

ABSTRACT
The ischemia-reperfusion (I/R) induced skin lesion has been identified as primary cause of pressure ulcer. Better understanding of the mechanism is required for new therapy development. Leucine rich repeat containing protein 19 (LRRC19) is a recently discovered transmembrane protein containing leucine-rich repeats and plays a role in immune response. To investigate the role of LRRC19 in pressure ulcers, mouse ulcer model was established with two cycles of I/R. The expression of LRRC19 was assessed during injury. siRNA mediated LRRC19 downregulation was applied to investigate the disease severity, immune cell infiltration and pro-inflammatory cytokines production. The primary skin fibroblasts were stimulated with IL-1β to dissect the molecular mechanism. LRRC19 was readily induced in I/R induced lesion site in a pattern mimicking the disease progress as measured by wound area. Knockdown of LRRC19 by siRNA significantly alleviated the disease severity and attenuated immune cell infiltration and pro-inflammatory cytokines production. In primary skin fibroblast model, siRNA knockdown of LRRC19 suppressed IL-1β mediated NFκB activation and its downstream cytokines production. LRRC19 was a novel factor for I/R-induced tissue damage by promoting NFκB dependent pro-inflammatory response. Our results supported that LRRC19 could be a potential therapeutic target for pressure ulcers.
KEYWORDS: Pressure ulcer, Ischemia–Reperfusion (I/R), LRRC19, NFκB pathway, inflammatory response
INTRODUCTION
Pressure ulcers are defined as a localized damage to the skin and/or underlying tissue that usually occur over a bony prominence as a result of pressure, or pressure in combination with shear and/or friction.1 These wounds are commonly associated with elderly, bedridden, and debilitated patients, spinal-cord injury and neuropathy, and patients undergoing major orthopedic reconstruction. Pressure ulcers are a significant problem in healthcare, not only affecting the quality of life, morbidity and mortality of patients but also in terms of healthcare costs.2 Pressure ulcers are becoming an increasingly common and serious problem for wheelchair users as well as hospitalized patients with acute care. Hospital-acquired pressure injuries occur in 3%–34% of hospitalized patients worldwide depending on the care institution and result in longer hospital stays, higher morbidity, increased human suffering and expenditure for patient care.3,4 And they are expected to increase in frequency as the population ages.
The conditions leading to the formation of pressure ulcers are considered to be multifactorial. Physical forces including pressure, shear forces, friction and tissue compression, malnutrition, and the lack of physical activity, are risk factors in the development of pressure ulcers.1, 4 The occurrence of cycles of ischemia–reperfusion (I/R) has been considered more recently to be a significant contributor to the pathogenesis of pressure ulcers.5, 6 I/R injury has been defined as cellular injury resulting from the reperfusion of blood to previously ischemic tissue.7 Those tissues deprived of blood supply during the ischemic episode reduce metabolism in an effort to preserve tissue function. The restoration of blood flow to oxygen and nutrient depleted tissues (i.e., the reperfusion event) can initiate a series of events due to an abrupt increase in oxygen-derived free-radicals, which exceed the capacity of the normal free-radical scavenging mechanisms. The cytotoxic activity of the excess free radicals leads to inflammation and severe dysregulation of the cell recruitment to the site of injury.7, 8 The detailed mechanism for pressure ulcers is believed to be multifaceted and more new players are awaiting to be discovered.
Leucine rich repeat containing protein 19 (LRRC19) belongs to the large protein family containing a signature leucine-rich repeats (LRRs) domain. LRR proteins including Toll-like receptor (TLR), NOD-like receptor (NLR), RIG-I-like receptor (RLR) subfamilies have well-characterized functions in the innate immune system that are similar in diverse organisms from plants to mammals.9, 10 However, the function and expression patterns of other unusual proteins in this family have not yet been fully elucidated. LRRC19 is a recently identified member that has four LRR motifs at its extracellular domain and is strongly expressed in kidney, spleen and intestine in mouse.11, 12 Expression of LRRC19 can induce multiple TLR ligand-mediated NFκB activation in 293T cells. In vivo animal model with LRRC19 knockout showed the impaired production of cytokine, chemokine and anti-microbial substances in kidney and gut tissues through downregulation of TRAF2/6-mediated NFκB and MAPK signaling pathways.11, 13 Recently a microarray based transcriptome study in rat pressure ulcer model has revealed the upregulation of LRRC19 gene associated with the lesion progress.14 Therefore, it would be intriguing to validate the change of LRRC19 and investigate its functional implication in pressure ulcer pathogenesis.
In the current study we established the connection of LRRC19 and pressure ulcers. Our results aimed to prove LRRC19 as a novel mediator for the inflammatory response associated with pressure ulcers.
RESULTS
LRRC19 expression is induced in skin lesion of pressure ulcer model mice
We first established the mouse model for I/R induced pressure ulcer in C57BL/6 strain (n = 10).5, 6 In this model, mice received the cyclic compression-release procedure on dorsal skin developed cutaneous wound resembling pressure ulcers. Based on the previous study,16 two cycles of IR delivered better wounding pattern than three cycles. Therefore, we chose two-cycles activation in our study. After two cycles of I/R, the average size of the wound area at the skin lesion skin site was measured (n = 10) to determine the wound-healing process at day 1, 3, 7, 10, and 14 post injury. Here, the condition before I/R injury applied was taken as day 0. The skin injury quickly increased to its full-blown stage at day 3 (which was normalized to 100%) and gradually recovered to about 20% of the peak at day 14 (Figure 1A). A qualitative assessment of I/R induced skin wounding manifestation was also shown in Supplementary Figure S1. At the same time, injured tissues from another cohort of mice after I/R were excised for total RNA and protein lysate extraction at the different time points post I/R. We examined the level of LRRC19 during the time course by RT-PCR and Western blotting. Interestingly, the RNA and protein level of LRRC19 were induced in a pattern mimicking that of wound area. The peak level of LRRC19 mRNA was observed at day 1 after I/R which was about 14 times of that observed at day 0 (normalized as 1). LRRC19 mRNA then slowly decreased from day 3 to day 14 (Figure 1B), which was consistent with the change of protein level (Figure 1C). The basal level of LRRC19 protein at day 0 was barely detectible in Western blot; however, it was dramatically induced to high level at day 1. Then it maintained similar high level at day 3, which was followed by gradual decrease to the low level from day 7 to day 14. These results demonstrated that LRRC19 is responsive to I/R induced skin damage and implicated in the pressure ulcers development.
FIGURE 1.
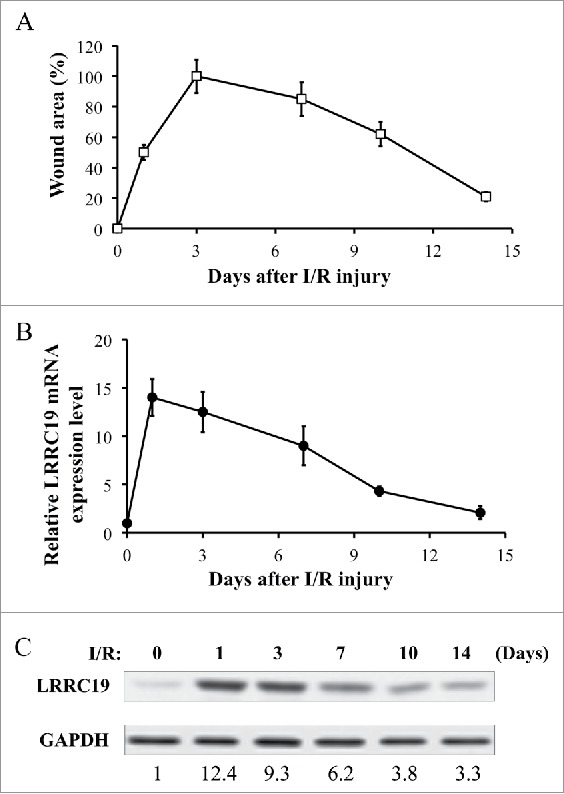
LRRC 19 expression is induced during I/R injury. A) The average size of the wound area in mice (n = 10) after I/R injury were measured at day 0, 1, 3, 7 10 and 14 post injury. The end of 2nd ischemia was assigned day 0. The size of the ulcer on day 3 was assigned a value of 100%. B) Realtime PCR analysis of LRRC19 mRNA expression in the I/R site. The end of 2nd ischemia was assigned day 0. Data are relative to mRNA level on day 0. Values were determined in n = 10 mice. C) Western blot analysis of LRRC19 protein expression at the wound site during I/R injury for the indicated time points. The end of 2nd ischemia was assigned day 0. The data are representative of n = 10 subjects. The band intensity of LRCC19 was normalized against loading control. The normalized band densitometry at day 0 was set as baseline of value 1.
Knocking down LRRC19 reduces pressure ulcer formation and decreases the inflammatory cell infiltration in wound area
To address the role of LRRC19 in I/R induced skin damage, we next examined thedisease progress in the condition of LRRC19 knockdown. With the method adopted from Charafeddine et al. with slight modification,17 we managed to achieve the effective knockdown of LRRC19 protein level in the injured skin tissue, as shown in Figure 2A. The siRNA control group consistently showed the gradual induction of LRRC19 from day 0 to day 3 post I/R, while LRRC19 siRNA group showed only minimal induction of LRRC19. The localized repression of LRRC19 expression was also confirmed by histological staining (Supplementary Figure S2). Next, we assessed the damage severity by wound area measurement. As shown in Figure 2B, the group (n = 10) with siRNA knockdown of LRRC19 showed greatly alleviated wound area throughout the process with statistical significance. Pathological analysis of the skin wound revealed that LRRC19 siRNA group showed dramatically reduced infiltration of MPO (myeloperoxidase) positive neutrophils (Figure 2C) and CD68 positive macrophages (Figure 2D) at day 1 and day 3 post I/R treatment compared to scramble siRNA controls. As reference, the representative images of histological staining were shown in Supplementary Figure S3. These results clearly suggested that LRRC19 contributes to the pressure ulcer pathogenesis and was required for inflammatory cell infiltration.
FIGURE 2.
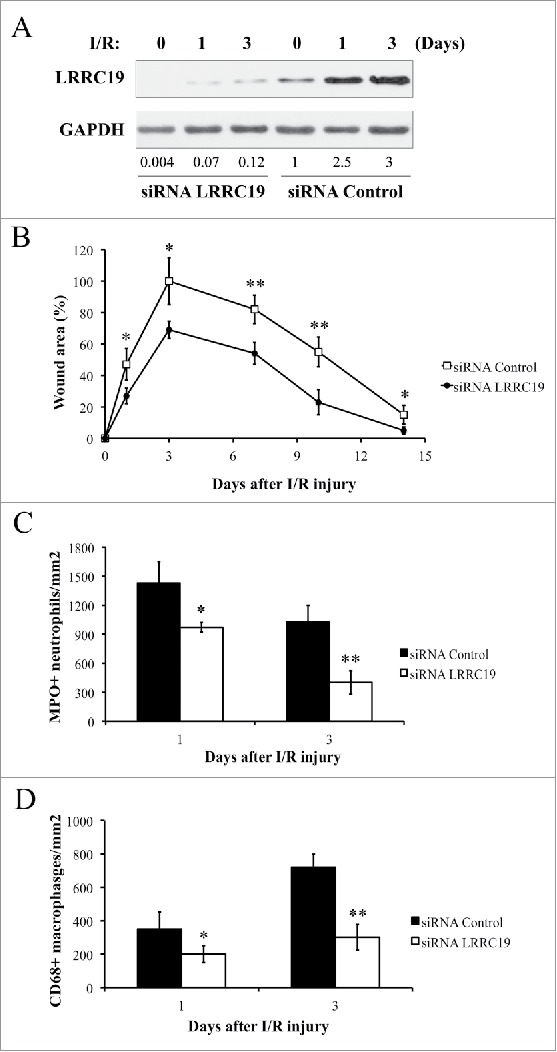
Knocking down LRRC19 reduces pressure ulcer formation and decreases the inflammatory cell infiltration in wound area. A) Western blot analysis of LRRC19 protein expression at the wound site during I/R injury. The wound area was topically applied with 100 pmol of either LRRC19 siRNA or control siRNA immediately after 1st ischemia delivery, followed by day 0, 1, 3, 6, 9, 12 after I/R injury induced. The end of 2nd ischemia was assigned day 0. The data are representative of n = 4 subjects for each group. The band intensity of LRCC19 was normalized against GAPDH. The normalized band densitometry of the siRNA control treated sample at day 0 was set as baseline of value 1. B) The average size of the wound area in mice (n = 6 for each group) treated with either LRRC19 or control siRNA during I/R injury were measured at day 0, 1, 3, 7 10 and 14 post injury. The end of 2nd ischemia was assigned day 0. The size of the ulcer in control group on day 3 after I/R was assigned a value of 100%. Immunohisochemistry was used to study the number of C) neutrophils (myeloperoxidase positive) and D) macrophages (CD68 positive) at the lesion site. Values were determined in six random microscopic fields (∼20,000 mm2) in n = 6 mice per group. The results are presented as cells per mm2. The data are presented as the mean ± SD (n = 4 for each time point). *p < 0.05, **p < 0.01, significantly different from control group.
Knockdown of LRCC19 suppresses pro-inflammatory cytokines production during I/R injury
Wound healing is a complex and dynamic biological process that involves the coordinated efforts of multiple cell types and is executed and regulated by numerous growth factors and cytokines.18 Poor wound healing is associated with sustained expression of proinflammatory cytokines, including TNFα (tumor necrosis factor-α), IL-1β (Interleukin 1β), IL6 (Interleukin 6).19 These cytokines could rapidly activate the neutrophils, macrophages to transmigrate across the endothelial cells barrier to the wound site.20 We next measured the production of these cytokines in the damaged tissues by ELISA. TNFα level at day 0 was comparable in both siRNA control and LRRC19 siRNA treated groups (∼2 pg/mg tissue), then it was induced to about 15 pg/mg tissue in the control group followed by returning to the similar level with that of day 0 (∼3 pg/mg tissue). LRRC19 siRNA treated group showed 40% less of TNFα at day 1 and 50% less at day 3 compared to controls (Figure 3A). IL-1β and IL6 showed prolonged induction by I/R damage at day 1 and day 3 which were 5–8 fold of the levels observed in day 0. Similarly, LRRC19 siRNA treated groups showed significantly reduced levels of IL-1β and IL6 at day 1, day3 compared to control group. The reduction was more dramatically observed for IL-1β than IL6: about 50% less of IL-1β was detected at day 1 and more than 70% reduction found in LRRC19 siRNA group compared to control group at day 3. For IL6, about 30% reduction of levels observed for both day 0 and day 3 LRRC19 siRNA groups compared with controls (Figure 3B & C). These results suggested that LRRC19 is required for production of those proinflammatory cytokines in I/R induced damaged site.
FIGURE 3.
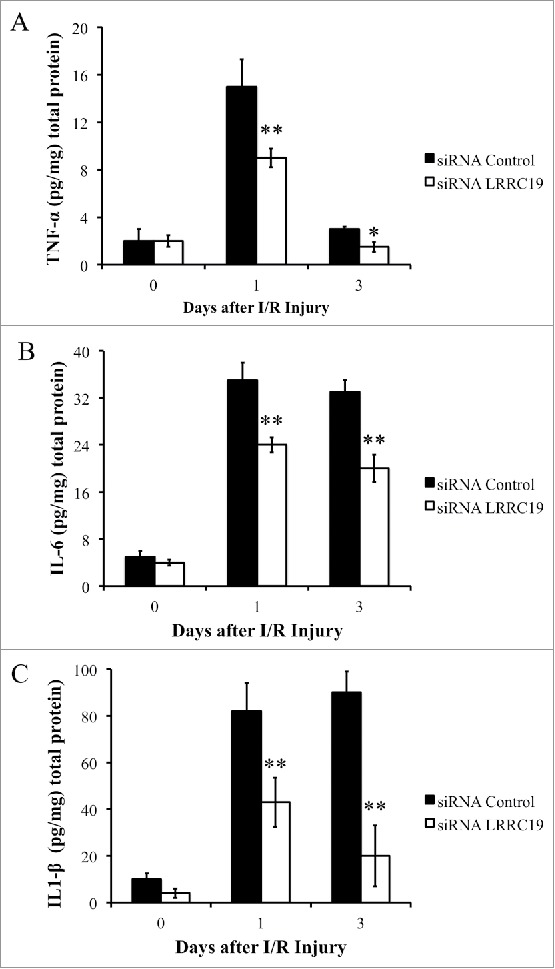
Knocking down LRRC19 suppresses proinflammatory cytokines production during I/R injury. ELISA analysis of A) TNFα, B) IL-6 and C) IL-1β at the wound site during I/R injury at the indicated time points. The wound area was topically applied with 100 pmol of either LRRC19 siRNA or control siRNA immediately after 1st ischemia delivery, followed by day 0, 1 after I/R injury induced. The end of 2nd ischemia was assigned day 0. The data are presented as the mean ± SD (n = 4). *p < 0.05, **p < 0.01, significantly different from control group measured on the same day.
Knockdown of LRCC1 suppresses NFκB signaling and proinflammatory cytokines production activated by IL-1β
As NFκB is the key modulator for inflammatory response and activated by TNFα and IL-1β stimulation,21 we next sought to test whether LRRC19 affected this pathway. We isolated the mouse primary dermal fibroblasts and knocked down the LRRC19 by siRNA. Then the cells were treated for IL-1β for different time points. Firstly, we checked the immediate activation of NFκB signaling with short time of IL-1β stimulation. As shown in Figure 4A, the components of NFκB signaling apparatus such as p65 and its inhibitor IκBα were found to be robustly phosphorylated by 10 and 20 min IL-1β stimulation in siRNA control cells, but their activation was almost abolished in LRRC19 siRNA cells. Once activated, NFκB can turn on some important downstream mediators of inflammation such as iNOS and COX-2, which act as part of the long term effectors.22 Indeed, long exposure to IL-1β induced the expression of COX-2 and iNOS in siRNA control cells. On the contrary, the induction was greatly attenuated in LRRC19 siRNA cells, which only showed marginal expression of them at 16 h stimulation with IL-1β (Figure 4B). To further validate LRRC19 playing a regulatory role in NFκB signaling, we also proceeded to measure the IL-6 and chemokine IL-8 production under IL-1β challenge. As shown in Figure 5A and 5B, IL-6 and IL-8 were markedly induced in control primary fibroblasts stimulated with IL-1β for 24 h. And in cells transfected with siRNA against LRRC19, the induction of IL-6 and IL-8 were greatly suppressed, suggesting LRRC19 positively regulating NFκB signaling axis. Taken together, we could conclude that LRRC19 plays a negative role for IL-1β mediated NFκB activation and its downstream effectors.
FIGURE 4.
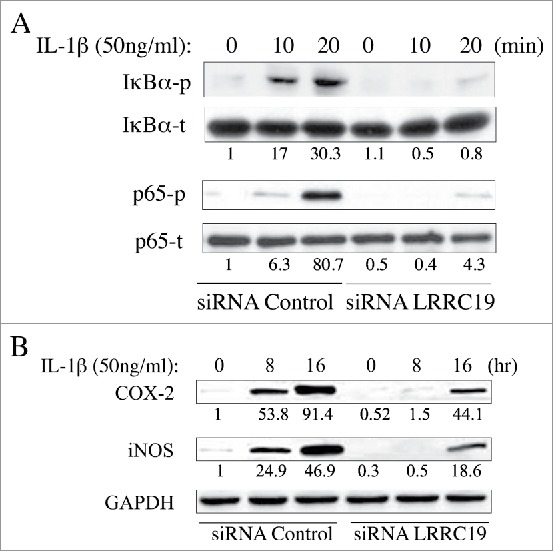
Knocking down LRRC19 suppresses NFκB signaling. Western blot analysis of NFκB signaling components. Primary dermal fibroblasts transfected with either scramble siRNA or LRRC19 siRNA were challenged with 50 ng/ml IL-1β for the indicated time points. Cell lysate after treatment was subjected to SDS-PAGE to determine the level of A) total p65, phospho-p65 (S536), total IkBα and phosphor-IkBα (S32); or the level of B) COX-2 and iNOS. The band intensity of each target protein was normalized against its corresponding loading control. The normalized band densitometry of the siRNA control transfected sample at day 0 was set as baseline of value 1. The data are representative of at least three independent experiments.
FIGURE 5.
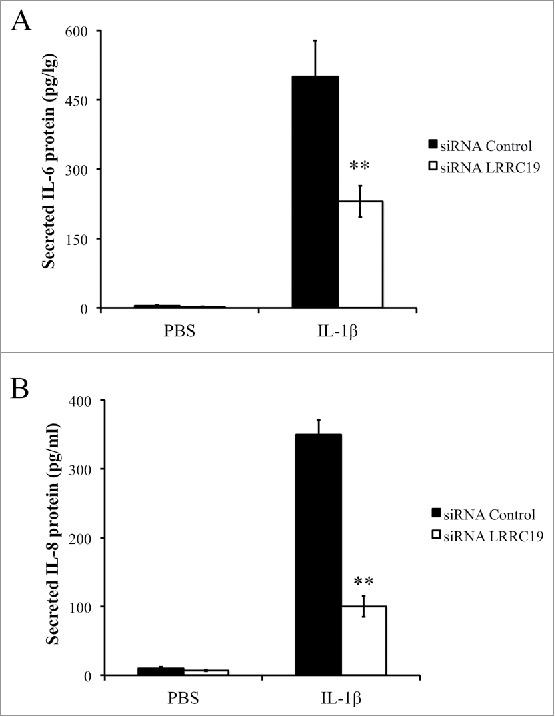
Knocking down LRRC19 reduces IL-1β mediated proinflammatory cytokines production. Primary dermal fibroblasts transfected with either scramble siRNA or LRRC19 siRNA were challenged with 50 ng/ml IL-1β or PBS for 24 h. The cell culture supernatant was then harvested to determine the concentrations of A) IL-6 and B) IL-8 by ELISA. The data are presented as the mean ± SD (n = 4). *p < 0.05, **p < 0.01, significantly different from PBS control group.
DISCUSSION
Pressure ulcers are common skin lesions frequently found in many hospitalized patients with immobility. They are significant sources of pain and distress, leading to the impairment of the quality of life of patients 23 Although it has long been considered that the pathogenesis of pressure ulcer is driven by multiple factors including chronic tissue ischemia, lack of physical activity, there has been increasing evidence that I/R is associated with the pathogenesis of pressure ulcers.5, 6 In this study, we identified LRRC19 as a new contributing factor in the well-established I/R caused pressure ulcer model. The basal level of LRRC19 in skin tissue was not high, but the induction of LRRC19 was robust and prompt. Interestingly, LRRC19 induction was in the similar pattern as seen in skin wound healing process, with LRRC19 reaching its peak ahead of wound area (Figure 1A&B). siRNA knockdown of LRRC19 in skin tissue would alleviate the tissue damage severity possibly through downregulation of infiltration of immune cells, secretion of pro-inflammatory cytokines (Figure 2&3). In a skin primary fibroblast model, siRNA knockdown of LRRC19 suppressed the activation of NFκB by IL-1β stimulation (Figure 4). These data suggested that LRRC19 promoted the tissue damage and became a potential therapeutic target for the disease.
LRRC19 is a newly identified transmembrane protein of the large protein family with leucine-rich repeats motif. It shares certain sequence homology with TLR3,4,11,12 at extracellular LRR domain; however, LRRC19 has no TIL domain at intracellular region, which is found in all TLR and IL-1R.12 This unique sequence trait suggests that LRRC19 may play different roles compared to conventional TLRs. Several earlier studies have demonstrated that LRRC19 is involved in bacteria clearance in kidney and intestine tissue and required for the activation of NFκB and MAPK signal pathways.11, 13 While in pressure ulcers it is generally thought that the necrotic cell after ischemia stress largely causes a different kind of inflammatory response which is not due to infections at least at early stage.24 Cutaneous I/R induces the recruitment of neutrophils and macrophages and the subsequent release of proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, and toxic oxygen-derived free radicals induces the apoptosis of skin fibroblasts and skin injury.25 Our result indicates that LRRC19 is readily induced during the I/R cause skin injury and recovery process, and it is required for the production of pro-inflammatory cytokines, infiltration of immune cells, and downstream NFκB activation. Prolonged inflammation is a major event that impairs wound healing and increases scarring in pressure ulcer model.26 Therefore, as suggested in our study, the beneficial effect of knocking down LRRC19 is strongly associated with its regulation in inflammation. In addition, the function of LRRC19 in this sterile inflammation model is largely consistent with what have been reported in the kidney and gut using LRRC19 knockout mouse model. As a presently favored model, the initiators of inflammation involved in pressure ulcers are the pro-inflammatory damage-associated molecular patterns that are normally intracellular and hidden by the plasma membrane.27 Apparently these stimulants are different from bacterial components found in kidney or gut infections, they share certain downstream innate immunity adaptor and signaling events, such as Myd88, TRAF2/6 mediated ubiquitination, and downstream NFκB activation.24, 28 In the late stage of pressure ulcer, the damage of skin integrity by tissue necrosis also undoubtedly leads to bacteria infection. Therefore, what we have observed in pressure ulcer model should resemble the findings of LRRC19 function in kidney infection. We could speculate that LRRC19 exerts its pro-inflammatory role through the regulation of TRAF2/6 mediated ubiquitination to activate NFκB pathway, which should be better examined in LRRC19 knockout mouse model in the future.
Noteworthy, we first report that LRRC19 could be up-regulated in skin tissue in the pathological progress of I/R induced pressure ulcer model. Previous reports of LRRC19 by Yang et al. focused on the kidney and intestine tissues with constitutively high expression level of LRRC19 and unveiled the important function of this novel LRR protein member.11-13 However, they overlooked that LRRC19 level could be induced in response to certain stress or stimuli. This mode of action is commonly observed for many stress proteins, such as the famous p53 protein and many acute phase response proteins induced by inflammation.29 Currently it is not clear how LRRC19 is transcriptionally induced by I/R, which will be addressed in future investigation. Our finding about LRRC19 inducibility warrants further study to identify new stimulation as well as tissue types for LRRC19 upregulation. One important scenario is I/R induced heart damage, which is caused by the pro-inflammatory response, associated with oxidative stress.30 According to Yang et. al, LRRC19 is not found in mouse heart tissue;12 however, it is worthwhile to investigate whether LRRC19 could be induced in heart tissue in response to I/R and be a potential target for myocardial infarction.
In conclusion, LRRC19 is induced in I/R caused pressure ulcer model and promotes pro-inflammatory response and tissue damage. Targeting LRRC19 offers a new avenue to treat pressure ulcers and other I/R caused diseases.
MATERIALS AND METHODS
Reagents
Recombinant mouse IL-1β (I5271) was purchased from Sigma-Aldrich. All antibodies against phospho-IκBα (Ser32) (14D4) (#2859, 1:1,000 in use), IκBα (44D4) (#4812, 1:1,000 in use), phospho-NFκB p65 (Ser536) (93H1) (#3033, 1:2,000 in use), NFκB p65 (D14E12) (#8242, 1:1,000 in use), Cox2 (#4842, 1:1,000 in use), iNOS (Mouse Specific) (#2982, 1:2,000 in use), GAPDH (D16H11) (#5174, 1:5,000 in use) were from Cell Signaling Technology. Myeloperoxidase (#PA5-16672, 1:500 in use) and CD68 (FA-11) (#14-0681-82, 1:500 in use) antibodies were from Thermo Fisher Scientific. Anti-LRRC19 antibody (ab106657, 1:800 in use) was from Abcam.
Establishment of pressure ulcer animal model
Animal experiments were performed according to the guidelines published by the Institute of Laboratory Animal Resources of National Research Council and animal care for this study was approved by the Institutional Animal Care and Use Committee of Cangzhou Central Hospital.
Pressure ulcer model was established following the procedure described in Stadler et al.5 Briefly, C57BL/6 mice were anesthetized with isoflurane inhalation. 3 cm × 2 cm rectangle of skin on the mouse back was shaved, cleaned and disinfected with 70% isopropanol. A template was used to mark the location of the magnetic plates to assure a consistent placement on each animal. The skin was gently pulled up and placed between 2 round ceramic magnetic plates that had 12 mm diameter and were 5.0 mm thick, with an average weight of 2.4 g and 1,000 G magnetic force (Magnetic Source, Castle Rock, CO). The resultant “pinch” procedure was designed to leave a 5.0-mm skin bridge between the two magnets. Two ischemia–reperfusion cycles were used in each animal to initiate decubitus ulcer formation. A single ischemia–reperfusion cycle consisted of a 16-h period of magnet placement followed by a release or rest period of 8 h. Animals were not immobilized, anesthetized, or otherwise treated during the ischemia–reperfusion cycles. The animals were allowed food and water ad ibitum.
Topical application of nanoparticle encapsulated siRNA
500 μl of Tetramethyl orthosilicate (TMOS) was hydrolyzed in the presence of 100 μl of 1 mM HCl by sonication on ice for about 15 minutes, until a single phase formed. The hydrolyzed TMOS (100 μl) was added to 900 μl of 100 pmol of siRNA (mouse LRRC19 or the negative control) solution containing 10 mM phosphate, pH 7.4. A gel was formed within 10 minutes. The gel was frozen at −80°C for 15 minutes and lyophilized. 100 pmol nanoparticle encapsulated LRRC19 siRNA or control siRNA was first applied on the lesion during the reperfusion period of first I/R cycle, followed by day 0, 3, 6, 9, 12 after I/R injury induced. Mice were randomly selected for euthanasia on designated days, and tissue from the wounds was excised for both PCR and histopathologic analysis.
Sequence of siRNA used was listed as below:
LRRC19 siRNA:
Sense: 5′-GAAUCUGCAAGGCAAUUUGAUACGC-3′
Anti-sense: 5′-GCGUAUCAAAUUGCCUUGCAGAUUCAG-3′
Scramble control:
Sense: 5′-GAAGCAGCACGACUUCUUCTT-3′
Anti-sense: 5′-GGAACGAUCUAGUCGACUCTT-3
Reverse Transcription Polymerase Chain Reaction (RT-PCR) assay
Total cellular RNA samples from skin tissues were extracted using the TrizolTM Reagent (Invitrogen, Carlsbad, CA, USA). RT-PCR was performed using the ThermoScript RT-PCR System (Invitrogen, USA). Primers used were listed as below:
LRRC19 F: (5′-TGCTATCAAGTGCCCAGTATG-3′)
LRRC19 R: (5′-TTTCTGCCTCATGCTCTTCC-3′)
ACTINβ F (5′-TCACCCACACTGTGCCCATCTACGA-3′)
ACTINβ R (5′-GGATGCCACAGGATTCCATACCCA-3′)
Cell culture
Mouse primary dermal fibroblasts cells were isolated from mouse skin as described 15 and maintained in Dulbecco's modified Eagle's medium (Invitrogen, USA) supplemented with 10% fetal bovine serum (Sigma, St. Louis, MO, USA), 100 U/ml penicillin and 100 μg/ml streptomycin in a 5% CO2 atmosphere at 37°C.
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixed paraffin-embedded rat infarct heart tissue sections. To expose target proteins, heat-induced epitope retrieval (HIER) was performed in sodium citrate buffer (pH 6.0) for 20 minutes at 100°C. Tissues were blocked in 10% normal goat serum for 20 minutes at room temperature and probed with a myeloperoxidase polyclonal antibody (# PA5-16672), anti-CD68 (#14-0681-82) at a dilution of 1:200 in 1% normal goat serum for 1hour at room temperature. Tissues were washed extensively with 1 × PBS. Detection was performed using a biotinylated goat anti-rabbit or anti-mouse IgG secondary antibody followed by an avidin-biotin complex reagent and DAB colorimetric substrate. The images were acquired with microscopy. The positive cells were counted in six random microscopic fields (∼20,000 mm2) for different groups of samples (n = 10). The results were presented as cells per mm2.
Measurement of cytokine levels by ELISA
Skin tissue was collected from mice with control siRNA or LRRC19 siRNA treatments during and after I/R injury. The skin tissue was then solubilized in the lysis buffer provided by the kit. Supernatant of lysate was collected by centrifugation for 20 min at 12,000 rpm, 4°C and they were stored at −80°C after snap freezing until further use. The level of cytokines was measured with ELISA kits from Thermo Fisher Scientific: TNFα (BMS607-3), IL-1β (BMS6002) and IL-6 (BMS603-2), following the instruction of manufacturer. For cell culture experiment, primary dermal fibroblasts transfected with either scramble siRNA or LRRC19 siRNA were challenged with 50 ng/ml IL-1β (#BMS332, Thermo Scientific) for 24h. The cell media were harvested and the concentration of IL-6 (M6000B, R&D Systems) and IL-8 (#D8000C, R&D Systems) in the culture supernatant were determined by ELISA following the instruction of manufacturer.
Western blotting
Total protein from cells or mouse tissues was extracted using RIPA buffer. Following solubilization, proteins were quantified using a BCA protein quantification kit (Thermo Scientific, Rockford, IL, USA). 40 micrograms of protein were loaded into each well and separated on a 10% SDS Tris-Glycine gel, then transferred onto polyvinylidene difluoride (PVDF) membranes (Invitrogen, USA) at a constant current of 250 mA in transfer buffer (50 mM Tris, pH 8.0, 0.192 M glycine, 20% (v/v) methanol), using a Bio-Rad Trans-Blot Cell (Bio-Rad, Hercules, CA, USA). The membranes were incubated for 1 h at room temperature in blocking buffer (TBS containing 5% non-fat milk), followed by an overnight incubation at 4°C with appropriate primary antibodies in blocking buffer. After three washes with TBS containing 0.05% Tween 20, strips were incubated for 1 h with corresponding HRP-conjugated secondary antibodies. Protein-antibody complexes were visualized using an ECL detection system as recommended by the manufacturer (Applygen Technologies, Beijing, China). Data were recorded using a Bio-Rad Gel Doc 2000 system. The quantification of protein bands was done by densitometry with ImageJ.
Statistical analysis
All experiments described here have been repeated at least three times. Results are presented as mean ± standard deviation (SD). Comparison of the data was performed using Student's t test. Significance was defined as p < 0.05. Statistical analysis was performed using SPSS software.
Supplementary Material
Funding Statement
None.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Hoogendoorn I, Reenalda J, Koopman B, Rietman JS. The effect of pressure and shear on tissue viability of human skin in relation to the development of pressure ulcers: a systematic review. Journal of tissue viability. 2017;26(3):157–71. doi: 10.1016/j.jtv.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Demarre L, Van Lancker A, Van Hecke A, Verhaeghe S, Grypdonck M, Lemey J, Annemans L, Beeckman D. The cost of prevention and treatment of pressure ulcers: A systematic review. International journal of nursing studies. 2015;52(11):1754–74. doi: 10.1016/j.ijnurstu.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Lyder CH. Pressure ulcer prevention and management. Jama. 2003;289(2):223–6. doi: [DOI] [PubMed] [Google Scholar]
- 4.Allman RM. Pressure ulcer prevalence, incidence, risk factors, and impact. Clinics in geriatric medicine. 1997;13(3):421–36. doi: [PubMed] [Google Scholar]
- 5.Stadler I, Zhang RY, Oskoui P, Whittaker MS, Lanzafame RJ. Development of a simple, noninvasive, clinically relevant model of pressure ulcers in the mouse. Journal of investigative surgery: the official journal of the Academy of Surgical Research. 2004;17(4):221–7. doi: 10.1080/08941930490472046. [DOI] [PubMed] [Google Scholar]
- 6.Peirce SM, Skalak TC, Rodeheaver GT. Ischemia-reperfusion injury in chronic pressure ulcer formation: a skin model in the rat. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2000;8(1):68–76. doi: [DOI] [PubMed] [Google Scholar]
- 7.Carden DL, Granger DN. Pathophysiology of ischaemia-reperfusion injury. The Journal of pathology. 2000;190(3):255–66. doi:. [DOI] [PubMed] [Google Scholar]
- 8.Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell metabolism. 2016;23(2):254–63. doi: 10.1016/j.cmet.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 9.Ng AC, Eisenberg JM, Heath RJ, Huett A, Robinson CM, Nau GJ, Xavier RJ. Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl):14631–8. doi: 10.1073/pnas.1000093107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Current opinion in structural biology. 2001;11(6):725–32. doi: [DOI] [PubMed] [Google Scholar]
- 11.Su X, Min S, Cao S, Yan H, Zhao Y, Li H, Chai L, Mei S, Yang J, Zhang Y, et al.. LRRC19 expressed in the kidney induces TRAF2/6-mediated signals to prevent infection by uropathogenic bacteria. Nature communications. 2014; 54434. doi: 10.1038/ncomms5434. [DOI] [PubMed] [Google Scholar]
- 12.Chai L, Dai L, Che Y, Xu J, Liu G, Zhang Z, Yang R. LRRC19, a novel member of the leucine-rich repeat protein family, activates NF-kappaB and induces expression of proinflammatory cytokines. Biochemical and biophysical research communications. 2009;388(3):543–8. doi: 10.1016/j.bbrc.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 13.Cao S, Su X, Zeng B, Yan H, Huang Y, Wang E, Yun H, Zhang Y, Liu F, Li W, et al.. The Gut Epithelial Receptor LRRC19 Promotes the Recruitment of Immune Cells and Gut Inflammation. Cell reports. 2016;14(4):695–707. doi: 10.1016/j.celrep.2015.12.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurose T, Hashimoto M, Ozawa J, Kawamata S. Analysis of Gene Expression in Experimental Pressure Ulcers in the Rat with Special Reference to Inflammatory Cytokines. PloS one. 2015;10(7):e0132622. doi: 10.1371/journal.pone.0132622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seluanov A, Vaidya A, Gorbunova V. Establishing primary adult fibroblast cultures from rodents. Journal of visualized experiments: JoVE. 2010;(44). doi: 10.3791/2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strong AL, Bowles AC, MacCrimmon CP, Lee SJ, Frazier TP, Katz AJ, Gawronska-Kozak B, Bunnell BA, Gimble JM. Characterization of a Murine Pressure Ulcer Model to Assess Efficacy of Adipose-derived Stromal Cells. Plastic and reconstructive surgery Global open. 2015;3(3):e334. doi: 10.1097/GOX.0000000000000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charafeddine RA, Makdisi J, Schairer D, O'Rourke BP, Diaz-Valencia JD, Chouake J, Kutner A, Krausz A, Adler B, Nacharaju P, et al.. Fidgetin-Like 2: A Microtubule-Based Regulator of Wound Healing. The Journal of investigative dermatology. 2015;135(9):2309–18. doi: 10.1038/jid.2015.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiological reviews. 2003;83(3):835–70. doi: 10.1152/physrev.00031.2002. [DOI] [PubMed] [Google Scholar]
- 19.Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. The Journal of investigative dermatology. 2007;127(3):514–25. doi: 10.1038/sj.jid.5700701. [DOI] [PubMed] [Google Scholar]
- 20.LeBert DC, Huttenlocher A. Inflammation and wound repair. Seminars in immunology. 2014;26(4):315–20. doi: 10.1016/j.smim.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harbor perspectives in biology. 2009;1(6):a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang C, Ling H, Zhang M, Yang Z, Wang X, Zeng F, Wang C, Feng J. Oxidative stress mediates chemical hypoxia-induced injury and inflammation by activating NF-kappab-COX-2 pathway in HaCaT cells. Molecules and cells. 2011;31(6):531–8. doi: 10.1007/s10059-011-1025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorecki C, Brown JM, Nelson EA, Briggs M, Schoonhoven L, Dealey C, Defloor T, Nixon J, European Quality of Life Pressure Ulcer Project g . Impact of pressure ulcers on quality of life in older patients: a systematic review. Journal of the American Geriatrics Society. 2009;57(7):1175–83. doi: 10.1111/j.1532-5415.2009.02307.x. [DOI] [PubMed] [Google Scholar]
- 24.Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nature medicine. 2007;13(7):851–6. doi: 10.1038/nm1603. [DOI] [PubMed] [Google Scholar]
- 25.Diegelmann RF. Excessive neutrophils characterize chronic pressure ulcers. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2003;11(6):490–5. doi: [DOI] [PubMed] [Google Scholar]
- 26.Qian LW, Fourcaudot AB, Yamane K, You T, Chan RK, Leung KP. Exacerbated and prolonged inflammation impairs wound healing and increases scarring. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2016;24(1):26–34. doi: 10.1111/wrr.12381. [DOI] [PubMed] [Google Scholar]
- 27.Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annual review of immunology. 2010; 28321–42. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kono H, Chen CJ, Ontiveros F, Rock KL. Uric acid promotes an acute inflammatory response to sterile cell death in mice. The Journal of clinical investigation. 2010;120(6):1939–49. doi: 10.1172/JCI40124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. The New England journal of medicine. 1999;340(6):448–54. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 30.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure. Journal of molecular and cellular cardiology. 2001;33(6):1065–89. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.