

Abstract
Feline immunodeficiency virus (FIV) is a naturally occurring Lentivirus causing acquired immunodeficiency syndrome in felines. It is considered a useful non-primate model to study HIV infection, and to test anti-HIV vaccine. Similarly to HIV, FIV enters cells via a mechanism involving a surface glycoprotein named gp36. C8 is a short synthetic peptide corresponding to the residues 770WEDWVGWI777 of gp36 membrane proximal external region (MPER). It elicits antiviral activity by inhibiting the fusion of the FIV and host cell membrane. C8 is endowed with evident membrane binding property, inducing alteration of the phospholipid bilayer and membrane fusion. The presence and the position of tryptophan residues in C8 are important for antiviral activity: the C8 derivative C6a, obtained by truncating the N-terminal 770WE771 residues, exhibits conserved antiviral activity, while the C8 derivative C6b, derived from the truncation of the C-terminal 776WI777, is nearly inactive. To elucidate the structural factors that induce the different activity profiles of C6a and C6b, in spite of their similarity, we investigated the structural behaviour of the two peptides in membrane mimicking environments. Conformational data on the short peptides C6a and C6b, matched to those of their parent peptide C8, allow describing a pharmacophore model of antiviral fusion inhibitors. This includes the essential structural motifs to design new simplified molecules overcoming the pharmacokinetic and high cost limitations affecting the antiviral entry inhibitors that currently are in therapy.
Introduction
37 million people are living with human immunodeficiency virus (HIV) infection (www.unaids.org). The so-called “highly active antiretroviral therapy” (HAART), [1–3] combinatorial use of 3 or 4 antiretroviral therapeutics (ARTs) determined reduction of the viral load and extension of the patients’ lives. [4, 5] However, for an increasing number of patients these medicines lost their efficacy due to the emergence of resistant HIV variants. Development of alternative drugs exhibiting broad and sustained antiretroviral activity against HIV-1 remains a pursued goal to date. [6]
In view of the development of new anti-HIV therapy targeting the single steps of HIV life cycle, thousands of molecules have been tested. Among these, many have been designed and screened as virus entry inhibitors. [6, 7] Nowadays, enfuvirtide is the only effective entry inhibitor approved for use as anti-HIV in humans. [8–10] It is used in combined therapy with other anti-HIV drugs, but owing to its peptide nature—responsible for poor pharmacokinetic and high economic cost-, it has a restricted clinical application.
A crucial event for the entry of HIV in host cell membranes is the conformational rearrangement of the envelope glycoprotein gp41. During the structural refolding of gp41, more than one conformational events synergically work to realise the fusion of virus and cell membranes: i) the C-terminal heptad repeats (CHR or HR2) come in close proximity with the N-terminal heptad repeats (NHR or HR1); ii) the extreme hydrophobic “membrane proximal external region” (MPER) is exposed to the host cell and interacts with lipid surface, destabilizing the lipid bilayer. [11–14] Many molecules have been screened as potential inhibitors of CHR-NHR interaction in gp41 [15–19]; others as potential inhibitors of the binding between MPER and the host cell membrane. [20]
Among the molecules tested for their ability to prevent the interaction of MPER with the host cell membrane, we have extensively studied C8, an octapeptide corresponding to the sequence 770–777 of MPER in gp36 glycoprotein of feline immunodeficiency virus (FIV). [21]
FIV is the pathogen of the acquired immunodeficiency syndrome (AIDS) in felines. [22–24] It exhibits biological properties very similar to HIV, and as such it is considered a useful non-primate model to study HIV infection, and to test anti-HIV vaccine [25–27] and drugs. Analogously to HIV, FIV [28] enters cells thanks to gp36, the envelope glycoprotein [29–31] showing the same structural and functional properties of gp41. [32–34] C8, including three equally spaced Trp residues in its sequence, elicits antiviral activity preventing the entry of the virus in the host cells. C8 is endowed with strong membrane binding property, and many evidence show that its antiviral action is based on its ability to prevent the interaction of gp36 MPER with the surface of the host cell. [35]
Previously, we extensively investigated the membrane active properties of C8 using different physical-chemical methodologies. [36] C8 was studied by circular dichroism (CD) and nuclear magnetic resonance (NMR) spectroscopy, showing β-turn conformations in micelle solution; the regularity of this structure varies with the charge of the micelle surface and the positioning of the Trp side chains on the lipid surface. [36–39] C8 was analysed by EPR and fluorescence spectroscopy and the results were validated by molecular dynamic calculations: in lipid vesicles of varying complexity and composition, C8 was able to disrupt phospholipid bilayers, reducing membrane thickness and inducing membrane fusion. [36–39]
Structure-activity relationship analysis on shorter synthetic derivatives of C8 revealed the importance of Trp residues for the antiviral activity. [37] Experiments performed to test the inhibition of the replication of primary FIV isolates in lymphoid cells showed that the fragment of C8 (C6a) deriving from the truncation of the C8 N-terminal residues 770WE771 (Fig 1), showed a twofold IC50 activity compared to C8 (C8: IC50 0.06 and 0.05 μg/ml; C6a: IC50 0.15 and 0.06 μg/ml) on the viral strains FIV-M2 and FIV-Pet, respectively. [35] On the contrary, the fragment of C8 (C6b) deriving from the truncation of the C8 C-terminal residues 776WI777 was nearly inactive, with IC50 values for FIV-M2 and FIV-Pet > 50 μg/ml. [35]
Fig 1. Graphic representation of the gp36 MPER domain of FIV coat glycoprotein.

The fragments relative to C8, C6a and C6b peptide sequences are highlighted.
In the present work, we report the conformational analysis of C6a and C6b by CD and NMR spectroscopy in micelle solution composed of dodecylphosphocholine (DPC)/sodium dodecyl sulphate (SDS) 90:10 M/M. On a higher dimension scale, we studied C6a and C6b using confocal microscopy imaging on vesicles characterized by different phospholipid compositions. [40] Our data allow identifying the conformational factors determining the different C6a and C6b membrane binding properties and hence the different antiviral activity profiles.
One major concern relative to the anti-HIV fusion inhibitors is their peptide chemical structure and the high number of residues. [41] A valuable quality of C8 and its derivatives is the small size. The structural features of C6a and C6b derived from our analysis, matched to those of their parent peptide C8, lead to describe a pharmacophore model for anti-HIV fusion inhibitors; this includes the essential structural motifs to design new simplified molecules overcoming the limitations affecting the entry inhibitors that currently are in clinical application.
Materials and methods
Peptide synthesis
The C8, C6a and C6b peptides were synthesized by the Fmoc solid phase synthesis method according to the previously published procedure. [36] For the confocal microscopy analysis, all peptides labelled with nitrobenzoxadiazole (NBD) fluorophore and the L-diamino propionic acid Fmoc-Dap(NBD)-OH) [40] were used in substitution of 770W for C6b and 773W for C6a. Labelled C8 peptide was prepared by synthesizing Ac-770Dap(NBD)EDWVGWI777-NH2, the C6b peptide was prepared by synthesizing Ac-770Dap(NBD)EDWVG775-NH2, and the C6a peptide was prepared by synthesizing Ac-772D-Dap(NBD)-VGWI777-NH2.
Confocal microscope Imaging of liposomes
The multilamellar lipid vesicles (MLVs) of DOPC, DOPG and DOPC/DOPG (90:10, 70:30, 30:70, and 10:90 M/M) were prepared by mixing appropriate amounts of lipids, dissolved in dichloromethane/methanol mixture (25 mM lipid concentration), in a round-bottom test tube. The total weight of the lipid for each sample was 1.0 mg. MLVs were prepared from lipid DOPC and DOPG solutions that were desiccated, dried overnight and hydrated in a 10 mM phosphate buffer at pH 7.4. MLVs were prepared in six different lipid compositions: DOPC only, DOPG only, DOPC/DOPG 90:10 M/M, DOPC/DOPG 70:30 M/M, DOPC/DOPG 30:70 M/M and DOPC/DOPG 10:90 M/M. MLVs, in the presence of NBD-labelled C8, NBD-labelled C6a and NBD-labelled C6b peptides, were prepared by adding during their formation, the labelled peptides at a concentration of 0.5 mM (lipid to peptide 50:1 M/M). Lipid solutions in the presence or absence of labelled peptides were used for microscopy analysis. [42]
A total of 10 μL of each solution, in the presence or absence of NBD-labelled C8, NBD-labelled C6a and NBD-labelled C6b peptides was taken and spotted onto a cover slip. Images were acquired as previously described [43, 44] on a laser scanning confocal microscope (LSM 510; Carl Zeiss MicroImaging) equipped with a plan Apo 63X, NA 1.4 oil immersion objective lens. For each field, both fluorescent and transmitted light images were acquired on separate photomultipliers and were analysed with Zeiss LSM 510 4.0 SP2 software. In samples in which different z-planes were distinguishable, a z-stack acquisition mode was performed to focus a single z-plane, as published previously. [45]
CD analysis
CD experiments were performed at 25°C on an 810-Jasco spectropolarimeter as an average of 4 scans with 10 nm/min scan speed, 4 s response time and 2 nm bandwidth, using a quartz cuvette with a path length of 1 mm, a measurement range from 190 to 260 nm (far UV), at temperature of 25°C. Throughout the measurements, the trace of the high-tension voltage was verified to be less than 700 V, which ensures the reliability of the obtained data.
Far UV CD spectrum of only mixed micelles (DPC/SDS 90:10 M/M (27 mM/3 mM)) in aqueous solution (pH 7.4, 10 mM phosphate buffer containing H2O) was acquired as reference, than 500 μM of C6a and C6b peptides were added to mixed micelles for the conformational analysis. [46] The processed curves of C6a and C6b peptides in mixed micelles were obtained by using Spectra Analysis tool of Jasco software. The CD curves of C6a and C6b peptides in mixed micelles were corrected for the solvent contribution by subtraction of CD reference spectrum and then final CD spectra were obtained after baseline correction and binomial smoothing. Those resulting spectra were used for the estimation of the secondary structure content using the algorithms CONTIN and SELCON from the DICHROWEB website. [47]
NMR analysis
Samples for the NMR experiments were prepared at 1 mM C6a and C6b concentrations, in a mixture of d38-DPC or d25-SDS 90:10 M/M (27 mM/3 mM) in aqueous solution (pH 7.4, 10 mM phosphate buffer containing H2O/D2O). [46] For the spin label experiments, the 5- and 16-doxylstearic acids were solubilized in dimethyl sulfoxide-d6 and then added to the samples.
The NMR spectra were recorded on a Bruker DRX-600 spectrometer. All samples had a recording of the 1D 1H homonuclear spectra in the Fourier mode, with quadrature detection and two-dimensional (2D) TOCSY and NOESY spectra in the phase–sensitive mode using quadrature detection in ω1 by time–proportional phase incrementation of the initial pulse. [48–50] Water signal was suppressed using WATERGATE pulse sequence experiments. [51] Data block sizes contained 2048 addresses in t2 and 512 equidistant t1 values. Prior to Fourier transformation, the time domain data matrices were multiplied by shifted sin2 functions in both dimensions. A mixing time of 70 ms was used for the TOCSY experiments. The NOESY experiments were run at 300 K with mixing times in the range of 100–300 ms. Qualitative and quantitative analyses of TOCSY and NOESY spectra were carried out using SPARKY software. [52]
NMR structure calculations
Peak volumes were translated into upper distance bounds with the CALIBA routine from the CYANA software package. [53] The requisite pseudo-atom corrections were applied for non-stereo specifically assigned protons at prochiral centres and for the methyl group. After discarding redundant and duplicated constraints, the final list of experimental constraints was used to generate a group of 100 structures by the standard CYANA protocol of simulated annealing in the torsion angle space (using 10.000 steps). No dihedral angle or hydrogen bond restraints were applied. The best 20 structures that had low target function values and small residual violations were refined by in vacuo minimization in the AMBER 1991 force field using the SANDER program of the AMBER 5.0 suite. [54, 55] In order to mimic the effect of solvent screening, all net charges were reduced to 20% of their true values. Additionally, a distance-dependent dielectric constant (ε = r) was used. The cut-off for non-bonded interactions was 12 Å. The NMR-derived upper bounds were imposed as semi-parabolic penalty functions, with force constants of 16 Kcal/mol Å2. The function was shifted to be linear when the violation exceeded 0.5 Å. The best 10 structures after minimization had AMBER energies ranging from -441.4 to -391.1 Kcal/mol. The final structures were analysed using AutoDock 4.2 software. [56]
Results
Confocal microscopy imaging
Confocal microscopy imaging of C6a and C6b was carried out in MLVs of different DOPC/DOPG compositions. To evaluate the behaviour of C6a and C6b in MLVs characterized by different membrane charge, we used MLVs composed of zwitterionic DOPC, negatively charged DOPG and DOPC/DOPG varying in composition (90:10; 70:30; 30:70; 10:90 M/M). The peptides were labelled with NBD fluorophore [40] to examine their localization on the vesicles. MLVs were imaged alone and in the presence of the labelled peptides. (Figs 2 and 3) In absence of peptide, MLVs assume spherical shape, with diameter ranging from 5 to 20 μm; addition of C6a (NBD773)W and C6b(NBD770)W induces fluorescence of the lipid membranes, indicating the localization of the peptides on the lipid surface. Figs 2 and 3 report confocal microscopy images of MLVs composed of DOPC/DOPG 90:10 M/M, in presence of NBD labelled C8, C6a and C6b peptides.
Fig 2. Confocal microscopy images of MLVs in presence of NBD labelled C8, C6a and C6b peptides.

MLVs are composed of DOPC/DOPG 90:10 M/M. Images were acquired on a laser scanning confocal microscope (LSM 510; Carl Zeiss MicroImaging) equipped with a plan Apo 63X, NA 1.4 oil immersion objective lens. For each field, both fluorescent and transmitted light images were acquired on separate photomultipliers and were analysed with Zeiss LSM 510 4.0 SP2 software.
Fig 3.

Confocal microscopy images of MLEVs in presence of NBD labelled C8 (left) and C6a (right) peptides. MLEVs vary in DOPC/DOPG composition moving from DOPC/DOPG 100:0 M/M (top) to DOPC/DOPG 0:100 M/M (bottom). Images were acquired on a laser scanning confocal microscope (LSM 510; Carl Zeiss MicroImaging) equipped with a plan Apo 63X, NA 1.4 oil immersion objective lens. For each field, both fluorescent and transmitted light images were acquired on separate photomultipliers and were analysed with Zeiss LSM 510 4.0 SP2 software.
Fig 2 shows that in MLVs composed of DOPC/DOPG 90:10 and including C6b, the diameter ranges between 5 and 10 μM; only sporadically membrane fusion occurs, generating MLVs up to 50 μM diameter. In the presence of the C6a peptide, highly dynamic budding and fusion events are observable: large spherical liposomes characterized by 5–20 μM diameter, often appear because of the impaired budding of the smaller vesicles. Notably, MLVs are interconnected through the formation of straight-chain tubular structures (Fig 3: DOPC, 90:10, 70:30 panels) that often extend throughout the entire field of view and beyond and are typically capped at both ends with large liposomal structures. These “tubulation” events occur in different z-planes, forming a crowded three-dimensional network of tubules and liposomes.
The frequency of tubule formation is high in DOPC and 90:10 DOPC/DOPG samples and decreases with the reduction of DOPC concentration, until nearly disappearing in the 10:90 DOPC/DOPG and fully DOPG samples. A similar pattern of tubulation and budding is shown by MLVs in the presence of C8 peptide (Figs 2 and 3), indicating that the residues 772D-I777, corresponding to the C6a sequence, have the structural requirements necessary for the membrane active property.
CD and NMR conformational analysis
The conformational behaviour of C6a and C6b was studied in mixed DPC/SDS (90:10 M/M) micelles using CD and NMR spectroscopy. [46] Micelle solutions are often used as a biomimetic membrane model to study the structural features of membranotropic molecules. They are made of surfactants, such as the zwitterionic DPC and the negatively charged SDS, at concentrations much higher than their critical micelle concentration (c.m.c.). These surfactants form spherical aggregates where the polar head groups are located on the surface and the hydrophobic tails point to the centre. Micelle solutions are ideal systems for solution CD and NMR spectroscopy, as they tumble sufficiently quickly to result in high-resolution spectral lines. [57, 58]
Fig 4 shows the far UV CD spectra of C6a and C6b recorded in DPC/SDS 90:10 M/M micelle solution. The shapes of the far UV CD spectra are consistent with the prevalence in C6a of regular turn-helix conformation, and non-canonical secondary structures in C6b. Quantitative evaluation of the CD spectra using CONTINN algorithm (DICHROWEB website) [47] confirm these indications.
Fig 4. Processed far UV CD spectra of C8, C6a and C6b peptides in DPC/SDS 90:10 M/M.

The CD spectra were acquired using a JASCO 810 spectropolarimeter at room temperature with a cell path length of 1 mm. The measurement range spans from 190 to 260 nm.
NMR spectra of C6a and C6b in the same mixed DPC/SDS micelle solution (90:10 M/M) were acquired using a Bruker DRX-600 spectrometer. 2D COSY, TOCSY and NOESY data were recorded to allow for chemical shift assignment, according to the conventional procedure of Wuthrich. [59] The proton chemical shifts are reported in the Supporting Information (S1 and S2 Tables). COSY, TOCSY and NOESY spectra were analysed using SPARKY software. [52] The sequential and medium-range connectivities observed in the NOESY spectra (S2 Fig) indicate the presence of folded structures in both the peptides. NOE data were transformed in interprotonic distances for the calculation of C6a and C6b NMR structures using CYANA software. [60] Fig 5 shows the NMR structure bundles of C6a and C6b. 20 structures out of 50 calculated were selected according to the lowest values of the target function. [60] The selected conformers were superimposed at level of the backbone heavy atoms showing 0.32 Å and 0.47 Å RMSD for C6a and C6b respectively. Analysis of the backbone dihedral angles by using PROMOTIF [61] indicates the prevalence in C6a of regular β-turn structures encompassing the residues 773W-W776 and stabilized by H-bonds between the carbonyl of 772D and the HN of 775G and 776W (22 out of 50 calculated structures) (Fig 5). Inspection of the side chains in C6a NMR structures evidences a parallel orientation of the Trp indolyl rings, to expose a large hydrophobic surface.
Fig 5. Low energy NMR models calculated for C8, C6a and C6b peptides in DPC/SDS 90:10 molar ratio.

The calculation was carried out using standard CYANA protocol of simulated annealing in torsion angle space.
Analysis of C6b NMR structure bundle indicates the prevalence of non-canonical secondary structures. The amino acid side chains form a globular amphipathic structure exposing, on one side, the negatively charged side chains of 771E and 772D, and on the other side the hydrophobic indolyl rings of 770W and 773W.
An estimation of the molecular surface, calculated according to the aptitude of the peptides to engage electrostatic/polar interaction with positively/negatively charged surfaces, or hydrophobic interactions with hydrophobic surfaces (Supplementary information, S3 Table) [56] indicates that C6a and C6b expose 286.2 Å2 and 220.5 Å2 hydrophobic surface respectively. A similar calculation for the NMR structure of the parent peptide C8, indicate that the extension of the hydrophobic surface of C8 is 288.3 Å2, very similar to that of C6a. Moreover, the molecular surfaces of C6a and C6b and of their parent peptide C8 shows several spots of electrostatically charged surfaces. According to molecular dynamic calculations performed by us, [38] these charged sites engage electrostatic interactions with the polar heads of the phospholipids. Hence, hydrophobic and electrostatic interactions are synergic to destabilize the phospholipid bilayer and modify membrane size and shape.
Spin-label studies
The NMR analysis in DPC/SDS 90:10 M/M micelle solutions was the starting point to investigate the positioning of the peptides with respect to the surface and interior of the micelle using 5-doxylstearic acid (5-DSA) and 16-doxylstearic acid (16-DSA) as paramagnetic probes. Both of these compounds contain a doxyl head group and a cyclic nitroxide with unpaired electrons that is bound to the aliphatic carbon chain at either position 5 or 16. NMR experiments were recorded in micelle solutions containing 5-DSA or 16-DSA. Paramagnetic probes are able to induce a broadening of the NMR signal and a decrease in the resonance intensities. Generally, the sites of the peptide that are closest to the NO· moiety are affected by the unpaired electrons, with an increase in nuclear relaxation rates and, thus, a decrease in proton signals. If the peptide is close to the surface, a decrease in intensity is observed in the 5-DSA spin-labelled micelles, but if the peptide penetrates the inner core of the micelle, a decrease in intensity is observed in the 16-DSA spin-labelled micelles. [46] 1D and 2D TOCSY spectra of the C6a and C6b peptides were recorded in DPC/SDS 90:10 M/M micelle solutions in the presence and absence of 5-DSA and 16-DSA while keeping all other conditions constant. A comparison of the 1D 1H NMR proton spectra of the C6a and C6b peptides recorded in the presence and absence of 5-DSA show a selective perturbation of Trp CHα-NH and Trp-indolyl proton signals in the presence of 5-DSA. Comparison of TOCSY spectra of C6a and C6b recorded in DPC/SDS 90:10 molar ratio micelle solutions in the presence of 5-DSA shows that the Trp Hε1 signals are decreased in intensities due to the presence of the spin label. This effect is nearly 60% in C6a and 30% in C6b. Backbone NHs of Trp are 30% and 15% decreased in intensities for C6a and C6b respectively. This effect is consistent with the localization of the peptide on the surface of micelles, as observed in the confocal microscopy imaging (Figs 2 and 3).
Discussion
C8 is a tryptophan-rich octa-peptide belonging to the MPER of gp36. [21, 35, 39] It exerts antiviral activity by inhibiting the fusion of the virus with the cell membrane. In membrane models of different complexity and composition, C8 destabilizes phospholipid bilayer and induces membrane fusion. [36–38] C8 includes three equally spaced Trp residues and several evidence suggest that its antiviral molecular mechanism consists in the inhibition of the interaction of gp36 MPER with the surface of the host cell. [35, 36] SAR studies on C8 derivatives indicated that the presence and the position of Trp residues are essential for the antiviral activity: C6a and C6b, resulting from the truncation of the N-terminus 770WE771 and the C-terminus 776WI777 respectively, differ in their activity profiles: C6a inhibits the replication of primary FIV isolates in lymphoid cells, whereas C6b is nearly inactive. C6a and C6b are structurally similar: i) both are fragments of C8; as shown in Fig 1, they are characterized by the same amino acid composition, except for one residue; ii) both include two out of the three Trp residues. To elucidate the structural factors causing the different biological activities of C6a and C6b, we performed CD and NMR conformational analysis in mixed micelle solutions composed of zwitterionic DPC and negatively charged SDS. On a larger scale, we analysed by confocal microscopy the effect of the two peptides on MLVs containing zwitterionic DOPC and negatively charged DOPG phospholipids. Confocal microscopy data confirmed the relationship between antiviral activity and the membrane active properties. The interaction with the vesicle surface of the antiviral active peptides C6a and C8, induce perturbation of the bilayer architecture and consequent modification of vesicle size and shape. On the contrary, DOPC/DOPG vesicles remain unmodified in their size and shape upon the action of C6b. Fig 3 shows a comparison of confocal microscopy images of MLVs with different DOPC/DOPG compositions, including either C6a or C8. Both, C6a and C8 induce membrane tubulation in a manner dependent on the DOPC/DOPG ratio. In MLVs characterized by zwitterionic phospholipid content less than 50%, the effect of C6a is an increase of vesicle size possibly consequent to vesicle fusion. In contrast, in MLVs having a zwitterionic phospholipid content greater than 50% (or in pure DOPC vesicles), C6a induces membrane deformation with the appearance of a complex network of membrane tubes. Interestingly, our data indicate that the effect of the peptide on the membrane is critically dependent on the properties of the phospholipid bilayer: this aspect is important and requires attention when considering the risk for virus resistance.
As previously reported, the conformations of C6a and C6b were studied in mixed zwitterionic DPC/SDS (90:10 M/M) micelles. Consistent with the observation of a high number of NOE effects in 2D NOESY spectra, the two peptides assume non-random coil but well-structured conformation in the micelle solution. This may be explained considering that they are confined to the surface of micelles and have limited conformational freedom. Analysis of the backbone dihedral angles reveals that C6a adopts regular β-turn structures involving the residues 773WVGW776 (S4 Table), while C6b assumes folded, but non-canonical, secondary structures. According to the amino acid sequences, we can infer that in C6a, the 774VG775 residues act as a flexible linker between 773W and 776W, favouring a regular β-turn conformation. In C6b, 771ED772 forms a more rigid linker between 770W and 773W, preventing the formation of regular secondary structures. Analysis of the most representative C6a and C6b NMR structures evidence that C6a is characterised by an elongated molecular shape, with the indolyl rings of 773W and 776W in a nearly parallel orientation (the planes that run through the 773W and 776W are at an angle of angle of 13.64°). These generate a hydrophobic molecular surface (Fig 6), calculated for 220.5 Å2. The negatively charged 772D side chain flanks the 773W indolyl ring directly in this molecular area. Contrarily to C6a, C6b has a globular shape, and the planes that run through the 770W and 773W indolyl rings are at an angle of 62°. The 220.5 Å2 molecular surface generated by these residues is smaller as compared to C6a. The negatively charged side chains of 771E and 772D are on the opposite side with respect to the tryptophan indolyl rings (Fig 6).
Fig 6. Molecular surface calculated as propensities of the different portions of the molecules to interact according to the specific surface properties.

Molecular surfaces of Trp residues are coloured in green-white: they account for the propensity of the peptides to engage hydrophobic interactions. Molecular surfaces of Asp residues are coloured in blue: they account for the propensity of the peptides to engage polar/electrostatic interactions.
Many peptide molecules have been studied as antiviral fusion inhibitors. However, those approved for antiviral therapy have high molecular weight, with complications deriving from the high economic cost and the difficulty of administration. C8 and its derivatives are small peptides. The comparison of the most representative conformations of C8 and C6a (Fig 7) indicates that it is possible to draw a pharmacophore model including the indolyl rings of the two Trp residues (which in C8 belong to 770W and 776W, and in C6a belong to 773W and 776W) and the side chain of 772D. This model may be the starting point to design new small peptide-peptidomimetic molecules endowed with improved pharmacodynamics/ pharmacokinetic properties.
Fig 7. Pharmacophore model of anti-FIV fusion inhibitors.
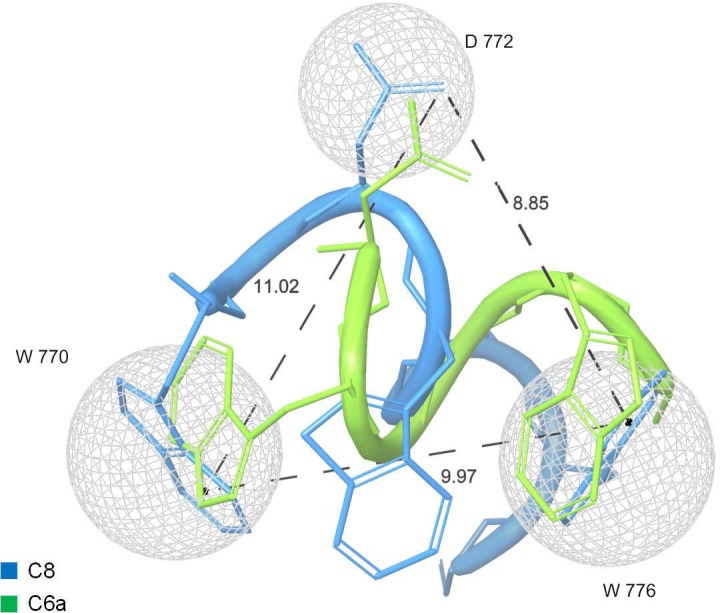
Overlapping of C8 (light blue) and C6a NMR representative models. Molecular surfaces (grey) surround the carboxylic moiety of 772D side chain and indolyl rings of 770W and 776W in C8, and 773W and 776W in C6a.
Using atomistic molecular simulations, [38] we previously demonstrated that the binding of C8- with phospholipid bilayers is mediated by i) hydrophobic interactions involving the Trp indolyl rings and the hydrophobic chains of phospholipids ii) salt bridge interactions established between the negatively charged amino acids (i.e., 771E and 772D) and the phospholipid zwitterionic polar heads. The pharmacophore shown in Fig 7 is consistent with this model of interaction for C6a and C8, but not for C6b; notably, the absence of the 770WE771 N-terminal residues may explain the 2 times less activity of C6a as compared to the C8 for FIV-M2. Accordingly, we can speculate that activity at vesicle surface and at cell membrane rely in the synergic molecular interactions involving Trp indolyl rings and negatively charged aminoacid: these induce destabilization of the bilayer, determining membrane tubes and membrane fusion.
Conclusions
C6a and C6b two derivatives of the C8 anti-FIV peptide, although very similar to their precursor show different activity profile: in particular, C6a preserves anti-viral activity, while C6b is nearly inactive. We analysed C6a and C6b in bio-membrane mimicking environments using confocal microscopy and CD and NMR spectroscopy. Our data provide additional evidence that common antiviral activity profiles correspond to similar membrane binding properties: actually C6a, similarly to C8, has the ability to destabilize membrane vesicles, producing complex network of membrane tubes. Conformational data show that the action of C6a and C8 at membrane level is mediated by interactions involving 772D and two Trp residues, which correspond to 770W and 776W in C8, and 773W and 776W in C6a. These residues define a pharmacophoric model that can be considered the basis to design simplified molecules, overcoming the pharmacokinetic and high cost limitations affecting the entry inhibitors currently used as anti-HIV therapeutics.
Supporting information
1H chemical shift of C6a in DPC/SDS micelle solution 90:10 M/M.
(DOC)
1H chemical shift of C6b in DPC/SDS micelle solution 90:10 M/M.
(DOCX)
Molecular surface values calculated as propensities to engage molecular interaction according to specific surface properties.
(DOCX)
Backbone dihedral angles of C6a and C6b in DPC/SDS 90:10 M/M micelle solution.
(DOCX)
Sequential and medium range connectivities collected in NOESY spectra of C6a and C6b in DPC/SDS 90:10 M/M.
(DOCX)
Low field region of the 600 MHz 1D proton spectra of C6a (a) and C6b (b) peptides in presence of spin labels. Low field region of the 600 MHz 1D proton spectra of C6a (a) and C6b (b) peptides recorded in DPC/SDS 90:10 M/M micelles solutions at 300 K in absence (blue) and in presence of 5-DSA and 16-DSA (red) at a concentration of one spin label per micelle.
(DOCX)
Acknowledgments
This work would not have been possible without the financial support of the Instruct-ERIC R&D Award APPID 300.
Data Availability
All relevant data are reported in the manuscript and in the Supporting Information files.
Funding Statement
The authors acknowledge the support and the use of resources of Instruct-ERIC. This work would not have been possible without the financial support of the Instruct-ERIC R&D Award APPID 300.
References
- 1.Yeni P. Update on HAART in HIV. J Hepatol. 2006;44:S100–S3. 10.1016/j.jhep.2005.11.021 [DOI] [PubMed] [Google Scholar]
- 2.Williams AB. New horizons: antiretroviral therapy in 1997. J Assoc Nurses AIDS Care. 1997;8(4):26–38. 10.1016/S1055-3290(97)80011-8 [DOI] [PubMed] [Google Scholar]
- 3.Aghokeng AF, Ayouba A, Mpoudi-Ngole E, Loul S, Liegeois F, Delaporte E, et al. Extensive survey on the prevalence and genetic diversity of SIVs in primate bushmeat provides insights into risks for potential new cross-species transmissions. Infect Genet Evol. 2010;10(3):386–96. 10.1016/j.meegid.2009.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. 1997;337(11):734–9. 10.1056/NEJM199709113371102 [DOI] [PubMed] [Google Scholar]
- 5.Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387(6629):188–91. 10.1038/387188a0 [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, Curreli F, Waheed AA, Mercredi PY, Mehta M, Bhargava P, et al. Dual-acting stapled peptides target both HIV-1 entry and assembly. Retrovirol. 2013;10(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Ac Sci. 1994;91(21):9770–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Badani H, Garry RF, Wimley WC. Peptide entry inhibitors of enveloped viruses: the importance of interfacial hydrophobicity. Biochim Biophys Acta. 2014;1838(9):2180–97. 10.1016/j.bbamem.2014.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, Bolognesi D. Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat Rev Drug Discov. 2004;3(3):215–25. 10.1038/nrd1331 [DOI] [PubMed] [Google Scholar]
- 10.He Y, Xiao Y, Song H, Liang Q, Ju D, Chen X, et al. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J Biol Chem. 2008;283(17):11126–34. 10.1074/jbc.M800200200 [DOI] [PubMed] [Google Scholar]
- 11.Bar S, Alizon M. Role of the ectodomain of the gp41 transmembrane envelope protein of human immunodeficiency virus type 1 in late steps of the membrane fusion process. J Virol. 2004;78(2):811–20. 10.1128/JVI.78.2.811-820.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doms RW, Moore JP. HIV-1 membrane fusion: targets of opportunity. J Cell Biol. 2000;151(2):F9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Rosa M, Zanfardino A, Notomista E, Wichelhaus TA, Saturnino C, Varcamonti M, Soriente A Novel promising linezolid analogues: rational design, synthesis and biological evaluation. Eur J Med Chem. 2013;69:779–85. 10.1016/j.ejmech.2013.09.035 [DOI] [PubMed] [Google Scholar]
- 14.Zwick MB, Saphire EO, Burton DR. gp41: HIV's shy protein. Nat Med. 2004;10(2):133–4. 10.1038/nm0204-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maselko M, Ward C, Pastey M. A RhoA-derived peptide inhibits human immunodeficiency virus-1 entry in vitro. Curr HIV Res. 2011;9(1):1–5. [DOI] [PubMed] [Google Scholar]
- 16.Thomas CJ, Casquilho-Gray HE, York J, DeCamp DL, Dai D, Petrilli EB, et al. A specific interaction of small molecule entry inhibitors with the envelope glycoprotein complex of the Junin hemorrhagic fever arenavirus. J Biol Chem. 2011;286(8):6192–200. 10.1074/jbc.M110.196428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basu A, Antanasijevic A, Wang M, Li B, Mills DM, Ames JA, et al. New small molecule entry inhibitors targeting hemagglutinin-mediated influenza a virus fusion. J Virol. 2014;88(3):1447–60. 10.1128/JVI.01225-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper DA, Lange JM. Peptide inhibitors of virus-cell fusion: enfuvirtide as a case study in clinical discovery and development. Lancet Infect Dis. 2004;4(7):426–36. 10.1016/S1473-3099(04)01058-8 [DOI] [PubMed] [Google Scholar]
- 19.Hrobowski YM, Garry RF, Michael SF. Peptide inhibitors of dengue virus and West Nile virus infectivity. J Virol 2005;2:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng G, Montero A, Gastaminza P, Whitten-Bauer C, Wieland SF, Isogawa M, et al. A virocidal amphipathic {alpha}-helical peptide that inhibits hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2008;105(8):3088–93. 10.1073/pnas.0712380105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D'Ursi AM, Giannecchini S, Esposito C, Alcaro MC, Sichi O, Armenante MR, et al. Development of antiviral fusion inhibitors: short modified peptides derived from the transmembrane glycoprotein of feline immunodeficiency virus. Chembiochem. 2006;7(5):774–9. 10.1002/cbic.200500390 [DOI] [PubMed] [Google Scholar]
- 22.Lee J, Malmberg JL, Wood BA, Hladky S, Troyer R, Roelke M, et al. Feline Immunodeficiency Virus Cross-Species Transmission: Implications for Emergence of New Lentiviral Infections. J Virol. 2017;91(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bendinelli M, Pistello M, Lombardi S, Poli A, Garzelli C, Matteucci D, et al. Feline immunodeficiency virus: an interesting model for AIDS studies and an important cat pathogen. Clin Microbiol Rev. 1995;8(1):87–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Troyer JL, Pecon-Slattery J, Roelke ME, Johnson W, VandeWoude S, Vazquez-Salat N, et al. Seroprevalence and genomic divergence of circulating strains of feline immunodeficiency virus among Felidae and Hyaenidae species. J Virol. 2005;79(13):8282–94. 10.1128/JVI.79.13.8282-8294.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luttge BG, Freed EO. FIV Gag: virus assembly and host-cell interactions. Vet Immunol Immunopathol. 2010;134(1–2):3–13. 10.1016/j.vetimm.2009.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burkhard MJ, Dean GA. Transmission and immunopathogenesis of FIV in cats as a model for HIV. Curr HIV Res. 2003;1(1):15–29. [DOI] [PubMed] [Google Scholar]
- 27.Elder JH, Lin YC, Fink E, Grant CK. Feline immunodeficiency virus (FIV) as a model for study of lentivirus infections: parallels with HIV. Curr HIV Res. 2010;8(1):73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giannecchini S, Bonci F, Pistello M, Matteucci D, Sichi O, Rovero P, et al. The membrane-proximal tryptophan-rich region in the transmembrane glycoprotein ectodomain of feline immunodeficiency virus is important for cell entry. Virol. 2004;320(1):156–66. [DOI] [PubMed] [Google Scholar]
- 29.Pancino G, Camoin L, Sonigo P. Structural analysis of the principal immunodominant domain of the feline immunodeficiency virus transmembrane glycoprotein. J Virol. 1995;69(4):2110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serres PF. Molecular mimicry between the trimeric ectodomain of the transmembrane protein of immunosuppressive lentiviruses (HIV-SIV-FIV) and interleukin 2. C R Acad Sci III. 2000;323(11):1019–29. [DOI] [PubMed] [Google Scholar]
- 31.Frey SC, Hoover EA, Mullins JI. Feline immunodeficiency virus cell entry. J Virol. 2001;75(11):5433–40. 10.1128/JVI.75.11.5433-5440.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barbato G, Bianchi E, Ingallinella P, Hurni WH, Miller MD, Ciliberto G, et al. Structural analysis of the epitope of the anti-HIV antibody 2F5 sheds light into its mechanism of neutralization and HIV fusion. J Mol Biol. 2003;330(5):1101–15. [DOI] [PubMed] [Google Scholar]
- 33.Suarez T, Nir S, Goni FM, Saez-Cirion A, Nieva JL. The pre-transmembrane region of the human immunodeficiency virus type-1 glycoprotein: a novel fusogenic sequence. FEBS Lett. 2000;477(1–2):145–9. [DOI] [PubMed] [Google Scholar]
- 34.Salzwedel K, West JT, Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol. 1999;73(3):2469–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giannecchini S, Di Fenza A, D'Ursi AM, Matteucci D, Rovero P, Bendinelli M. Antiviral activity and conformational features of an octapeptide derived from the membrane-proximal ectodomain of the feline immunodeficiency virus transmembrane glycoprotein. J Virol. 2003;77(6):3724–33. 10.1128/JVI.77.6.3724-3733.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scrima M, Di Marino S, Grimaldi M, Campana F, Vitiello G, Piotto SP, et al. Structural features of the C8 antiviral peptide in a membrane-mimicking environment. Biochim Biophys Acta. 2014;1838(3):1010–8. 10.1016/j.bbamem.2013.12.010 [DOI] [PubMed] [Google Scholar]
- 37.D'Errico G, Vitiello G, D'Ursi AM, Marsh D. Interaction of short modified peptides deriving from glycoprotein gp36 of feline immunodeficiency virus with phospholipid membranes. Eur Biophys J. 2009;38(7):873–82. Epub 2009/05/06. 10.1007/s00249-009-0454-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merlino A, Vitiello G, Grimaldi M, Sica F, Busi E, Basosi R, et al. Destabilization of lipid membranes by a peptide derived from glycoprotein gp36 of feline immunodeficiency virus: a combined molecular dynamics/experimental study. J Phys Chem B. 2012;116(1):401–12. 10.1021/jp204781a [DOI] [PubMed] [Google Scholar]
- 39.Oliva R, Del Vecchio P, Stellato MI, D'Ursi AM, D'Errico G, Paduano L, et al. A thermodynamic signature of lipid segregation in biomembranes induced by a short peptide derived from glycoprotein gp36 of feline immunodeficiency virus. Biochim Biophys Acta. 2015;1848(2):510–7. 10.1016/j.bbamem.2014.10.017 [DOI] [PubMed] [Google Scholar]
- 40.Dufau I, Mazarguil H. Design of a fluorescent amino acid derivative usable in peptide synthesis. Tetrahedron Letter. 2000;41(32):6063–6. [Google Scholar]
- 41.Yi HA, Fochtman BC, Rizzo RC, Jacobs A. Inhibition of HIV Entry by Targeting the Envelope Transmembrane Subunit gp41. Curr HIV Res. 2016;14(3):283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaiser E, Colescott R, Bossinger C, Cook P. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem. 1970;34(2):595–8. [DOI] [PubMed] [Google Scholar]
- 43.Amodio G, Sasso E, D'Ambrosio C, Scaloni A, Moltedo O, Franceschelli S, et al. Identification of a microRNA (miR-663a) induced by ER stress and its target gene PLOD3 by a combined microRNome and proteome approach. Cell biology and toxicology. 2016;32(4):285–303. 10.1007/s10565-016-9335-z [DOI] [PubMed] [Google Scholar]
- 44.Ranieri R, Ciaglia E, Amodio G, Picardi P, Proto MC, Gazzerro P, et al. N6-isopentenyladenosine dual targeting of AMPK and Rab7 prenylation inhibits melanoma growth through the impairment of autophagic flux. Cell Death Differ. 2018;25(2):353–67. 10.1038/cdd.2017.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fasano D, Parisi S, Pierantoni GM, De Rosa A, Picillo M, Amodio G, et al. Alteration of endosomal trafficking is associated with early-onset parkinsonism caused by SYNJ1 mutations. Cell Death Dis. 2018;9(3):385 10.1038/s41419-018-0410-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zerbe O. BioNMR in drug research methods and principles in medicinal chemistry. 2003. [Google Scholar]
- 47.Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32(Web Server issue):W668–73. 10.1093/nar/gkh371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piantini U, Sorensen O, Ernst RR. Multiple quantum filters for elucidating NMR coupling networks. J Am Chem Soc. 1982;104(24):6800–1. [Google Scholar]
- 49.Bax A, Davis DG. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J Magn Reson 1985;65(2):355–60. [Google Scholar]
- 50.Jeener J, Meier B, Bachmann P, Ernst R. Investigation of exchange processes by two‐dimensional NMR spectroscopy. J Chem Physics. 1979;71(11):4546–53. [Google Scholar]
- 51.Piotto M, Saudek V, Sklenář V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biom NMR. 1992;2(6):661–5. [DOI] [PubMed] [Google Scholar]
- 52.Goddard T, Kneller D. SPARKY 3. University of California, San Francisco: 2004. [Google Scholar]
- 53.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program D1. J Mol Biol. 1997;273(1):283–98. 10.1006/jmbi.1997.1284 [DOI] [PubMed] [Google Scholar]
- 54.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, DeBolt S, et al. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Com Physics Comm. 1995;91(1–3):1–41. [Google Scholar]
- 55.Case DA, Pearlman DA, Caldwell JW, Cheatham III TE, Wang J, Ross WS, et al. AMBER 7. University of California, San Francisco: 2002. [Google Scholar]
- 56.Rizvi SM, Shakil S, Haneef M. A simple click by click protocol to perform docking: AutoDock 4.2 made easy for non-bioinformaticians. EXCLI J. 2013;12:831–57. [PMC free article] [PubMed] [Google Scholar]
- 57.Mannhold R, Kubinyi H, Folkers G, Zerbe O. BioNMR in drug research: John Wiley & Sons; 2006. [Google Scholar]
- 58.Grimaldi M, De Rosa M, Di Marino S, Scrima M, Posteraro B, Sanguinetti M, Soriente A, D'Ursi AM. Synthesis of new antifungal peptides selected against Cryptococcus Neoformans. Biorg Med Chem. 2010;18(22):7985–90. [DOI] [PubMed] [Google Scholar]
- 59.Wüthrich K. NMR with Proteins and Nucleic Acids. Europhys News. 1986;17(1):11–3. [Google Scholar]
- 60.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–78. 10.1385/1-59259-809-9:353 [DOI] [PubMed] [Google Scholar]
- 61.Hutchinson EG, Thornton JM. PROMOTIF—a program to identify and analyze structural motifs in proteins. Protein Sci. 1996;5(2):212–20. 10.1002/pro.5560050204 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H chemical shift of C6a in DPC/SDS micelle solution 90:10 M/M.
(DOC)
1H chemical shift of C6b in DPC/SDS micelle solution 90:10 M/M.
(DOCX)
Molecular surface values calculated as propensities to engage molecular interaction according to specific surface properties.
(DOCX)
Backbone dihedral angles of C6a and C6b in DPC/SDS 90:10 M/M micelle solution.
(DOCX)
Sequential and medium range connectivities collected in NOESY spectra of C6a and C6b in DPC/SDS 90:10 M/M.
(DOCX)
Low field region of the 600 MHz 1D proton spectra of C6a (a) and C6b (b) peptides in presence of spin labels. Low field region of the 600 MHz 1D proton spectra of C6a (a) and C6b (b) peptides recorded in DPC/SDS 90:10 M/M micelles solutions at 300 K in absence (blue) and in presence of 5-DSA and 16-DSA (red) at a concentration of one spin label per micelle.
(DOCX)
Data Availability Statement
All relevant data are reported in the manuscript and in the Supporting Information files.