

Summary
The immune system plays a role in the pathogenesis of non‐alcoholic steatohepatitis (NASH) underlying hepatocyte injury and fibrosis progression at all disease stages. Oral administration of anti‐CD3 monoclonal antibody (mAb) has been shown in preclinical studies to be an effective method for systemic immune modulation and alleviates immune‐mediated disorders without T cell depletion. In the present review, we summarize the concept of the oral administration of humanized anti‐CD3 mAb in patients with NASH and discuss the potential of this treatment to address the current requirements of treatments for NASH. Recently published preclinical and clinical data on oral administration of anti CD3 are discussed. Human trials have shown that the oral administration of anti‐CD3 in healthy volunteers, patients with chronic hepatitis C virus (HCV) infection and patients with NASH and type 2 diabetes is safe and well tolerated, as well as biologically active. Oral anti‐CD3 induces regulatory T cells, suppresses the chronic inflammatory state associated with NASH and exerts a beneficial effect on clinically relevant parameters. Foralumab is a fully human anti‐CD3 mAb that has recently been shown to exert a potent anti‐inflammatory effect in humanized mice. It is being developed for treatment of NASH and primary biliary cholangitis (PBC). Oral administration of anti CD3 may provide an effective therapy for patients with NASH.
Keywords: anti CD3, NAFLD, NASH, oral tolerance, treatment
Introduction
Non‐alcoholic fatty liver disease (NAFLD) affects 20–40% of the population. Its active form, non‐alcoholic steatohepatitis (NASH), is characterized by hepatocyte injury, liver inflammation and progression of fibrosis 1. NASH has emerged as an important cause of chronic liver disease and hepatocellular carcinoma (HCC) worldwide. The immune system plays an important role in the pathogenesis of NASH and underlies the hepatocyte injury and fibrosis progression in all disease stages. The oral administration of anti‐CD3 monoclonal antibody (mAb) is a method developed for systemic immune modulation by the induction of regulatory T cells (Tregs) 2. In the present review, we summarize the preclinical and clinical data which support the concept of oral administration of anti‐CD3 mAb as a novel immunomodulatory treatment for NASH and type 2 diabetes (T2D).
The role of the immune system in the pathophysiology of NASH
NAFLD, the most common form of chronic liver disease, encompasses a histological spectrum ranging from simple steatosis to NASH 3. NASH is characterized by lobular inflammation and hepatocellular ballooning, and it may be associated with liver fibrosis, thus leading to cirrhosis and its complications, as well as to liver malignancy 4. NASH is likely to become the most common indication for liver transplantation during the next decade 3. Weight reduction of 10% by dietary restriction and regular exercise are sufficient to reverse NASH, but are difficult to maintain 1. Thus, NASH is currently a major therapeutic challenge. Several drugs are currently being developed to treat this disorder, targeting different disease‐related pathways 1, 4, 5. The modulation of nuclear transcription factors, targeting lipotoxicity and oxidative stress, the modulation of cellular energy homeostasis and metabolism and the inflammatory response are potential therapeutic targets being explored 3.
In NASH, a combination of environmental factors, host genetics and gut microbiota are associated with the accumulation of lipids in the liver, thus resulting in lipotoxicity, which triggers hepatocyte cell death, liver inflammation, fibrosis and pathological angiogenesis. NASH can progress further to liver cirrhosis and eventually to hepatocellular carcinoma 6. The immune system in NASH is being recognized increasingly to contribute to the pathogenic mechanisms of NASH and as a potential therapeutic target 7. The low‐grade inflammatory state present in obesity promotes the progression of NAFLD to NASH. Augmented hepatic steatosis is accompanied by aberrant intrahepatic inflammation and exacerbated hepatocellular injury 8. Lipotoxicity and the associated chronic inflammation associated with metabolic dysregulation or ‘metaflammation’ follow the chronic metabolic stress that occurs during prolonged nutrient excess or obesity 9. The lipid influx can exceed the adipose tissue storage capacity and result in the accumulation of deleterious lipid species in the liver and muscle 9. These lipids and the associated generation of signalling intermediates interfere with immune regulation, thus leading to a vicious cycle of immune‐metabolic dysregulation.
The immune system participates in this process, and disturbances in the cells constituting both the innate and adaptive immune systems in the liver, pancreas, muscle and adipose tissue are observed in NASH. The infiltration of different subsets of innate and adaptive immune cells, such as monocytes, T lymphocytes and neutrophils to the liver and in‐situ activation of Kupffer cells and stellate cells, which underlie the progression of liver damage 8. The involvement of adaptive immunity in adipose tissue inflammation has been shown in obesity and NASH 8, 10. In the initial phase, the fat‐resident macrophages secrete chemokines, which recruit CD4+/CD8+ T lymphocytes and natural killer (NK) T cells to the adipose tissue which, in turn, enhance macrophage activation and proinflammatory mediator release 11. Both macrophages and lymphocytes represent the most frequent inflammatory infiltrates of NASH liver 12. In NASH liver biopsy sections, the portal tract infiltrates are dominated by CD8(+) lymphocytes 13. Hepatic T helper type 17 (Th17) cell infiltration is found in NASH 14. Also, interleukin (IL)‐17 secretion exacerbates hepatic steatosis and inflammation, whereas IL‐17 neutralization attenuates lipopolysaccharide (LPS)‐induced liver injury. IL‐17A−/− mice were resistant to the development of NASH 15, 16. B cells play a role in NASH pathogenesis via T cell activation and proinflammatory cytokine secretion 17. An imbalance of Th17/Treg cells is involved in the progression of NAFLD 18, 19. Both NK and NK T cells participate in the immune cascade leading to NASH 20. Tregs play a critical role in regulating inflammatory processes in NASH 21. Compared with healthy controls, a lower frequency of Tregs and higher frequencies of interferon (IFN)‐γ+ and IL‐4+ cells were detected among CD4+ T cells of peripheral blood in NASH 16.
Gut‐derived endotoxins and oxidative stress activate innate‐dependent immune pathways 22. Activation of the innate immune system and increased release of proinflammatory cytokines and other mediators play a role in various stages of disease progression. Inflammatory mediators, including those that are derived from adipose tissue and the liver, are involved in the cascade of inflammation, fibrosis and eventually tumorigenesis 7. A cytokine imbalance is associated with Th17 differentiation, fibrogenesis and steatohepatitis 19.
Consequently, paths at the intersection of lipid metabolism and immune function are potential therapeutic targets for NASH 23, 24, 25. Strategies to enhance the resolution of inflammation show promise to reverse both the early and advanced stages of fibrosis in liver disease 3.
Immune modulation using anti‐CD3 monoclonal antibodies
T cells recognize antigens through the T cell receptor (TCR), which is associated with the CD3 molecule 26. The CD3/TCR complex is present on the surfaces of all T cells and is involved in antigen recognition and signal transduction. Whereas TCR chains are subject to gene rearrangement and are variable, CD3 chains are invariable and are not antigen‐specific 27. The first murine anti‐CD3 mAb [immunoglobulin (Ig)G2a] was approved in 1985 (Muromonab, OKT3, Ortho Kung T3; Orthoclone®) and was used to treat allograft rejection in kidney transplantation, on the basis of its potent immunosuppressive effects via the depletion of T cells 28, 29.
The induction of tolerance by the enteral administration of anti‐CD3 mAb, termed oral tolerance, which in contrast to immunosuppression is an active state defined as the absence of a pathogenic immune response despite the concomitant presence of autoantigens 30, 31. This type of antigen‐directed tolerance remains intact for long periods after treatment withdrawal. In mice prone to developing autoimmune diabetes, pancreatic islet β cell tolerance has been induced through this method 32. The mechanism has been shown to be associated with the generation of Tregs 33. Parenteral OKT3 has been administered in clinical trials to patients with multiple sclerosis (MS), inflammatory bowel disease (IBD) and rheumatoid arthritis (RA) 28, 34, 35, 36, 37, 38, 39, 40, 41.
However, murine anti‐CD3 mAb is associated with toxicity, thus significantly limiting its clinical use. Adverse reactions occurring during the first 24 h of anti‐CD3 infusion are referred to as infusion‐related reactions (IRRs). IRRs also include acute hypersensitivity reactions such as anaphylactic reactions. Murine anti‐CD3 mAb stimulates an extensive release of cytokines within the initial hours after the first administration. When these reactions are associated with a significant release of cytokines, the constellation of signs and symptoms is referred to as cytokine release syndrome (CRS), which is the main mechanism of anti‐CD3 mAb toxicity 30, 31, 42, 43. CRS occurs after the first dose of antibody infusion and correlates with the capacity of the Fc of the anti‐CD3 mAbs to bind to Fc gamma receptors (FcγRs) on leucocytes or complement (C1q) 44, 45. This cascade causes the release of proinflammatory cytokines, including tumour necrosis factor (TNF)‐α and IFN‐γ, into the circulation 46, 47, 48. CRS is amplified by the capacity of the Fc portion of anti‐CD3 mAb to interact with Fc receptors present on the surfaces of other white blood cells, such monocytes, macrophages, NK cells and B cells. Furthermore, the efficacy of the repeated administration of murine anti‐CD3 mAb is decreased as a result of neutralizing antibodies produced against the rodent portion of the mAb 49, 50.
Humanized forms of several CD3 antibodies were used in several trials, including teplizumab (huOKT3γ1) 51, 52, 53, otelixizumab 54, 55 (aglycosylated rat YTH12.5) and visilizumab 36, 38 (HuM291). Modifications in the Fc region of anti‐CD3 mAb have been made to decrease T cell activation and cytokine release and to improve the antibody tolerability. These modifications are associated with the induction of regulatory cells, production of IL‐10 or transforming growth factor (TGF)‐β and partial exhaustion of T cells 56, 57, 58, 59. A tolerable intravenous dose of teplizumab, an anti‐CD3 mAb that is mutated to decrease Fc receptor binding, has been found to decrease C‐peptide loss in T1D 2 years after diagnosis 60. This treatment induces partial CD8 T cell exhaustion, decreases the expression of genes associated with immune activation and increases the expression of genes associated with T cell differentiation and regulation 51, 58. Short‐term anti‐CD3 using the humanized antibodies Teplizumab and Otelixizumab has been shown to mitigate the long‐term deterioration in insulin production and to improve the metabolic control, thus decreasing the need for exogenous insulin by preserving β cell function in subjects with autoimmune type 1 diabetes (T1D) 34, 39, 41, 50. However, these adjustments do not eliminate the need for intravenous administration and the potential for cell activation.
Oral administration of anti‐CD3 monoclonal antibody as a potent immunomodulatory agent: preclinical and clinical studies
Whereas intravenous administration of anti‐CD3 acts via transient depletion of the activated effector T cells, oral anti‐CD3 mAbs act by the induction of Tregs, thus causing the immunomodulation of the CD3/TCR complex and decreasing common unwanted adverse effects associated with parenteral administration, such as CRS 61. Oral anti‐CD3 mAb, unlike its intravenous counterparts, affects the gut immune system and mesenteric lymph nodes (MLNs), thus promoting Treg activity without inducing generalized immunosuppression 62. This mode of therapy uses the gut immune system and underlying lamina propria for the generation of immune signals, thereby inducing a favourable systemic immune response 23, 24.
The induction of regulatory cells has been described following oral administration of anti‐CD3 mAb in mice and in humanized mice, following migration of the antibodies to the gut wall 63. The oral administration of anti‐CD3 mAb in experimental autoimmune encephalitis, a model of multiple sclerosis, induces CD4+CD25– latency‐associated peptide (LAP)+ T cells that exhibit regulatory properties 64, 65. Similarly, oral anti‐CD3 mAb suppresses low‐dose streptozotocin‐induced and non‐obese diabetic (NOD) diabetes models of T1D through the induction of IL‐10‐secreting CD4+CD25–LAP+ regulatory cells, thus decreasing T cell proliferation and IFN‐γ and IL‐17 production and increasing TGF‐β production 66, 67, 68. Oral anti‐CD3 is beneficial in animal models of colitis 69 and atherosclerosis 70.
Orally and intranasally administered anti‐CD3 have been found to suppress autoantibody production in a mouse lupus model 71, 72. An increased release of IL‐10 in the serum has been demonstrated and suggested to account for the protective effects of systemically administered anti‐CD3 mAbs 59, 73.
In humans, in a healthy volunteer clinical trial, different dosages of the murine anti‐CD3 mAb OKT3 were orally administered daily for 5 days. Treatment was well tolerated without any adverse reactions. There were no changes in the CD3 lymphocyte count and no human anti‐mouse antibodies detected 30 days post‐oral treatment, thus suggesting that the orally administered anti‐CD3 mAb was not absorbed systemically. The biological activity has also been documented. Oral OKT3 increased immunophenotypical markers for regulatory T cells such as CD25hi, forkhead box protein 3 (FoxP3) and cytotoxic T lymphocyte antigen (CTLA)‐4 and up‐regulated the anti‐inflammatory CD4+CD25+ and CD8+CD25+ T lymphocyte subsets 74. Oral OKT3 suppressed Th1 and Th17 responses, increased TGF‐β/IL‐10 expression and decreased IL‐23/IL‐6 expression by dendritic cells, and affected the IgG repertoire as measured by antigen arrays.
In a blind placebo‐controlled clinical trial, the mouse anti‐CD3 mAb OKT3 was orally administered daily for 30 days to patients with chronic hepatitis C virus (HCV) who were non‐responders to IFN and ribavirin. OKT3 was found to be safe and well tolerated. A decreased viral load in all treatment groups and a decrease in liver enzyme levels in the low‐ and high‐dose groups along with increases in the Treg levels was shown 75.
Together, results from these preclinical and clinical studies indicate that orally administered anti‐CD3 mAb is a new method for treating autoimmune and inflammatory diseases with promising effects on efficacy and few treatment‐related adverse events. The oral administration of anti‐CD3 mAb overcomes many of the obstacles encountered with parenteral use. Orally administered anti‐CD3 is not absorbed systemically and its mode of action is primarily through activating mucosal immunity. It is a novel method for the induction of tolerance without causing detectable side effects such as CRS, immunogenicity or generalized immunosuppression.
Immunotherapy for NASH by oral administration of anti‐CD3
During the process of liver damage, tissue repair pathways are activated to restore tissue and metabolic homeostasis 23. Inflammatory response induces hepatocyte damage and can generate irreversible liver damage, fibrosis and carcinogenesis. Inflammatory genes are over‐expressed in NASH. Increased expression of genes that regulate inflammation in patients with NAFLD and NASH have been noted in the subcutaneous adipose tissue (SAT), visceral adipose tissue (VAT) and phenotypes of adipose tissue macrophages (ATMs) in obese patients 76. Liver injury is associated with the secretion of proinflammatory factors by Kupffer cells, NK T cells 77, hepatic stellate cells, sinusoidal endothelial cells, dendritic cells (DC), NK cells, monocytes and lymphocytes, which develop in response to injury or damage to the liver 23. Cytokines, chemokines, lipid messengers and reactive oxygen species promote the apoptotic or necrotic demise of hepatocytes 78. A cycle of inflammation and cell death is generated by dying hepatocytes, releasing damage‐associated molecular patterns (DAMPs) that bind to evolutionarily conserved pattern recognition receptors (PAMPs) to activate cells of the innate immune system and stimulate the inflammatory process 78. Because the immune system plays a role in the pathogenesis of NASH, various treatments are being developed to target directly or indirectly the relevant inflammatory pathways 23, 79.
Because the immune system plays a role in the pathogenesis of NASH, oral anti‐CD3 mAb has been tested in preclinical and clinical trials of NASH and T2D. In the leptin‐deficient model of fatty liver and insulin resistance, oral anti‐CD3 mAb induces LAP+ Tregs and NK T cell‐dependent immunoregulation 80. The treatment decreases pancreatic hyperplasia, hepatic fat accumulation and muscle inflammation and is associated with the upregulation of TGF‐β and IL‐10 80.
The oral administration of murine OKT3 mAb has been tested in a blind placebo‐controlled dose‐dependent clinical trial in patients with biopsy‐proven NASH and T2DM. The drug was well tolerated without any treatment‐related adverse reactions 81. No suppression of CD3+ lymphocytes was found. OKT3 induced Tregs with increases in the CD4+CD25+FoxP3+ population at day 14 in the treatment groups as well as increased TGF‐ β secretion. An increase in CD4+LAP+ and CD4+CD25+LAP+ lymphocytes was observed. OKT3 decreased AST levels in all treatment groups. A decrease in the fasting plasma glucose levels was observed in all groups, whereas the oral glucose tolerance test was improved in the high‐dose group. Serum anti‐CD3 OKT3 levels were not detectable. Human anti‐mouse antibodies were detected in the circulatory system in only the high‐dose group.
The preclinical and clinical data demonstrate that oral administration of anti‐CD3 mAbs has the potential to suppress the inflammatory process in NASH and T2d via the promotion of Tregs and secretion of anti‐inflammatory cytokines, without immunosuppression. The data support its use as a method for immunomodulation to alleviate the liver damage associated with NASH. The lack of significant absorption suggests that the activity of oral anti‐CD3 mAb is restricted to its interactions with gut‐resident T cells.
Foralumab
Foralumab (TZLS‐0401; known formerly as NI‐0401) is a fully human monoclonal antibody that binds to the epsilon (ε) chain of the CD3/TCR complex present at the surface of all peripheral T cells. An amino acid sequence for both heavy and light chains has been identified from the translation of the nucleotide sequence to encode foralumab. It is composed of two heavy chains with an IgG1 constant region and two light chains with a kappa constant region. The heavy chain constant regions are mutated at two amino acid positions, 234 and 235 61, 82.
When bound to its target, foralumab triggers calcium flux and modulates the CD3/TCR complex, thus causing its transient removal from the cell surface. The combination of the two point mutations introduced into the Fc portion of foralumab results in the abrogation of the binding to FcγRs and Clq and consequently eliminates T cell proliferation and the release of cytokines, including TNF‐α and IFN‐γ, in vitro. It does not cross‐react with CD3 molecules expressed by T cells of other species. After a single intravenous injection, foralumab modulates human CD3 epsilon expression in a dose‐dependent manner, and more than 80% of the cell surface protein is removed within 24 h. This treatment leads to an 80% decrease in the number of circulating T cells when it is administered at a saturating dose.
Two clinical trials using parenteral foralumab have been conducted: a Phase I/IIa randomized, double‐blind, placebo‐controlled and dose‐escalation study in subjects with moderate to severe active Crohn’s disease (CD) and an open‐label, dose‐titration, multi‐centre study for the treatment of biopsy‐proven, acute cellular renal allograft rejection. Sixty‐four subjects with active Crohn’s disease and 11 subjects with acute cellular renal allograft rejection were treated in these studies. These studies have shown that the short‐term safety profile of foralumab is similar to that reported with the parenteral use of other anti‐CD3 antibodies. The most common adverse events were IRRs and CRS. In most cases, these symptoms were mild (66%) and were reported after the first two infusions. The number of affected patients and severity of symptoms tended to increase with the increasing dose level. Patients who received premedication with steroids had mild or no IRRs, and CRS was also decreased. No anti‐drug antibodies were detected, as the antibody is fully humanized (unpublished).
Oral OKT3, a mAb specific for human CD3, had equivalent effects in transgenic NOD mice expressing the human CD3 epsilon chain, thus serving as a preclinical model for testing human CD3‐specific mAb 68. Decreased proliferation of splenocytes from mice fed with anti‐CD3 mAbs has been reported and found to be similar to the findings in humanized mice.
A humanized mouse model [NOD/severe combined immunodeficient (SCID) IL‐2γc–/–] reconstituted with human haematopoietic stem cells has been developed to study and identify mechanisms of action relevant to human diseases to treat autoimmune and inflammatory diseases for patients being treated with mAb and immune modulators 73, 83, 84.
This humanized mouse model has been used to evaluate the efficacy of the oral administration of foralumab in allograft survival and its impact on human T cells in vivo 82. Foralumab was well tolerated and superior in preventing xenogeneic skin graft rejection compared with control human IgG. Foralumab passes intact through the stomach and small bowel. The therapeutic effect was not due to systemic T cell depletion, and no overt changes in the phenotype of T cells were found. Foralumab decreased the proliferative responses of CD8+ cells, increased serum IL‐10 levels and decreased the release of TNF‐α. Additionally, a dose dependency was shown; a modest increase in CD4+LAP+ T cells has been reported. At the highest dose, evidence of cell activation and signs of cytokine release was found; however, this dose is 10‐fold higher than the intravenous dose of teplizumab 83, 87. Only a minor amount of foralumab is absorbed systemically, and the serum levels of foralumab have been found to vary among mice, thus reflecting the ‘leakiness’ of the gut in the NOD background and some degree of graft‐versus‐host response in this model. The median level observed was approximately sixfold lower than the peak levels of teplizumab in children and adults with T1D administered with the drug intravenously 41. Previous preclinical and clinical studies of orally administered anti‐CD3 mAb have indicated that the drug is not absorbed systemically and induces tolerance through the promotion of Tregs 64, 74, 80, 85, 86, 87, 88. In these animal studies biological activity of orally administered foralumab was comparable to or better than orally administered OKT3. The data suggest that orally administered foralumab may modulate the systemic immune responses of T cells via targeting the TCR of T lymphocytes in the gut, thus providing therapeutic benefits in treating autoimmune diseases without the occurrence of potential adverse events.
Figure 1 shows the proposed mechanism of action for the oral administration of foralumab in patients with NASH and T2D. The current understanding of the requirements for drugs being developed for NASH is that they must be suitable for long‐term treatment, and thus have a high safety profile. These compounds must be able to be combined with other drugs, which act on different disease‐associated pathways, enabling combination therapy. Preferably, drugs for NASH should have the ability to target all stages of disease, and to alleviate some of the concomitant disorders such as diabetes.
Figure 1.
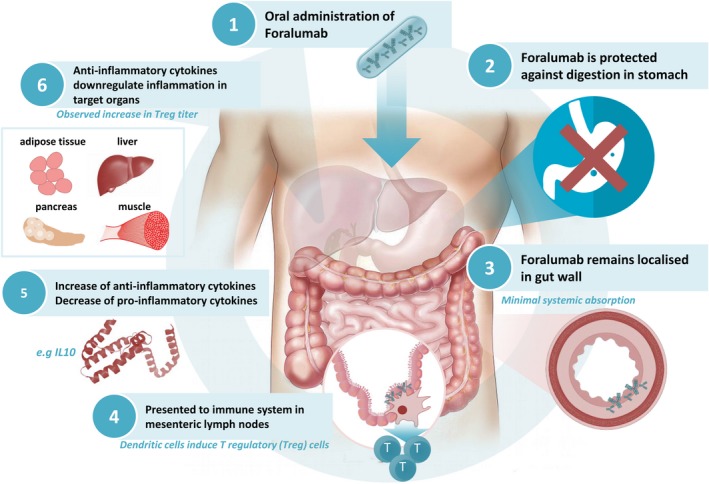
Schematic presentation of the mechanism of action of foralumab in non‐alcoholic steatohepatitis (NASH). After the oral administration of anti‐CD3 monoclonal antibody (mAb), the antibody is established in the small bowel and mesenteric lymph nodes. Foralumab, a specific anti‐CD3 epsilon mAb, binds T lymphocytes in the gut and modulates the CD3/T cell receptor (TCR complex), thus eliminating T cell proliferation and the release of proinflammatory cytokines. Binding leads to tolerogenic cross‐talk between dendritic cells and lymphocytes in the gut‐associated lymphoid tissue and mesenteric lymph nodes, thus promoting systemic regulatory T cells and the secretion of anti‐inflammatory cytokines which alleviate the chronic inflammatory state in target organs in NASH.
Oral immunotherapy using anti‐CD3 mAb has been shown to target systemic inflammation, with promising clinical results and without systemic immunosuppression 75, 81. The gut‐immune system is targeted by this mode of therapy, thereby generating gut associated immune signals that then affect the systemic immune response. This systemic response may affect both the fibrotic and inflammatory mechanisms associated with chronic liver diseases 23, 24, 79.
Currently, two clinical trials using the oral administration of foralumab are being considered. The objective of the first study is to determine the safety and efficacy of foralumab in patients with NASH and T2DM (NCT03291249). The second study aims to evaluate its safety and efficacy in patients with primary biliary cholangitis (PBC) or primary sclerosing cholangitis (PSC).
In summary, chronic inflammation underlies the pathogenesis of various stages of NASH and the development of cirrhosis and tumorigenesis. Preclinical and clinical data from the oral administration of anti‐CD3 mAb support the notion that foralumab may address the requirements of therapies for NASH, owing to its relatively high safety profile, its potential for targeting all stages of disease and its ability to be combined with other drugs for NASH. The data from the upcoming clinical trials using foralumab in NASH and T2D should shed light on the potential of this treatment.
Disclosure
Y. I. is the CMO of Tiziana Life Sciences, and consultant for Immune Pharmaceuticals, Immuron, Teva, Enzo Biochem, Protalix, Therapix, Nasvax, JTI and Natural Shield; Kunwar Shailubhai is CEO and CSO of Tiziana LifeSciences company. A. S. is President of Sanyal Biotechnology and has stock options in Genfit, Akarna, Tiziana, Indalo, Durect. He has served as a consultant to AbbVie, Astra Zeneca, Nitto Denko, Ardelyx, Conatus, Nimbus, Amarin, Salix, Tobira, Takeda, Fibrogen, Jannsen, Gilead, Boehringer, Lilly, Zafgen, Novartis, Pfizer, Immuron, Exhalenz and Genfit. He has been an unpaid consultant to Intercept, Echosens, Immuron, Galectin, Fractyl, Syntlogic, Novo Nordisk, Affimune, Chemomab, Nordic Bioscience and Bristol Myers Squibb. His institution has received grant support from Gilead, Salix, Tobira, Bristol Myers, Shire, Intercept, Merck, Astra Zeneca, Malinckrodt, Cumberland and Novartis. He receives royalties from Elsevier and UptoDate.
Acknowledgement
This work was supported in part by a grant from The Roman‐Epstein Research Foundation (Y. I.).
References
- 1. Wong VW, Chitturi S, Wong GL et al. Pathogenesis and novel treatment options for non‐alcoholic steatohepatitis. Lancet Gastroenterol Hepatol 2016; 1:56–67. [DOI] [PubMed] [Google Scholar]
- 2. da Cunha AP, Weiner HL. Induction of immunological tolerance by oral anti‐CD3. Clin Dev Immunol 2012; 2012:425021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Musso G, Cassader M, Gambino R. Non‐alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov 2016; 15:249–74. [DOI] [PubMed] [Google Scholar]
- 4. Perazzo H, Dufour JF. The therapeutic landscape of non‐alcoholic steatohepatitis. Liver Int 2017; 37:634–47. [DOI] [PubMed] [Google Scholar]
- 5. Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non‐alcoholic fatty liver disease. Gut 2017; 66:180–90. [DOI] [PubMed] [Google Scholar]
- 6. Povero D, Feldstein AE. Novel molecular mechanisms in the development of non‐alcoholic steatohepatitis. Diabetes Metab J 2016; 40:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vonghia L, Francque S. Cross talk of the immune system in the adipose tissue and the liver in non‐alcoholic steatohepatitis: pathology and beyond. World J Hepatol 2015; 7:1905–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nati M, Haddad D, Birkenfeld AL et al. The role of immune cells in metabolism‐related liver inflammation and development of non‐alcoholic steatohepatitis (NASH). Rev Endocr Metab Disord 2016; 17:29–39. [DOI] [PubMed] [Google Scholar]
- 9. Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res 2016; 57:2099–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Magee N, Zou A, Zhang Y. Pathogenesis of nonalcoholic steatohepatitis: interactions between liver parenchymal and nonparenchymal cells. Biomed Res Int 2016; 2016:5170402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sell H, Habich C, Eckel J. Adaptive immunity in obesity and insulin resistance. Nat Rev Endocrinol 2012; 8:709–16. [DOI] [PubMed] [Google Scholar]
- 12. Brunt EM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 2010; 7:195–203. [DOI] [PubMed] [Google Scholar]
- 13. Gadd VL, Skoien R, Powell EE et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014; 59:1393–405. [DOI] [PubMed] [Google Scholar]
- 14. Tang Y, Bian Z, Zhao L et al. Interleukin‐17 exacerbates hepatic steatosis and inflammation in non‐alcoholic fatty liver disease. Clin Exp Immunol 2011; 166:281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harley IT, Stankiewicz TE, Giles DA et al. IL‐17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014; 59:1830–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rau M, Schilling AK, Meertens J et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol 2016; 196:97–105. [DOI] [PubMed] [Google Scholar]
- 17. Winer DA, Winer S, Chng MH et al. B Lymphocytes in obesity‐related adipose tissue inflammation and insulin resistance. Cell Mol Life Sci 2014; 71:1033–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He B, Wu L, Xie W et al. The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol 2017; 18:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paquissi FC. Immune imbalances in non‐alcoholic fatty liver disease: from general biomarkers and neutrophils to interleukin‐17 axis activation and new therapeutic targets. Front Immunol 2016; 7:490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhan YT, An W. Roles of liver innate immune cells in nonalcoholic fatty liver disease. World J Gastroenterol 2010; 16:4652–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Świderska M, Jaroszewicz J, Stawicka A et al. The interplay between Th17 and T‐regulatory responses as well as adipokines in the progression of non‐alcoholic fatty liver disease. Clin Exp Hepatol 2017; 3:127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagata K, Suzuki H, Sakaguchi S. Common pathogenic mechanism in development progression of liver injury caused by non‐alcoholic or alcoholic steatohepatitis. J Toxicol Sci 2007; 32:453–68. [DOI] [PubMed] [Google Scholar]
- 23. Ilan Y. Review article: novel methods for the treatment of non‐alcoholic steatohepatitis – targeting the gut immune system to decrease the systemic inflammatory response without immune suppression. Aliment Pharmacol Ther 2016; 44:1168–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ilan Y. Oral immune therapy: targeting the systemic immune system via the gut immune system for the treatment of inflammatory bowel disease. Clin Transl Immunol 2016; 5:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ilan Y. Immune therapy for nonalcoholic steatohepatitis: are we there yet? J Clin Gastroenterol 2013; 47:298–307. [DOI] [PubMed] [Google Scholar]
- 26. Call ME, Wucherpfennig KW. Molecular mechanisms for the assembly of the T cell receptor–CD3 complex. Mol Immunol 2004; 40:1295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malissen B, Wegener AM, Hoeveler A et al. Molecular dissection of the T‐cell receptor/CD3 complex. Immunol Ser 1993; 59:29–40. [PubMed] [Google Scholar]
- 28. Woodle ES, Xu D, Zivin RA et al. Phase I trial of a humanized, Fc receptor nonbinding OKT3 antibody, huOKT3gamma1(Ala‐Ala) in the treatment of acute renal allograft rejection. Transplantation 1999; 68:608–16. [DOI] [PubMed] [Google Scholar]
- 29. Friend PJ, Hale G, Chatenoud L et al. Phase I study of an engineered aglycosylated humanized CD3 antibody in renal transplant rejection. Transplantation 1999; 68:1632–7. [DOI] [PubMed] [Google Scholar]
- 30. Chatenoud L. CD3‐specific antibody‐induced active tolerance: from bench to bedside. Nat Rev Immunol 2003; 3:123–32. [DOI] [PubMed] [Google Scholar]
- 31. Chatenoud L. CD3 antibody treatment stimulates the functional capability of regulatory T cells. Novartis Found Symp 2003; 252:279–86; discussion: 286–90. [PubMed] [Google Scholar]
- 32. Vallera DA, Carroll SF, Brief S et al. Anti‐CD3 immunotoxin prevents low‐dose STZ/interferon‐induced autoimmune diabetes in mouse. Diabetes 1992; 41:457–64. [DOI] [PubMed] [Google Scholar]
- 33. Takiishi T, Cook DP, Korf H et al. Reversal of diabetes in NOD mice by clinical‐grade proinsulin and IL‐10‐secreting lactococcus lactis in combination with low‐dose anti‐CD3 depends on the induction of Foxp3‐positive T cells. Diabetes 2017; 66:448–59. [DOI] [PubMed] [Google Scholar]
- 34. Daifotis AG, Koenig S, Chatenoud L et al. Anti‐CD3 clinical trials in type 1 diabetes mellitus. Clin Immunol 2013; 149:268–78. [DOI] [PubMed] [Google Scholar]
- 35. Utset TO, Auger JA, Peace D et al. Modified anti‐CD3 therapy in psoriatic arthritis: a phase I/II clinical trial. J Rheumatol 2002; 29:1907–13. [PubMed] [Google Scholar]
- 36. Baumgart DC, Targan SR, Dignass AU et al. Prospective randomized open‐label multicenter phase I/II dose escalation trial of visilizumab (HuM291) in severe steroid‐refractory ulcerative colitis. Inflamm Bowel Dis 2010; 16:620–9. [DOI] [PubMed] [Google Scholar]
- 37. Plevy S, Salzberg B, Van Assche G et al. A phase I study of visilizumab, a humanized anti‐CD3 monoclonal antibody, in severe steroid‐refractory ulcerative colitis. Gastroenterology 2007; 133:1414–22. [DOI] [PubMed] [Google Scholar]
- 38. Sandborn WJ, Colombel JF, Frankel M et al. Anti‐CD3 antibody visilizumab is not effective in patients with intravenous corticosteroid‐refractory ulcerative colitis. Gut 2010; 59:1485–92. [DOI] [PubMed] [Google Scholar]
- 39. Keymeulen B, Vandemeulebroucke E, Ziegler AG et al. Insulin needs after CD3‐antibody therapy in new‐onset type 1 diabetes. N Engl J Med 2005; 352:2598–608. [DOI] [PubMed] [Google Scholar]
- 40. Herold KC, Gitelman SE, Masharani U et al. A single course of anti‐CD3 monoclonal antibody hOKT3{gamma}1(Ala‐Ala) results in improvement in C‐peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 2005; 54:1763–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Herold KC, Hagopian W, Auger JA et al. Anti‐CD3 monoclonal antibody in new‐onset type 1 diabetes mellitus. N Engl J Med 2002; 346:1692–8. [DOI] [PubMed] [Google Scholar]
- 42. Chatenoud L. The immune response against therapeutic monoclonal antibodies. Immunol Today 1986; 7:367–8. [DOI] [PubMed] [Google Scholar]
- 43. Chatenoud L, Jonker M, Villemain F et al. The human immune response to the OKT3 monoclonal antibody is oligoclonal. Science 1986; 232:1406–8. [DOI] [PubMed] [Google Scholar]
- 44. Vossen AC, Tibbe GJ, Kroos MJ et al. Fc receptor binding of anti‐CD3 monoclonal antibodies is not essential for immunosuppression, but triggers cytokine‐related side effects. Eur J Immunol 1995; 25:1492–6. [DOI] [PubMed] [Google Scholar]
- 45. Xue Y, Castanos‐Velez E, Biberfeld P et al. Anti‐CD3 induced thymocyte apoptosis in vivo require the antibody Fc domain. Scand J Immunol 2000; 51:441–6. [DOI] [PubMed] [Google Scholar]
- 46. Chatenoud L, Bach JF. Monoclonal antibodies to CD3 as immunosuppressants. Semin Immunol 1990; 2:437–47. [PubMed] [Google Scholar]
- 47. Ferran C, Dy M, Merite S et al. Reduction of morbidity and cytokine release in anti‐CD3 MoAb‐treated mice by corticosteroids. Transplantation 1990; 50:642–8. [DOI] [PubMed] [Google Scholar]
- 48. Ferran C, Bluestone J, Bach JF et al. In vivo T lymphocyte activation induced in mice following the injection of anti‐CD3 monoclonal antibody. Transplant Proc 1990; 22:1922–3. [PubMed] [Google Scholar]
- 49. Alegre ML, Lenschow DJ, Bluestone JA. Immunomodulation of transplant rejection using monoclonal antibodies and soluble receptors. Dig Dis Sci 1995; 40:58–64. [DOI] [PubMed] [Google Scholar]
- 50. Chatenoud L, Waldmann H. CD3 monoclonal antibodies: a first step towards operational immune tolerance in the clinic. Rev Diabet Stud 2012; 9:372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tooley JE, Vudattu N, Choi J et al. Changes in T‐cell subsets identify responders to FcR‐nonbinding anti‐CD3 mAb (teplizumab) in patients with type 1 diabetes. Eur J Immunol 2016; 46:230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vudattu NK, Herold KC. Treatment of new onset type 1 diabetes with teplizumab: successes and pitfalls in development. Expert Opin Biol Ther 2014; 14:377–85. [DOI] [PubMed] [Google Scholar]
- 53. Sherry N, Hagopian W, Ludvigsson J et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1‐year results from a randomised, placebo‐controlled trial. Lancet 2011; 378:487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guglielmi C, Williams SR, Del Toro R et al. Efficacy and safety of otelixizumab use in new‐onset type 1 diabetes mellitus. Expert Opin Biol Ther 2016; 16:841–6. [DOI] [PubMed] [Google Scholar]
- 55. MacDonald A, Ambery P, Donaldson J et al. Subcutaneous administration of otelixizumab is limited by injection site reactions: results of an exploratory study in type 1 diabetes mellitus patients. Exp Clin Endocrinol Diabetes 2016; 124:288–93. [DOI] [PubMed] [Google Scholar]
- 56. Belghith M, Bluestone JA, Barriot S et al. TGF‐beta‐dependent mechanisms mediate restoration of self‐tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med 2003; 9:1202–8. [DOI] [PubMed] [Google Scholar]
- 57. You S, Leforban B, Garcia C et al. Adaptive TGF‐beta‐dependent regulatory T cells control autoimmune diabetes and are a privileged target of anti‐CD3 antibody treatment. Proc Natl Acad Sci USA 2007; 104:6335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Long SA, Thorpe J, DeBerg HA et al. Partially exhausted CD8 T cells are associated with clinical response to teplizumab in new‐onset type 1 diabetes. Sci Immunol 2016; 1:eaai7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bisikirska B, Colgan J, Luban J et al. TCR stimulation with modified anti‐CD3 mAb expands CD8 T cell population and induces CD8CD25 Tregs. J Clin Invest 2005; 115:2904–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hagopian W, Ferry RJ Jr, Sherry N et al. Teplizumab preserves C‐peptide in recent‐onset type 1 diabetes: two‐year results from the randomized, placebo‐controlled Protege trial. Diabetes 2013; 62:3901–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kuhn C, Weiner HL. Therapeutic anti‐CD3 monoclonal antibodies: from bench to bedside. Immunotherapy 2016; 8:889–906. [DOI] [PubMed] [Google Scholar]
- 62. Weiner HL, da Cunha AP, Quintana F et al. Oral tolerance. Immunol Rev 2011; 241:241–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Esplugues E, Huber S, Gagliani N et al. Control of TH17 cells occurs in the small intestine. Nature 2011; 475:514–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ochi H, Abraham M, Ishikawa H et al. Oral CD3‐specific antibody suppresses autoimmune encephalomyelitis by inducing CD4± CD25– LAP± T cells. Nat Med 2006; 12:627–35. [DOI] [PubMed] [Google Scholar]
- 65. Ochi H, Abraham M, Ishikawa H et al. New immunosuppressive approaches: oral administration of CD3‐specific antibody to treat autoimmunity. J Neurol Sci 2008; 274:9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ishikawa H, Ochi H, Chen ML et al. Inhibition of autoimmune diabetes by oral administration of anti‐CD3 monoclonal antibody. Diabetes 2007; 56:2103–9. [DOI] [PubMed] [Google Scholar]
- 67. Zhang X, Izikson L, Liu L et al. Activation of CD25(±)CD4(±) regulatory T cells by oral antigen administration. J Immunol 2001; 167:4245–53. [DOI] [PubMed] [Google Scholar]
- 68. Kuhn C, Rezende RM, da Cunha AP et al. Mucosal administration of CD3‐specific monoclonal antibody inhibits diabetes in NOD mice and in a preclinical mouse model transgenic for the CD3 epsilon chain. J Autoimmun 2017; 76:115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Forster K, Goethel A, Chan CW et al. An oral CD3‐specific antibody suppresses T‐cell‐induced colitis and alters cytokine responses to T‐cell activation in mice. Gastroenterology 2012; 143:1298–307. [DOI] [PubMed] [Google Scholar]
- 70. Sasaki N, Yamashita T, Takeda M et al. Oral anti‐CD3 antibody treatment induces regulatory T cells and inhibits the development of atherosclerosis in mice. Circulation 2009; 120:1996–2005. [DOI] [PubMed] [Google Scholar]
- 71. Wu HY. Induction of mucosal tolerance in SLE: a sniff or a sip away from ameliorating lupus? Clin Immunol 2009; 130:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wu HY, Quintana FJ, Weiner HL. Nasal anti‐CD3 antibody ameliorates lupus by inducing an IL‐10‐secreting CD4± CD25‐ LAP± regulatory T cell and is associated with down‐regulation of IL‐17± CD4± ICOS± CXCR72± follicular helper T cells. J Immunol 2008; 181:6038–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Waldron‐Lynch F, Henegariu O, Deng S et al. Teplizumab induces human gut‐tropic regulatory cells in humanized mice and patients. Sci Transl Med 2012; 4:118ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ilan Y, Zigmond E, Lalazar G et al. Oral administration of OKT3 monoclonal antibody to human subjects induces a dose‐dependent immunologic effect in T cells and dendritic cells. J Clin Immunol 2010; 30:167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Halota W, Ferenci P, Kozielewicz D et al. Oral anti‐CD3 immunotherapy for HCV‐nonresponders is safe, promotes regulatory T cells and decreases viral load and liver enzyme levels: results of a phase‐2a placebo‐controlled trial. J Viral Hepatol 2015; 22:651–7. [DOI] [PubMed] [Google Scholar]
- 76. du Plessis J, van Pelt J, Korf H et al. Association of adipose tissue inflammation with histologic severity of nonalcoholic fatty liver disease. Gastroenterology 2015; 149:635–48.e14. [DOI] [PubMed] [Google Scholar]
- 77. Kaur S, Venktaraman G, Jain M et al. Recent trends in antibody‐based oncologic imaging. Cancer Lett 2012; 315:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Brenner C, Galluzzi L, Kepp O et al. Decoding cell death signals in liver inflammation. J Hepatol 2013; 59:583–94. [DOI] [PubMed] [Google Scholar]
- 79. Ilan Y. Compounds of the sphingomyelin‐ceramide‐glycosphingolipid pathways as secondary messenger molecules: new targets for novel therapies for fatty liver disease and insulin resistance. Am J Physiol Gastrointest Liver Physiol 2016; 310:G1102–17. [DOI] [PubMed] [Google Scholar]
- 80. Ilan Y, Maron R, Tukpah AM et al. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc Natl Acad Sci USA 2010; 107:9765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lalazar G, Mizrahi M, Turgeman I et al. Oral administration of OKT3 MAb to patients with NASH, promotes regulatory T‐cell induction, and alleviates insulin resistance: results of a phase IIa blinded placebo‐controlled trial. J Clin Immunol 2015; 35:399–407. [DOI] [PubMed] [Google Scholar]
- 82. Ogura M, Deng S, Preston‐Hurlburt P et al. Oral treatment with foralumab, a fully human anti‐CD3 monoclonal antibody, prevents skin xenograft rejection in humanized mice. Clin Immunol 2017; 183:240–6. [DOI] [PubMed] [Google Scholar]
- 83. Herold KC, Gitelman SE, Willi SM et al. Teplizumab treatment may improve C‐peptide responses in participants with type 1 diabetes after the new‐onset period: a randomised controlled trial. Diabetologia 2013; 56:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Waldron‐Lynch F, Deng S, Preston‐Hurlburt P et al. Analysis of human biologics with a mouse skin transplant model in humanized mice. Am J Transplant 2012; 12:2652–62. [DOI] [PubMed] [Google Scholar]
- 85. Herold KC, Gitelman SE, Ehlers MR et al. Teplizumab (anti‐CD3 mAb) treatment preserves C‐peptide responses in patients with new‐onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013; 62:3766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wu HY, Maron R, Tukpah AM et al. Mucosal anti‐CD3 monoclonal antibody attenuates collagen‐induced arthritis that is associated with induction of LAP± regulatory T cells and is enhanced by administration of an emulsome‐based Th2‐skewing adjuvant. J Immunol 2010; 185:3401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wu HY, Center EM, Tsokos GC et al. Suppression of murine SLE by oral anti‐CD3: inducible CD4±CD25‐LAP± regulatory T cells control the expansion of IL‐17± follicular helper T cells. Lupus 2009; 18:586–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Slavin AJ, Maron R, Weiner HL. Mucosal administration of IL‐10 enhances oral tolerance in autoimmune encephalomyelitis and diabetes. Int Immunol 2001; 13:825–33. [DOI] [PubMed] [Google Scholar]