

Abstract
Endotoxin induces a variety of proinflammatory mediators and plays a crucial role in kidney inflammation. The receptor tyrosine kinase, Mer, diminishes renal inflammation by attenuating inflammatory responses. We previously reported that Mer is predominantly expressed on glomerular endothelial cells (GECs) and that Mer deficiency is associated with increased renal inflammation when mice are challenged with nephrotoxic serum. We consequently hypothesized that Mer signaling down-regulates LPS-driven inflammatory responses in GECs. To test this hypothesis, primary GECs were isolated from the kidneys of Mer-KO and wild-type (WT) control mice. LPS treatment induced Akt and STAT3 activation along with Bcl-xl up-regulation in wild-type GECs; these responses were all increased in Mer-deficient GECs. In addition, STAT1 and ERK1/2 upregulation and activation were observed in Mer-KO GECs exposed to LPS. In contrast, expression of the inhibitory signaling molecule, SOCS-3, was much higher in LPS-stimulated wild-type than Mer-deficient GECs. Deficiency of Mer was also associated with significantly increased NF-κB expression and activation. These observations indicate that Mer functions as an intrinsic feedback inhibitor of inflammatory mediator-driven immune responses in GECs during kidney injury and suggest a new therapeutic strategy for glomerular diseases.
One-sentence Summary:
Mer signaling intrinsically inhibits glomerular endothelial inflammation
Introduction
Glomerulonephritis (GN) encompasses a group of immune-mediated disorders that cause inflammation within the glomerulus and other compartments in the kidney. GN remains a leading cause of end-stage renal disease, yet treatment is still very limited [1, 2]. GN pathogenesis is also complex. Glomerular endothelial cells (GECs) have an active role in the glomerular response to injury [3]. Altered GEC function has been associated with increased leukocyte recruitment and increased angiogenesis. GEC injury was commonly present in a cohort of type 2 diabetes patients with macroalbuminuria and GEC abnormalities were more closely associated with increasing urine albumin excretion [4]. GECs express a subset of Toll-like receptors (TLRs) and can respond to intrinsic damage-associated molecular patterns (DAMPS) [5]. However, the mechanism by which GEC dysfunction results in albuminuria is poorly understood.
TAM (Tyro-3, Axl, and Mer) receptor tyrosine kinases are essential regulators of immune homeostasis that dampen the activation of immune cells and restore tissue function by promoting tissue repair and clearance of apoptotic cells [6–8]. TAM-dependent pathways act as a negative feedback mechanism that suppresses inflammation [7]. Consistent with this, deficient TAM signaling has been linked to the pathogenesis of autoimmune, inflammatory, and infectious diseases [9–11]. One of the TAM receptor tyrosine kinases, Mer, has been particularly associated with an anti-inflammatory effect. Mer signaling participates in a novel inhibitory pathway in TLR activation [12–14] and a Mer signaling pathway in macrophages that involves PI3K/Akt and NF-κB down-regulates proinflammatory signals [15, 16]. In addition, Mer-KO mice have been shown to be hypersensitive to LPS [17]. Our previous work showed that Mer down-regulates renal inflammation, with consequent increased susceptibility to anti-glomerular basement membrane (GBM)-mediated kidney damage in Mer-deficient mice [18, 19]. Consistent with this, Mer is highly expressed in the glomerulus and is upregulated upon kidney inflammation [18, 20]. However, the molecular basis of the Mer-mediated signaling pathway involved in downregulation of inflammatory responses in glomerular endothelial cells has not been established.
In the present study, we extend our previous work by investigating the inhibitory role of Mer on LPS-induced inflammatory signaling pathways in GECs. After defining the expression pattern of Mer on cell subsets in mouse kidney, we compared LPS signaling in isolated primary GECs from wild-type (WT) and Mer-deficient mice. Results of these studies provide a mechanism for the anti-inflammatory effects of Mer by showing strong Mer expression in GECs that is upregulated by LPS, as well as increased STAT1, STAT3, Akt, Bcl-2, and ERK1/2 expression and activation and decreased SOCS-3 expression in GECs in the absence of Mer.
Materials and methods
Animals and reagents
WT and Mer-KO mice on a C57BL/6 background were housed under specific pathogen-free conditions at the animal facilities of the University of Cincinnati. All animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee.
LPS (Escherichia coli lipopolysaccharide, 055:B5) was purchased form Sigma Aldrich (St. Louis, MO, USA). The antibodies used in this study were all purchased from Cell Signaling Technology (Boston, MA, USA), unless otherwise stated in the text.
Immunofluorescent staining
Mouse kidneys were snap-frozen in liquid nitrogen. Sections (4μm) were fixed in methanol and blocked with 5% BSA. Anti-mouse antibodies (Abs) (1:100 dilution) were added and incubated with sections. Secondary Abs were added after wash as indicated in the figure legends. Images were acquired using a Nikon E1000 fluorescent microscope (Melville, NY, USA) equipped with camera and analyzed with a bioquantification software system (Bioquant TCW 98, Nashville, TN).
Flow cytometry
Renal cortex of kidney samples was isolated from healthy WT and Mer-KO mice. Single cell suspensions were obtained as previously described [18]. Cells were then incubated with Fc blocker (2.4G2) for 30 minutes and stained for an endothelial cell marker (anti-CD31), a mesangial cell marker (anti-smooth muscle actin), a podocyte marker (anti-nephrin), a neutrophil marker (anti-Gr-1), or a macrophage marker (CD11b), respectively. Mer expression on cell subsets was detected with biotin-labeled anti-Mer antibody from R&D Systems (Minneapolis, MN, USA) and visualized with PE-conjugated streptavidin (BD Biosciences, San Jose, CA, USA). Cell samples were analyzed with a BD FACS Canto flow cytometer (BD Biosciences), using FlowJo software 10.2 (San Carlos, CA, USA).
Isolation and culture of primary glomerular endothelial cells
Mer-KO mice were first genotypically confirmed by conventional PCR using whole tail lysate (Fig. 1A) [17]. Whole kidneys from healthy WT and Mer-KO mice were then surgically removed under sterile conditions. Primary GECs were isolated and prepared to achieve 95–99% purity at the Cell Biologics company (Chicago, IL, USA). Cells were then aliquoted and stored in liquid nitrogen. Tissue culture flasks and plates were coated with gelatin at 37°C for 1 hr. GECs were cultured in the endothelial basal medium supplied by Cell Biologics. Mer deficiency in GECs was further confirmed by Western Blot (Fig. 1A). For LPS treatment experiments, GECs were cultured in 6-well plates one day before stimulation at a density of 0.5×106 cells per well. Cells were washed once with fresh medium the next day and a range of concentrations of LPS was added. Three days later, cells were harvested for Western blot analysis. Culture supernatants were centrifuged for 20 minutes at 15,000 rpm to remove debris and stored at −80°C.
Figure 1. Mer is expressed in mouse glomeruli.
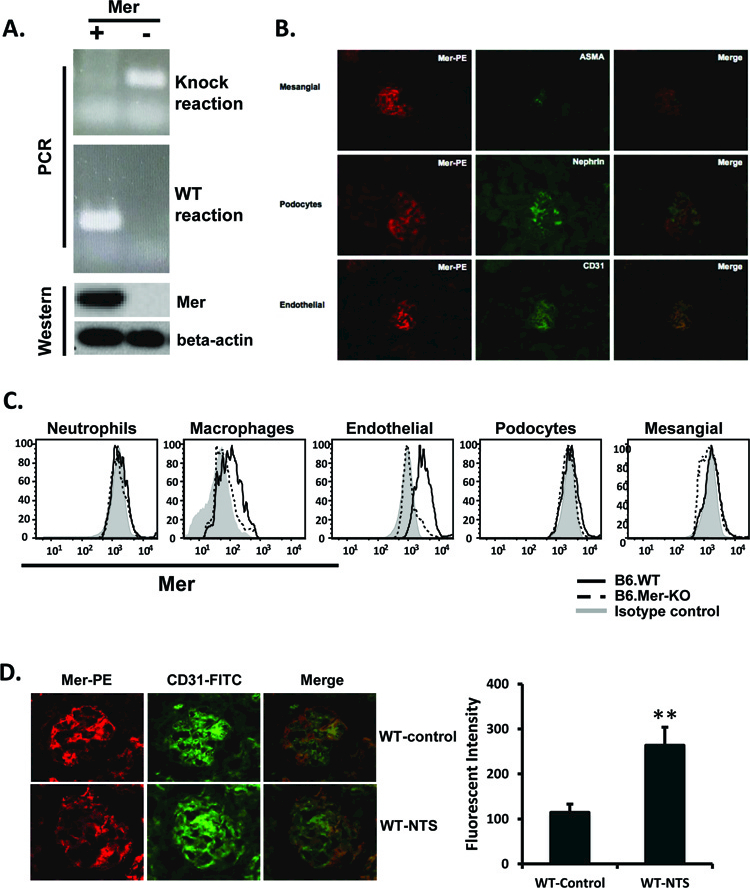
A. Mouse tail lysate was obtained and genotyping PCR was performed as previously described [17]. Western blotting (WB) analysis of Mer expression on primary GECs was also performed (bottom row). B. Kidney sections (4 μm) obtained from C57BL/6 mice were stained with biotin-conjugated anti-mouse Mer and visualized with PE-streptavidin. Glomerular cell subsets were stained with specific markers as indicated in the figure. C. Kidneys from B6. WT and B6.Mer-KO mice were cut into <1mm pieces and subjected to collagenase I and DNase digestion. After EDTA incubation, single cell suspensions were stained for specific markers. Neutrophils were gated for Gr-1 and CD11b double positivity; macrophages were gated on CD11b+ and Gr-1- populations; glomerular mesangial cells, endothelial cells, and podocytes were gated according to surface antigens, as described in Methods. D. Kidney sections were obtained from WT and nephrotoxic serum (NTS)-injected mice [19] and stained with anti-Mer and anti-CD31 antibodies. Data are representative of two repeats. **p<0.01.
Protein array analysis
The cytokine profile of GECs stimulated with LPS was measured using the Proteome Profiler Array from R&D Systems according to the manufacturer’s guidelines. In brief, cell culture supernatants were centrifuged to remove particles. A mouse cytokine array membrane was blocked for one hour at room temperature and incubated with a detecting antibody/sample mix overnight at 4°C. The membrane was then washed and incubated with Streptavidin-HRP. Cytokine protein levels were visualized with Chemi Reagent Mix.
Western blotting
Following stimulation with LPS at the indicated concentrations, cells were washed in cold PBS and lysed in RIPA buffer (Santa Cruz Biotechnology, Dallas, TX, USA) with proteinase and phosphatase inhibitors (Roche, Indianapolis, IN, USA). Proteins were separated by SDS-PAGE, transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA) and incubated with primary antibodies (rabbit antibodies against mouse cell signaling molecules as indicated in figures, and subsequently with secondary antibodies labeled with horseradish peroxidase (HRP), both in blocking buffer (1% dry milk in buffer). Proteins were visualized with enhanced chemiluminescence (Fisher Scientific, Hampton, NH, USA).
Statistics
Western blot data were analyzed using ImageJ software (NIH, Bethesda, MD, USA). Intensity differences between groups were tested using the Mann-Whitney U test. Data are shown as median with interquartile range. All statistical analyses were carried out using Microsoft Excel or Prism software (GraphPad, San Diego, CA, USA). Results were considered significant at *p < 0.05, **p<0.01, ***p<0.001.
Results
Mer is primarily expressed on glomerular endothelial cells.
We previously reported that Mer protects against renal inflammation [18, 19]. Consistent with this, renal Mer expression, which was originally demonstrated by Northern blot [20, 21], is highest in both mice and humans in the kidney [19–21]. We confirmed renal expression of Mer protein by Western blot [19] and by immunofluorescent staining (Fig. 1B). We also stained a single cell suspension of kidney cells with antibodies that are specific for renal cell subtypes and identified glomerular endothelial cells as the primary Mer-bearing cells (Fig. 1C). Kidney resident macrophages also expressed low levels of surface Mer (Fig. 1C). Increased Mer expression was previously reported in the kidney of nephritic mice as compared to the kidney of control mice [18]. Figure 1D shows that glomerular Mer continues to be predominantly expressed by endothelial cells following treatment of mice with NTS.
Mer inhibits LPS-induced expression and activation of Akt and ERK1/2 in GECs.
LPS induces proliferation and survival through expression and activation of Akt and ERK1/2 in many cells, including endothelial cells [22]. Although TAM receptors inhibit LPS-induced signals [8], the ability of Mer to directly inhibit LPS signaling in GECs has not been studied. To address this, we studied the effects of LPS 72 hr post-incubation, which is the time of maximal LPS-induced cellular activation [23]. LPS increased total and phosphorylated Akt protein in WT primary GECs; however, this was seen to a considerably greater extent in Mer-deficient than in WT GECs (Fig. 2A and B). Total and phospho-ERK were both considerably increased in Mer-deficient GECs, but were not significantly increased in WT GECs (Fig. 2C and D). Consistent with our previous findings [19], p38MAP kinase remained undetectable in primary GECs regardless of Mer expression (data not shown). Taken together, these data suggest an inhibitory function of Mer in regulating LPS-mediated Akt and ERK1/2 expression and activation.
Figure 2. Mer inhibits LPS-induced Akt and ERK1/2 expression and phosphorylation.
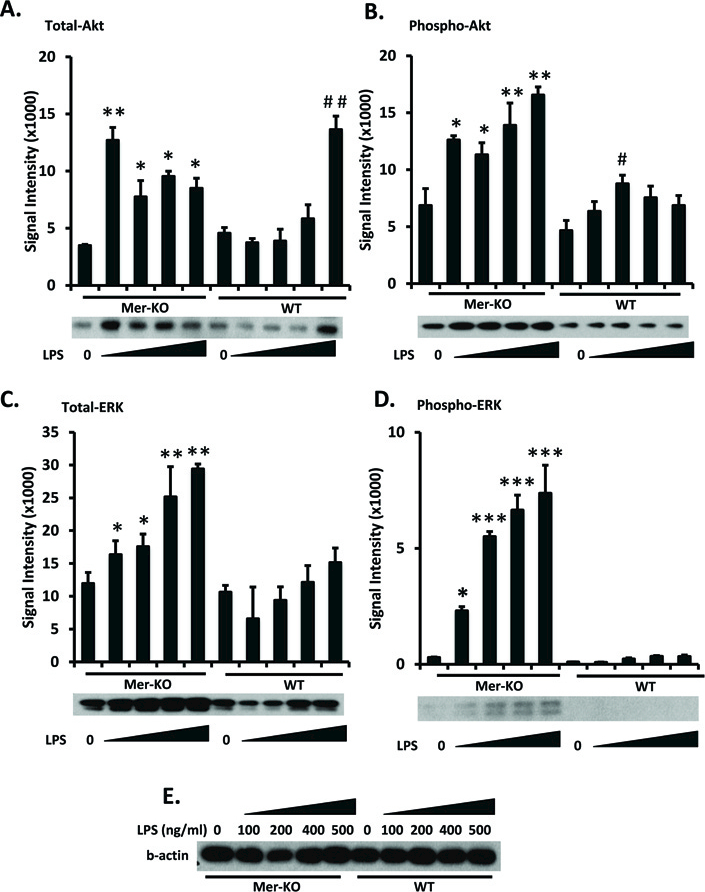
Primary GECs were isolated as described in Methods. WT and Mer-KO GECs were cultured in 6-well plates at a density of 0.5×106 cells per well overnight. GECs were then treated with a range of concentrations of LPS for 3 days. Cells were then lysed and the LPS-downstream signaling proteins, Akt (A) and ERK1/2 (C), and their phosphorylated forms (B and D) were analyzed by Western blotting with specific antibodies. E. Loading controls (β-actin). Experiments were repeated independently at least three times. Densitometry analyses to quantify the intensity of the indicated proteins are shown. Representative blots were shown below each bar graph. Values are the median and interquartile range. The Mann-Whitney test was used to determine statistical significance. *p<0.05, **p<0.01. ***p<0.001, Mer-KO LPS treated versus untreated; ##p<0.01, WT LPS versus WT untreated.
Mer suppresses LPS-induced Bcl expression.
LPS-induced TLR4 activation triggers increased expression of cell survival genes, including Bcl family proteins [24]. Baseline Bcl-2 levels were significantly higher in Mer-deficient than in WT GECs and were increased by LPS stimulation to a relatively small, but significant extent in both (Fig. 3 A and B). Increased baseline level of Bcl-2 expression in the Mer-deficient GECs may reflect a surveillance function of Mer on GECs to keep GECs quiescent under pressure and constant contact with stimuli, such as cell debris and immunoglobulin. A surveillance function of Mer on GECs may also explain the maintenance of a higher level of Mer on GECs. Baseline Bcl-6 levels were also higher in Mer-deficient than in WT GECs and did not increase in response to LPS; in fact, relatively low, but not high doses of LPS decreased Bcl-6 levels in Mer-deficient GECs (Fig. 3A and C). In contrast, baseline Bcl-xl levels were lower in Mer-deficient than in WT GECs, but increased to a greater extent in the Mer-deficient than in the WT cells in response to high levels of LPS, so that Bcl-xl levels in LPS-stimulated WT and Bcl-xl GECs were comparable (Fig 3A and D).
Figure 3. Enhanced Bcl-2 expression in Mer-KO GECs.
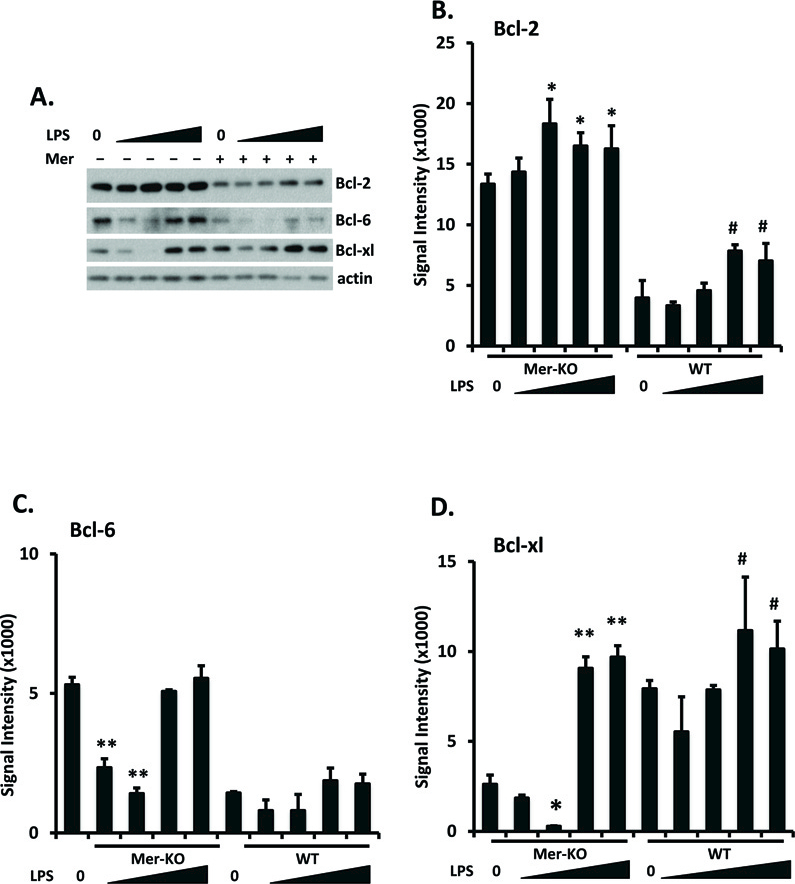
Primary GECs were prepared, cultured, and LPS-treated as in figure 2. Bcl-2, Bcl-6, and Bcl-xl expression was analyzed by Western immunoblotting. Representative blots were shown in A. Total amounts of protein loading were indicated by β-actin. Experiments were repeated independently at least three times. Densitometry analyses to quantify the intensity of the indicated proteins are shown in B-D. Values are the median and interquartile range. The Mann-Whitney test was used to determine statistical significance. *p<0.05, **p<0.01, Mer-KO LPS treated versus untreated; #p<0.05, WT LPS versus WT untreated.
Mer-mediated suppression is associated with SOCS upregulation and STAT and NF-κB downregulation.
In macrophages, Mer signaling suppresses LPS-induced STAT1 activation by increasing SOCS-1 and SOCS-3 protein synthesis [25]. We explored this pathway in mouse GECs by quantitating total SOCS and STAT proteins and phosphorylated STAT proteins. Indeed, Mer deficiency increased baseline total STAT1 and STAT3 levels; these levels were not increased or only modestly increased by LPS stimulation (Fig. 4 A, B, and D). Baseline levels of activated (phospho) STAT1 and STAT3 were relatively unaffected by Mer deficiency, but less LPS was required to increase STAT3 activation in Mer-deficient than in WT GECs (Fig. 4 A and C) and LPS only increased STAT1 activation in the Mer-deficient cells (Fig. 4A and E).
Figure 4. Mer inhibits STAT expression and phosphorylation in LPS-treated GECs.
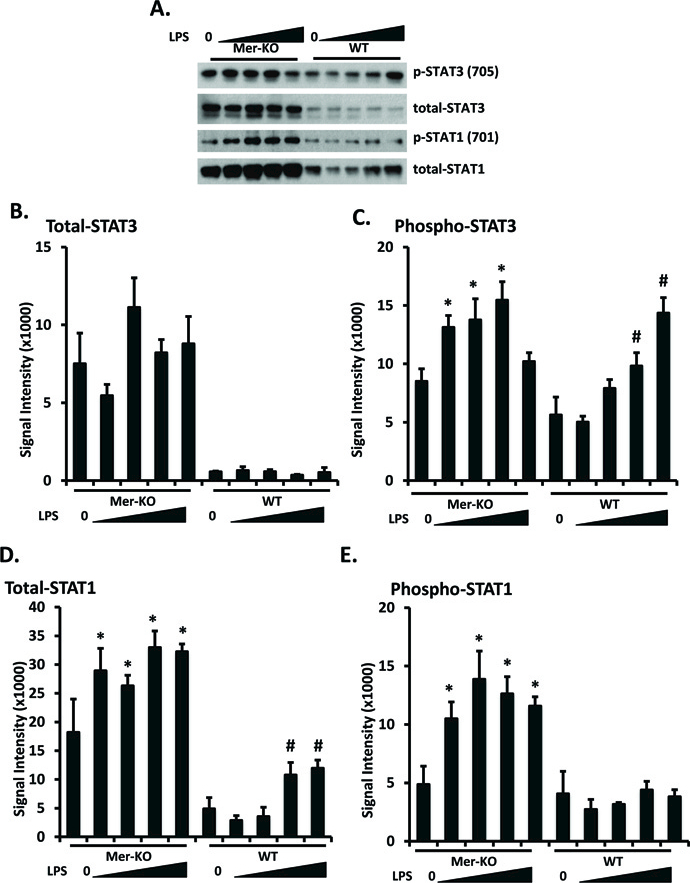
Primary GECs were prepared, cultured, and LPS-treated as in figure 2. STAT1 and STAT3 expression and phosphorylation were analyzed by Western immunoblotting. Representative blots were shown in A. Experiments were repeated independently at least three times. Densitometry analyses to quantify the intensity of total STAT1 and STAT3 are shown in B and C. Values are the median and interquartile range. The Mann-Whitney test was used to determine statistical significance. *p<0.05, **p<0.01, Mer-KO LPS treated versus untreated; ##p<0.01, ###p<0.001, WT LPS versus WT untreated.
Rothlin et al revealed a SOCS-mediated STAT inhibitory pathway in macrophages that was deficient when all three TAM receptors were absent [8]. We hypothesized that Mer suppression of STAT activation in GECs might similarly be due to SOCs proteins. Consistent with this hypothesis, much greater expression of SOCS-3 was detected in LPS-treated WT than Mer-deficient GECs; even the smallest dose of LPS tested was sufficient to elicit this difference (Fig. 5). SOCS-1 expression was not detected in any samples studied (data not shown). Taken together, the data shown here are consistent with the hypothesis that Mer suppresses LPS-induced STAT1/3 activation, at least in part, by activating SOCS-3.
Figure 5. LPS-induced upregulation of SOCS3 expression in GECs is Mer-dependent.
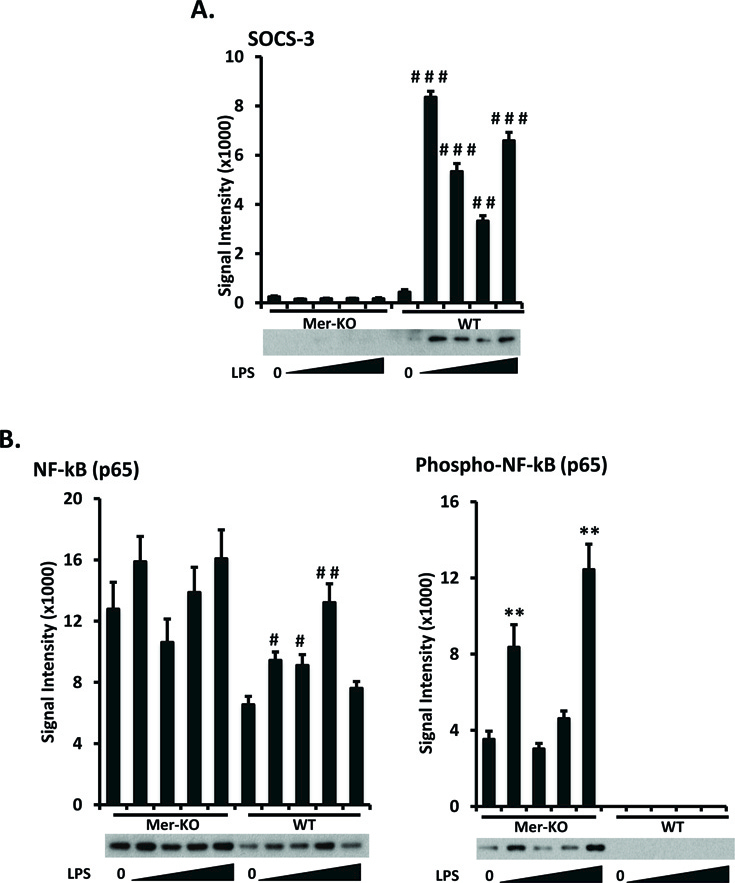
Primary GECs were prepared, cultured, and LPS-treated as in figure 2. A. Expression levels of SOCS3 were analyzed by Western immunoblotting. B. The expression and activation (phosphorylation) levels of NF-κB p65 subunit were analyzed and quantified. Representative Western blot images are shown. Experiments were repeated independently at least three times. Densitometry analyses to quantify the intensity of SOCS3 and NF-κB p65 are shown. Values are the median and interquartile range. **p<0.01, Mer-KO LPS treated versus untreated; #p<0.05, ##p<0.01, ###p<0.001, WT LPS-treated versus untreated, Mann-Whitney U test.
TAM kinases were reported to suppress the effect of LPS by targeting the PI3K/Akt pathway, leading to the inhibition of NF-κB [26, 27]. To investigate whether Mer-mediated inhibitory pathway involves NF-κB activation in the GECs, we measured expression levels of the NF-κB P65 subunit in cell lysates and compared levels of the activated/phosphorylated subunit in WT and Mer-KO GECs. A significantly decreased level of NF-κB P65 activation was revealed in the WT GECs as compared to similarly treated Mer-KO GECs (Fig. 5B). Similar to Bcl-2 and Bcl-6 expression (Fig. 3), baseline level of NF-κB expression and activation in the Mer deficient GECs were also increased (Fig. 5B). Data presented here reflect a central role for Mer in inhibiting kidney inflammation.
Mer decreases secretion of some cytokines and chemokines by LPS-stimulated GECs
Mer deficiency in mice results in significantly enhanced cytokine and chemokine expression in the kidney when experimental nephritis is induced [18]. To investigate whether some of these cytokines/chemokines are directly regulated by Mer-expressing GECs, a Proteome Profiler array was employed that measured the levels of 40 cytokines and chemokines secreted into a GEC culture supernatant. Ten cytokines and chemokines were upregulated by LPS in GECs, and Mer disruption significantly modulated 3 of them (Fig. 6A). MCP-1 is an important regulator of monocyte/macrophages migration and infiltration [28]. Surprisingly, LPS activation significantly decreased GEC MCP-1 secretion (Fig. 6B). However, there was significantly less of a decrease in Mer-deficient than in WT LPS-stimulated GECs. (Fig. 6B). CXCL-1 (also known as KC in mice) plays a pivotal role in recruiting and activating neutrophils at tissue sites [29]. This is relevant because we observed considerable neutrophil infiltration in the kidneys of nephritic Mer-KO mice [18]. Here, we confirmed our previous observation that LPS stimulation significantly enhances CXCL-1 secretion by GECs. The amount of CXCL-1 secreted tended to be slightly higher with LPS-stimulated Mer-deficient than WT GECs (Fig. 6C), but the difference was not statistically significant at the LPS dose tested. M-CSF can stimulate macrophage proliferation and cytokine secretion. Endothelial cells activated by LPS are known to secrete M-CSF [30]. Although LPS-stimulated WT and Mer-KO GECs both secreted considerable M-CSF, over twice as much was secreted by the Mer-deficient cells (Fig. 6D). LPS also induced a marked increase in IL-6 secretion, which was not affected by Mer deficiency at the LPS dose tested (Fig. 6E).
Figure 6. Measurement of cytokine/chemokine expression in primary GECs treated with LPS.
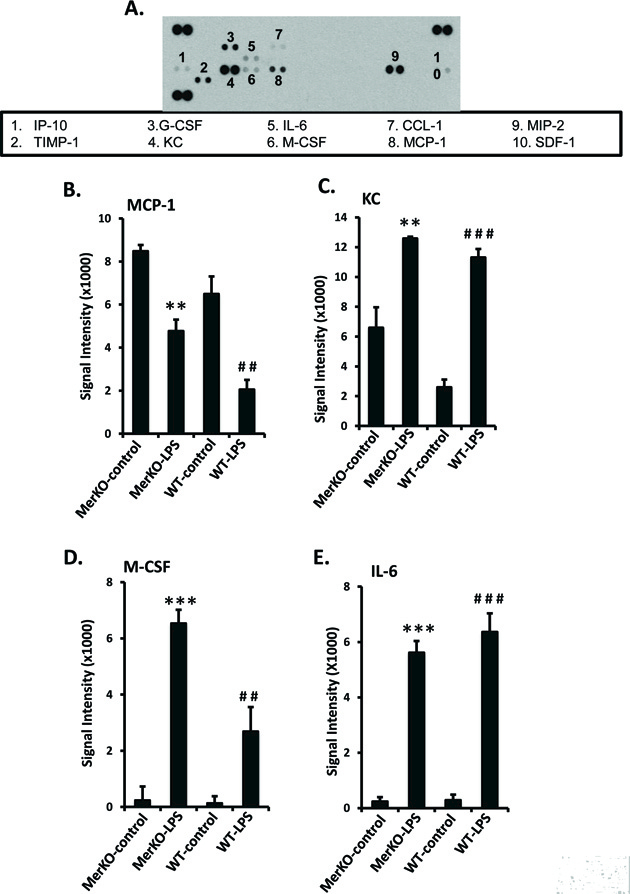
Primary GECs were prepared, cultured, and LPS-treated as in Fig. 2. Culture supernatants were incubated with the R&D Systems Proteome Profiler Array according to the manufacturer’s guidelines. Representative dot plot results are shown in A. Positive and negative controls are as indicated. The density of each dot was quantified with ImageJ software. Experiments were repeated twice using different cultures. Values are the median and interquartile range. **p<0.01, **p<0.001, Mer-KO LPS treated versus untreated; ##p<0.01, ###p<0.001, WT LPS-treated versus untreated, Mann-Whitney U test.
Discussion
Our previous in vivo data provided evidence of Mer expression and activation in the kidney before and after nephrotoxic serum injection, respectively [18, 19]. Moreover, these data suggested that Mer is a feedback inhibitor of inflammation-driven immune response during nephritis [19]. Observations that Mer mRNA and protein expression are highest in the kidney suggested that Mer might contribute importantly to homeostasis and protection from inflammation in this organ [20, 21]. In this study, we identified glomerular endothelial cells as the principal Mer expressing cells in the kidney. Previously, the immuoregulatory role of Mer has been mainly studied in macrophages and dendritic cells [31–35], although Mer has been reported to have a negative regulatory effect on LPS-induced lung inflammation [12]. To our knowledge, ours is the first evidence of intrinsic Mer regulation of inflammation associated with GECs.
TAM-dependent pathways lie at the intersection of the innate and the adaptive immune systems, where they provide inhibitory feedback that is required to dampen inflammation [8]. The loss of function of these three receptors results in a broad-spectrum autoimmune disease with lymphocyte infiltration into all tissues [10]. Although most immunological phenotypes are visible or exacerbated only when all three receptors are simultaneously ablated [6], a unique deficiency in Mer, but neither of the other TAM receptors, is sufficient to induce autoimmune disease [9]. The essential role of TAM kinase activation in dampening inflammation was, in fact, initially discovered in Mer-KO mice. Mer-KO mice are hyper-sensitive to inflammatory stimuli, which cause them to overproduce pro-inflammatory cytokines; this causes these mice to die in response to even low doses of LPS [17]. The constitutively high expression of Mer on glomerular endothelium may be necessary to clear apoptotic debris and maintain immune homeostasis. The large quantity of Mer on GECs should allow Mer to quickly down-regulate GEC inflammation.
Data from the current study demonstrate that Mer inhibits the inflammatory effects of LPS on GECs by targeting multiple downstream molecules. LPS induces several downstream pathways in GECs, including Akt and ERK activation [22]. Mer inhibits both events: dampening Akt phosphorylation, and completely blocking ERK phosphorylation. A second possible pathway involved in Mer-mediated inhibition of LPS signaling is the inhibition of NF-κB and upregulation of SOCS proteins (Fig. 5). STAT3-activation in macrophages is largely negatively regulated by intracellular levels of SOCS3. Our data support this notion, in that an association between SOCS3 upregulation and STAT1/3 downregulation was observed in the WT GECs. Mer-KO mice died from sub-lethal dose of LPS due to cytokine storm (mainly TNF-α), indicating a suppressive role of Mer in mediating inflammatory cytokine production [17]. The increased TNF-α production was associated with increased NF-κB activity [17]. The suppressive role of Mer in LPS-induced inflammatory responses also involves the inhibition of NF-κB (Fig. 5B). NF-κB was found to increase Bcl-2 transcription 10-fold [36]. Decreased Bcl-2 in WT GECs incubated with LPS may be a result of Mer-mediated NF-κB inhibition. Taken together, Mer-mediated inhibition of NF-κB may down-regulate several signaling molecules, including TNF-α, Bcl-2, Bcl-xl, et al. The net effect of Mer-mediated inhibition may depend on the cell type and stimulus involved.
High Mer expression on GECs may be one mechanism that allows these cells to suppress inflammation. This is essential, inasmuch as GECs are key components of the glomerular filtration unit that is critical for maintaining glomerular basement membrane integrity, which, in turn, is required to maintain blood pressure and intravascular volume stability. A deeper understanding of the regulatory pathways in glomerular endothelial cells may facilitate the development of novel therapies to avoid the pathological events associated with endothelial injury during nephritis.
Acknowledgements
This study was supported by the U.S. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, grant K01 DK095067 (to W.H.S.) and the University of Cincinnati, College of Medicine, Dean Funds (W.H.S.). We thank Dr. Philip L. Cohen for critical reading and discussion of the manuscript.
Abbreviations:
- Bcl
B cell lymphoma
- ERK
extracellular signal-regulated kinases
- GEC
glomerular endothelial cell
- GN
glomerular nephritis
- IL-6
interleukin-6
- LPS
lipopolysaccharide
- MCP-1
monocyte chemotactic protein-1
- NTS
nephrotoxic serum
- PI3K
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- SOCS
suppressor of cytokine signaling
- STAT
signal transducer and activator of transcription
- TAM
Tyro-3, Axl and Mer
- TLR
Toll-like receptor
Footnotes
Conflict of interest Disclosure
The authors have no financial conflict of interest.
References
- 1.Chadban SJ and Atkins RC (2005) Glomerulonephritis. Lancet 365, 1797–806. [DOI] [PubMed] [Google Scholar]
- 2.Hricik DE, Chung-Park M, Sedor JR (1998) Glomerulonephritis. N Engl J Med 339, 888–99. [DOI] [PubMed] [Google Scholar]
- 3.Fu J, Lee K, Chuang PY, Liu Z, He JC (2015) Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am J Physiol Renal Physiol 308, F287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weil EJ, Lemley KV, Mason CC, Yee B, Jones LI, Blouch K, Lovato T, Richardson M, Myers BD, Nelson RG (2012) Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int 82, 1010–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mulay SR, Kulkarni OP, Rupanagudi KV, Migliorini A, Darisipudi MN, Vilaysane A, Muruve D, Shi Y, Munro F, Liapis H, Anders HJ (2013) Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1beta secretion. J Clin Invest 123, 236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paolino M and Penninger JM (2016) The Role of TAM Family Receptors in Immune Cell Function: Implications for Cancer Therapy. Cancers (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemke G and Rothlin CV (2008) Immunobiology of the TAM receptors. Nat Rev Immunol 8, 327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G (2007) TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131, 1124–36. [DOI] [PubMed] [Google Scholar]
- 9.Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA (2002) Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med 196, 135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Q, Gore M, Zhang Q, Camenisch T, Boast S, Casagranda F, Lai C, Skinner MK, Klein R, Matsushima GK, Earp HS, Goff SP, Lemke G (1999) Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 398, 723–8. [DOI] [PubMed] [Google Scholar]
- 11.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK (2001) Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–11. [DOI] [PubMed] [Google Scholar]
- 12.Choi JY, Park HJ, Lee YJ, Byun J, Youn YS, Choi JH, Woo SY, Kang JL (2013) Upregulation of Mer receptor tyrosine kinase signaling attenuated lipopolysaccharide-induced lung inflammation. J Pharmacol Exp Ther 344, 447–58. [DOI] [PubMed] [Google Scholar]
- 13.Lee YJ, Lee SH, Youn YS, Choi JY, Song KS, Cho MS, Kang JL (2012) Preventing cleavage of Mer promotes efferocytosis and suppresses acute lung injury in bleomycin treated mice. Toxicol Appl Pharmacol 263, 61–72. [DOI] [PubMed] [Google Scholar]
- 14.Zhang B, Fang L, Wu HM, Ding PS, Xu K, Liu RY (2016) Mer receptor tyrosine kinase negatively regulates lipoteichoic acid-induced inflammatory response via PI3K/Akt and SOCS3. Mol Immunol 76, 98–107. [DOI] [PubMed] [Google Scholar]
- 15.Cummings CT, Deryckere D, Earp HS, Graham DK (2013) Molecular pathways: MERTK signaling in cancer. Clin Cancer Res 19, 5275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guttridge KL, Luft JC, Dawson TL, Kozlowska E, Mahajan NP, Varnum B, Earp HS (2002) Mer receptor tyrosine kinase signaling: prevention of apoptosis and alteration of cytoskeletal architecture without stimulation or proliferation. J Biol Chem 277, 24057–66. [DOI] [PubMed] [Google Scholar]
- 17.Camenisch TD, Koller BH, Earp HS, Matsushima GK (1999) A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol 162, 3498–503. [PubMed] [Google Scholar]
- 18.Shao WH, Zhen Y, Rosenbaum J, Eisenberg RA, McGaha TL, Birkenbach M, Cohen PL (2010) A protective role of Mer receptor tyrosine kinase in nephrotoxic serum-induced nephritis. Clin Immunol 136, 236–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhen Y, Priest SO, Shao WH (2016) Opposing Roles of Tyrosine Kinase Receptors Mer and Axl Determine Clinical Outcomes in Experimental Immune-Mediated Nephritis. J Immunol 197, 2187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graham DK, Bowman GW, Dawson TL, Stanford WL, Earp HS, Snodgrass HR (1995) Cloning and developmental expression analysis of the murine c-mer tyrosine kinase. Oncogene 10, 2349–59. [PubMed] [Google Scholar]
- 21.Graham DK, Dawson TL, Mullaney DL, Snodgrass HR, Earp HS (1994) Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ 5, 647–57. [PubMed] [Google Scholar]
- 22.Dauphinee SM and Karsan A (2006) Lipopolysaccharide signaling in endothelial cells. Lab Invest 86, 9–22. [DOI] [PubMed] [Google Scholar]
- 23.Lu YC, Yeh WC, Ohashi PS (2008) LPS/TLR4 signal transduction pathway. Cytokine 42, 145–51. [DOI] [PubMed] [Google Scholar]
- 24.Guo J and Friedman SL (2010) Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair 3, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bode JG, Ehlting C, Haussinger D (2012) The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell Signal 24, 1185–94. [DOI] [PubMed] [Google Scholar]
- 26.Alciato F, Sainaghi PP, Sola D, Castello L, Avanzi GC (2010) TNF-alpha, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J Leukoc Biol 87, 869–75. [DOI] [PubMed] [Google Scholar]
- 27.Eken C, Martin PJ, Sadallah S, Treves S, Schaller M, Schifferli JA (2010) Ectosomes released by polymorphonuclear neutrophils induce a MerTK-dependent anti-inflammatory pathway in macrophages. J Biol Chem 285, 39914–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deshmane SL, Kremlev S, Amini S, Sawaya BE (2009) Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29, 313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawant KV, Poluri KM, Dutta AK, Sepuru KM, Troshkina A, Garofalo RP, Rajarathnam K (2016) Chemokine CXCL1 mediated neutrophil recruitment: Role of glycosaminoglycan interactions. Sci Rep 6, 33123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clinton SK, Underwood R, Hayes L, Sherman ML, Kufe DW, Libby P (1992) Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am J Pathol 140, 301–16. [PMC free article] [PubMed] [Google Scholar]
- 31.Rahman ZS, Shao WH, Khan TN, Zhen Y, Cohen PL (2010) Impaired apoptotic cell clearance in the germinal center by Mer-deficient tingible body macrophages leads to enhanced antibody-forming cell and germinal center responses. J Immunol 185, 5859–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Behrens EM, Gadue P, Gong SY, Garrett S, Stein PL, Cohen PL (2003) The mer receptor tyrosine kinase: expression and function suggest a role in innate immunity. Eur J Immunol 33, 2160–7. [DOI] [PubMed] [Google Scholar]
- 33.Hilliard BA, Zizzo G, Ulas M, Linan MK, Schreiter J, Cohen PL (2014) Increased expression of Mer tyrosine kinase in circulating dendritic cells and monocytes of lupus patients: correlations with plasma interferon activity and steroid therapy. Arthritis Res Ther 16, R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gohlke PR, Williams JC, Vilen BJ, Dillon SR, Tisch R, Matsushima GK (2009) The receptor tyrosine kinase MerTK regulates dendritic cell production of BAFF. Autoimmunity 42, 183–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yi Z, Li L, Matsushima GK, Earp HS, Wang B, Tisch R (2009) A novel role for c-Src and STAT3 in apoptotic cell-mediated MerTK-dependent immunoregulation of dendritic cells. Blood 114, 3191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Catz SD and Johnson JL (2001) Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene 20, 7342–51. [DOI] [PubMed] [Google Scholar]