

Abstract
Hispidulin is a naturally occurring flavone known to have various Central nervous system (CNS) activities. Proposed synthetic approaches to synthesizing hispidulin have proven unsatisfactory due to their low feasibility and poor overall yields. To solve these problems, this study developed a novel scheme for synthesizing hispidulin, which had an improved overall yield as well as more concise reaction steps compared to previous methods reported. Additionally, using the same synthetic strategy, d-labelled hispidulin was synthesized to investigate its metabolic stability against human liver microsome. This work may produce new chemical entities for enriching the library of hispidulin-derived compounds.
Keywords: hispidulin, total synthesis, flavone, microsome stability, deuterium-labelled compound
1. Introduction
Flavonoids, a group of polyphenolic compounds, occur ubiquitously in plants. These compounds have generally been categorized into several classes, including chalcones, flavonols, flavones, isoflavones, flavan-3-ols, neoflavones, flavanones and anthocyanins, based on their chemical structures [1]. In addition to the diverse structures, they also have many biological properties, such as antioxidative [2], anti-inflammatory [3], antimicrobial [4], anticonvulsant [5], antidepressant [6] and anticancer [7] activities. These facts indicate the important role of flavonoid compounds.
Our recent clinical case report described a patient with a refractory chronic motor tic disorder who dramatically improved after taking the leaf juice of a local herb, Clerodendrum inerme (L.) Gaertn (CI) [8]. The ethanol extract of CI leaf alleviated methamphetamine-induced hyperlocomotion (MIH) as a mouse model of motor tic [9]. Using a bioassay-guided purification procedure in this animal model, we further isolated the constituents from CI leaf extract to identify a flavone hispidulin (6-methoxy-4′,5,7-trihydroxyflavone) to be the main active ingredient (Figure 1) [9]. Hispidulin significantly attenuated MIH and even in amounts up to 100 mg/kg was incapable of affecting spontaneous locomotor activity or performance in mice [9]. Importantly, hispidulin had no hit on human ether-à-go-go-related gene (hERG) channels, an undesirable target in drug development [10].
Figure 1.
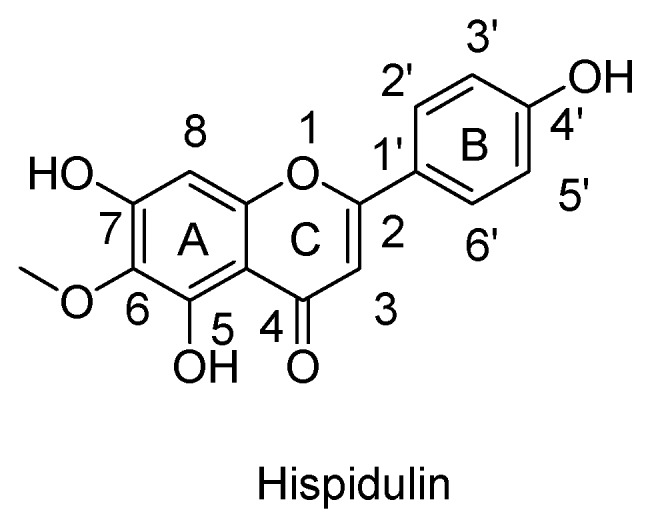
The chemical structure of hispidulin.
Hispidulin is widely distributed in Asteraceae [11,12,13,14] and some of the Lamiaceae [15]. Further assays for screening towards 92 target proteins associated with Central nervous system (CNS) diseases indicated that hispidulin formed strong bonds with GABAA receptors (IC50 = 0.73−1.78 μM) and inhibited catecholamine-O-methyl-transferase (COMT) (IC50 1.32 μM) [10]. By acting as a positive allosteric modulator (PAM), it enhanced cerebellar α6GABAA receptor activity. Studies revealed some C6-substituted flavones are identified to be PAMs of GABAA receptors [16,17]. The results of a structure–activity relationship (SAR) study indicated the C6 substituent in flavones greatly contributes to activity [10].
Because of its interesting biological activity, several research groups have developed synthetic approaches to hispidulin [18,19,20,21,22,23]. For example, Shen and coworkers developed a strategy for semisynthesis of hispidulin in seven reaction steps by using a naturally occurring scutellarin (Scheme 1) [22]. Although this method is concise and has an overall yield as high as 10.7% [22], the researchers showed that, upon the large-scale synthesis of compound 1, the protection of the catechol moiety of scutellarein using dichlorodiphenylmethane at 175 °C failed [20,23]. A seven-step synthesis route developed by Lin and coworkers solved this problem but reduced the overall yield to 7.1% (Scheme 1) [20]. Zhang and coworkers then developed a scheme that only required four reaction steps (Scheme 1). Nonselective MOM (methoxymethyl) protection of scutellarein caused the overall yield of the synthesis of hispidulin to be decreased (6.3%) [23]. Despite the satisfactory overall yield of these strategies, the tedious purification procedure required to isolate scutellarin from plants limits their use for large-scale preparation of hispidulin.
Scheme 1.

Synthesis of hispidulin starting from scutellarin.
Kavvadias and coworkers developed a method synthesizing hispidulin from 2,4,6-trihydroxyacetophenone compound 6 in nine reaction steps (Scheme 2). However, the overall yield of this method is very limited (1.1%) [18]. We recently developed a feasible and reproducible approach for synthesizing hispidulin (Scheme 3) [21]. This method slightly improved the overall yield due to the low yield of Friedel–Crafts acetylation of compound 8 as well as unsatisfactory regioselective MOM protection of compound 9. These facts motivated us to investigate an efficient and high-yield route to synthesize hispidulin.
Scheme 2.

Synthesis of hispidulin by Kavvadias and coworkers.
Scheme 3.

Synthesis of hispidulin in our previous study.
Deuterium is a stable isotope of hydrogen. Because deuterium has a stronger chemical bond with carbon than hydrogen, deuterium-labelled compounds can affect the absorption, distribution, metabolism and toxicology of their counterpart compound [24,25]. The Food and Drug Administration (FDA) recently approved the first deuterated drug, deutetrabenazine, for treating involuntary writhing movements or chorea in Huntington’s disease [26]. Deuterium incorporation of tetrabenazine markedly increased its half-life and area under the curve (AUC) in plasma [27]. Study showed that the pig caecal microflora metabolized hispidulin to scutellarein due to O-demethylation at the C6 position [28]. This result suggested that replacement of OCH3 of this position of hispidulin using OCD3 may reduce its metabolic liability. Therefore, this study replaced C6-OCH3 in hispidulin with C6-OD3 to investigate whether such modification allows enhancement of the metabolic stability. Notably, introducing deuterium into hispidulin leads to a new chemical entity (NCE) that meets the criteria for a 505(b) (2) patent application [27].
The new hispidulin synthesis scheme developed in this study is more feasible compared to all methods previously reported. In particular, it had the highest overall yield, which may help to synthesize C6-OCH3-containing hispidulin derivatives. The same strategy can also be used to synthesize d-labelled hispidulin. The microsome stability of hispidulin and its deuterium counterpart were compared.
2. Results and Discussion
2.1. Retrosynthetic Analysis of Hispidulin
Figure 2 shows the retrosynthetic analysis to successfully synthesize hispidulin. Hispidulin can be produced from compound I via debenzylation and oxidative cyclization. Compound I is derived from compound II and commercially available compound III, which are used to conduct Claisen–Schmidt condensation and deprotection of MOM (methoxymethoxy). Compound II in turn can be prepared from compound IV via methylation. Compound IV is considered to be the key intermediate that possesses two different protecting groups as well as acetyl and hydroxy moieties. Selective Bayer–Villiger reaction of compound V provided compound IV. Compound V is prepared from compound VI via Stille coupling. Benzylation and regioselective iodination of compound VII gives compound VI. Starting from commercially available compound 7, Compound VII is synthesized via selective MOM protection.
Figure 2.

Retrosynthetic analysis of hispidulin.
2.2. Synthesis of Hispidulin
Scheme 4 shows a synthetic approach originally developed for hispidulin. Using compound 6 as the starting material, selective MOM protection towards its C4- and C6-OH gave compound 11. First, N-bromosuccinimide (NBS) was used for bromination of C3 of compound 11; however, only undesirable C3 and C5 dibromination occurred. Alternatively, compound 11, when reacted with I2 in the presence of Lewis acid CF3CO2Ag, provided selectively iodinated product 12 in the high yield (90%). The experimental result resulted from the difference of steric hindrance between C3 and C5 upon using the more bulky reagent I2. Benzylation of compound 12 generated compound 13. Next, several reported oxidation methods were used [29,30,31] in attempts to convert the iodide group of compound 13 into phenol. These oxidation catalysts included CuI, Cu and Pd, as shown in Table 1. Reaction using Pd2dba3 coupled with the ligand 2-di-tert-butylphosphino-2′,4′,6′-triisopropylbiphenyl (tBuXPhos) gave compound 14, but its yield was poor (27%). Despite the use of microwave irradiation, the yields were incapable of being improved. Instead, Claisen–Schmidt condensation of compound 13, reacted with 4-(benzyloxy)benzaldehyde prior to selective deprotection of MOM group in the presence of HCl, gave chalcone 15. The selective deprotection reaction can be manipulated by decreasing the reaction time to avoid further MOM deprotection of C4. Oxidative cyclization of compound 15 in the presence of catalytic I2 provided flavone 16. Notably, excess I2 caused an undesirable retro-aldol reaction that gave C4- and C6-deprotected compound 13. Treatment of catalytic Pd(PPh3)4 and tributyl(1-ethoxyvinyl)tin converted compound 16 to compound 17 through the Stille reaction. Baeyer–Villiger oxidation followed by basic hydrolysis of compound 17 by using H2O2 or m-chloroperoxybenzoic acid (MCPBA) [32] failed to give expected product 18.
Scheme 4.

Initial attempt for the synthesis of hispidulin. Reagents and conditions: (a) MOMCl, DIPEA, CH2Cl2, 0 °C, 64%; (b) CF3COOAg, I2, CH2Cl2, 0 °C, 90%; (c) K2CO3, BnBr, DMF, 0 °C, 99%; (d) Pd2dba3, tBuXPhos, KOH, dioxane–H2O, 90 °C, 27%; (e) (1) BnOPhCHO, KOH, EtOH, H2O, 0 °C; (2) c-HCl, MeOH, THF, 0 °C, 77%; (f) I2, DMSO, 120 °C, 32%; (g) tributyl(1-ethoxyvinyl)tin, Pd(PPh3)4, dioxane, 100 °C, 61%.
Table 1.
Attempts towards the oxidation of aryl halide 13.
| Entry | Catalyst | Ligand | Base | Solvent | T (°C) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | CuI | 1,10-Phenanthroline | KOH | DMSO–H2O | 100 | trace |
| 2 | Cu | - | NaOH | H2O | 100 | trace |
| 3 | Cu | - | NaOH | DMSO–H2O | 100 | trace |
| 4 | Pd2dba3 | tBuXPhos | KOH | Dioxane–H2O | 100 | 27 |
| 5 | Pd2dba3 | tBuXPhos | KOH | Dioxane–H2O | 70 | 14 |
| 6 | Pd2dba3 | tBuXPhos | KOH | Dioxane–H2O | 100 1 | 26 |
| 7 | Pd2dba3 | tBuXPhos | KOH | Dioxane–H2O | 100 2 | 23 |
| 8 | Pd2dba3 | tBuXPhos | KOH | DMF | 100 1 | trace |
1 Microwave reaction: 100 W, 20 min; 2 microwave reaction: 120 W, 20 min.
Thus, we developed a new synthesis (Scheme 5) for hispidulin using the retrosynthetic analysis as shown in Figure 2. First, 2,4,6-trihydroxybenzaldehyde 7 reacted with MOMCl gave the bis(methoxymethoxy)-protected compound 20. Regioselective iodination of compound 20 prior to reaction with BnBr produced compound 21. The chemical structure of compound 21 was confirmed by rotating frame nuclear Overhauser effect spectroscopy (ROESY) spectrum. Figure 3 shows the key correlation of H-5 (δH 6.84) to H-2′″ (δH 3.52), H-2″″ (δH 3.54), H-1′″ (δH 5.29) and H-1″″ (δH 5.32); and H-1′ (δH 10.30) to H-2″″ (δH 3.54), H-1″ (δH 4.99) and H-1″″ (δH 5.32) (Supplementary Materials). Stille coupling of compound 21 produced compound 22. Table 2 shows how tributyl(1-ethoxyvinyl)tin was used to optimize Stille coupling. First, catalyst Pd(PPh3)4 was used in dioxane at 100 °C. The reaction had a satisfactory yield (68%), but the reaction time was up to 30 h. Replacement of the solvent by toluene led to the reaction time decreasing to 24 h. Further experiments using palladium catalysts such as PdCl2(PPh3)2 and Pd(dppf)Cl2 in dioxane or toluene showed that PdCl2(PPh3)2 significantly improved the yield and decreased the reaction time. In particular, PdCl2(PPh3)2 coupled with toluene not only gave the highest yield, but also had the lowest reaction time. Baeyer–Villiger oxidation and basic hydrolysis of compound 22 afforded compound 14. Methylation of compound 14 using CH3I gave compound 23a. Claisen–Schmidt condensation of compound 23a with 4-(benzyloxy)benzaldehyde prior to MOM deprotection produced chalcone 24a. Cyclization of compound 24a in the presence of catalytic I2 generated flavone 25a. Although debenzylation of compound 25a catalyzed by 10% Pd–C did not yield hispidulin, a reaction using BCl3 at −80 °C successfully converted compound 25a into hispidulin. The chemical structure of hispidulin was identified as judged by 2D-NMR analyses. Figure 4 shows the key correlation in the ROESY spectrum of hispidulin that 5-OH (δH 13.07) was correlated to 6-OMe′″ (δH 3.74) and H-8 (δH 6.59) was correlated to H-2′ and H-6′ (δH 7.92). Additionally, the HMBC spectrum showed that 5-OH (δH 13.07) correlated to C-5 (δC 152.8), C-6 (δC 131.4), C-7 (δC 157.3), C-9 (δC 152.4) and C-10 (δC 104.1); H-8 (δH 13.07) correlated to C-6 (δC 131.4), C-7 (δC 157.3), C-9 (δC 152.4) and C-10 (δC 104.1); 6-OMe-H (δH 3.74) correlated to C-6 (δC 131.4) (Supplementary Materials). The 1H- and 13C-NMR data of synthesized hispidulin were similar to those of hispidulin previously isolated (Supplementary Materials) [33].
Scheme 5.

Synthesis of hispidulin and d-hispidulin. Reagents and conditions: (a) MOMCl, DIPEA, CH2Cl2, 0 °C, 80%; (b) (1) CF3COOAg, I2, CH2Cl2, 0 °C; (2) K2CO3, BnBr, DMF, 0 °C, 89%; (c) tributyl(1-ethoxyvinyl)tin, PdCl2(PPh3)2, toluene, 100 °C, 83%; (d) (1) MCPBA, CH2Cl2, 0 °C; (2) 10% NaOH(aq), MeOH, 68%; (e) 23a: CH3I, K2CO3, acetone, 56 °C, 92%; 23b: CD3I, K2CO3, acetone, 56 °C, 93%; (f) (1) BnOPhCHO, KOH, EtOH, H2O, 0 °C; (2) c-HCl, MeOH, THF, 0 °C; 24a: 92%, 24b: 81%; (g) I2, DMSO, 120 °C, 25a: 93%, 25b: 79%; (h) 1M BCl3 in hexane; CH2Cl2, −80 °C, hispidulin: 85%, d-hispidulin: 80%.
Figure 3.

Key rotating frame nuclear Overhauser effect spectroscopy (ROESY) correlations of compound 21.
Table 2.
Optimization of reaction condition for Stille coupling of compound 21.
| Entry | Catalyst | Solvent | Yield (%) | Reaction Time (h) |
|---|---|---|---|---|
| 1 | Pd(PPh3)4 | Dioxane | 68 | 30 |
| 2 | Pd(PPh3)4 | Toluene | 70 | 24 |
| 3 | PdCl2(PPh3)2 | Dioxane | 73 | 20 |
| 4 | PdCl2(PPh3)2 | Toluene | 83 | 10 |
| 5 | Pd(dppf)Cl2 | Dioxane | 34 | 78 |
| 6 | Pd(dppf)Cl2 | Toluene | 53 | 43 |
Figure 4.

Key ROESY and HMBC correlations of hispidulin.
2.3. Synthesis of d-Hispidulin
The synthesis of d-hispidulin is described in Scheme 5. Methylation of compound 14 using CD3I gave compound 23b. The same method to hispidulin was used to synthesize d-labelled hispidulin starting from compound 23b. Due to the absence of a proton signal of the CD3O group in the 1H-NMR spectra of the d-containing intermediate compounds 23b, 24b, 25b and d-hispidulin, these compound structures were identified depending on the 13C-NMR spectra without 1H decoupling and the mass technique. The 13C-NMR spectra revealed a characteristic multiplet splitting pattern of the 13C signal for the CD3O group in compounds 23b, 24b, 25b and d-hispidulin. The mass spectra also supported chemical structures of these d-labelled compounds. All synthesized compounds had an estimated purity of at least 98% as determined by HPLC analysis (Supplementary Materials).
2.4. Comparison of Hispidulin Synthesis Methods
Strategies for synthesizing hispidulin can be classified as semisynthesis and total synthesis strategies. The starting material used in most semisynthetic methods is scutellarin, which is a natural product. Table 3 shows that the semisynthesis routes had fewer reaction steps compared to the total synthesis methods; however, they need tedious isolation procedures for scutellarin, which limits the scale for further chemical modification. Furthermore, their overall yields are only 6.3–10.7% [20,22,23]. For total synthesis, Kavvadias and coworkers developed a nine-step synthesis approach. The starting material used in this method is commercially available 2,4,6-trihydroxyacetophenone. Although this method solves the issue of the source for starting material, its drawback is low overall yield [18]. We previously developed a feasible route of hispidulin synthesis that has an overall yield comparable to that of the method developed by Kavvadias and coworkers [21]. This present study further made the reaction steps more concise. In particular, the synthetic scheme showed the highest overall yield of all approaches to synthesize hispidulin.
Table 3.
Comparison of hispidulin synthesis methods.
| Research Group | Synthesis Route | Reaction Steps | Overall Yield (%) |
|---|---|---|---|
| Shen and coworkers | Semisynthesis | Seven | 10.7 |
| Lin and coworkers | Semisynthesis | Seven | 7.1 |
| Zhang and coworkers | Semisynthesis | Four | 6.3 |
| Kavvadias and coworkers | Total synthesis | Nine | 1.1 |
| Chao and coworkers | Total synthesis | Ten | 1.6 |
| This study | Total synthesis | Eight | 26.9 |
2.5. Human Liver Microsome Stability
Metabolic stability is associated with susceptibility of compounds to biotransformation. Metabolic half-life (t1/2) and intrinsic clearance (CLint) was compared between hispidulin and d-hispidulin by testing these synthesized compounds in a human liver microsome stability assay. The study revealed that FDA-approved deuterated agent deutetrabenazine had a t1/2 (8.6 h) superior to tetrabenazine (4.8 h). In addition to t1/2, the AUC of deutetrabenazine (542 ng·hr/mg) was also higher than that of its counterpart compound (261 ng·hr/mg) [27]. In contrast, the experimental results indicated hispidulin and d-hispidulin had no significant difference in t1/2 and CLint (Table 4), which suggested that the C6-OMe of hispidulin is resistant to be modified by the human liver microsome. The metabolic site of hispidulin in the human liver microsome is worthy of further study.
Table 4.
Human liver microsome stability of hispidulin and d-hispidulin.
| Compound | Half-Life (min) | CLint 1 (mL/min/mg Protein) |
|---|---|---|
| Hispidulin | 46 | 0.0298 |
| d-Hispidulin | 43 | 0.0325 |
| Testosterone | 19 | 0.0727 |
1 Intrinsic clearance (CLint) was calculated based on CLint = k/P, where k is the elimination rate constant and P is the protein concentration in the incubation.
The experiments showed that this method of synthesizing hispidulin and its d-labelled derivative is highly feasible. Specifically, it increases overall yield compared to previous methods. The t1/2 and intrinsic clearance of these two compounds were identified as well. Overall, this synthetic route can be applied to produce 6-OMe-containing hispidulin derivatives as new chemical entities for investigating their biological activities.
3. Experimental Section
3.1. General Information
The NMR spectra (1H- and 13C-NMR, ROESY, HSQC and HMBC) were obtained with a Bruker AV500 using standard pulse programs. The MS data were recorded with a Finnigan Mat TSQ-7000 mass spectrometer (HR–ESI–MS) (Thermo, Ringoes, NJ, USA). The HPLC was performed on a C18 column (150 mm × 4.6 mm, Ascentis) by using an L-2130 pump (Hitachi, Ibaraki, Japan) and a UV/vis L-2420 detector (Hitachi, Ibaraki, Japan). The column chromatography was performed on silica gel (70-230 mesh, Merck, Darmstadt, Germany). All TLC analyses were performed on silica gel plates (KG60-F254, Merck, Darmstadt, Germany). Reagents and materials were used without further purification and chemicals were purchased from ACROS (Geel, Belgium). Dry dichloromethane was distilled from CaH2 under nitrogen atmosphere. MOMCl was acquired from TCI (Tokyo, Japan), and 2,4,6-trihydroxybenzaldehyde was purchased from Alfa Aesar (Heydham, UK).
3.2. Chemistry
2-Hydroxy-4,6-bis(methoxymethoxy)benzaldehyde (20). To a solution of compound 7 (10 g, 64.9 mmol) in CH2Cl2 (200 mL) was added DIPEA (28.3 mL, 162.2 mmol). The resulting mixture was stirred for 10 mins in an ice-bath under N2. MOMCl (10.8 mL, 142.7 mmol) was added dropwise to the reaction mixture by addition funnel. The reaction mixture was warmed to room temperature (rt) and stirred for 3 h. The mixture was concentrated in vacuo. The residue was diluted with EtOAc (100 mL) and washed with distilled H2O (3 × 80 mL). The organic layer was dried over Na2SO4, filtered and removed in vacuo. The residue was purified by silica gel chromatography (EtOAc:n-hexane = 1:9) to give compound 20 (12.3 g, 80%), a light-yellow microcrystalline powder; 1H-NMR (CDCl3, 300 MHz) δ 12.29 (1H, s), 10.16 (1H, s), 6.25 (1H, d, J = 2.1 Hz), 6.23 (1H, d, J = 2.1 Hz), 5.24 (2H, s), 5.18 (2H, s), 3.51 (3H, s), 3.47 (3H, s); 13C-NMR (CDCl3, 125 MHz) δ 192.1, 165.6, 165.5, 161.2, 106.9, 96.6, 94.6, 94.1, 94.0, 56.6, 56.5; HR–ESI–MS m/z 243.0858 [M + H]+ (calcd. for C11H15O6, 243.0863).
2-Benzyloxy-3-iodo-4,6-bis(methoxymethoxy)benzaldehyde (21). To a mixture of compound 20 (7.2 g, 29.6 mmol) and CF3CO2Ag (7.8 g, 35.5 mmol) in CH2Cl2 (150 mL) at 0 °C was added I2 (8.3 g, 32.5 mmol) in CH2Cl2 (200 mL) dropwise by an addition funnel over 2 h. The reaction mixture was warmed to rt and stirred for 3 h. Then, saturated Na2S2O3(aq) (50 mL) was added and the mixture was washed with distilled H2O (3 × 100 mL). The organic layer was dried over Na2SO4, filtered and removed in vacuo. The residue was dissolved in DMF (100 mL) and then K2CO3 (7.2 g, 51.8 mmol) was added. Benzyl bromide (460 μL, 3.9 mmol) was added dropwise to the mixture by an addition funnel at 0 °C. The resulting solution was warmed to rt and stirred for 3 h. The mixture was diluted with EtOAc (300 mL) and washed with distilled H2O (3 × 00 mL). The organic layer was dried over Na2SO4, filtered and removed in vacuo. The residue was purified by silica gel chromatography (EtOAc:n-hexane = 1:5) to give compound 21 (12.1 g, 89%), a white microcrystalline powder; 1H-NMR (CDCl3, 300 MHz) δ 10.30 (1H, s), 7.66 (2H, dd, J = 1.8, 8.1 Hz), 7.45–7.36 (3H, m), 6.84 (1H, s), 5.32 (2H, s), 5.29 (2H, s), 4.99 (2H, s), 3.54 (3H, s), 3.52 (3H, s); 13C-NMR (CDCl3, 125 MHz) δ 187.2, 161.9, 161.8, 161.6, 136.1, 129.0, 128.5, 128.4, 115.5, 98.2, 95.2, 94.9, 78.5, 56.7; HR–ESI–MS m/z 459.0294 [M + H]+ (calcd. for C18H20O6I, 459.0299).
3-Acetyl-2-benzyloxy-4,6-bis(methoxymethoxy)benzaldehyde (22). To a mixture of compound 21 (5 g, 10.9 mmol) and PdCl2(PPh3)2 (766 mg, 1.1 mmol) in toluene (200 mL) was added tributyl(1-ethoxyvinyl)tin (5.5 mL, 16.4 mmol). The resulting solution was heated to 100 °C and stirred for 12 h. After cooling to rt, the reaction mixture was acidified with 1 M HCl (50 mL) and stirred for 30 min. The mixture was diluted with EtOAc (200 mL) and washed with distilled H2O (3 × 100 mL). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (EtOAc:n-hexane = 1:4) to give compound 22 (3.5 g, 83%), a yellow microcrystalline powder; 1H-NMR (CDCl3, 300 MHz) δ 10.37 (1H, s), 7.48 (2H, dd, J = 1.8, 7.8 Hz), 7.41–7.33 (3H, m), 6.80 (1H, s), 5.29 (2H, s), 5.24 (2H, s), 4.96 (2H, s), 3.53 (3H, s), 3.49 (3H, s), 2.43 (3H, s); 13C-NMR (CDCl3, 125 MHz) δ 200.9, 187.3, 162.1, 159.3, 158.4, 136.1, 129.0, 128.8, 128.6, 128.5, 128.4, 121.8, 114.3, 97.5, 95.0, 94.5, 79.1, 56.8, 32.5; HR–ESI–MS m/z 375.1432 [M + H]+ (calcd. for C20H23O7, 375.1438).
2-Benzyloxy-3-hydroxy-4,6-bis(methoxymethoxy)acetophenone (14). To a solution of 70% MCPBA (6.8 g, 27.6 mmol) in dry CH2Cl2 (100 mL) at 0 °C was added compound 22 (3.4g, 9.2 mmol) in CH2Cl2 (100 mL) dropwise by an addition funnel over 1 h. The resulting solution was warmed to rt and stirred for 8 h. Then, saturated Na2S2O3(aq) (30 mL) was added and the reaction mixture was washed with distilled H2O (3 × 100 mL). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was dissolved in MeOH (80 mL) and 10% NaOH(aq) (60 mL) was added. The resulting solution was stirred at rt for 1.5 h. The mixture was diluted with EtOAc (250 mL) and washed with distilled H2O (3 × 100 mL). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (EtOAc:n-hexane = 1:4) to give compound 14 (2.3 g, 68%), a light-yellow oil; 1H-NMR (CDCl3, 300 MHz) δ 7.44 (2H, dd, J = 1.8, 8.1 Hz), 7.40–7.32 (3H, m), 6.76 (1H, s), 5.61 (1H, s), 5.21 (2H, s), 5.08, (2H, s), 5.06 (2H, s), 3.53 (3H, s), 3.46 (3H, s), 2.45 (3H, s); 13C-NMR (CDCl3, 125 MHz) δ 201.3, 146.6, 146.2, 143.2, 136.9, 134.9, 128.5, 128.3, 122.1, 100.4, 96.0, 95.8, 76.3, 56.6, 56.3, 32.6; HR–ESI–MS m/z 363.1431 [M + H]+ (calcd. for C19H23O7, 363.1438).
2-Benzyloxy-3-methoxy-4,6-bis(methoxymethoxy)acetophenone (23a). To a mixture of compound 14 (587 mg, 1.6 mmol) and K2CO3 (1.1 g, 8.1 mmol) in acetone (20 mL) was added CH3I (0.5 mL, 8.1 mmol). The resulting solution was heated to 56 °C and stirred for 5 h. The reaction mixture was concentrated in vacuo, diluted with EtOAc (100 mL) and washed with distilled H2O (3 × 50 mL). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (EtOAc:n-hexane = 1:5) to give compound 23a (558 mg, 92%), a white microcrystalline powder; 1H-NMR (CDCl3, 300 MHz) δ 7.42 (2H, dd, J = 2.0, 8.3 Hz), 7.40–7.32 (3H, m), 6.76 (1H, s), 5.23 (2H, s), 5.11 (2H, s), 5.07 (2H, s), 3.85 (3H, s), 3.53 (3H, s), 3.46 (3H, s), 2.38 (3H, s); 13C-NMR (CDCl3, 125 MHz) δ 200.6, 151.8, 149.4, 149.2, 137.9, 136.7, 128.1, 128.0, 127.7, 121.4, 99.6, 95.0, 94.9, 76.0, 60.8, 56.0, 55.9, 32.1; HR–ESI–MS m/z 377.1587 [M + H]+ (calcd. for C20H25O7, 377.1595).
2-Benzyloxy-3-[2H3]-methoxy-4,6-bis(methoxymethoxy)acetophenone (23b). Following the procedure as described for compound 23a, reaction of compound 14 (1.8 g, 4.8 mmol), K2CO3 (3.3 g, 24.2 mmol), CD3I (1.5 mL, 24.2 mmol) in acetone (40 mL) gave compound 23b (1.7 g, 93%), a white microcrystalline powder; 1H-NMR (CDCl3, 300 MHz) δ 7.42 (2H, dd, J = 2.1, 8.4 Hz), 7.39–7.31 (3H, m), 6.76 (1H, s), 5.23 (2H, s), 5.11 (2H, s), 5.07 (2H, s), 3.53 (3H, s), 3.46 (3H, s), 2.38 (3H, s); 13C-NMR (CDCl3, 125 MHz) δ 200.6, 151.8, 149.4, 149.2, 137.8, 136.7, 128.1, 128.0, 127.7, 121.4, 99.6, 95.0, 94.9, 76.0, 56.0, 55.9, 32.1; HR–ESI–MS m/z 380.1775 [M + H]+ (calcd. for C20H22D3O7, 380.1783).
(E)-1-(2-Benzyloxy-4,6-dihydroxy-3-methoxyphenyl)-3-(4-benzyloxyphenyl)prop-2-en-1-one (24a). To a solution of 23a (545 mg, 1.45 mmol) and 4-benzyloxybenzaldyde (615 mg, 2.9 mmol) in EtOH (20 mL) was added KOH (813 mg, 14.5 mmol) in EtOH–H2O (3 mL:3 mL) dropwise by addition funnel at 0 °C over 30 min. The resulting solution was warmed to rt and stirred for 24 h. The mixture was diluted with distilled H2O (50 mL) and washed with EtOAc (3 × 50 mL). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. MeOH–THF (14.5 mL:14.5 mL) and 12 M HCl (0.7 mL) was added to the residue at 0 °C. The resulting solution was warmed to rt and stirred for 8 h. The reaction mixture was diluted with distilled H2O (50 mL) and washed with EtOAc (3 × 50 mL). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (EtOAc:n-hexane = 1:4) to give 24a (641 mg, 92%), a microcrystalline powder; 1H-NMR (DMSO-d6, 300 MHz) δ 12.92 (1H, s), 10.52 (1H, s), 7.59 (2H, s), 7.48–7.45 (2H, m), 7.44–7.37 (5H, m), 7.32–7.29 (5H, m), 6.94 (2H, d, J = 8.8 Hz), 6.21 (1H, s), 5.16 (2H, s), 5.06 (2H, s), 3.77 (3H, s); 13C-NMR (DMSO-d6, 125 MHz) δ 192.0, 160.2, 159.6, 157.7, 153.1, 143.0, 136.7, 136.6, 134.4, 130.3, 128.5, 128.4, 128.3, 128.2, 128.0, 127.8, 127.4, 124.6, 115.2, 109.0, 99.7, 75.7, 69.4, 60.7; HR–ESI–MS m/z 483.1795 [M + H]+ (calcd. for C30H27O6, 483.1802).
(E)-1-(2-Benzyloxy-4,6-dihydroxy-3-[2H3]methoxyphenyl)-3-(4-benzyloxyphenyl)prop-2-en-1-one (24b). According to the procedure as described for compound 24a, reaction of compound 23b (1.0 g, 2.6 mmol), 4-benzyloxybenzaldyde (1.1 g, 5.3 mmol) and KOH (1.5 g, 26.4 mmol) in EtOH (60 mL) followed by treatment of 12 M HCl (1.3 mL) and MeOH–THF (26.4 mL:26.4 mL) gave compound 24b (1.0 g, 81%), a yellow microcrystalline powder; 1H-NMR (DMSO-d6, 300 MHz) δ 12.95 (1H, s), 10.54 (1H, s), 7.59 (2H, s), 7.48–7.45 (2H, m), 7.44–7.36 (5H, m), 7.33–7.29 (5H, m), 6.94 (2H, d, J = 8.8 Hz), 6.22 (1H, s), 5.16 (2H, s), 5.06 (2H, s); 13C-NMR (DMSO-d6, 125 MHz) δ 192.0, 160.2, 159.7, 157.7, 153.2, 143.0, 136.7, 136.6, 134.4, 130.3, 128.5, 128.4, 128.3, 128.2, 128.0, 127.8, 127.4, 124.6, 115.2, 109.0, 99.7, 75.7, 69.4; HR–ESI–MS m/z 486.1983 [M + H]+ (calcd. for C30H24D3O6, 486.1990).
4′-Benzyloxy-6-methoxy-5-benzyloxy-7-hydroxyflavone (25a). To a solution of compound 24a (598 mg, 1.2 mmol) in dry DMSO (100 mL) was added I2 (32 mg, 0.1 mmol) in DMSO (3 mL) dropwise by syringe. The resulting solution was heated to 120 °C, and stirred for 2 h. After cooling to rt, saturated Na2S2O3(aq) (10 mL) was added to the reaction mixture. The mixture was diluted with EtOAc (100 mL) and wash with distilled H2O (3 × 50 mL). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (EtOAc:n-hexane = 1:3) to give 25a (555 mg, 93%), a yellow microcrystalline powder; 1H-NMR (DMSO-d6, 300 MHz) δ 10.76 (1H, s), 7.98 (2H, d, J = 8.9 Hz), 7.61 (2H, d, J = 7.0 Hz), 7.48 (2H, d, J = 7.0 Hz), 7.44–7.34 (6H, m), 7.18 (2H, d, J = 8.9 Hz), 6.91 (1H, s), 6.67 (1H, s), 5.22 (2H, s), 5.00 (2H, s), 3.75 (3H, s); 13C-NMR (DMSO-d6, 125 MHz) δ 175.8, 160.9, 160.1, 156.1, 153.8, 150.7, 139.6, 137.6, 136.6, 128.5, 128.3, 128.1, 128.0, 127.8, 123.4, 115.3, 111.4, 106.0, 100.1, 75.6, 69.5, 60.9; HR–ESI–MS m/z 481.1637 [M + H]+ (calcd. for C30H25O6, 481.1646).
4′-Benzyloxy-6-[2H3]methoxy-5-benzyloxy-7-hydroxyflavone (25b). Following the procedure as described for compound 25a, reaction of compound 24b (849 mg, 1.7 mmol) with I2 (44 mg, 0.2 mmol) in DMSO (120 mL) gave compound 25b (664 mg, 79%), a yellow microcrystalline powder; 1H-NMR (DMSO-d6, 300 MHz) δ 10.75 (1H, s), 7.98 (2H, d, J = 8.9 Hz), 7.61 (2H, d, J = 6.8 Hz), 7.48 (2H, dd, J = 1.8, 8.4 Hz), 7.44–7.34 (6H, m), 7.18 (2H, d, J = 8.9 Hz), 6.91 (1H, s), 6.67 (1H, s), 5.22 (2H, s), 5.01 (2H, s); 13C-NMR (DMSO-d6, 125 MHz) δ 175.8, 160.9, 160.1, 156.1, 153.7, 150.7, 139.5, 137.6, 136.6, 128.5, 128.3, 128.1, 128.0, 127.8, 123.4, 115.3, 111.4, 106.0, 100.1, 75.6, 69.5; HR–ESI–MS m/z 484.1827 [M + H]+ (calcd. for C30H22D3O6, 484.1834).
Hispidulin. To a solution of compound 25a (301 mg, 0.6 mmol) in dry CH2Cl2 (40 mL) was added 1 M BCl3 (2.5 mL, 2.5 mmol) in dry CH2Cl2 (5 mL) dropwise by syringe at −78 °C over 20 min. The resulting solution was stirred for 1 h. The reaction mixture was diluted with distilled H2O (50 mL) and washed with EtOAc (3 × 50 mL). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel chromatography (EtOAc:n-hexane = 1:2) to give hispidulin (160 mg, 85%), a yellow microcrystalline powder; 1H-NMR (DMSO-d6, 500 MHz) δ 13.07 (1H, s, 5-OH), 10.73 (1H, s, 7-OH), 10.38 (1H, s, 4′-OH), 7.92 (2H, d, J = 8.9 Hz, H-2′, H-6′), 6.92 (2H, d, J = 8.9 Hz, H-3′, H-5′), 6.77 (1H, s, H-3), 6.59 (1H, s, H-8), 3.74 (3H, s, 6-OMe); 13C-NMR (DMSO-d6, 125 MHz) δ 182.2 (C-4), 163.9 (C-2), 161.2 (C-4′), 157.3 (C-7), 152.8 (C-5), 152.4 (C-9), 131.4 (C-6), 128.5 (C-2’, C-6′), 121.2 (C-1), 116.0 (C-3′, C-5′), 104.1 (C-10), 102.4 (C-3), 94.3 (C-8), 60.0 (6-OMe); HR–ESI–MS m/z 301.0702 [M + H]+ (calcd. for C16H13O6, 301.0707).
d-Hispidulin. Following the procedure as described for hispidulin, reaction of compound 25b (536 mg, 1.1 mmol) in CH2Cl2 (75 mL) with 1 M BCl3 (4.4 mL, 4.4 mmol) in CH2Cl2 (8.8 mL) gave d-hispidulin (268 mg, 80%), a yellow microcrystalline powder; 1H-NMR (DMSO-d6, 500 MHz) δ 13.07 (1H, s, 5-OH), 10.70 (1H, s, 7-OH), 10.36 (1H, s, 4′-OH), 7.91 (2H, d, J = 8.9 Hz, H-2′, H-6′), 6.92 (2H, d, J = 8.9 Hz, H-3′, H-5′), 6.76 (1H, s, H-3), 6.58 (1H, s, H-8); 13C-NMR (DMSO-d6, 125 MHz) δ 182.1 (C-4), 163.8 (C-2), 161.2 (C-4’), 157.2 (C-7), 152.8 (C-5), 152.4 (C-9), 131.3 (C-6), 128.5 (C-2′, C-6′), 121.2 (C-1), 116.0 (C-3′, C-5′), 104.1 (C-10), 102.4 (C-3), 94.2 (C-8); HR–ESI–MS m/z 304.0888 [M + H]+ (calcd. for C16H10D3O6, 304.0895).
3.3. Human Liver Microsome Stability Assay
Mixed-gender human liver microsomes (Lot # 1210347) were purchased from XenoTech. The reaction mixture minus NADPH was prepared as described below. The test compounds were added into the reaction mixture at a final concentration of 1 μM. A separate reaction with the control compound, testosterone, was run simultaneously with the reactions with the test compounds. An aliquot of the reaction mixture (without cofactor) was equilibrated in a shaking water bath at 37 °C for 3 min. After addition of cofactor to initiate the reaction, the mixture was incubated in a shaking water bath at 37 °C. Aliquots (100 μL) were withdrawn at 0, 10, 20, 30 and 60 min for the test compounds and testosterone. The reaction was terminated by immediately combining the tested compounds and testosterone samples with 400 μL of ice-cold 50/50 acetonitrile (ACN)/H2O containing 0.1% formic acid and internal standard. The samples were then mixed and centrifuged to precipitate proteins. All samples were assayed by LC-MS/MS using electrospray ionization. The peak area response ratio (PARR) to internal standard was compared to the PARR at time 0 to determine the percent remaining at each time point. The values for half-life (t1/2) and intrinsic clearance (CLint) of the tested compounds were determined by Absorption System Corp. Half-life calculated using GraphPad software was fitted to a single-phase exponential decay equation.
Acknowledgments
We gratefully acknowledge the financial support from the Ministry of Science and Technology (MOST105-2320-B-038-024-MY3 to W.-J. Huang, MOST 105-2311-B-038-001 to K.-C. Hsu and MOST105-2325-B-002-004 to L.-C. Chiou).
Supplementary Materials
The following are available online. 1H- and 13C-NMR spectra and HPLC chromatogram of all compounds synthesized; 1H- and 13C-NMR spectra comparison of experimental and reported hispidulin; ROESY spectra of compound 21; and ROESY, HMQC and HMBC spectra of final products hispidulin and d-hispidulin.
Author Contributions
Liang-Chieh Chen, Kai-Cheng Hsu, Hui-Ju Tseng and Wei-Jan Huang conceived and designed the experiments; Liang-Chieh Chen and Hui-Ju Tseng performed the experiments; Liang-Chieh Chen, Kai-Cheng Hsu and Wei-Jan Huang analyzed the data; Liang-Chieh Chen, Lih-Chu Chiou, Kai-Cheng Hsu and Wei-Jan Huang wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 12, 32, 33, 34, 35, 36, 37, 38, 41, 42, 43, 44, 45a, 45b, 46a, 46b, 47a, 47b, hispidulin and d-hispidulin are available from the authors.
References
- 1.Grassi D., Desideri G., Ferri C. Flavonoids: Antioxidants against atherosclerosis. Nutrients. 2010;2:889–902. doi: 10.3390/nu2080889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pietta P.G. Flavonoids as antioxidants. J. Nat. Prod. 2000;63:1035–1042. doi: 10.1021/np9904509. [DOI] [PubMed] [Google Scholar]
- 3.Serafini M., Peluso I., Raguzzini A. Flavonoids as anti-inflammatory agents. Proc. Nutr. Soc. 2010;69:273–278. doi: 10.1017/S002966511000162X. [DOI] [PubMed] [Google Scholar]
- 4.Cushnie T.P., Lamb A.J. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Agents. 2005;26:343–356. doi: 10.1016/j.ijantimicag.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cardenas-Rodriguez N., Gonzalez-Trujano M.E., Aguirre-Hernandez E., Ruiz-Garcia M., Sampieri A., III, Coballase-Urrutia E., Carmona-Aparicio L. Anticonvulsant and antioxidant effects of Tilia americana var. mexicana and flavonoids constituents in the pentylenetetrazole-induced seizures. Oxid. Med. Cell. Longev. 2014;2014:329172. doi: 10.1155/2014/329172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guan L.P., Liu B.Y. Antidepressant-like effects and mechanisms of flavonoids and related analogues. Eur. J. Med. Chem. 2016;121:47–57. doi: 10.1016/j.ejmech.2016.05.026. [DOI] [PubMed] [Google Scholar]
- 7.Ravishankar D., Rajora A.K., Greco F., Osborn H.M. Flavonoids as prospective compounds for anti-cancer therapy. Int. J. Biochem. Cell Biol. 2013;45:2821–2831. doi: 10.1016/j.biocel.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Fan P.C., Huang W.J., Chiou L.C. Intractable chronic motor tics dramatically respond to Clerodendrum inerme (L) Gaertn. J. Child Neurol. 2009;24:887–890. doi: 10.1177/0883073808331088. [DOI] [PubMed] [Google Scholar]
- 9.Huang W.J., Lee H.J., Chen H.L., Fan P.C., Ku Y.L., Chiou L.C. Hispidulin, a constituent of Clerodendrum inerme that remitted motor tics, alleviated methamphetamine-induced hyperlocomotion without motor impairment in mice. J. Ethnopharmacol. 2015;166:18–22. doi: 10.1016/j.jep.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Liao Y.H., Lee H.J., Huang W.J., Fan P.C., Chiou L.C. Hispidulin alleviated methamphetamine-induced hyperlocomotion by acting at alpha6 subunit-containing GABAA receptors in the cerebellum. Psychopharmacology. 2016;233:3187–3199. doi: 10.1007/s00213-016-4365-z. [DOI] [PubMed] [Google Scholar]
- 11.Cui B., Lee Y.H., Chai H., Tucker J.C., Fairchild C.R., Raventos-Suarez C., Long B., Lane K.E., Menendez A.T., Beecher C.W., et al. Cytotoxic sesquiterpenoids from Ratibida columnifera. J. Nat. Prod. 1999;62:1545–1550. doi: 10.1021/np990260y. [DOI] [PubMed] [Google Scholar]
- 12.Iwashina T., Kamenosono T., Ueno T. Hispidulin and nepetin 4 -glucosides from Cirsium oligophyllum. Phytochemistry. 1999;51:1109–1111. doi: 10.1016/S0031-9422(99)00178-8. [DOI] [Google Scholar]
- 13.Flamini G., Antognoli E., Morelli I. Two flavonoids and other compounds from the aerial parts of Centaurea bracteata from Italy. Phytochemistry. 2001;57:559–564. doi: 10.1016/S0031-9422(01)00066-8. [DOI] [PubMed] [Google Scholar]
- 14.Fullas F., Hussain R.A., Chai H.B., Pezzuto J.M., Soejarto D.D., Kinghorn A.D. Cytotoxic constituents of Baccharis gaudichaudiana. J. Nat. Prod. 1994;57:801–807. doi: 10.1021/np50108a017. [DOI] [PubMed] [Google Scholar]
- 15.Kavvadias D., Monschein V., Sand P., Riederer P., Schreier P. Constituents of sage (Salvia officinalis) with in vitro affinity to human brain benzodiazepine receptor. Planta Med. 2003;69:113–117. doi: 10.1055/s-2003-37712. [DOI] [PubMed] [Google Scholar]
- 16.Hall B.J., Karim N., Chebib M., Johnston G.A., Hanrahan J.R. Modulation of ionotropic GABA receptors by 6-methoxyflavanone and 6-methoxyflavone. Neurochem. Res. 2014;39:1068–1078. doi: 10.1007/s11064-013-1157-2. [DOI] [PubMed] [Google Scholar]
- 17.Ren L., Chan W.M., Wang F., Xu Z., Zhao C., Mat W.K., Chai Y., Wong J.T., Tsang S.Y., Xue H. Effects of flavone 6-substitutions on GABAA receptors efficacy. Eur. J. Pharmacol. 2011;670:121–129. doi: 10.1016/j.ejphar.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 18.Kavvadias D., Sand P., Youdim K.A., Qaiser M.Z., Rice-Evans C., Baur R., Sigel E., Rausch W.D., Riederer P., Schreier P. The flavone hispidulin, a benzodiazepine receptor ligand with positive allosteric properties, traverses the blood-brain barrier and exhibits anticonvulsive effects. Br. J. Pharmacol. 2004;142:811–820. doi: 10.1038/sj.bjp.0705828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Z.H., Li N.G., Wang Z.J., Tang Y.P., Dong Z.X., Zhang W., Zhang P.X., Gu T., Wu W.Y., Yang J.P., et al. Synthesis and biological evaluation of methylated scutellarein analogs based on metabolic mechanism of scutellarin in vivo. Eur. J. Med. Chem. 2015;106:95–105. doi: 10.1016/j.ejmech.2015.10.039. [DOI] [PubMed] [Google Scholar]
- 20.Lin H., Zhang W., Dong Z.X., Gu T., Li N.G., Shi Z.H., Kai J., Qu C., Shang G.X., Tang Y.P., et al. A new and practical synthetic method for the synthesis of 6-O-methyl-scutellarein: One metabolite of scutellarin in vivo. Int. J. Mol. Sci. 2015;16:7587–7594. doi: 10.3390/ijms16047587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chao S.W., Su M.Y., Chiou L.C., Chen L.C., Chang C.I., Huang W.J. Total Synthesis of Hispidulin and the Structural Basis for Its Inhibition of Proto-oncogene Kinase Pim-1. J. Nat. Prod. 2015;78:1969–1976. doi: 10.1021/acs.jnatprod.5b00324. [DOI] [PubMed] [Google Scholar]
- 22.Shen M.Z., Shi Z.H., Li N.G., Tang H., Shi Q.P., Tang Y.P., Yang J.P., Duan J.A. Efficient Synthesis of 6-O-methyl-scutellarein from Scutellarin via Selective Methylation. Lett. Org. Chem. 2013;10:733–737. doi: 10.2174/15701786113109990046. [DOI] [Google Scholar]
- 23.Zhang W., Dong Z.X., Gu T., Li N.G., Zhang P.X., Wu W.Y., Yu S.P., Tang Y.P., Yang J.P., Shi Z.H. A new and efficient synthesis of 6-O-methylscutellarein, the major metabolite of the natural medicine scutellarin. Molecules. 2015;20:10184–10191. doi: 10.3390/molecules200610184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katsnelson A. Heavy drugs draw heavy interest from pharma backers. Nat. Med. 2013;19:656. doi: 10.1038/nm0613-656. [DOI] [PubMed] [Google Scholar]
- 25.Gant T.G. Using deuterium in drug discovery: Leaving the label in the drug. J. Med. Chem. 2014;57:3595–3611. doi: 10.1021/jm4007998. [DOI] [PubMed] [Google Scholar]
- 26.Mullard A. FDA approves first deuterated drug. Nat. Rev. Drug Discov. 2017;16:305. doi: 10.1038/nrd.2017.89. [DOI] [PubMed] [Google Scholar]
- 27.Tung R.D. Deuterium medicinal chemistry comes of age. Future Med. Chem. 2016;8:491–494. doi: 10.4155/fmc-2016-0032. [DOI] [PubMed] [Google Scholar]
- 28.Labib S., Hummel S., Richling E., Humpf H.U., Schreier P. Use of the pig caecum model to mimic the human intestinal metabolism of hispidulin and related compounds. Mol. Nutr. Food Res. 2006;50:78–86. doi: 10.1002/mnfr.200500144. [DOI] [PubMed] [Google Scholar]
- 29.Xu H.J., Liang Y.F., Cai Z.Y., Qi H.X., Yang C.Y., Feng Y.S. CuI-nanoparticles-catalyzed selective synthesis of phenols, anilines, and thiophenols from aryl halides in aqueous solution. J. Org. Chem. 2011;76:2296–2300. doi: 10.1021/jo102506x. [DOI] [PubMed] [Google Scholar]
- 30.Anderson K.W., Ikawa T., Tundel R.E., Buchwald S.L. The selective reaction of aryl halides with KOH: Synthesis of phenols, aromatic ethers, and benzofurans. J. Am. Chem. Soc. 2006;128:10694–10695. doi: 10.1021/ja0639719. [DOI] [PubMed] [Google Scholar]
- 31.Chen J.M., Yuan T.J., Hao W.Y., Cai M.Z. Simple and efficient Cul/PEG-400 system for hydroxylation of aryl halides with potassium hydroxide. Catal. Commun. 2011;12:1463–1465. doi: 10.1016/j.catcom.2011.06.002. [DOI] [Google Scholar]
- 32.Kantevari S., Yempala T., Yogeeswari P., Sriram D., Sridhar B. Synthesis and antitubercular evaluation of amidoalkyl dibenzofuranols and 1H-benzo[2,3]benzofuro[4,5-e][1,3]oxazin-3(2H)-ones. Bioorg. Med. Chem. Lett. 2011;21:4316–4319. doi: 10.1016/j.bmcl.2011.05.054. [DOI] [PubMed] [Google Scholar]
- 33.Hase T., Ohtani K., Kasai R., Yamasaki K., Picheansoonthon C. Revised Structure for Hortensin, a Flavonoid from Millingtonia-Hortensis. Phytochemistry. 1995;40:287–290. doi: 10.1016/0031-9422(95)00206-M. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.