

SUMMARY
Resilience and vulnerability to neuropsychiatric disorders are linked to molecular changes underlying excitability that are still poorly understood. Here, we identify glycogen-synthase kinase 3β (GSK3β) and voltage-gated Na+ channel Nav1.6 as regulators of neuroplasticity induced by environmentally enriched (EC) or isolated (IC) conditions—models for resilience and vulnerability. Transcriptomic studies in the nucleus accumbens from EC and IC rats predicted low levels of GSK3β and SCN8A mRNA as a protective phenotype associated with reduced excitability in medium spiny neurons (MSNs). In vivo genetic manipulations demonstrate that GSK3β and Nav1.6 are molecular determinants of MSN excitability and that silencing of GSK3β prevents maladaptive plasticity of IC MSNs. In vitro studies reveal direct interaction of GSK3β with Nav1.6 and phosphorylation at Nav1.6T1936 by GSK3β. A GSK3β-Nav1.6T1936 competing peptide reduces MSNs excitability in IC, but not EC rats. These results identify GSK3β regulation of Nav1.6 as a biosignature of MSNs maladaptive plasticity.
In Brief
Scala et al. show how vulnerability to reward-related behaviors associates with maladaptive plasticity of medium spiny neurons through phosphorylation of the voltage-gated Na+ channel Nav1.6 by the enzyme GSK3β.
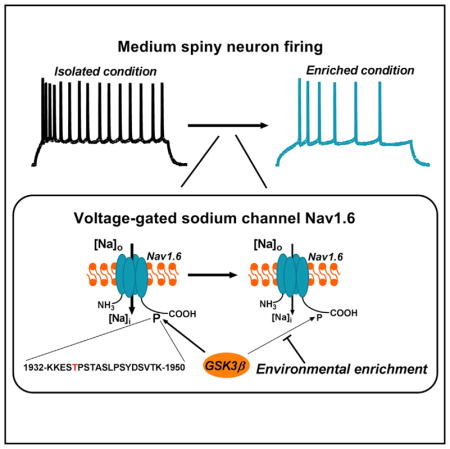
INTRODUCTION
The ability of a neuron to fire action potentials is an intrinsic property of the cell that depends on the configuration of ion channels. Neurons adapt their firing by activating or inhibiting specific signaling mechanisms in response to the environment, setting cellular landscapes that can ultimately protect against or predispose to a disease state (Beck and Yaari, 2008; Camp, 2012). Medium spiny neurons (MSNs) in the nucleus accumbens (NAc) are a highly vulnerable population of cells whose number, morphology, and pattern of firing have been associated with depression-like behaviors, addiction, and neurodegeneration (Bessa et al., 2013; Francis et al., 2015; Kourrich et al., 2015; Roselli and Caroni, 2015; Wallace et al., 2009). However, how maladaptive changes of neuronal firing in these cells are mechanistically linked to modifications of ion channels is poorly understood.
We are investigating differential rearing that promotes either resilience (enriched condition) or vulnerability (isolated condition) to reward-related neuropsychiatric disorders as a way to identify mechanisms underlying ion channel plasticity of MSNs and provide targets for preventive or disease modifying therapies. Environmental enrichment is a non-drug, non-genetic, and non-surgical manipulation that produces resilience to addiction-related and depression-like behaviors in rodents (Green et al., 2002, 2010; Lehmann and Herkenham, 2011; Russo et al., 2012). Compared to rats reared in an isolated condition (IC), rats reared during a critical developmental period (P; 21–50) in an enriched condition (EC), with constant access to novelty, social contact, and exercise, show a protective behavioral phenotype for addiction and depression largely encoded by coordinated changes in gene expression of CREB signaling, ΔFosB, and the retinoic acid pathway (Green et al., 2002, 2003, 2010; Lichti et al., 2014; Zhang et al., 2014, 2016a, 2016b) in the NAc shell.
Recent proteomic and transcriptomic studies (Fan et al., 2013a, 2013b; Lichti et al., 2014; Zhang et al., 2016b) have pointed to other targets previously associated with synaptic wealth and neuroplasticity, such as Wnt/β-catenin (Ataman et al., 2008; Chen et al., 2006), brain-derived neurotrophic factor (BDNF) (Graham et al., 2007; Namekata et al., 2012), and D1, D2, and D3 dopamine receptors (Dunleavy et al., 2013; Lebel et al., 2009; Salles et al., 2013; Urs et al., 2012), as targets of behavioral resilience to addiction and depression-like disorders, indicating that the rearing environment might directly affect electrical properties of neurons. The objective of the present work is to identify molecular mechanisms underlying adaptation of neuronal firing in response to resilience and vulnerable states of MSNs in the NAc. Here, we investigated the role of GSK3β, a center stage kinase in the biology and pharmacology of mood disorders (Jope and Roh, 2006; Li and Jope, 2010), in regulating intrinsic firing of MSNs in the NAc and report a molecular mechanism responsive to the EC/IC paradigm that depends upon GSK3β and the voltage-gated Na+ channel Nav1.6 in MSNs. Premise for this mechanism is supported by in vitro evidence of protein:protein interaction, phosphorylation and functional regulation of Nav1.6 by GSK3β. In native conditions, a peptide that uncouples GSK3β from the Nav1.6 target and prevents Nav1.6 T1936 phosphorylation restores maladaptive firing of MSNs in IC rats (i.e., vulnerable condition) while sparing effects of firing in EC rats (i.e., protected condition). These results might provide insights into the mechanisms of cell vulnerability in the context of reward-related neuropsychiatric disorders.
RESULTS
The Effect of Environmental Enrichment and Social Isolation on NAc Transcriptome and MSNs Neuronal Excitability
To uncover molecular mechanisms controlling intrinsic excitability in the reward circuit, we interrogated a previous large-scale differential transcriptomic dataset derived from the NAc of EC or IC rats (Zhang et al., 2016b). An IPA (ingenuity pathway analysis) bioinformatic analysis revealed a significant decrease of the canonical PI3K/Akt/GSK3β signaling pathway in EC compared to IC rats (Figure 1A, -log (p value) = 4.01). Furthermore, SCN8A mRNA, which codes for the Nav1.6 α subunit, was found to be decreased in the aforementioned pathway (Figure 1A). The degree of mRNA regulation is shown in the inset of Figure 1A. A GSEA (gene set enrichment analysis) also identified regulation of the Reactome_PI3K_AKT_Activation pathway in EC versus IC rats (Figure 1B, normalized enrichment score [NES] = −1.59, p = 0.038). Significant changes in GSK3β, but not SCN8A, mRNA level in EC compared to IC rats were also found with RT-PCR (Figure S1). Because GSK3β regulates neuronal excitability (Hsu et al., 2015) and has direct effects on Nav channels (James et al., 2015), we postulated that GSK3β and Nav1.6 might be part of a pathway that controls plasticity of MSNs in response to vulnerability and resilience. To test this, we characterized intrinsic firing and persistent sodium current (INaP, a functional signature of NAc MSNs that controls intrinsic excitability) in MSNs from IC and EC rats. Whole-cell patch-clamp recordings in acute NAc slices revealed that MSNs from EC rats exhibited a marked decrease in neuronal excitability (Figures 1D and 1E) when compared to MSNs from IC rats (Figures 1C and 1E). Input-output curves showed a dramatic reduction in intrinsic firing across all stimulating current steps, resulting in 17.6 ± 1.8 evoked action potentials (APs; at 180 pA current step) in IC (n = 20) versus 9.7 ± 1.2 evoked APs (n = 27) in EC MSNs (p < 0.005 with Student’s t test; Figure 1E). Voltage-clamp recordings showed a decrease in the amplitude of ramp-induced INaP in EC (Figure 1G) compared to that of IC (Figure 1F) rats. Normalized INaP for EC MSNs was −2.7 ± 0.4 pA/pF, n = 17 versus −4.7 ± 0.9 pA/pF, n = 11 in IC MSNs (p < 0.05, Mann-Whitney test, Figure 1H). Thus, isolation and enrichment differ with respect to intrinsic firing and INaP of MSNs.
Figure 1. Unbiased Transcriptomic Screening Identifies GSK3β and SCN8A (Nav1.6) as EC-Sensitive Protecting Genes in the NAc.

(A) Custom IPA pathway depicting EC/IC-regulated transcripts; RNA-seq analysis (top right inset) of GSK3β and SCN8A mRNA in EC versus IC conditions (Zhang et al., 2016b).
(B) GSEA enrichment plot of PI3K/Akt/GSK3 Reactome pathway and corresponding heatmap in EC versus IC conditions (Zhang et al., 2016b).
(C–E) Representative traces of APs in MSNs from IC rats (C), EC rats (D), and input-output curves (E).
(F–H) Representative traces of MSN INaP from IC rats (F), EC rats (G), and bar graph (H). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.005 with Student’s t test. #p < 0.05 with Mann-Whitney test.
MSNs Intrinsic Firing and Persistent Na+ Current Are Regulated by GSK3 Level and Activity
To investigate a possible mechanistic link between the GSK3β signaling pathway and Nav1.6, we utilized a recently designed and validated AAV-shGSK3β-GFP vector (Crofton et al., 2017) and a previously validated AAV-shControl-GFP vector that does not target any known rat transcript (Benzon et al., 2014; Crofton et al., 2017; Hommel et al., 2003; Zhang et al., 2016b) in order to knock down GSK3β in the NAc of rats in standard pair-housing conditions. After stereotaxic injection of the AAV-shGSK3β-GFP or AAV-shControl-GFP vectors, intrinsic firing and INaP in MSNs were studied between the two groups using whole-cell patch-clamp recordings. We found a significant decrease in the number of evoked APs in AAV-shGSK3β-GFP-positive NAc MSNs compared to AAV-shControl (Figures 2A–2C). The effect was consistent across almost the entire input-output curve (Figure 2C) with an average number of APs of 19.2 ± 1.6 in AAV-shControl (n = 24) compared to 10.8 ± 1.2 APs (n = 31) in AAV-shGSK3β-GFP positive NAc MSNs at a representative 180 pA current step (p < 0.01 with Student’s t test, Figure 2C). This reduction in intrinsic firing was accompanied by a significant decrease in INaP amplitude in AAV-shGSK3β MSNs (Figure 2E) compared to AAV-shControl MSNs (Figure 2D). The normalized INaP in AAV-shControl MSNs was −4.4 ± 0.6 pA/pF (n = 9) versus −2.3 ± 0.2 pA/pF (n = 8) in AAV-shGSK3β MSNs (p < 0.05, Student’s t test; Figure 2F). We then postulated that if silencing of GSK3β reduced MSN activity, then increased expression of GSK3 might lead to opposing phenotypes. To test this hypothesis, we conducted parallel current and voltage clamp studies in the GSK3α21A/21A/β/9A/9A knockin mouse (GSK3-KI), in which GSK3 lacks inhibitory phosphorylation by Akt, resulting in a constitutively high level of GSK3 enzyme activity. This animal model has been used to recapitulate behavioral traits of mood disorders including depression (McManus et al., 2005; Polter et al., 2010). In MSNs of GSK3-KI mice, both firing and INaP were significantly increased compared to wild-type control animals (Figures 2G–2L). The number of APs at current step of 180 pA in wild-type MSNs was 21.6 ± 1.2 (n = 9) versus 31.8 ± 2.1 APs (n = 8) in GSK3-KI MSNs (p < 0.005; Student’s t test, Figure 2I); normalized INaP in wild-type MSNs was −2.5 ± 0.5 pA/pF (n = 6) versus −5.4 ± 0.6 pA/pF (n = 8) in GSK3-KI MSNs (p < 0.01; Student’s t test; Figure 2L). Thus, changes in the levels of GSK3 activity in MSNs leads to coupled, bidirectional modulation of intrinsic firing and INaP. We then posited whether in vivo silencing of GSK3β would be sufficient to prevent maladaptive firing and increase in INaP of MSNs from IC rats. To test this hypothesis, AAV-shGSK3β-GFP or AAV-shControl-GFP vectors were stereotaxically injected in rats at the beginning of the IC protocol. We found a significant decrease in the number of evoked APs in MSNs of IC rats injected with AAV-shGSK3β compared to AAV-shControl (Figures 3A–3C). The effect was consistent and significant across the input-output curve starting from 100 pA current step (Figure 3C) with an average number of APs of 24.4 ± 1.6 in IC AAV-shControl MSNs (n = 22) compared to 16.1 ± 2.2 APs (n = 18) in IC AAV-shGSK3β-GFP positive MSNs at a representative 180 pA current step (p < 0.01 with Student’s t test, Figure 3C). Along with reduction in intrinsic firing, we also found a significant decrease in INaP amplitude in IC AAV-shGSK3β MSNs (Figure 3E) compared to IC AAV-shControl MSNs (Figure 3D). The normalized INaP in IC AAV-shControl MSNs was −2.5 ± 0.1 pA/pF (n = 4) versus −1.3 ± 0.2 pA/pF (n = 5) in IC AAV-shGSK3β MSNs (p < 0.01, Student’s t test; Figure 3F). We concluded that GSK3β is a key determinant of plasticity of intrinsic firing in MSNs of the NAc.
Figure 2. Intrinsic Firing and Na+-Persistent Current of MSNs Are Bi-directionally Controlled by the GSK3 Pathway.

(A–C) Representative traces of APs in NAc MSNs from shControl rats (A), shGSK3β rats (B), and input-output curves (C).
(D–F) Representative traces of MSN INaP from shControl rats (D), shGSK3β rats (E), and bar graph (F).
(G–I) Representative traces of APs in NAc MSNs from wild-type mice (G), GSK3 knockin mice (H), and input-output curves (I).
(J–L) Representative traces of MSN INaP from wild-type mice (J), GSK3 KI mice (K), and bar graph (L). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.005 with Student’s t test.
Figure 3. In Vivo Genetic Silencing of GSK3β Prevents Maladaptive Plasticity of MSNs.

(A–C) Representative traces of APs MSNs from shControl rats (A), shGSK3β-expressing IC rats (B), and corresponding input-output curves (C).
(D–F) Representative traces of MSNs INaP from shControl rats (D), shGSK3β IC rats (E), and bar graph (F). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01 with Student’s t test.
Nav1.6 Is a Molecular Determinant of MSNs Intrinsic Firing and INaP
If GSK3 and Nav1.6 are part of a converging molecular mechanism, then silencing SCN8A in MSNs should mimic the effects of AAV-shGSK3β. Confocal imaging confirmed expression of Nav1.6 in the NAc and its localization at the axon initial segment of neurons that visually corresponded to MSNs (Figures 4A and 4B) (Ali et al., 2018). Nav1.6 co-localized with Ankirin G (Ank)—a marker of the axon initial segment with a high degree of green/red channel correlation (Manders’ overlap coefficient = 0.8030 ± 0.0466, n = 20 axonal initial segments, Figure 4C). Nav1.6 silencing in NAc was achieved using a newly designed and validated AAV-shSCN8A-GFP vector. As hypothesized, a significant decrease in intrinsic firing and INaP amplitude was found in MSNs expressing AAV-shSCN8A compared to AAV-shControl (Figures 4D–4I). At a representative 180 pA current step, the number of APs in AAV-shControl expressing MSNs was 19.2 ± 1.6 (n = 24) versus 10.9 ± 2.4 APs (n = 19) in AAV-shSCN8A-GFP (p < 0.01; Student’s t test; Figure 4F), while normalized INaP was −4.4 ± 0.6 pA/pF (n = 9) in AAV-shControl versus −1.9 ± 0.1 pA/pF (n = 7) in AAV-shSCN8A expressing MSNs (p < 0.05; Student’s t test; Figure 4I).
Figure 4. Nav1.6 Is a Molecular Determinant of Intrinsic Firing in MSNs.

(A) Confocal imaging of Nav1.6 (green), NeuN (blue), and ankyrin-G (red) at the axonal initial segment of neurons in the NAc.
(B) Zoom inset of (A), arrows indicate starting and end points of axon initial segment (AIS). Scale bar, 1 μm.
(C) Profile of Nav1.6 (green) channels and Ank (red) immunofluorescence intensity line scans along the AIS region in MSN.
(D–F) Representative traces of APs in MSNs from shControl (D), shSCN8A (E), and corresponding input-output curves (F).
(G–I) Representative traces of MSNs from shControl (G) and shSCN8A (F), respectively, and bar graphs (I). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.005; Student’s t test.
We showed that INaP current is significantly reduced upon silencing of GSK3β, but these changes may be occurring indirectly as a result of long-term homeostatic remodeling of the NAc circuitry (AAV-shGSK3β and AAV-shControl were injected 2.5–3 weeks prior to recordings to allow for maximal transduction and knock down in neurons) rather than a direct functional modulation of Nav1.6 by GSK3β. To address this, we tested whether a brief (1–2 hr) pharmacological treatment of wild-type NAc slices with specific inhibitors of the Akt-GSK3 signaling pathway, such as Akt inhibitor, triciribine (30 μM) or GSK3 inhibitor, CHIR99021 (2 μM), could mimic the effect of GSK3-KI or in vivo GSK3β genetic silencing in MSNs. We found that the triciribine-treated group showed a significant increase in MSNs firing compared to DMSO control group (Figures S2A and S2B) with 16.2 ± 0.6 (n = 23) APs in control versus 21.5 ± 0.9 APs (n = 12) in triciribine-treated cells (p < 0.005; ANOVA followed by Dunnett’s multiple comparisons test at injected current step 125 pA; Figure S2D). Accordingly, normalized INaP for DMSO-treated MSNs was −2.9 ± 0.3 pA/pF (n = 13) versus −4.3 ± 0.7 pA/pF (n = 9) for triciribine-treated MSNs (p < 0.05; ANOVA followed by Dunnett’s multiple comparisons test; Figures S2E, S2F, and S2H). As expected, a marked decrease in evoked neuronal firing was found in MSNs in the CHIR99021-treated group compared to DMSO (Figures S2A and S2C) with 16.2 ± 0.6 (n = 23) APs in control versus 12.2 ± 1.0 APs (n = 17) in CHIR99021-treated MSNs (p < 0.01 with ANOVA followed by Dunnett’s multiple comparisons test at injected current step 125 pA; Figure S2D). Similarly, normalized INaP for DMSO-treated MSNs was −2.9 ± 0.3 pA/pF (n = 13) versus −2.0 ± 0.3 pA/pF (n = 10) for CHIR99021-treated MSNs (p < 0.05; Kruskal-Wallis with uncorrected Dunn test; Figures S2E, S2G, and S2H).
In Vitro Studies of Functional Interaction between Nav1.6 and GSK3
Combined, the aforementioned results provide strong evidence for GSK3β and Nav1.6 as essential determinants of intrinsic firing and neuronal excitability in MSNs but do not provide a mechanistic model for how the two proteins might functionally interact. In western blot analyses, we found that the total level of GSK3β and Nav1.6 in the NAc were not significantly different in EC compared to IC rats (Figure S3), suggesting that functional changes in the activity of the two proteins might contribute to the observed phenotypes. To test this hypothesis, we isolated Nav1.6-encoded transient currents from HEK293 cells stably expressing the Nav1.6 channel α subunit using whole-cell patch clamp techniques. We found that cells treated for 1–2 hr with either GSK3 inhibitor XIII (30 μM) or CHIR-99021 (20 μM) exhibited a significantly reduced peak transient current (INa+) density compared to DMSO controls (Figures 5A–5D). At −10 mV, DMSO control cells exhibited a peak INa+ density of −72.6 ± 6.5 pA/pF (n = 18) that was significantly reduced (p < 0.01; one-way ANOVA post hoc Bonferroni test) to −44.9 ± 5.5 pA/pF (n = 12) and −34.7 ± 4.8 pA/pF (n = 12) with GSK3 inhibitor XIII or CHIR99021 treatment, respectively (Figure 5E). Treatments had no effect on V½ of INa+ activation but significantly shifted V½ of steady-state inactivation leftward, indicating effects of the inhibitors on Nav1.6 channel availability (Table S1). To further validate our in vitro studies, HEK293-Nav1.6 cells were transfected with either scrambled small interfering RNA (siRNA) or GSK3-siRNA (Figures 5F–5H). This treatment confirmed a reduced peak INa+ density and leftward shift in V½ of steady-state inactivation in the treated group compared to scrambled siRNA control (Figure 5I; Table S1). We then posited that overexpression of GSK3β in these cells could exert an effect on Nav1.6 function leading to opposite phenotypes to the ones observed upon silencing of the kinase. Transient overexpression of a GSK3β-IRES-GFP construct (James et al., 2015) increased peak INa+ density compared to cells transfected with IRES-GFP control (Figures S4A–S4D). At −10 mV, IRES-GFP cells exhibited a peak INa+ density of −40.6 ± 8.3 pA/pF (n = 15) that was significantly increased in GSK3β-IRES-GFP cells to −97.1 ± 11.4 pA/pF (n = 13, p < 0.01; Student’s t test; Figure S4D). Overexpression of GSK3β-IRES-GFP also led to a significant shift in the V½ of INa+ of activation leftward and changes in the slope of steady-state inactivation that could be attributed to increased channel activity and availability (Table S1).
Figure 5. In Vitro Studies of Functional Interaction between Nav1.6-Encoded Currents and GSK3.

(A–C) Representative traces of transient INa recorded from Nav1.6-HEK293 cells treated with DMSO (A), CHIR 99021 (B), and GSK3 inhibitor XIII (C).
(D and E) Nav1.6 current-voltage relationship of DMSO or CHIR 99021 or GSK3 inhibitor XIII treatment (D), and peak current density at −10 mV voltage step (E).
(F and G) Representative traces of transient INa recorded from Nav1.6-HEK293 cells treated with siScramble (F) and siGSK3 (G).
(H and I) Nav1.6 current-voltage relationship of siScramble versus siGSK3 (H), and peak current density at −10 mV voltage step (I).
(J) Representative SPR sensorgram of GSK3β binding to Nav1.6 C-tail.
(K) SPR fitting curve of GSK3β binding with Nav1.6 C-tail.
(L) Higher energy collisional dissociation (HCD) fragmentation spectrum of the phosphopeptide EStPSTASLPSYDSVTK, encompassing residues 1934–1950 of the Nav1.6 C terminus. The presence of non-phosphorylated b2 (theoretical m/z of 217.08, observed m/z of 217.08) and y14 (theoretical m/z of 1,452.72, observed m/z of 1,452.72) ions along with phosphorylated b3 (theoretical m/z of 398.10, observed m/z of 398.10) confirms T1936 as the site of phosphorylation. The parent ion was fragmented with a co-eluting peptide within the ±2 Da window (inset) resulting in the mixed spectrum shown here. t, phosphothreonine; *, fragment with loss of phosphoric acid). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01; Student’s t test.
The intracellular C-terminal tail of Nav channels is rich in predicted and validated phosphorylation sites and is a well-known region for protein:protein interactions (Berendt et al., 2010; Onwuli and Beltran-Alvarez, 2016). Thus, we posited that the Nav1.6 C-terminal tail might include a substrate of GSK3β and that the two molecules might be part of a protein complex. Direct binding of these two proteins as well as phosphorylation of the Nav1.6 C-terminal tail by GSK3 could mediate the functional interaction between GSK3β and the Nav1.6 channel. The premise for direct phosphorylation was based on our previous work showing that the amino acid residue T1966 of the similar Nav1.2 C-tail is a GSK3 target (James et al., 2015) and that the same putative GSK3 phosphorylation site residue is conserved in the Nav1.6 C-tail sequence at T1936. To assess direct binding, we used surface plasmon resonance (SPR) of purified proteins and found that GSK3β binds to the C-terminal region of Nav1.6 at a nanomolar range (Figure 5J) with an estimated binding affinity (KD) of 165.4 nM (Figure 5K). To verify phosphorylation, a 19-mer Nav1.6 C-tail peptide fragment 1932-KKESTPSTASLPSYDSVTK-1950 surrounding T1936 was used for in vitro phosphorylation studies and post hoc LC-MS/MS validation. Acquired MS2 spectra unambiguously demonstrated phosphorylation of the trypsinized fragment 1934-ESTPSTASLPSYDSVTK-1950 at T1936 (Figure 5L), confirming the C-tail of Nav1.6 as a GSK3 phosphorylation site.
Nav1.6-Based Peptide Modulates Neuronal Excitability in IC and GSK3 KI MSNs with No Effect in EC MSNs
We next sought to determine whether the T1936 phosphorylation at Nav1.6 C-tail by GSK3β is the mechanism by which MSNs differentially responded to the IC and EC paradigms. To address this, NAc slices from EC or IC rats were treated for 1–2 hr with 10 μM 19-mer wild-type (WT-pep) or T1938A mutant (Mut-pep) Nav1.6 C-tail peptides conjugated with rhodamine at their C termini. We found that the WT-pep (Figure 6B), but not its Mut-pep (Figure 6A), reduces neuronal excitability and persistent INaP in IC MSNs. Specifically, the number of evoked action potentials at 180 pA current step in IC MSNs treated with WT-pep was 13.3 ± 1.9, n = 11 versus 19.5 ± 1.3 APs, n = 12 in IC MSNs treated with Mut-pep (p < 0.05 with Student’s t test, Figure 6C). Similarly, INaP amplitude was also significantly reduced in IC MSNs treated with WT-pep (Figure 6E) compared to IC MSNs treated with Mut-pep (Figure 6D). Normalized INaP in IC MSNs treated with WT-pep was −1.6 ± 0.2 pA/pF, n = 8 versus −3.6 ± 0.8 pA/pF, n = 7, in IC MSNs treated with Mut-pep (p < 0.05 with Student’s t test, Figure 6F). As additional validation of the peptides’ efficacy both WT-pep and Mut-pep were compared to a scrambled peptide (Scrmb-pep), and MSNs excitability and INaP were measured. Experiments with Scrmbpep further validated the capability of WT-pep to effectively reduce both MSN firing and INaP, and validated the ineffectiveness of the Mut-pep in modulating neuronal excitability (Figures S5A–S5H).
Figure 6. Nav1.6-Based Peptide Restores Maladaptive Plasticity in IC Rats and GSK3 Knockin Mice.

(A–C) Representative traces of APs in NAc MSN from IC rats treated with Mut-pep (A), WT-pep (B), and input-output curves (C).
(D–F) Representative traces of INaP in NAc MSNs from IC rats treated with Mut-pep (D), WT-pep (E), and bar graph (F).
(G–I) Representative traces of APs in NAc MSN from GSK3-KI with Mut-pep (G), WT-pep (H), and input-output curves (I).
(J–L) Representative traces of INaP in NAc MSN from GSK3-KI treated with Mut-pep (J) and WT-pep (K), respectively, and bar graph (L).
(M–O) Representative traces of APs in NAc MSN from EC rats treated with Mut-pep (M) and WT-pep (N), and input-output curves (O).
(P–R) Representative traces of INaP in NAc MSN from EC rats treated with Mut-pep (P), WT-pep (Q), and bar graph (R). Data are represented as mean ± SEM.
*p < 0.05, **p < 0.01, ***p < 0.005 with Student’s t test or one-way ANOVA with Bonferroni or Dunnett’s post hoc test.
Consistently, we found a significant decrease in firing of GSK3-KI mice NAc MSN treated with WT-pep (Figure 6H) compared to Mut-pep (Figure 6G). For instance, the number of APs at a current step of 180 pA in GSK3-KI MSNs treated with WT-pep was 18.7 ± 2, n = 7 versus 27.5 ± 1.6 APs, n = 6 in GSK3-KI MSNs treated with Mut pep (p < 0.01 with Student’s t test, Figure 6I). Furthermore, we found a significant decrease in normalized INaP in GSK3-KI MSNs treated with WT-pep (Figure 6K) compared to GSK3-KI MSNs treated with Mut-pep (Figure 6J). Normalized INaP for GSK3-KI MSNs treated with WT-pep was −2.4 ± 0.3 pA/pF, n = 6 versus −3.9 ± 0.5 pA/pF, n = 4 for GSK3-KI MSNs treated with Mut-pep (p < 0.05 with Student’s t test, Figure 6L). We next tested whether the same treatment with the C-tail Nav1.6 19-mer mimetic peptide would be effective in modifying adaptive firing observed in EC MSNs (Figures 1D and 1E). Notably, we found that neither Mut-pep (Figure 6M) nor WT-pep (Figure 6N) were able to modify neuronal excitability and persistent INaP in EC MSNs. Specifically, the number of evoked action potentials at 180 pA current step in EC MSNs treated with WT-pep was 7.3 ± 1.2, n = 10 versus 5.4 ± 1.4 APs, n = 11 in EC MSNs treated with Mut-pep (p = 0.3 with Student’s t test, Figure 6O). Similarly, INaP amplitude was also not significantly affected in EC MSNs treated with WT-pep (Figure 6Q) compared to EC MSNs treated with Mut-pep (Figure 6P). Normalized INaP in EC MSNs treated with WT-pep was −2.3 ± 0.2 pA/pF, n = 8 versus −2.5 ± 0.6 pA/pF, n = 7, in EC MSNs treated with Mut-pep (p = 0.72; Student’s t test, Figure 6R).
DISCUSSION
Previous studies have established the validity of the EC/IC paradigm as a model of resilience or vulnerability toward depression-and addiction-related behavior (Green et al., 2002, 2010; Zhang et al., 2014). Here, we used the EC/IC paradigm to investigate mechanisms underlying neuroadaptive changes at the cellular level, focusing specifically on molecular mechanisms that could account for adaptive and maladaptive plasticity of intrinsic firing of MSNs in the NAc. Transcriptomic analysis of NAc in EC and IC rats revealed that resilient rats (ECs) had lower levels of mRNA coding for GSK3β and Nav1.6 channel compared to the vulnerable rats (ICs). Using a combination of in vitro and ex vivo studies and in vivo genetic silencing we show a form of neuroadaptation of intrinsic firing in NAc MSNs that develops in response to the EC/IC manipulation and can be prevented by GSK3β silencing. Under these behavioral paradigms, we observed a significant decrease in NAc MSNs intrinsic excitability in EC compared to IC animals that we linked to GSK3β-dependent functional modulation of firing through the Nav1.6 channel. This is supported by our mechanistic studies that uncovered a direct interaction between GSK3β and the Nav1.6 C-tail, GSK3β-dependent phosphorylation of Nav1.6 at T1936, and GSK3β-dependent modulation of Nav1.6-mediated currents and channel availability.
Phosphorylation has been implicated as a key regulator of Nav channels activity (Berendt et al., 2010; Scheuer, 2011). For example, cyclic AMP (cAMP)-dependent kinase (PKA), protein kinase C (PKC) and casein kinase 2 (CK2) all modulate Nav channel gating, availability, and trafficking through direct interaction or via binding to specific signaling complexes (Bréchet et al., 2008; Hien et al., 2014; Liu et al., 2010; Scheuer and Catterall, 2006; Tan et al., 2014; Wu et al., 2012). Specifically in striatal MSNs, phosphorylation via cAMP-PKA regulates Nav currents with opposite outcomes in D1-versus D2-type MSNs (Hu et al., 2005; Nishi et al., 1997; Schiffmann et al., 1995, 1998). The vast majority of these functionally relevant phosphorylation sites are present within the intracellular loops of Nav channels (Baek et al., 2011; Cantrell and Catterall, 2001; Cantrell et al., 2002). Here, we discovered a GSK3β phosphorylation site of Nav1.6 at the C-tail highlighting the importance of this intracellular domain in regulating channel function. Furthermore, we present biophysical evidence that GSK3β directly binds to Nav1.6 C-tail that is a new finding in light of no previously described direct interactions between GSK3β and CNS ion channels.
We hypothesize that GSK3β is a part of a signaling complex critical for Nav1.6 channel internalization and function. GSK3β binds to Nav1.6 C-tail and phosphorylates it at the T1936 residue—a site upstream of the PPxY recognition motif that allows Nav1.6 to be internalized through the NEDD4-2 ubiquitin-system (Gasser et al., 2010). It is possible that the reduction in Na+ current amplitudes we observed following silencing or pharmacological inhibition of GSK3 in Nav1.6-HEK293 cells and MSNs results from a cross-talk between GSK3β phosphorylation and NEDD4-dependent ubiquitination. In addition to Na+ current amplitudes, we show that inhibition of GSK3 (by pharmacological or siRNA means) impacts channel availability shifting the V1/2 of steady-state inactivation to more hyperpolarized values. This phenotype might also result from signaling cross-talk between GSK3β and other interactors at the channel C-tail such as calmodulin or intracellular fibroblast growth factor 14 (FGF14), which are both constituents of the Nav channel interactome in the brain (Wildburger et al., 2015), bind to the Nav1.6 C-tail (Reddy Chichili et al., 2013; Shavkunov et al., 2013), and regulate steady-state inactivation of the channel (Herzog et al., 2003; Laezza et al., 2007, 2009; Shavkunov et al., 2013; Yan et al., 2017). In the case of FGF14, cross-talk is supported by recent data demonstrating that GSK3β phosphorylates FGF14 at S226 (Hsu et al., 2017), corroborating the idea that GSK3β might be part of a multilayer signaling complex that controls Nav1.6 channel function directly and through protein:protein interactions.
We have previously reported that GSK3β phosphorylates Nav1.2 at T1966, which corresponds to the T1936 residue of Nav1.6 (James et al., 2015). While GSK3β inhibition enhances Nav1.2 current density and channel availability (James et al., 2015), it has the opposite effects on Nav1.6 currents (Figure 5). Previous studies have shown that Nav1.6 and Nav1.2 channels are differentially distributed in the brain and functionally specialized at the subcellular level (Chen et al., 2008). Nav1.2 is enriched at the proximal region of the AIS and throughout dendrites contributing to action potential backpropagation (Hu et al., 2009) that is critical for spike-time-dependent plasticity, while Nav1.6 is expressed more distally at the AIS, mediating forward propagation of action potentials and repetitive firing (Osorio et al., 2010; Royeck et al., 2008). Therefore, it is conceivable that GSK3β could promote sustained forward firing through Nav1.6 stimulation, while changing the rules of induction of spike-time-dependent plasticity triggered by Nav1.2-mediated back-propagating action potentials. This bi-directional and isoform-specific modulation of Na+ currents could be part of a global regulation of neuronal homeostasis that builds on known roles of GSK3 in maintaining neuronal polarity and modulating synaptic transmission and turnover of synaptic spines (Hur and Zhou, 2010; Kim et al., 2011; Ochs et al., 2015).
In this study, we demonstrate that Nav1.6 and GSK3β are determinants of neuronal excitability in MSNs. Nav channels were among the first discovered downstream targets of different G-protein coupled receptors involved in the regulation of MSN excitability including dopaminergic D1- and D2-receptors (Carrillo-Reid et al., 2009; D’Ascenzo et al., 2009; Hu et al., 2005; Schiffmann et al., 1995; Surmeier and Kitai, 1993). These receptors modulate fast transient and persistent Na+ currents that control neuronal excitability by altering action potential threshold, repetitive firing, bistable properties, and synchronous network activity of striatal MSNs (Carrillo-Reid et al., 2009; D’Ascenzo et al., 2009; Schiffmann et al., 1995). It is conceivable that functional modulation of Nav1.6 by GSK3β would directly impact these MSNs properties exerting powerful control over the entire NAc circuit.
Our studies show that in vivo genetic silencing of either Nav1.6 or GSK3β leads to suppression of INaP, a signature of the Nav1.6 channel, and intrinsic excitability in MSNs. These phenotypes were consistent with short exposure of MSNs to GSK3 inhibitors, suggesting that the effects induced by inhibition of the kinase are short-term and mediated by direct regulation at the protein level (as opposed to long-term indirect homeostatic changes). Opposite phenotypes (increased INaP and intrinsic firing) were observed in MSNs treated with the Akt inhibitor triciribine or MSNs from the GSK3 KI animal model that constitutively expresses an active form of GSK3. These bi-directional changes seen in AAV-sh-GSK3β and in the knock in animal model as well as by means of pharmacological inhibition of Akt-GSK3 signaling pathway are evidence of pathway specificity conserved across different models. In addition, recent studies show that GSK3β silencing suppresses intrinsic firing of tonically active neurons (TAN) in the NAc, presumably driven by loss of IHCN currents, and with no phenotypes attributable to Nav channel deficits (Crofton et al., 2017). Taken together, these findings support the notion that despite being ubiquitously expressed, GSK3β exerts a direct, cell-type-specific effect on neuronal firing (Scala et al., 2015) that could potentially open new avenues for drug discovery efforts targeting this enzyme.
Understanding the molecular mechanisms of MSNs resilience and vulnerability is critical for early disease intervention and prevention. Intrinsic excitability is an early marker of vulnerability in response to alcohol and drug abuse (Marty and Spigelman, 2012; Mu et al., 2010), chronic stress (Francis et al., 2015), and prolonged social isolation (Green et al., 2010; Wallace et al., 2009). Previous studies identified signaling pathways that are linked to MSNs resilience and vulnerability to social defeat stress (Christoffel et al., 2011; Francis et al., 2017; Vialou et al., 2010; Wilkinson et al., 2011). Yet, the understanding of how changes in gene expression translate into functional outcomes of MSNs is still limited, especially for both social isolation and environmental enrichment paradigms. In this study, we show that the GSK3-Nav1.6 complex contributes to MSN excitability under normal conditions (in pair-housed wild-type rodents) and to mal-adaptive firing that develops in IC conditions and can be prevented by in vivo GSK3β silencing. A dominant-negative Nav1.6-T1936 peptide restores aberrant activity (INaP and firing) in MSNs from IC and GSK3 KI animals, but had no effects in EC conditions, suggesting that high level of phosphorylation at T1936 is a bio-signature of MSNs vulnerability. Overall, limiting GSK3 phosphorylation of T1936 might represent a strategy to prevent maladaptive firing and drive resilience of MSNs at early disease stages associated with the dopamine reward pathway (Beaulieu, 2012; Del’Guidice and Beaulieu, 2010; Freyberg et al., 2010; Golpich et al., 2015; Li and Gao, 2011).
EXPERIMENTAL PROCEDURES
Detailed experimental procedures for molecular biology, cell culture, in vivo and in vitro protein expression knock down, protein biochemistry and biophysics, peptide synthesis, and mass spectrometry are provided in the Supplemental Experimental Procedures, as well as expanded methods for electrophysiology.
Animals
Rats
Male Sprague-Dawley rats (Harlan Laboratories, Houston), 21 days of age, were divided into two conditions (isolated condition and enriched condition), n = 12. For the IC group, the rats were separated one rat per cage in standard polycarbonate cages with no access to social contact or novelty, whereas EC rats were housed together with novel toys changed every day. In addition, some experiments were done in pair-housed rats. The pair-housing condition was chosen as an intermediate condition (Crofton et al., 2015), thus, regulation could be seen in either direction, and to increase relevance beyond the differential rearing literature. Food and water were freely available for rats and all rats were maintained in a controlled environment (temperature, 22°C; relative humidity, 50%; and 12 hr light/dark cycles) for 30 days prior to experiments.
Mice
A colony of GSK3α21A/21A/β/9A/9A (provided by Dr. Dario Alessi, College of Life Sciences, University of Dundee, UK) was maintained at the University of Texas Medical Branch vivarium; 1- to 3-month-old GSK3α21A/21A/β/9A/9A and age-matched C57/BL6J control male mice were used in this study. Most of the experiments involving animals were performed at the University of Texas Medical Branch, except for GSK3 inhibitor experiment in rat brain slices, which was conducted at the Catholic University in Rome. The University of Texas Medical Branch operates in compliance with the United States Department of Agriculture Animal Welfare Act, the NIH Guide for the Care and Use of Laboratory Animals, and American Association for Laboratory Animal Science, Institutional Animal Care and Use Committee-approved protocols. The Ethics Committee of the Catholic University complied with Italian and USA Ministry of Health guidelines and national laws, and European Union guidelines on animal research. All surgical procedures and experiments conformed to the NIH Guide for the Care and Use of Laboratory Animals and approved by The University of Texas Medical Branch Institutional Animal Care and Use Committee.
Electrophysiology
Whole Cell Patch Clamp in Slices
Brain slices were transferred to a submerged recording chamber and continuously perfused with regular artificial cerebrospinal fluid (aCSF) bubbled with 95% O2 and 5% CO2 (pH 7.4). The flow rate was kept at 1.5 mL/min, and bath temperature was maintained at 30°C–32°C by an inline solution heater and temperature controller (TC-344B, Warner Instruments, Hamden, CT, USA). Whole-cell patch-clamp recordings were performed using Axopatch 200B and Multiclamp 700B amplifiers. Somatic recording from visually identified MSNs were performed with pipettes (resistance of 3–5 MΩ) filled with internal solution containing (in mM): 145 K-gluconate, 2 MgCl2, 0.1 EGTA, 2 Na2ATP, and 10 HEPES (pH 7.2 with KOH; 290 mOsm). Access resistance (Ra) was monitored throughout the recording and was typically <25 MΩ. Data acquisition and stimulation were performed with a Digidata 1322A Series interface and pClamp 9 software (Molecular Device). Data were filtered at 2 kHz, digitized at 20 kHz, and were analyzed offline with pClamp 10 software. To measure MSN intrinsic firing 20 μM of NBQX, 100 μM of DL-AP5, and 20 μM of bicuculline were added to regular aCSF in order to prevent glutamatergic and GABAergic synaptic transmissions, respectively.
Whole-Cell Patch Clamp in Nav1.6-HEK293 Cells
Recordings were performed at room temperature (20°C–22°C) using an Axo-patch 200A or Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). Borosilicate glass pipettes with resistance of 3.5–5 MΩ were made using a Narishige PC-10 vertical Micropipette Puller (Narishige International, East Meadow, NY). The recording solutions were as follows: extracellular (mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 10 glucose, pH 7.3; and intracellular: 130 CH3O3SCs, 1 EGTA, 10 NaCl, 10 HEPES, pH 7.3. Membrane capacitance and series resistance were estimated by the dial settings on the amplifier. Capacitive transients and series resistances were compensated electronically by 70%–80% and cells exhibiting a series resistance of 25 MΩ or higher were excluded from the analysis. Data were acquired at 20 kHz and filtered at 5 kHz prior to digitization and storage. All experimental parameters were controlled using Clampex 7 or 9 software (Molecular Devices) and interfaced to the electrophysiological equipment using a Digidata 1200 or 1322A analog-digital interface (Molecular Devices).
Quantification and Statistical Analysis
Statistics were calculated in Prism 6 (GraphPad Software, San Diego, CA, USA). For comparison of two groups, significance was tested with unpaired, two-sided Student’s t tests. For multiple comparisons, two-sided one-way ANOVA with Bonferroni or Dunnett’s post hoc tests were used. All data before comparisons were tested for normal distribution. Non-parametric Mann-Whitney test was used if data did not pass normality testing. Data are presented as mean ± SEM. The level of significance is listed in the figure legends for each experimental group. Secondary analysis of transcriptomic data was performed using the Ingenuity Pathway Analysis Canonical Pathway Analysis (build current for 7-29-2016) (Zhang et al., 2016b). Additionally, gene set enrichment analysis (GSEA) was performed on transcriptomic expression data from drug-naive EC and IC rats using the C2 curated gene sets version 4 (Broad Institute, MA, USA) (Zhang et al., 2016b).
Supplementary Material
Highlights.
Low GSK3β and Nav1.6 mRNA levels were observed under resilient (EC) conditions
GSK3β and Nav1.6 are molecular determinants of MSNs intrinsic excitability
GSK3β phosphorylates Nav1.6 C-tail at T1936 and modulates channel activity
There is a direct interaction between GSK3β and the Nav1.6 channel C-tail
Acknowledgments
This work was supported by NIH grants R01 MH095995 (F.L.), R01MH111107 (F.L.), R01 DA029091 (T.A.G.), T32 DA007287 (E.J.C.); a Jeane B. Kempner Post-doctoral Fellowship Award (O.F.); NIH grants T32ES007254 (T.J.), R01DK106229 (J.D.H.), UL1TR001439 (J.D.H.), KL2TR001441 (J.D.H.), and T32DA07287 (A.E.S.); and UTMB Bench Tutorials Program: Scientific Research and Design (NIH/National Institute of Environmental Health Sciences/Department of Health and Human Services grant P30ES006676/ES) (H.E.).
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the data reported in this paper is GEO: GSE88736 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE88736; https://www.mendeley.com/sign-in/?routeTo=https%3A%2F%2Fapi.mendeley.com%2Foauth%2Fauthorize%3Fredirect_uri%3Dhttps%253A%252F%252Fdata.mendeley.com%252Fauth%252Fcallback%26scope%3Dall%26state%3D642335%26response_type%3Dcode%26client_id%3D1025).
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, five figures, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.062.
DECLARATION OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Conceptualization, F.S., M.N.N., E.J.C., Y.Z., T.A.G., and F.L.; Methodology, F.S., M.N.N., E.J.C., A.K.S., Y.Z., O.F., B.C.C., N.C.W., T.F.J., M.A.A., T.K.A., H.E., C.G., J.M.K., A.E.S., J.D.H., C.F.L., J.S.R., M.D., T.A.G., and F.L.; Writing –Original Draft, M.N.N., F.S., and F.L.; Writing – Review & Editing, F.S., M.N.N., E.J.C., O.F., T.A.G., and F.L.; Funding Acquisition, T.G.A. and F.L; Resources, C.G., M.D., T.G.A., and F.L.; Supervision, M.D., T.A.G., and F.L.
References
- Ali SR, Liu Z, Nenov MN, Folorunso O, Singh AK, Scala F, Chen H, James TF, Alshammari M, Panova-Elektronova NI, et al. Functional modulation of voltage-gated sodium channels by a FGF14-based peptidomimetic. ACS Chem Neurosci. 2018 doi: 10.1021/acschemneuro.7b00399. Published online February 6, 2018. [DOI] [PMC free article] [PubMed]
- Ataman B, Ashley J, Gorczyca M, Ramachandran P, Fouquet W, Sigrist SJ, Budnik V. Rapid activity-dependent modifications in synaptic structure and function require bidirectional Wnt signaling. Neuron. 2008;57:705–718. doi: 10.1016/j.neuron.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek JH, Cerda O, Trimmer JS. Massspectrometry-based phosphoproteomics reveals multisite phosphorylation on mammalian brain voltage-gated sodium and potassium channels. Semin Cell Dev Biol. 2011;22:153–159. doi: 10.1016/j.semcdb.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM. A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J Psychiatry Neurosci. 2012;37:7–16. doi: 10.1503/jpn.110011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Yaari Y. Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci. 2008;9:357–369. doi: 10.1038/nrn2371. [DOI] [PubMed] [Google Scholar]
- Benzon CR, Johnson SB, McCue DL, Li D, Green TA, Hommel JD. Neuromedin U receptor 2 knockdown in the paraventricular nucleus modifies behavioral responses to obesogenic high-fat food and leads to increased body weight. Neuroscience. 2014;258:270–279. doi: 10.1016/j.neuroscience.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendt FJ, Park KS, Trimmer JS. Multisite phosphorylation of voltage-gated sodium channel alpha subunits from rat brain. J Proteome Res. 2010;9:1976–1984. doi: 10.1021/pr901171q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessa JM, Morais M, Marques F, Pinto L, Palha JA, Almeida OF, Sousa N. Stress-induced anhedonia is associated with hypertrophy of medium spiny neurons of the nucleus accumbens. Transl Psychiatry. 2013;3:e266. doi: 10.1038/tp.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bréchet A, Fache MP, Brachet A, Ferracci G, Baude A, Irondelle M, Pereira S, Leterrier C, Dargent B. Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J Cell Biol. 2008;183:1101–1114. doi: 10.1083/jcb.200805169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp AJ. Intrinsic neuronal excitability: a role in homeostasis and disease. Front Neurol. 2012;3:50. doi: 10.3389/fneur.2012.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell AR, Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Tibbs VC, Yu FH, Murphy BJ, Sharp EM, Qu Y, Catterall WA, Scheuer T. Molecular mechanism of convergent regulation of brain Na(+) channels by protein kinase C and protein kinase A anchored to AKAP-15. Mol Cell Neurosci. 2002;21:63–80. doi: 10.1006/mcne.2002.1162. [DOI] [PubMed] [Google Scholar]
- Carrillo-Reid L, Tecuapetla F, Vautrelle N, Hernández A, Vergara R, Galarraga E, Bargas J. Muscarinic enhancement of persistent sodium current synchronizes striatal medium spiny neurons. J Neurophysiol. 2009;102:682–690. doi: 10.1152/jn.00134.2009. [DOI] [PubMed] [Google Scholar]
- Chen J, Park CS, Tang SJ. Activity-dependent synaptic Wnt release regulates hippocampal long term potentiation. J Biol Chem. 2006;281:11910–11916. doi: 10.1074/jbc.M511920200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yu FH, Sharp EM, Beacham D, Scheuer T, Catterall WA. Functional properties and differential neuromodulation of Na(v)1.6 channels. Mol Cell Neurosci. 2008;38:607–615. doi: 10.1016/j.mcn.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffel DJ, Golden SA, Dumitriu D, Robison AJ, Janssen WG, Ahn HF, Krishnan V, Reyes CM, Han MH, Ables JL, et al. IκB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci. 2011;31:314–321. doi: 10.1523/JNEUROSCI.4763-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofton EJ, Zhang Y, Green TA. Inoculation stress hypothesis of environmental enrichment. Neurosci Biobehav Rev. 2015;49:19–31. doi: 10.1016/j.neubiorev.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofton EJ, Nenov MN, Zhang Y, Scala F, Page SA, McCue DL, Li D, Hommel JD, Laezza F, Green TA. Glycogen synthase kinase 3 beta alters anxiety-, depression-, and addiction-related behaviors and neuronal activity in the nucleus accumbens shell. Neuropharmacology. 2017;117:49–60. doi: 10.1016/j.neuropharm.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ascenzo M, Podda MV, Fellin T, Azzena GB, Haydon P, Grassi C. Activation of mGluR5 induces spike afterdepolarization and enhanced excitability in medium spiny neurons of the nucleus accumbens by modulating persistent Na+ currents. J Physiol. 2009;587:3233–3250. doi: 10.1113/jphysiol.2009.172593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del’Guidice T, Beaulieu JM. Psychotropic drugs and the involvement of the Akt/GSK3 signalling pathway in mental illnesses. Med Sci (Paris) 2010;26:647–651. doi: 10.1051/medsci/2010266-7647. [DOI] [PubMed] [Google Scholar]
- Dunleavy M, Provenzano G, Henshall DC, Bozzi Y. Kainic acid-induced seizures modulate Akt (SER473) phosphorylation in the hippocampus of dopamine D2 receptor knockout mice. J Mol Neurosci. 2013;49:202–210. doi: 10.1007/s12031-012-9927-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Li D, Lichti CF, Green TA. Dynamic proteomics of nucleus accumbens in response to acute psychological stress in environmentally enriched and isolated rats. PLoS ONE. 2013a;8:e73689. doi: 10.1371/journal.pone.0073689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Li D, Zhang Y, Green TA. Differential phosphoproteome regulation of nucleus accumbens in environmentally enriched and isolated rats in response to acute stress. PLoS ONE. 2013b;8:e79893. doi: 10.1371/journal.pone.0073689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis TC, Chandra R, Friend DM, Finkel E, Dayrit G, Miranda J, Brooks JM, Iñiguez SD, O’Donnell P, Kravitz A, Lobo MK. Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol Psychiatry. 2015;77:212–222. doi: 10.1016/j.biopsych.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis TC, Chandra R, Gaynor A, Konkalmatt P, Metzbower SR, Evans B, Engeln M, Blanpied TA, Lobo MK. Molecular basis of dendritic atrophy and activity in stress susceptibility. Mol Psychiatry. 2017;22:1512–1519. doi: 10.1038/mp.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyberg Z, Ferrando SJ, Javitch JA. Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am J Psychiatry. 2010;167:388–396. doi: 10.1176/appi.ajp.2009.08121873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser A, Cheng X, Gilmore ES, Tyrrell L, Waxman SG, Dib-Hajj SD. Two Nedd4-binding motifs underlie modulation of sodium channel Nav1.6 by p38 MAPK. J Biol Chem. 2010;285:26149–26161. doi: 10.1074/jbc.M109.098681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golpich M, Amini E, Hemmati F, Ibrahim NM, Rahmani B, Mohamed Z, Raymond AA, Dargahi L, Ghasemi R, Ahmadiani A. Glycogen synthase kinase-3 beta (GSK-3β) signaling: implications for Parkinson’s disease. Pharmacol Res. 2015;97:16–26. doi: 10.1016/j.phrs.2015.03.010. [DOI] [PubMed] [Google Scholar]
- Graham DL, Edwards S, Bachtell RK, DiLeone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10:1029–1037. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- Green TA, Gehrke BJ, Bardo MT. Environmental enrichment decreases intravenous amphetamine self-administration in rats: dose-response functions for fixed- and progressive-ratio schedules. Psychopharmacology (Berl) 2002;162:373–378. doi: 10.1007/s00213-002-1134-y. [DOI] [PubMed] [Google Scholar]
- Green TA, Cain ME, Thompson M, Bardo MT. Environmental enrichment decreases nicotine-induced hyperactivity in rats. Psychopharmacology (Berl) 2003;170:235–241. doi: 10.1007/s00213-003-1538-3. [DOI] [PubMed] [Google Scholar]
- Green TA, Alibhai IN, Roybal CN, Winstanley CA, Theobald DE, Birnbaum SG, Graham AR, Unterberg S, Graham DL, Vialou V, et al. Environmental enrichment produces a behavioral phenotype mediated by low cyclic adenosine monophosphate response element binding (CREB) activity in the nucleus accumbens. Biol Psychiatry. 2010;67:28–35. doi: 10.1016/j.biopsych.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RI, Liu C, Waxman SG, Cummins TR. Calmodulin binds to the C terminus of sodium channels Nav1.4 and Nav1.6 and differentially modulates their functional properties. J Neurosci. 2003;23:8261–8270. doi: 10.1523/JNEUROSCI.23-23-08261.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hien YE, Montersino A, Castets F, Leterrier C, Filhol O, Vacher H, Dargent B. CK2 accumulation at the axon initial segment depends on sodium channel Nav1. FEBS Lett. 2014;588:3403–3408. doi: 10.1016/j.febslet.2014.07.032. [DOI] [PubMed] [Google Scholar]
- Hommel JD, Sears RM, Georgescu D, Simmons DL, DiLeone RJ. Local gene knockdown in the brain using viral-mediated RNA interference. Nat Med. 2003;9:1539–1544. doi: 10.1038/nm964. [DOI] [PubMed] [Google Scholar]
- Hsu WC, Nenov MN, Shavkunov A, Panova N, Zhan M, Laezza F. Identifying a kinase network regulating FGF14:Nav1.6 complex assembly using splitluciferase complementation. PLoS ONE. 2015;10:e0117246. doi: 10.1371/journal.pone.0117246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu WJ, Wildburger NC, Haidacher SJ, Nenov MN, Folorunso O, Singh AK, Chesson BC, Franklin WF, Cortez I, Sadygov RG, et al. PPARgamma agonists rescue increased phosphorylation of FGF14 at S226 in the Tg2576 mouse model of Alzheimer’s disease. Exp Neurol. 2017;295:1–17. doi: 10.1016/j.expneurol.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XT, Dong Y, Zhang XF, White FJ. Dopamine D2 receptor-activated Ca2+ signaling modulates voltage-sensitive sodium currents in rat nucleus accumbens neurons. J Neurophysiol. 2005;93:1406–1417. doi: 10.1152/jn.00771.2004. [DOI] [PubMed] [Google Scholar]
- Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12:996–1002. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539–551. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James TF, Nenov MN, Wildburger NC, Lichti CF, Luisi J, Vergara F, Panova-Electronova NI, Nilsson CL, Rudra JS, Green TA, et al. The Nav1.2 channel is regulated by GSK3. Biochim Biophys Acta. 2015;1850:832–844. doi: 10.1016/j.bbagen.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Roh MS. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr Drug Targets. 2006;7:1421–1434. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YT, Hur EM, Snider WD, Zhou FQ. Role of GSK3 Signaling in Neuronal Morphogenesis. Front Mol Neurosci. 2011;4:48. doi: 10.3389/fnmol.2011.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Calu DJ, Bonci A. Intrinsic plasticity: an emerging player in addiction. Nat Rev Neurosci. 2015;16:173–184. doi: 10.1038/nrn3877. [DOI] [PubMed] [Google Scholar]
- Laezza F, Gerber BR, Lou JY, Kozel MA, Hartman H, Craig AM, Ornitz DM, Nerbonne JM. The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J Neurosci. 2007;27:12033–12044. doi: 10.1523/JNEUROSCI.2282-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laezza F, Lampert A, Kozel MA, Gerber BR, Rush AM, Nerbonne JM, Waxman SG, Dib-Hajj SD, Ornitz DM. FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol Cell Neurosci. 2009;42:90–101. doi: 10.1016/j.mcn.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel M, Patenaude C, Allyson J, Massicotte G, Cyr M. Dopamine D1 receptor activation induces tau phosphorylation via cdk5 and GSK3 signaling pathways. Neuropharmacology. 2009;57:392–402. doi: 10.1016/j.neuropharm.2009.06.041. [DOI] [PubMed] [Google Scholar]
- Lehmann ML, Herkenham M. Environmental enrichment confers stress resiliency to social defeat through an infralimbic cortex-dependent neuroanatomical pathway. J Neurosci. 2011;31:6159–6173. doi: 10.1523/JNEUROSCI.0577-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Gao WJ. GSK-3β activity and hyperdopamine-dependent behaviors. Neurosci Biobehav Rev. 2011;35:645–654. doi: 10.1016/j.neubiorev.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Jope RS. Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacology. 2010;35:2143–2154. doi: 10.1038/npp.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichti CF, Fan X, English RD, Zhang Y, Li D, Kong F, Sinha M, Andersen CR, Spratt H, Luxon BA, Green TA. Environmental enrichment alters protein expression as well as the proteomic response to cocaine in rat nucleus accumbens. Front Behav Neurosci. 2014;8:246. doi: 10.3389/fnbeh.2014.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Q, Su Y, Bao L. Prostaglandin E2 promotes Na1.8 trafficking via its intracellular RRR motif through the protein kinase A pathway. Traffic. 2010;11:405–417. doi: 10.1111/j.1600-0854.2009.01027.x. [DOI] [PubMed] [Google Scholar]
- Marty VN, Spigelman I. Effects of alcohol on the membrane excitability and synaptic transmission of medium spiny neurons in the nucleus accumbens. Alcohol. 2012;46:317–327. doi: 10.1016/j.alcohol.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Moyer JT, Ishikawa M, Zhang Y, Panksepp J, Sorg BA, Schlüter OM, Dong Y. Exposure to cocaine dynamically regulates the intrinsic membrane excitability of nucleus accumbens neurons. J Neurosci. 2010;30:3689–3699. doi: 10.1523/JNEUROSCI.4063-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekata K, Harada C, Guo X, Kimura A, Kittaka D, Watanabe H, Harada T. Dock3 stimulates axonal outgrowth via GSK-3β-mediated microtubule assembly. J Neurosci. 2012;32:264–274. doi: 10.1523/JNEUROSCI.4884-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs SM, Dorostkar MM, Aramuni G, Schön C, Filser S, Pöschl J, Kremer A, Van Leuven F, Ovsepian SV, Herms J. Loss of neuronal GSK3β reduces dendritic spine stability and attenuates excitatory synaptic transmission via β-catenin. Mol Psychiatry. 2015;20:482–489. doi: 10.1038/mp.2014.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onwuli DO, Beltran-Alvarez P. An update on transcriptional and post-translational regulation of brain voltage-gated sodium channels. Amino Acids. 2016;48:641–651. doi: 10.1007/s00726-015-2122-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio N, Cathala L, Meisler MH, Crest M, Magistretti J, Delmas P. Persistent Nav1.6 current at axon initial segments tunes spike timing of cerebellar granule cells. J Physiol. 2010;588:651–670. doi: 10.1113/jphysiol.2010.183798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polter A, Beurel E, Yang S, Garner R, Song L, Miller CA, Sweatt JD, McMahon L, Bartolucci AA, Li X, Jope RS. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology. 2010;35:1761–1774. doi: 10.1038/npp.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy Chichili VP, Xiao Y, Seetharaman J, Cummins TR, Sivaraman J. Structural basis for the modulation of the neuronal voltage-gated sodium channel NaV1.6 by calmodulin. Sci Rep. 2013;3:2435. doi: 10.1038/srep02435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselli F, Caroni P. From intrinsic firing properties to selective neuronal vulnerability in neurodegenerative diseases. Neuron. 2015;85:901–910. doi: 10.1016/j.neuron.2014.12.063. [DOI] [PubMed] [Google Scholar]
- Royeck M, Horstmann MT, Remy S, Reitze M, Yaari Y, Beck H. Role of axonal NaV1.6 sodium channels in action potential initiation of CA1 pyramidal neurons. J Neurophysiol. 2008;100:2361–2380. doi: 10.1152/jn.90332.2008. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Murrough JW, Han MH, Charney DS, Nestler EJ. Neurobiology of resilience. Nat Neurosci. 2012;15:1475–1484. doi: 10.1038/nn.3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salles MJ, Hervé D, Rivet JM, Longueville S, Millan MJ, Girault JA, Mannoury la Cour C. Transient and rapid activation of Akt/ GSK-3β and mTORC1 signaling by D3 dopamine receptor stimulation in dorsal striatum and nucleus accumbens. J Neurochem. 2013;125:532–544. doi: 10.1111/jnc.12206. [DOI] [PubMed] [Google Scholar]
- Scala F, Fusco S, Ripoli C, Piacentini R, Li Puma DD, Spinelli M, Laezza F, Grassi C, D’Ascenzo M. Intraneuronal Aβ accumulation induces hippocampal neuron hyperexcitability through A-type K(+) current inhibition mediated by activation of caspases and GSK-3. Neurobiol Aging. 2015;36:886–900. doi: 10.1016/j.neurobiolaging.2014.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuer T. Regulation of sodium channel activity by phosphorylation. Semin Cell Dev Biol. 2011;22:160–165. doi: 10.1016/j.semcdb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuer T, Catterall WA. Control of neuronal excitability by phosphorylation and dephosphorylation of sodium channels. Biochem Soc Trans. 2006;34:1299–1302. doi: 10.1042/BST0341299. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Lledo PM, Vincent JD. Dopamine D1 receptor modulates the voltage-gated sodium current in rat striatal neurones through a protein kinase A. J Physiol. 1995;483:95–107. doi: 10.1113/jphysiol.1995.sp020570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann SN, Desdouits F, Menu R, Greengard P, Vincent JD, Vanderhaeghen JJ, Girault JA. Modulation of the voltage-gated sodium current in rat striatal neurons by DARPP-32, an inhibitor of protein phosphatase. Eur J Neurosci. 1998;10:1312–1320. doi: 10.1046/j.1460-9568.1998.00142.x. [DOI] [PubMed] [Google Scholar]
- Shavkunov AS, Wildburger NC, Nenov MN, James TF, Buzhdygan TP, Panova-Elektronova NI, Green TA, Veselenak RL, Bourne N, Laezza F. The fibroblast growth factor 14·voltage-gated sodium channel complex is a new target of glycogen synthase kinase 3 (GSK3) J Biol Chem. 2013;288:19370–19385. doi: 10.1074/jbc.M112.445924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Kitai ST. D1 and D2 dopamine receptor modulation of sodium and potassium currents in rat neostriatal neurons. Prog Brain Res. 1993;99:309–324. doi: 10.1016/s0079-6123(08)61354-0. [DOI] [PubMed] [Google Scholar]
- Tan ZY, Priest BT, Krajewski JL, Knopp KL, Nisenbaum ES, Cummins TR. Protein kinase C enhances human sodium channel hNav1.7 resurgent currents via a serine residue in the domain III-IV linker. FEBS Lett. 2014;588:3964–3969. doi: 10.1016/j.febslet.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs NM, Snyder JC, Jacobsen JP, Peterson SM, Caron MG. Deletion of GSK3β in D2R-expressing neurons reveals distinct roles for β-arrestin signaling in antipsychotic and lithium action. Proc Natl Acad Sci USA. 2012;109:20732–20737. doi: 10.1073/pnas.1215489109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Robison AJ, Laplant QC, Covington HE, 3rd, Dietz DM, Ohnishi YN, Mouzon E, Rush AJ, 3rd, Watts EL, Wallace DL, et al. DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat Neurosci. 2010;13:745–752. doi: 10.1038/nn.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DL, Han MH, Graham DL, Green TA, Vialou V, Iñiguez SD, Cao JL, Kirk A, Chakravarty S, Kumar A, et al. CREB regulation of nucleus accumbens excitability mediates social isolation-induced behavioral deficits. Nat Neurosci. 2009;12:200–209. doi: 10.1038/nn.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildburger NC, Ali SR, Hsu WC, Shavkunov AS, Nenov MN, Lichti CF, LeDuc RD, Mostovenko E, Panova-Elektronova NI, Emmett MR, et al. Quantitative proteomics reveals protein-protein interactions with fibroblast growth factor 12 as a component of the voltage-gated sodium channel 1.2 (nav1.2) macromolecular complex in Mammalian brain. Mol Cell Proteomics. 2015;14:1288–1300. doi: 10.1074/mcp.M114.040055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MB, Dias C, Magida J, Mazei-Robison M, Lobo M, Kennedy P, Dietz D, Covington H, 3rd, Russo S, Neve R, et al. A novel role of the WNT-dishevelled-GSK3β signaling cascade in the mouse nucleus accumbens in a social defeat model of depression. J Neurosci. 2011;31:9084–9092. doi: 10.1523/JNEUROSCI.0039-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DF, Chandra D, McMahon T, Wang D, Dadgar J, Kharazia VN, Liang YJ, Waxman SG, Dib-Hajj SD, Messing RO. PKCε phosphorylation of the sodium channel NaV1.8 increases channel function and produces mechanical hyperalgesia in mice. J Clin Invest. 2012;122:1306–1315. doi: 10.1172/JCI61934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Wang C, Marx SO, Pitt GS. Calmodulin limits pathogenic Na+ channel persistent current. J Gen Physiol. 2017;149:277–293. doi: 10.1085/jgp.201611721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Crofton EJ, Li D, Lobo MK, Fan X, Nestler EJ, Green TA. Overexpression of DeltaFosB in nucleus accumbens mimics the protective addiction phenotype, but not the protective depression phenotype of environmental enrichment. Front Behav Neurosci. 2014;8:297. doi: 10.3389/fnbeh.2014.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Crofton EJ, Fan X, Li D, Kong F, Sinha M, Luxon BA, Spratt HM, Lichti CF, Green TA. Convergent transcriptomics and proteomics of environmental enrichment and cocaine identifies novel therapeutic strategies for addiction. Neuroscience. 2016a;339:254–266. doi: 10.1016/j.neuroscience.2016.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kong F, Crofton EJ, Dragosljvich SN, Sinha M, Li D, Fan X, Koshy S, Hommel JD, Spratt HM, et al. Transcriptomics of environmental enrichment reveals a role for retinoic acid signaling in addiction. Front Mol Neurosci. 2016b;9:119. doi: 10.3389/fnmol.2016.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.