 AML mouse model, a recent publication proposed that different SETD2 levels might manifest in diverging phenotypes. While homozygous Setd2 loss resulted in strongly delayed MLL-AF9-induced leukemogenesis, heterozygous Setd2 deletion caused accelerated disease development and chemotherapy resistance.9 This is particularly interesting as leukemia patients harboring heterozygous SETD2 mutations often exhibit increased genomic complexity, therapy resistance and relapse, indicating that SETD2 might act as a haplo-insufficient tumor suppressor.10
While the exact role of SETD2 in MLL-rearranged leukemia remains elusive, these recent studies demonstrate its important function in this disease. Further work will be required to achieve a deeper understanding of the functional implications of SETD2-activity and -mutations in AML.
Funding Statement
This work was supported by the FP7 People: Marie-Curie Actions [289611];H2020 European Research Council [636855/StG];Österreichische Forschungsförderungsgesellschaft [857935];
References
- 1.Mertens F, Johansson B, Fioretos T, Mitelman F The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15(6):371–381. [DOI] [PubMed] [Google Scholar]
- 2.Meyer C, Burmeister T, Gröger D, Tsaur G, Fechina L, Renneville A, Sutton R, Venn NC, Emerenciano M, Pombo-de-Oliveira MS, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32:273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slany RK. The molecular mechanics of mixed lineage leukemia. Oncogene. 2016;35:5215–5223. doi: 10.1038/onc.2016.30. [DOI] [PubMed] [Google Scholar]
- 4.McDaniel SL, Strahl BD.. Shaping the cellular landscape with Set2/SETD2 methylation. Cell Mol Life Sci. 2017;18:3317–3334. doi: 10.1007/s00018-017-2517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu X, He F, Zeng H, Ling S, Chen A, Wang Y, Yan X, Wei W, Pang Y, Cheng H, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet. 2014;46:287–293. doi: 10.1038/ng.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33:661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skucha A, Ebner J, Schmollerl J, Roth M, Eder T, Cesar-Razquin A, Stukalov A, Vittori S, Muhar M, Lu B, et al. MLL-fusion-driven leukemia requires SETD2 to safeguard genomic integrity. Nat Commun. 2018;9:1983. doi: 10.1038/s41467-018-04329-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mar BG, Chu SH, Kahn JD, Krivtsov AV, Koche R, Castellano CA, Kotlier JL, Zon RL, McConkey ME, Chabon J, et al. SETD2 alterations impair DNA damage recognition and lead to resistance to chemotherapy in leukemia. Blood. 2017;130:2631–2641. doi: 10.1182/blood-2017-03-775569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K, Neuberg DS, Sinha AU, Sallan SE, Silverman LB, et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun. 2014;5:3469. doi: 10.1038/ncomms5972. [DOI] [PMC free article] [PubMed] [Google Scholar]