

ABSTRACT
Loss-of-function mutations of the ß-cell ATP-sensitive potassium channels (KATP) cause the most common and severe form of congenital hyperinsulinism (KATPHI), a disorder of ß-cell function characterized by severe hypoglycemia. Children with KATPHI are typically unresponsive to medical therapy and require pancreatectomy for intractable hypoglycemia. We tested the hypothesis that inhibition of insulin receptor signaling may prevent hypoglycemia in KATPHI. To test this hypothesis, we examined the effect of an antibody allosteric inhibitor of the insulin receptor, XMetD, on fasting plasma glucose in a mouse model of KATPHI (SUR-1−/− mice). SUR-1−/− and wild-type mice received twice weekly intraperitoneal injections of either XMetD or control antibody for 8 wks. Treatment with XMetD significantly decreased insulin sensitivity, and increased hepatic glucose output and fasting plasma glucose. These findings support the potential use of insulin receptor antagonists as a therapeutic approach to control the hypoglycemia in congenital hyperinsulinism.
KEYWORDS: hyperinsulinism, hypoglycemia, pancreas, insulin, beta cell
Introduction
Congenital hyperinsulinism (HI) is a genetic disorder of pancreatic β-cell function characterized by failure to suppress insulin secretion in the setting of hypoglycemia, resulting in severe hypoglycemia that can cause brain damage or death if inadequately treated. Loss-of-function mutations of the ATP-sensitive potassium channels (composed of two subunits: Kir6.2 and SUR-1) are responsible for the most common and severe form of hyperinsulinism (KATPHI). Children with KATPHI present shortly after birth with severe hypoglycemia and require glucose infusion rates up to four times higher than physiologic requirement to maintain normal plasma glucose concentrations. 1 Diazoxide, the mainstay of medical therapy for hyperinsulinism, suppresses insulin by promoting the opening of the β-cell KATP channel and is ineffective in patients with KATPHI. Thus, most of these children require pancreatectomy to control the hypoglycemia. Children with a focal form of the disease can be cured by limited pancreatic resection, but for children with the diffuse form, a near-total pancreatectomy only partially controls the hypoglycemia 2 and results in insulin-requiring diabetes later in life. 3 , 4 There are no comprehensive published studies describing the natural history of the disease; however, there is evidence that the severity of the disease ameliorates with age. 5 Age-related changes on insulin sensitivity may contribute to the amelioration of the hypoglycemia overtime. 6 Thus, we hypothesize that modulating insulin responsiveness at the level of the insulin receptor (INSR) may be a novel mechanism for treating congenital hyperinsulinism.
In this study, we tested the hypothesis that inhibition of INSR signaling may prevent hypoglycemia in KATPHI. For this purpose we employed XMetD [also known as XOMA358 (X358)], a human anti-INSR IgG2 monoclonal antibody, discovered by XOMA (US) LLC. XMetD is a negative allosteric modulator (NAM) of the INSR that primarily inhibits INSR activation both in vitro and in vivo in a dose-dependent manner. 7 In cultured cells XMetD was shown to markedly antagonize insulin-dependent INSR autophosphorylation and downstream metabolic effects, including AKT phosphorylation and glucose transport. In vivo, XMetD increased plasma glucose concentrations in normal mice and prevented insulin-induced hypoglycemia in mice receiving insulin through the insertion of sustained-release insulin implants. 7 In a Phase 1 clinical study, a single infusion of XMetD resulted in a dose-dependent reduction in insulin sensitivity in healthy adults. 8 We have now examined the effect of XMetD on fasting plasma glucose and other parameters of glucose and energy metabolism in a mouse model of KATPHI, the SUR-1−/− mice. 9 , 10
Results
Twenty 10–12 week old male SUR-1 −/− and wild-type control mice underwent a baseline evaluation 1 and 2 weeks prior to initiation of treatment, including body weight, fasting and fed plasma glucose and fasting plasma insulin, followed by randomization to treatment with XMetD or control antibody (KLH2G2) (Figure 1).
Figure 1.

Experimental Paradigm. Twenty male wild-type and 20 SUR-1 −/− mice were randomized to treatment with XMetD or control antibody. Fasting plasma glucose (FPG) and fasting insulin (FI) were measured weekly before and during the first 3 weeks of treatment. Body composition and energy expenditure were measured after 6 weeks of treatment (n = 5 per group) and a hyperinsulinemic euglycemic clamp was performed at 8 weeks (n = 5 per group).
Baseline evaluation
As previously reported, 11 fasting plasma glucose concentrations were significantly lower in SUR-1 −/− mice compared to wild-type controls (Figure 2A). In contrast, fed plasma glucose was significantly higher in SUR1−/− mice compared to wild-type controls (Figure 2B). Fasting plasma insulin was not significantly different in SUR1−/− and wild-type mice (P ≥0.4) (Figure 2C). At baseline, SUR1−/− mice were slightly but significantly heavier than wild-type control mice (Figure 3).
Figure 2.

Fasting and Fed Plasma Glucose and Plasma Insulin. (A) Fasting plasma glucose (in mg/dL) in wild-type mice treated with control antibody (n = 10), wild-type mice treated with XMetD (n = 10), SUR-1 −/− mice treated with control antibody (n = 10), SUR-1 −/− mice treated with XMetD (n = 10). * P≤0.001: wild-type vs. SUR-1 −/−; # P≤0.005 SUR-1 −/− control vs. SUR-1 −/− XMetD. (B) Fed plasma glucose (in mg/dL) in wild-type mice treated with control antibody (n = 10), wild-type mice treated with XMetD (n = 10), SUR-1 −/− mice treated with control antibody (n = 10), SUR-1 −/− mice treated with XMetD (n = 10). * P≤0.05: wild-type vs. SUR-1 −/−; # P≤0.01 SUR-1 −/− control vs. SUR-1 −/− XMetD; ^ P≤0.03 wild-type control vs. wild-type XMetD (C) Fasting plasma insulin levels (in ng/mL) in wild-type mice treated with control antibody (n = 10), wild-type mice treated with XMetD (n = 10), SUR-1 −/− mice treated with control antibody (n = 10), SUR-1 −/− mice treated with XMetD (n = 10). # P≤0.0004 SUR-1 −/− control vs. SUR-1 −/− XMetD; ^ P≤0.0004 wild-type control vs. wild-type XMetD.
Figure 3.

Body Weight. Body weight (in g) in wild-type mice treated with control antibody (n = 10), wild-type mice treated with XMetD (n = 10), SUR-1 −/− mice treated with control antibody (n = 10), SUR-1 −/− mice treated with XMetD (n = 10). * P≤0.02 wild-type vs. SUR-1 −/−.
Effect of treatment on feeding behavior and weight gain
There was no significant effect of XMetD on body weight gain in either SUR1−/− mice or wild-type mice (P > 0.2) (Figure 3). Food and water consumption, measured in 5 mice/group during a 24 hrs period on week 6 of treatment, was likewise not different among the treatment groups (data not shown).
Effect of treatment on glucose metabolism
Fasting plasma glucose was significantly different among the groups during the first 3 weeks of treatment (P ≤ 0.004). Fasting plasma glucose was significantly higher in XMetD-treated SUR1−/− mice compared to control-treated SUR1−/− mice week 1–3 (P ≤ 0.005) and not different from the control-treated wild-type mice (P ≥ 0.2). In wild-type mice, XMetD treatment increased fasting plasma glucose, but this effect was not statistically significant when adjusted for the multiple comparisons (P ≥ 0.2) (Figure 2A). Fed plasma glucose was significantly different among the groups during the first 3 weeks of treatment (P ≤ 0.003) (Figure 2B). Fed plasma glucose was significantly higher in XMetD-treated SUR1−/− mice compared to control-treated SUR1−/− mice week 1 and 3 (P ≤ 0.01), but not significantly different on week 2 of treatment (P = 0.5). In wild-type mice, XMetD treatment significantly increased fed plasma glucose week 1–3 (P ≤ 0.03) (Figure 2B).
Fasting plasma insulin was significantly different among the groups week 1–3 of treatment (P ≤ 0.0001). Fasting plasma insulin was significantly higher in XMetD-treated SUR1−/− and wild-type mice compared to control-treated mice week 1–3 (P ≤ 0.0004) (Figure 2C).
A glucose tolerance test was perfomed on week 3 of treatment. After an overnight fast, mice were given an intraperitoneal (i.p.) load of glucose (2 g/kg). Plasma glucose and insulin concentrations were measured at time 0 and every 30 min for 2 hrs. Plasma glucose was significantly different among the treatment groups during the glucose tolerance test at all time points (P ≤ 0.04) (Figure 4A). As previously reported, SUR1−/− mice have significantly higher glucose excursion in response to a glucose load than wild-type mice. 11 Plasma glucose was significantly higher in XMetD-treated SUR1−/− compared to control-treated SUR1−/− mice at time 0 (P = 0.005), but was not different in response to a glucose load (P > 0.9 at all time points). In wild-type mice, plasma glucose was not significantly different in XMetD-treated compared to control-treated mice at time 0 and at any time point after the glucose load (P>0.06). Plasma insulin was significantly higher at all times in XMetD-treated SUR1−/− and wild-type mice compared to control-treated mice (P ≤ 0.002) (Figure 4B).
Figure 4.
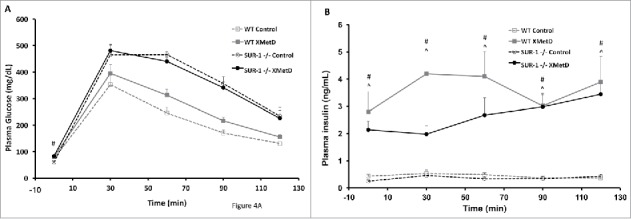
Glucose Tolerance Test. (A) Intraperitoneal glucose tolerance (2g/kg) in wild-type mice treated with control antibody (n = 10), wild-type mice treated with XMetD (n = 10), SUR-1 −/− mice treated with control antibody (n = 10), SUR-1 −/− mice treated with XMetD (n = 10). # P = 0.005 SUR-1 −/− control vs. SUR-1 −/− XMetD (B) Insulin secretion in response to an i.p. glucose load (2g/kg) in in wild-type mice treated with control antibody (n = 10), wild-type mice treated with XMetD (n = 10), SUR-1 −/− mice treated with control antibody (n = 10), SUR-1 −/− mice treated with XMetD (n = 10). # P≤0.002 SUR-1 −/− control vs. SUR-1 −/− XMetD; ^ P≤0.002 wild-type control vs. wild-type XMetD.
To assess the effect of treatment on insulin sensitivity, an insulin tolerance test was performed on week 4 of treatment. After a 6 hr fast, mice were given an i.p. injection of insulin (1 unit). Plasma glucose concentration was measured at time 0 and every 10 min for 30 min. The test was terminated earlier in mice that developed significant hypoglycemia. If needed, mice were rescued with an i.p. bolus of dextrose. Plasma glucose levels decreased in all groups in response to insulin (Figure 5). The slope of the decline was significantly different among the groups: WT control = 4.7 ± 1.7; WT XMetD = 3.9 ± 2.3; SUR1−/− control = 4.9 ± 0.9; SUR1−/−XMetD = 3.5 ± 0.8 (P = 0.03). The slope of the decline was significantly smaller in XMetD-treated SUR1−/− mice compared to control-treated SUR1−/− mice (P < 0.005). In wild-type mice, the difference was not statistically significant (P = 0.2).
Figure 5.

Insulin Tolerance Test. Plasma glucose in response to an insulin (1 U i.p.) in wild-type mice treated with control antibody (n = 10), wild-type mice treated with XMetD (n = 10), SUR-1 −/− mice treated with control antibody (n = 10), SUR-1 −/− mice treated with XMetD (n = 10). # Slope of decline: 4.9 ± 0.9 in SUR-1 −/− control vs. 3.5 ± 0.8 in SUR-1 −/− XMetD, P≤0.005.
Effect of treatment on energy metabolism
After 5 weeks of treatment mice were transferred from the housing facility to the mouse phenotyping facility for further studies. At week 6 of treatment, body composition and energy expenditure were assessed (n = 5 for each group). Lean and fat body mass was not significantly different among the groups (P = 0.5) (data not shown). There was no difference in oxygen consumption (VO2), respiratory quotient (RQ = VCO2/VO2), metabolic rate (3.815+1.232xRQ)xVO2), and total and ambulatory activity between the XMetD and control-treated SUR1−/− and wild-type mice during the light or dark cycle (Table 1). However, there was a small difference in oxygen consumption between wild-type and SUR1−/−. In control-treated mice, the average VO2 during the light and dark cycle was signficantly higher in SUR1−/− compared to wild-type mice. Respiratory quotient was not different between SUR1−/− and wild-type mice. However, basal metabolic rate (calculated as the average of the 5 lowest during a 24 hrs period. 12 ) was significantly lower in SUR1−/− compared to wild-type mice (P < 0.04).
Table 1.
Metabolic Parameters.
| Metabolic parameters | WT Control | WT XMetD | SUR1−/−Control | SUR1−/− XMetD | P value |
|---|---|---|---|---|---|
| Body weight (g) | 28.6 ± 1.9 | 27.2 ± 1.4 | 28.6 ± 1.8 | 28.6 ± 2.7 | ANOVA: P = 0.6 |
| 12 hr light – VO2 (mL/hr) | 77.9 ± 9.2 | 76.2 ± 11.7 | 92.6 ± 8.0 | 92.8 ± 14.3 | ANOVA: P = 0.04 |
| SUR1−/−control vs. | |||||
| WTcontrol: P = 0.03 | |||||
| 12 hr dark – VO2 (mL/hr) | 92.6 ± 12.4 | 88.8 ± 11.2 | 107.4 ± 3.8 | 107.5 ± 19.5 | ANOVA: P = 0.07 |
| SUR1−/−control vs. | |||||
| WTcontrol: P = 0.04 | |||||
| 12 hr light – VCO2 (mL/hr) | 65.2 ± 9.3 | 62.6 ± 11.8 | 77.7 ± 9.1 | 75.1 ± 13.0 | ANOVA: P = 0.12 |
| 12 hr dark – VCO2 (mL/hr) | 81.6 ± 12.5 | 77.7 ± 11.8 | 95.1 ± 4.6 | 90.7 ± 16.9 | ANOVA: P = 0.14 |
| 12 hr light – RQ | 0.83 ± 0.02 | 0.82 ± 0.04 | 0.84 ± 0.03 | 0.80 ± 0.02 | ANOVA: P = 0.4 |
| 12 hr dark – RQ | 0.88 ± 0.03 | 0.87 ± 0.03 | 0.88 ± 0.03 | 0.84 ± 0.04 | ANOVA: P = 0.25 |
| Total Activity | 1072 ± 202 | 1507 ± 195 | 1174 ± 270 | 1264 ± 391 | ANOVA: P = 0.12 |
| Ambulatory Activity | 511 ± 131 | 795 ± 171 | 576 ± 183 | 632 ± 220 | ANOVA: P = 0.12 |
| 12 hr light – Metabolic Rate (mL/hr) | 377.5 ± 46.4 | 367.7 ± 59.0 | 449.2 ± 41.6 | 446.6 ± 70.7 | ANOVA: P = 0.06 |
| 12 hr dark – Metabolic Rate (mL/hr) | 453.9 ± 62.5 | 434.7 ± 57.4 | 526.8 ± 18.9 | 521.8 ± 94.5 | ANOVA: P = 0.08 |
| Basal Metabolic Rate (mL/hr) | 304.2 ± 42.5 | 306.5 ± 43.5 | 383.9 ± 41.2 | 393.9 ± 31.9 | ANOVA: P = 0.004 |
| SUR1−/−control vs. | |||||
| WTcontrol: P = 0.04 | |||||
| Fat mass (g) | 1.8 ± 0.5 | 1.4 ± 0.4 | 1.9 ± 1.1 | 1.3 ± 0.6 | ANOVA: P = 0.5 |
| Lean mass (g) | 25.8 ± 1.3 | 24.8 ± 1.7 | 25.6 ± 1.2 | 26.4 ± 2.2 | ANOVA: P = 0.5 |
VO2: oxygen consumption; VCO2: carbon diaoxide excretion; RQ: respiratory quotient. Data presented as mean ± SD; n = 5/group.
Effect of treatment on hepatic and peripheral glucose metabolism
A hyperinsulinemic-euglycemic clamp was performed on week 8–9 (n = 5 for each group) (Figure 6). Baseline hepatic glucose production was not significantly different among the treatment groups (P = 0.12) (Figure 6A). However, as would be expected, hepatic glucose production was lower in SUR1−/− mice compared to wild-type mice. During the hyperinsulinemic-euglycemic clamp, hepatic glucose production was significantly different among the treatment groups (P = 0.03). Hepatic glucose production was higher in XMetD-treated SUR1−/− and wild-type mice compared to control-treated mice, but the difference was not statistically significant when adjusted for multiple comparisons (P = 0.07, for SUR1−/− mice; P = 0.6 for wild-type mice) (Figure 6B).
Figure 6.
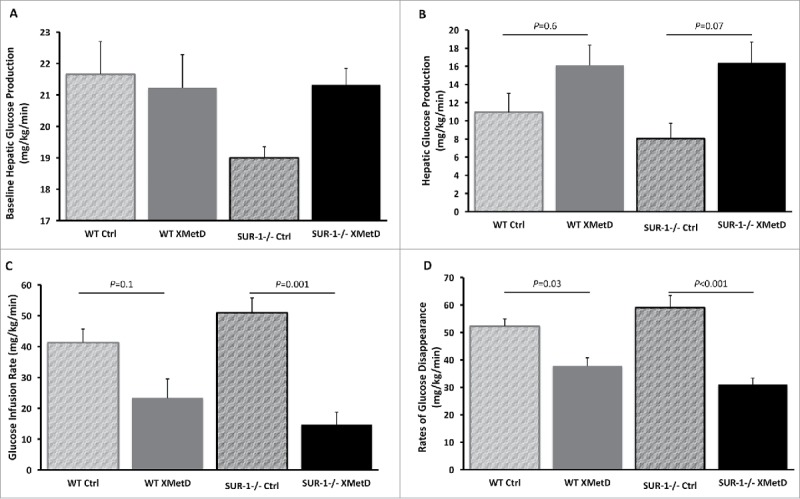
Hyperinsulinemic-Euglycemic Clamp. (A) Baseline hepatic glucose production in wild-type mice treated with control antibody (n = 5), wild-type mice treated with XMetD (n = 5), SUR-1 −/− mice treated with control antibody (n = 5), SUR-1 −/− mice treated with XMetD (n = 5); ANOVA: P = 0.12. (B) Hepatic glucose production during hyperinsulinemic-euglycemic clamp in wild-type mice treated with control antibody (n = 5), wild-type mice treated with XMetD (n = 5), SUR-1 −/− mice treated with control antibody (n = 5), SUR-1 −/− mice treated with XMetD (n = 5), ANOVA: P = 0.03: SUR-1 −/− control vs. SUR-1 −/− XMetD. P = 0.07; wild-type control vs. wild-type XMetD, P = 0.6. (C) Glucose infusion rate to maintain euglycemia during hyperinsulinemic-euglycemic clamp in wild-type mice treated with control antibody (n = 5), wild-type mice treated with XMetD (n = 5), SUR-1 −/− mice treated with control antibody (n = 5), SUR-1 −/− mice treated with XMetD (n = 5), ANOVA: P = 0.0003: SUR-1 −/− control vs. SUR-1 −/− XMetD. P = 0.001; wild-type control vs. wild-type XMetD, P = 0.1. (D) Rates of glucose disappearance during hyperinsulinemic-euglycemic clamp in wild-type mice treated with control antibody (n = 5), wild-type mice treated with XMetD (n = 5), SUR-1 −/− mice treated with control antibody (n = 5), SUR-1 −/− mice treated with XMetD (n = 5), ANOVA: P<0.0001: SUR-1 −/− control vs. SUR-1 −/− XMetD. P<0.001; wild-type control vs. wild-type XMetD, P = 0.03.
Glucose infusion rate (GIR) during the clamp was significantly different among the treatment groups (P = 0.0003). GIR was significantly lower in XMetD-treated compared to control-treated SUR1−/− mice (P = 0.001). In contrast, GIR was not significantly different in XMetD-treated and control-treated wild-type mice (P = 0.1) (Figure 6C).
Rates of glucose disappearance (Rd, whole body glucose uptake) during the clamp was significantly different among the treatment groups (P < 0.0001). Rd was significantly lower in XMetD-treated SUR1−/− and wild-type mice compared to control-treated mice (P < 0.001 SUR1−/− XMetD vs. SUR1−/− control; P = 0.03 wild-type XMetD vs. wild-type control) (Figure 6D).
Muscle glucose uptake was significantly different among the treatment groups (P = 0.01). Muscle glucose uptake trended lower in XmetD-treated SUR1−/− mice compared to control-treated mice, but the difference was not statistically significant when adjusted for the multiple comparisons (SUR1−/− XMetD = 216 ± 81.8 nmol/g/min vs. SUR1−/− control = 289.4 ± 84.3 nmol/g/min, P = 1; wild-type XMetD = 417. 4 ± 87.7 nmol/g/min vs. wild-type control = 336.2 ± 81.2 nmol/g/min, P = 0.9). White adipose glucose uptake was not significantly different among the treatment groups (P = 0.9): SUR1−/− XMetD = 20.6 ± 12.1 nmol/g/min vs. SUR1−/− control = 23.8 ± 9.9 nmol/g/min; wild-type XMetD = 24.5 ± 16.7 nmol/g/min vs. wild-type control = 21.5 ± 8.9 nmol/g/min.
Discussion
Congenital hyperinsulinism due to inactivating mutations in the KATP channel is a devastating disease that is frequently unresponsive to available medical therapies. Many children with this condition require pancreatectomy for intractable hypoglycemia, which provides only marginal control of the hypoglycemia. 1 In children with diffuse hyperinsulinism, a 98% pancreatectomy provides only marginal control of the hypoglycemia. 2 Thus, there is a significant unmet medical need for safer and more effective treatments for KATPHI. Herein, we tested the hypothesis that a novel therapeutic approach targeting the insulin receptor may ameliorate the hypoglycemia and potentially prevent the need for pancreatectomy. Our results offer strong proof-of-concept for this therapeutic approach.
It has been previously shown that XMetD, a NAM of the INSR, ameliorates hypoglycemia in mice receiving exogenous insulin. 7 In healthy volunteers, a single dose of XMetD resulted in a significant elevation of postprandial plasma glucose concentration and a significant attenuation of the decrease in plasma glucose induced by insulin in the setting of an insulin tolerance test. 8 Our study extends these findings in a mouse model relevant to the human condition of endogenous hyperinsulinemic hypoglycemia. Although SUR-1 −/− mice are relatively normoglycemic in the fed state, 9 , 10 they develop hypoglycemia with fasting, which allowed us to test the potential for an insulin receptor allosteric inhibitor to normalize fasting plasma glucose in the absence of functional KATP channels. Our studies confirmed previous reports highlighting the phenotype of the SUR-1 −/− mice. 9 , 10 Compared to wild-type littermates, SUR-1 −/− mice are more hypoglycemic when fasted and more hyperglycemic when glucose loaded, and the glucose intolerance is the result of impaired glucose-stimulated insulin secretion.
The results of our study demonstrate that inhibition of insulin signaling by XMetD corrects fasting hypoglycemia in SUR1−/−mice without significantly impairing glucose tolerance. This is in contrast to findings in normal human subjects in whom XMetD did not have a persistent effect on fasting plasma glucose. Although there was significant compensatory ß-cell response and insulin concentration was significantly higher in both the SUR-1 −/− and wild-type mice treated with XMetD, hepatic insulin sensitivity was significantly reduced, accounting for the effect on fasting plasma glucose.
We quantitatively assessed the effect of the antibody on energy and glucose metabolism. There were no significant effects of XMetD on metabolic rate, which is not surprising since the antibody does not cross the blood-brain barrier. Interestingly, the lack of functional KATP channels clearly affects energy metabolism, and we show that oxygen consumption and basal metabolic rates are higher in SUR1−/−mice compared to wild-type mice. Genetic and electrophysiological evidence have shown that KATP channels are present in selective hypothalamic neurons. 13 , 14 It is thus not surprising that the lack of functional KATP channels may lead to changes in energy homeostasis. Investigation of the precise mechanisms by which KATP channels regulate energy homeostasis is beyond the scope of this study.
There are a number of limitations to our work. First, for this proof-of-concept study we used the SUR1−/− mouse as a model of KATPHI, which exhibits all the expected features resulting from inactivating KATP channels, but the mice have milder fasting hypoglycemia compared to humans. This mouse model has been previously used by our group for proof-of-concept studies of other potential therapies for hyperinsulinism. 11 that were successfully translated to clinical studies in affected individuals. 15 Second, the relatively short treatment period did not allow us to determine if the effects of treatment are sustained over longer period of times. Finally, as XMetD is a humanized antibody, it is possible that the effects on mice may have been altered by an immunogenicity response, although this seems less likely given the lasting effects on hepatic glucose metabolism.
In summary, we have shown that XMetD significantly raises fasting plasma glucose levels in SUR-1−/− mice through a direct effect on insulin sensitivity. Importantly, treatment with XMetD did not affect weight gain and was well-tolerated. These findings have significant translational application, given the lack of effective medical therapies for children with congenital hyperinsulinism due to KATP mutations. Indeed, the effect of XMetD on individuals with congenital or acquired hyperinsulinemic hypoglycemia is being evaluated (ClinicalTrials.gov: NCT02604485 and NCT02772718). The use of insulin receptor antagonists to control hypoglycemia in congenital hyperinsulinism could have a beneficial effect on morbidity and long-term outcome in this patient population.
Materials and Methods
Animals
The generation and genotyping of SUR-1 −/− mice were previously described. 10 Mice are maintained in our mouse colony in a C57Bl/6 genetic background. Wild-type C57Bl/6 control mice were acquired from Jackson Laboratories. Male, ten to thirteen week old, SUR-1 −/− and wild-type control mice were used in all experiments. Mice were maintained on a 12:12-h light-dark cycle and were fed a standard rodent chow diet. All procedures were approved and carried out according to the Children's Hospital of Philadelphia Institutional Animal Care and Use Committee guidelines.
Antibodies
The discovery and characteristics of the allosteric monoclonal antibody XMetD have been previously described. 7 Briefly, allosteric modulating antibodies targeting the human insulin receptor (hINSR) were identified by panning naïve human antibody phage display libraries using the recombinant extracellular domain of the hINSR complexed to insulin. Antibodies binding the hINSR-insulin complex were identified by fluorescence-activated cell sorting of bacterial periplasmic extracts. The identifed antibody was reformatted to fully human IgG2 antibodies. An IgG anti-KLH (KLH2G2) was used as negative control. XMetD and KLH2G2 antibodies were provided by XOMA Corporation. A loading dose of 20 mg/kg followed by a maintenance dose of 5 mg/kg twice weekly of XMetD and KLH2G2 were administered i.p. to SUR-1 −/− and wild-type mice for 8 weeks.
Glucose homeostasis
Random fed plasma glucose and morning fasting (16 hrs) plasma glucose and plasma insulin concentrations were measured at baseline and weekly for the first 3 weeks of treatment. Glucose tolerance testing was carried after a 16 hour fast by i.p. administration of 2g/kg of dextrose. For insulin tolerance testing, mice received 1 unit of insulin i.p. after a 6 hour fast.
After 5 weeks the mice were then transferred to the phenotyping core of the University of Pennsylvania Diabetes Research Center and a hyperinsulinemic-euglycemic clamp was performed as previously described. 16 Briefly, an indwelling catheter was inserted in the right internal jugular vein under sodium pentobarbital anesthesia and extended to the right atrium. Four days after recovery of pre-surgery weight and habituation in restrainers, the mice were fasted for 6 hours (0800-1300), tail plasma glucose was measured and a bolus injection of 5 μCi of [3-3H] glucose was administered intravenously, followed by a continuous intravenous infusion at 0.05 μCi min−1. Baseline glucose kinetics was measured for 120 min followed by hyperinsulinemic clamp for 120 min. A priming dose of regular insulin (16 mU kg−1, Humulin; Eli Lilly) was given intravenously, followed by a continuous infusion of 2.5 mU kg−1 min−1. A variable intravenous infusion of 20% dextrose was infused to attain plasma glucose concentrations of 120–140 mg/dL. Blood samples were drawn at 10-minute intervals for the determination of plasma glucose. At the end of the clamp, 10 μCi 2-deoxy-D-[1-14C] glucose was injected to estimate glucose uptake. The mice were euthanized, and liver, perigonadal adipose tissue, and soleous and gastrocnemius muscles were excised, frozen immediately in liquid nitrogen and stored at −80°C for analysis of glucose uptake. Rates of whole body glucose uptake and basal glucose turnover were measured as the ratio of the [3H] glucose infusion rate (d.p.m) to the specific activity of plasma glucose. Hepatic glucose production (HGP) during the clamp was measured by substracting the GIR from the whole body glucose uptake (Rd). Under basal (fasted) conditions, Rd = HGP.
Energy homeostasis
Body weight was measured at baseline and weekly during the treatment period. Food and water consumption were measured during a 24 hrs period on week 6 of treatment. Energy expenditure was measured by indirect calorimetry using a comprehensive laboratory animal monitoring system (Columbus Instruments). Body composition was analyzed using NMR (Echo Medical Systems).
Assays
Plasma glucose concentrations were measured using a hand-held glucose meter (NOVA, Nova Biomedical). Plasma insulin was measured by ELISA (ALPCO; catalogue #80-INSMS-E01).
Statistical analysis
Data presented are mean ± SD. Normality was assessed using skewness and kurtosis test. Normally distributed data was compared using ANOVA. Posthoc Bonferroni procedure was applied to compare specific groups of interest. Kruskal-Wallis test was used to compared not-normally distributed data. Differences were considered significant at P < 0.05. Analysis was performed using Stata (StataCorp LLC).
Grants
This study was supported by a grant from XOMA Corporation to DDDL, and by the University of Pennsylvania Diabetes Research Center National Institutes of Health grant P30DK19525.
Funding Statement
This work was supported by the XOMA Corporation.
Disclosure of potential conflicts of interest
This study was supported by a grant from XOMA Corporation to DDDL. JC, IDG, KJ, and PR are or were employed by XOMA Corporation.
References
- 1.Lord K, De Leon DD. Monogenic hyperinsulinemic hypoglycemia: current insights into the pathogenesis and management. Int J Pediatr Endocrinol. 2013;2013:3 doi: 10.1186/1687-9856-2013-3. PMID:23384201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lord K, Dzata E, Snider KE, Gallagher PR, De Leon DD. Clinical presentation and management of children with diffuse and focal hyperinsulinism: a review of 223 cases. J Clin Endocrinol Metab. 2013;98:E1786–1789 doi: 10.1210/jc.2013-2094. PMID:24057290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beltrand J, Caquard M, Arnoux JB, Laborde K, Velho G, Verkarre V, Rahier J, Brunelle F, Nihoul-Fekete C, Saudubray JM, et al.. Glucose metabolism in 105 children and adolescents after pancreatectomy for congenital hyperinsulinism. Diabetes Care. 2012;35:198–203 doi: 10.2337/dc11-1296. PMID:22190679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lord K, Radcliffe J, Gallagher PR, Adzick NS, Stanley CA, De Leon DD. High risk of diabetes and neurobehavioral deficits in individuals with surgically treated hyperinsulinism. J Clin Endocrinol Metab. 2015;100:4133–9 jc20152539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazor-Aronovitch K, Gillis D, Lobel D, Hirsch HJ, Pinhas-Hamiel O, Modan-Moses D, Glaser B, Landau H. Long-term neurodevelopmental outcome in conservatively treated congenital hyperinsulinism. Eur J Endocrinol. 2007;157:491–7 doi: 10.1530/EJE-07-0445. PMID:17893264. [DOI] [PubMed] [Google Scholar]
- 6.Bloch CA, Clemons P, Sperling MA. Puberty decreases insulin sensitivity. J Pediatr. 1987;110:481–7 doi: 10.1016/S0022-3476(87)80522-X. PMID:2950219. [DOI] [PubMed] [Google Scholar]
- 7.Corbin JA, Bhaskar V, Goldfine ID, Issafras H, Bedinger DH, Lau A, Michelson K, Gross LM, Maddux BA, Kuan HF, et al.. Inhibition of insulin receptor function by a human, allosteric monoclonal antibody: a potential new approach for the treatment of hyperinsulinemic hypoglycemia. MAbs. 2014;6:262–72 doi: 10.4161/mabs.26871. PMID:24423625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson KW, Neale A, Gordon A, Roessig J, Bezwada P, Vukelich S, Goldfine I., Rubin P. Attenuation of insulin action by an allosteric insulin receptor antibody in healthy volunteers. J Clin Endocrinol Metab. 2017;102:3021–8 doi: 10.1210/jc.2017-00822. PMID:28605468. [DOI] [PubMed] [Google Scholar]
- 9.Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J. Sur1 knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J Biol Chem. 2000;275:9270–7 doi: 10.1074/jbc.275.13.9270. PMID:10734066. [DOI] [PubMed] [Google Scholar]
- 10.Shiota C, Larsson O, Shelton KD, Shiota M, Efanov AM, Hoy M, Lindner J, Kooptiwut S, Juntti-Berggren L, Gromada J, et al.. Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem. 2002;277:37176–83 doi: 10.1074/jbc.M206757200. PMID:12149271. [DOI] [PubMed] [Google Scholar]
- 11.De Leon DD, Li C, Delson MI, Matschinsky FM, Stanley CA, Stoffers DA. Exendin-(9-39) corrects fasting hypoglycemia in SUR-1-/- mice by lowering cAMP in pancreatic beta-cells and inhibiting insulin secretion. J Biol Chem. 2008;283:25786–93 doi: 10.1074/jbc.M804372200. PMID:18635551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Speakman JR. Measuring energy metabolism in the mouse – theoretical, practical, and analytical considerations. Front Physiol. 2013;4:34 doi: 10.3389/fphys.2013.00034. PMID:23504620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seino S, Miki T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol. 2003;81:133–76 doi: 10.1016/S0079-6107(02)00053-6. PMID:12565699. [DOI] [PubMed] [Google Scholar]
- 14.Aguilar-Bryan L, Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev. 1999;20:101–35 PMID:10204114. [DOI] [PubMed] [Google Scholar]
- 15.Calabria AC, Li C, Gallagher PR, Stanley CA, De Leon DD. GLP-1 receptor antagonist exendin-(9-39) elevates fasting blood glucose levels in congenital hyperinsulinism owing to inactivating mutations in the ATP-sensitive K+ channel. Diabetes. 2012;61:2585–91 doi: 10.2337/db12-0166. PMID:22855730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varela GM, Antwi DA, Dhir R, Yin X, Singhal NS, Graham MJ, Crooke RM, Ahima RS. Inhibition of ADRP prevents diet-induced insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2008;295:G621–628 doi: 10.1152/ajpgi.90204.2008. PMID:18669627. [DOI] [PMC free article] [PubMed] [Google Scholar]