

Abstract
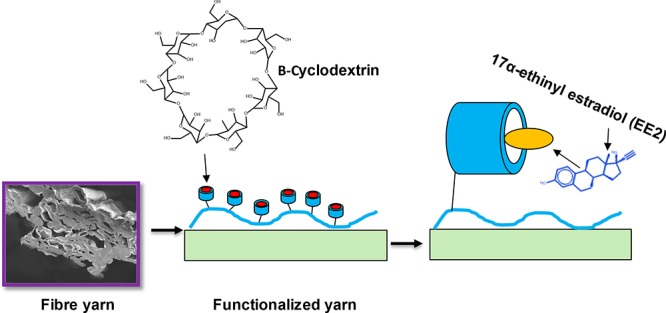
A wood based yarn platform for capturing pharmaceutical molecules from water was developed. Cellulose fiber yarns were modified with cyclodextrins, and the capture of 17α-ethinyl estradiol (EE2), a synthetic estrogen hormone used as contraceptive, from water was tested. The yarns were prepared by spinning a deep eutectic solution (DES) of cellulose in choline chloride-urea. Despite their high porosity and water sorption capacity (5 g/g), the spun fiber yarns displayed high wet strength, up to 60% of that recorded in dry condition (128 MPa with 17% strain at break). Cyclodextrin irreversible attachment on the yarns was achieved with adsorbed chitosan and the conjugation reactions and capture of EE2 by the cyclodextrin-modified cellulose were confirmed via online detection with Surface Plasmon Resonance (SPR). The facile synthesis of the bioactive yarns and EE2 binding capacity from aqueous matrices (as high as 2.5 mg/g) indicate excellent prospects for inexpensive platforms in disposable affinity filtration. The study presents a strategy to produce a wood fiber based yarn to be used as a platform for human and veterinary pharmaceutical hormone capture.
Introduction
Besides their advantages as far as carbon neutrality, wood-based cellulose materials have an extraordinarily large application potential due to their cost, biodegradability, and recyclability. They are suitable for manufacturing both long-lasting and disposable products that compete favorably against oil-based materials.1,2 Cellulosic fibers and fibrils are widely utilized in applications that include paper and board, films, filters, absorbents, and nonwoven materials for hygiene and home care.3 Because lignocellulosics typically are hydrophilic, they tend to swell when in contact with water and swell extensively, especially if they are present in open structures. This is due to the hydroxyl groups of cellulose, which form hydrogen bonds with water molecules.4 It is thus reasonable to state that the extent of water sorption is mainly limited by the number of the accessible hydroxyl groups.5−7 In contrast to native fibers and fibrils, cellulosic man-made filaments, for example, produced by regeneration,8 have a dominant dense, crystalline cellulose structure that limits water uptake.6 Recently, we reported a method to produce yarns by spinning native cellulose fibers dispersed in a deep eutectic solvent (DES).9 This method combined the water absorbing character of wood fibers with the characteristic one-dimensional structures in regenerated cellulose filaments.
Global awareness of environmental risks posed by estrogen based human and veterinary pharmaceutical hormone residues in sewage waters has raised during recently.10,11 Since conventional water purification systems are not capable to remove these residues, they end up in rivers, lakes, and seas. This is a major problem given the fact that synthetic hormone steroids have numerous negative consequences in humans, livestock, and wildlife.12 One of the most persistent steroid hormones, 17α-ethinyl estradiol (EE2), is widely used in contraceptive pills. The concentrations of the EE2 in river waters in Europe and the US have been reported to reach levels of 0.35 and 831 ng/L, respectively.13,14 Estrogen hormones have been utilized in pharmaceutical veterinary operations to increase the meat production, and thus, the estrogen hormone (E1 (estrone), E2 (estradiol), and E3 (estridiol)) levels up to 75 μg/L in the sewage waters have been reported.
The capture of estrogen hormones from water can be carried out by using affinity binding approach. Antiethinyl estradiol antibodies has been reported to bind specifically EE2 from water matrices.15−20 However, the use of antibodies in large volume applications has been difficult due the ethical and economic implications. An alternative to antibodies are cyclodextrins that are known to remove organic micropollutants from water.21 Cyclodextrins are ring shaped glucose-based structures with hydrophobic cavities that have been used for the solubilization of hydrophobic molecules in water in a wide range of applications (pharmacy, food, chemistry, chromatography, catalysis, biotechnology, agriculture, cosmetics, hygiene, medicine, textiles, and the environment).22−24 Since cyclodextrins only display hydroxyl functional groups on their outer surfaces, chemical modification is required covalent binding to the hydroxyl groups of cellulose.25 A large number of chemistries to immobilize cyclodextrins onto cellulose have been reported.26 Typically, these methods are unable to control the orientation of the cyclodextrin on the surfaces of the given substrate. Thus, there is a need for new avenues to covalently immobilize cyclodextrins on cellulose surfaces in an oriented manner, for example, by utilizing primary amine-containing molecules as a molecular anchor. An advancement in this direction was the use of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO)-mediated oxidation to develop carboxylate groups from the C6-hydroxyls of cyclodextrins.27 This opens the possibility for cationic biopolymers, such as chitosan, which can conveniently add amine groups onto the cellulosic materials,28,29 taking advantage of its glucosamine units and the strong affinity between the two polysaccharides.30,31
In this work, the water absorption properties of cellulose fiber yarns, their high wet strength and affinity to chitosan, were combined with the immobilization of TEMPO-oxidized cyclodextrins for the capture of 17α-ethinyl estradiol from aqueous matrices (Scheme 1). The carboxylation of the β-cyclodextrin was investigated by 1H NMR and Fourier transform infrared (FTIR) measurements. The immobilization reactions and EE2 capture were confirmed with surface plasmon resonance (SPR). The studies indicated that carboxylated β-cyclodextrin can be conjugated onto the cellulose surface by using chitosan as an anchor, and the cyclodextrin-functionalized surface was produced a five-fold increase in EE2 capture when compared to the reference system, cyclodextrin-free, chitosan-modified cellulose. The proposed platform represents an advancement toward the development of cellulose materials that are able to selectively remove harmful hormone estrogens from water.
Scheme 1. EE2 Hormone-Capturing Fiber Yarns Obtained by DES Solution Regeneration and Synthesized by First Adsorbing Chitosan Followed by Covalent Conjugation of Carboxylated Cyclodextrins.

Cyclodextrin moieties were able to bind EE2 hormones non-specifically from water matrices.
Experimental Section
Materials
Bleached pine chemical pulp fibers (Metsä Pulp, Finland) were used for fiber yarn manufacture. For experiments with SPR, bleached birch pulp fibers (Metsä Pulp, Finland) were used to prepare cellulose nanofibril (CNF) surfaces. β-Cyclodextrin (#C4767, purity >97%), choline chloride (#C1879, purity >98%), TEMPO (#214000, 2,2,6,6-tetramethyl-1-piperidinyloxy, free radical, purity 98%), urea (#U5378, purity >98%), PAA (#181285, poly(acrylic acid) , Mv ≈ 450 kg/mol), EDC (#03450, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride, purity >98%), NHS (#130672, N-hydroxysuccinimide, purity 98%), and EE2 (#E4876, 17α-ethinyl estradiol, purity >98%) were obtained from Sigma-Aldrich, Finland. Gold-coated sensors for surface plasmon resonance (SPR) were obtained from Bionavis Ltd., Finland. All other chemicals used in this study were laboratory grade. Water was double purified by a Milli-Q device.
Methods
Preparation of Fiber Yarn by Using the DES Method
The cellulose fiber yarn used in this study was prepared by using a deep eutectic solvent (DES) as a spinning medium, as described elsewhere.9 Briefly, never dried cellulose fibers were first washed to sodium form by lowering the pH of the suspension (to pH 2 with hydrochloric acid). After one-hour acid treatment, the fibers were washed several times with deionized water followed by titration into the sodium form by addition of NaHCO3. Finally, the excess sodium hydrogen carbonate was removed by washing the fibers with Milli-Q water. Bleached pine fibers were washed with water and acetone using filtration cycles followed by acetone evaporation in a vacuum oven at 40 °C.
The spinning medium, DES, was prepared by mixing choline chloride and urea (1:2 ratio) by using known procedures.32 In a 1-L reactor vessel, the washed cellulose fibers were mixed overnight into DES under constant stirring (4.5 w-% cellulose fibers content, 100 °C). When a uniform spinning dope was achieved, PAA (10 w-%) was added by utilizing a speed mixer (FlackTek Inc., UK) under constant 1600 rpm for 10 min in vacuum. The fiber yarn was prepared by using a laboratory scale spinning device by using ethanol as coagulant. The spinning rate was kept constant, ∼1.4 mL/min, and the nozzle diameter was 0.63 mm. After spinning, the prepared fiber yarn was kept in fresh ethanol for two hours to remove the DES solvent and the yarn was dried in ambient conditions. The fiber yarn, carrying 10% PAA, was cross-linked by using a laboratory oven (140 °C for 30 min) following the procedure presented earlier by us.33 The detailed procedure and analyses of the fiber yarn properties can be found in Tenhunen et al.9
Mechanical Strength of Fiber Yarn in Dry and Wet Conditions
The mechanical strength of the fiber yarn was measured by utilizing an Instron Universal Testing Instrument (model 33R4204) operating in tensile mode, with a 100 N static load cell attached. A strain rate of 2 mm min–1 was applied to each specimen (average dimensions ∼20.00 mm length, ∼100 μm diameter), with samples conditioned at 23 °C and 50% relative humidity for at least 88 h prior to analysis. Results presented are the average of five repetitions.
Water Absorption Properties of Fiber Yarn
Swelling of the fiber yarn was tested by utilizing the gravimetric equilibrium swelling test. First, 3 cm long fiber yarns were kept in a desiccator overnight followed by weighting (w0). The weighted samples were then placed into Milli-Q-water for two hours. After swelling, the samples were shortly wiped with a blotting paper and reweighted (w1). The water swelling capacity of the fiber yarn was calculated from seven repetitions.
The amount of loosely bound water in the swollen fiber yarn was tested by following the ISO 17190–6 standard. A preweighed fiber yarn pad was placed into a centrifugal sack, which was subsequently immersed in Milli-Q-water for 30 min. The loose water in the fiber yarn pad was removed by utilizing a centrifugal acceleration of 250g. Then the sample was reweighed and the average water retention capacity was calculated from ten parallel measurements.
The water absorption capacity under compression was measured following the ISO 17190–7 standard. A preweighed fiber yarn pad was allowed to swell in Milli-Q-water under 0.3 mechanical pressure of 0.3 psi for 60 min. Then the sample was weighed and the water swelling capacity under load was calculated from two parallel repetitions.
Optical and Scanning Electron Microscopy Imaging
Swelling of the fiber yarn in water was observed utilizing a Nikon H550S optical microscope with a 40× TU Plan Fluor objective. The fiber yarn was taped from the ends onto a microscope glass. A spot at the yarn surface was imaged following the addition of a droplet of Milli-Q-water on the yarn. When the swelling of the yarn progressed, microscope imaging was performed. The thickness of the fiber yarn was calculated from the microscopy images by utilizing the Nikon imaging software. The measurement was repeated five times to calculate the average value for the swelling.
The SEM imaging was performed by utilizing a Merlin FE-SEM (Carl Zeiss NTS GmbH, Germany) with gold sputter coating. First, all samples were fastened onto aluminum specimen stubs with double-sided carbon adhesive discs. Then the samples were coated with a thin layer of gold to prevent charging of sample surfaces under electron beam. The imaging was performed using the 5 keV electron energy with the secondary electron detector. The size of all SEM images was fixed at 2048 × 1536 pixels.
Selective TEMPO-Oxidation of β-Cyclodextrin
β-Cyclodextrin was first selectively carboxylated by using neutral TEMPO-NaClO-NaClO2 oxidation.34 One gram of β-cyclodextrin was dissolved in 90 mL of sodium phosphate buffer (0.05M, pH 6.8). Then 0.016 g of TEMPO (0.1 mmol/g) and 1.13 g of sodium chlorite (80%, 10 mmol) were dissolved into the cyclodextrin solution. The TEMPO-oxidation was initialized by an addition of sodium hypochlorite in the reaction buffer (1.0 mmol/g concentration in the reaction solution). The reaction was allowed to proceed in a closed bottle under laboratory conditions (23 °C) for 19.5 h. The carboxylate β-CD was purified by dialyzing against Milli-Q-water in a 100–500 Da dialysis membrane tube (MWCO 500 Da, Spectra/Por, Spectrum laboratories) until no changes in conductivity of the dialyze water were observed. The purified TEMPO-oxidized β-cyclodextrin (referred as TO-CD) was dehydrated by using a rotary evaporator and subsequent lyophilization via freeze-drying. The dry TO-CD powder was stored in a desiccator until use. As a reference, alkaline TEMPO-NaBr-NaClO reaction (room temperature, TEMPO 0.13 mmol/g, NaBr 4.7 mmol/g, NaClO 5.65 mmol/g, pH 10, oxidation time 30 min)35 was tested on the carboxylation of CD. The alkaline TEMPO-oxidized CD was purified using the same procedure used with neutral TEMPO-mediated oxidation.
Immobilization of TEMPO-Oxidized β-Cyclodextrin (TO-CD) on Fiber Yarn
The fiber yarn was first aminated by adsorbing chitosan from 50 mM NaOAC at pH 5 at a concentration of 0.5 g/L. The yarn was placed in a large volume of the chitosan solution and it was allowed to react for one hour. The chitosan-modified fiber yarn was purified with the 50 mM NaOAC buffer at pH 5 several times to remove unabsorbed chitosan. For covalent conjugation of TO-CD to the chitosan-modified fiber yarn, aqueous EDC/NHS coupling was utilized, which catalyzed the formation of amide bond between carboxyl and primary amine groups.36 Then 0.025 g of TO-CD, 0.06 g of EDC, and 0.106 g of NHS were dissolved into 50 mL of 50 mM NaOAc buffer at pH 5. Then the chitosan-modified yarn was placed into the reaction medium, and the solution was allowed to react overnight in room atmosphere. The TO-CD decorated fiber yarn (referred as CD-fiber yarn) was first washed several times with 50 mM NaOAc buffer at pH 5 and then with Milli-Q-water to remove unreacted chemicals. The CD-fiber yarn was removed from water by evaporation in ambient conditions. The prepared, dry functional fiber yarn was stored in room conditions.
Preparation of CNF Thin Films for SPR Experiments
CNF thin films were spin coated onto gold coated SPR sensors by using PEI anchoring.37 CNF were mechanically disintegrated from bleached birch pulp with sequential Masuko grinding (six passes) and microfluidization (10 passes) treatment. The individual cellulose nanofibrils were then produced by using tip ultrasonication (0.148w-% CNF in Milli-Q-water, 400W tip sonicator, Brandon 450 Digital Sonifier, Branson Ultrasonics, Danbury, USA) with a 10 min treatment time at 25% amplitude. The CNF fibril bundles were removed by using centrifugation at 10 400 rpm for 45 min followed by collecting fibrils from the supernatant by manual pipetting. The collected cellulose nanofibrils were then spin-coated onto PEI coated gold SPR sensors by using a spin coater (WS-650SX-6NPP, Laurell Technologies, PA, USA, 3000 rpm and 90 s spinning time).
Surface Plasmon Resonance (SPR) To Verify TO-CD Conjugation onto Cellulose and EE2 Capture by CD-Modified Cellulose
Conjugation of TO-CD onto cellulose by using chitosan as a molecule anchor was investigated in real-time by using a multiparametric surface plasmon resonance instrument MP-SPR Navi210A (Oy BioNavis Ltd., Finland). The measurements were carried out with gold-covered SPR sensors carrying spin-coated cellulose nanofibrils (CNF) that were utilized as a model for the surface of the fiber yarns. The thickness of the adsorbed layer was calculated based on the change of the SPR angle by using eq 1:38
| 1 |
where Δangle is the change in the MP-SPR angle, ld is a characteristic evanescent electromagnetic field decay length, assumed to be 0.37 of the light wavelength (240 nm), m is the sensitivity factor for the sensor obtained after calibration of the MP-SPR (109.94°/RIU), na is the refractive index of the adsorbed substance, and n0 is the refractive index of the bulk solution. The refractive indices utilized in the estimations were 1.5 for chitosan, 1.45 for TO-CD, and 1.623 for EE2. The adsorbed mass of chitosan was calculated from the thickness estimation by using specific mass density of chitosan (1.77 g/cm3 for chitosan). All SPR measurements were carried out at 23 °C with a 10 μL/min flow rate. All sample points were in duplicates, at least. The CNF-coated SPR sensors were kept in an oven (80 °C) for 10 min to ensure the fibrils attach to the PEI surface. The CNF-coated sensors were stored in a desiccator, and prior to use in SPR studies, they were stabilized overnight in Milli-Q-water.
The SPR studies were conducted by allowing 50 mM NaOAc buffer at pH 5 to flow over the sensor surfaces until no change in the SPR signal was observed. The 0.5 g/L chitosan in buffer (50 mM NaOAc at pH 5) was allowed to adsorb onto the CNF surface for 20 min followed by rinsing with buffer (50 mM NaOAc at pH 5) to remove nonadsorbed chitosan. The TO-CD was conjugate onto the chitosan modified CNF surface by allowing 0.5 g/L TO-CD (50 mM NaOAc at pH 5) with 0.06 g EDC and 0.106 g NHS to flow over the sensor surface for 20 min. The EDC and NHS contents were two- and six-fold, respectively, to the theoretical amount of the carboxyl groups in a TO-CD molecule. The unreacted TO-CD molecules were removed from the SPR chamber by allowing the buffer (50 mM NaOAc buffer at pH 5) to flow for 30 min following the buffer exchange into 50 mM phosphate buffer at pH 7.4. The 17α-ethinyl estradiol (EE2) binding with the conjugated CD was tested by allowing 2 μg/mL EE2 (50 mM phosphate at pH 7.4) to adsorb onto chitosan modified CNF surface with and without conjugated TO-CD. The binding time was 20 min followed by rinsing with buffer (50 mM phosphate at pH 7.4). After SPR measurements, the sensor surfaces were shortly washed with Milli-Q-water and stored in a desiccator prior to analysis with atomic force microscopy (AFM).
Colloidal Probe Microscopy (AFM) Imaging of CD-Modified CNF Surfaces
Surface topography of the CNF surfaces, before and after TO-CD conjugation in the SPR, was characterized by an atomic force microscope (AFM+, Anasys Instruments, Santa Barbara, USA). The imaging was carried out by using the tapping mode with silicon cantilevers that were obtained from μMash (tip radius 8 nm). The image size was constant 5 × 5 μm2, and at least three different spots on each sensor were investigated. No image processing except flattening was utilized.
Fourier Transform Infrared Reflectance (FTIR) Microscopy Analyses
The chemical features of β-cyclodextrin with and without neutral TEMPO-oxidation was characterized with a Thermo Scientific Nicolet iS50 FT-IR spectrometer with an ATR diamond (Thermo Scientific, USA). Also, the chemistry of the fiber yarn was characterized prior and after conjugation of cyclodextrin via chitosan attachment. All spectra were obtained from 32 scans with a resolution of 4 cm–1 and transmission mode by using the wavelength range from 400–4000 cm–1.
1H-Nuclear Magnetic Resonance Spectroscopy for Characterizing Carboxylation of TEMPO-Oxidized Cyclodextrin
The chemical structure of the β-cyclodextrin before and after neutral TEMPO-oxidation was characterized by 1H NMR spectroscopy. NMR spectra were recorded with a Bruker AVANCE III 500 NMR spectrometer with a magnetic flux density of 11.7 T, equipped with a 5 mm BB(F)O double resonance probe head. Samples were dissolved in D2O, with concentration of 30 mg/mL. All spectra were recorded at 22 °C with 32 scans for each spectrum and using a 30-degree flip angle rf-pulse for excitation. The delay between successive scans was 1.5 s, signal acquisition time was 2.0 s, and the spectral width 8 kHz. The spectra were processed with TopSpin 3.5 software.
Capture of EE2 Hormone from Water Solution with Cyclodextrin-Functionalized Fiber Yarn (CD-Fiber Yarn) by UV–vis Spectroscopy
The ability of the CD-fiber yarn to remove EE2 hormone from water was measured by a Lambda 900 UV/vis/NIR spectrometer (PerkinElmer, USA). The 2.5 cm long pieces of the CD-fiber yarn were swelled in 50 mM phosphate buffer at pH 7.4 for overnight to prevent the swelling effect during the measurement. The EE2 solutions in 50 mM phosphate buffer at pH 7.4 were prepared with the concentrations of 0, 0.1875, 0.375, 0.75, and 1.25 μg/mL. The prepared EE2 solutions were applied in measurement cuvettes (volume 3 mL) and their light absorbances at 280 nm were measured. Then swollen CD-fiber yarn samples were placed into the cuvettes (one piece of the CD-fiber yarn per cuvette) and the capture was allowed to progress for 30 min. Then the yarns were removed from the cuvettes, and the absorbance at 280 nm was remeasured. The decrease in the light intensity is proportional to the binding of EE2 from the solution. As a reference, absorbent measurements were carried out with chitosan modified fiber yarn without coupled CD. All sample points were duplicated.
Results
Structural and Water Absorption Properties of Fiber Yarn
The fiber yarn was manufactured by the utilizing the dry-jet wet spinning approach with choline chloride/urea, deep eutectic solvent (DES), as a spinning medium. The details of the yarn manufacturing have been presented previously by Tenhunen et al.9 When wood fibers in the DES solution have been forced through a narrow nozzle, they tend to orientate mainly into the parallel orientation (Figure 1a). Similar behavior takes also place when regenerated cellulose or CNF filaments are produced.39,40 The spun yarn was coagulated in pure ethanol, an antisolvent for cellulose fibers and poly(acrylic acid) (PAA). When all DES was removed from the fiber yarn, it was dried in air followed by cross-linking in an oven.33 The produced yarn had a porous inner structure, seen as open fiber lumens and cavities between the cellulose fibers (Figure 1b). The thickness of the produced fiber yarn was approximately 320 μm. The thickness of the yarn was significantly larger compared to that of typical regenerated cellulose filaments due to the dimensions of used cellulose fibers.8 On the SEM images was evidence of polymeric ribbons between the fiber surfaces (Figure 1c,d). The actual chemical content of the ribbons is not clear, but it can be speculated that the added cross-linker, PAA, formed polymeric ribbons onto the fiber surfaces and physical bridges between the cellulose fibers.
Figure 1.

SEM images of the (a) surface and (b) cross-section of the fiber yarn. SEM images of the possible poly(acrylic acid) (PAA) ribbons (c) on a cellulose fiber and (d) in the interface between cellulose fibers.
Cellulose fiber materials have typically poor wet strength properties since the material bonding takes place through hydrogen bonds, which are disrupted by water.41 The conditioned dry strength of the fiber yarn was approximately 128 ± 16 MPa that was reduced to approximately 65 ± 9 MPa when the yarn was kept in water. Therefore, it is evident that that the PAA cross-links the fiber structure. Moreover, the wet elongation of the fiber yarn was smaller compared to dry elongation (10 ± 1 vs 17 ± 4%, respectively), conceivably due to the PAA bridging between fibers. The mechanical properties of the produced fiber yarn were lower compared to that of reported for regenerated cellulose and CNF filaments.8,39,42−45 The reason for this observation is the lower specific surface area of wood fibers that leads to smaller number of hydrogen bonds and the porous inner structure.
The free swelling capacity of the prepared fiber yarn was approximately 4.9 ± 0.8 g/g. The measured value is significantly higher compared to that reported for cotton and regenerated viscose rayon, lyocell, and modal fibers (0.31–0.33, 0.36, 0.22, and 0.17 g/g, respectively).46−49 The water in the fiber yarn was loosely bound, which can be seen in the lower water retention capacity (1.7 ± 0.2 g/g) when centrifugation was applied. However, the value is almost twice to that of neat cellulose fibers, which highlights the effect of PAA.4 The manufactured fiber yarn resisted compression significantly since the swelling under compression of the yarn pad was 9 ± 1 g/g. The fiber yarn expanded significantly when it came into contact with water (Figure 2). The thickness of the fiber yarn increased approximately 166% (dry and wet thicknesses of 324 ± 25 and 539 ± 50 μm, respectively) when a droplet of water was applied on the yarn. This correlates with the cross-section area change of 276% (the fiber yarn cross-section was assumed to be a circle). The most probable reason for the observed large expansion is the contribution of PAA, a known water superabsorbent. The observed expansion potential is significantly higher than that of regenerated cellulose filaments. As an example, lyocell filaments have been reported to expand approximately 30% when kept in water.50
Figure 2.

Optical microscopy images of a fiber yarn (a) before and (b) after placing a water droplet on the yarn.
Carboxylation of β-Cyclodextrin with Neutral TEMPO-Oxidation
Both neutral and alkaline TEMPO-mediated oxidation chemistries were applied for the selective carboxylation of CD molecules. The recorded FTIR spectra (Figure 3a) reveled clearly that both TEMPO-oxidation approaches successfully installed carboxyl moieties on CD molecules. This can be seen from the appearance of two new peaks at 1600 and 1411 cm–1 wavenumbers, assigned to the characteristic peaks of carboxylic acid salts.51 Compared to the neutral oxidation, the alkaline TEMPO-oxidation caused more extensive carboxylation; the peak intensity after alkaline oxidation at 1600 and 1411 1/cm were larger compared to that after neutral oxidation. However, on the basis of the FTIR analysis, it is not possible to speculate if the ring structure of CD was damaged during the carboxylation.
Figure 3.

(a) FTIR spectra of the β-cyclodextrin before and after TEMPO-oxidation with alkaline and neutral TEMPO-mediated oxidation. (b) 1H-NMR spectra of β-cyclodextrin before and after TEMPO-oxidation. (i) nonoxidated cyclodextrin. (ii) TEMPO-oxidized cyclodextrin, neutral conditions. (iii) TEMPO-oxidized cyclodextrin, alkaline conditions.
The 1H NMR measurements were carried out to verify that TEMPO-oxidation did not break the ring structure of CD. Figure 3b shows the spectra from a reference nonoxidized b-cyclodextrin sample, and TO-CD with oxidation carried out in neutral and alkaline conditions. The signals from nonoxidized CD are in an agreement with earlier reports,52 and resolved to the extent that the J-couplings can be readily observed. The relative intensity of the signal from H6 at 3.77 ppm decreased in the spectrum of the TO-CD, indicating disappearance of the CH2–group due to the oxidation. However, the 1H NMR spectrum of the TO-CD oxidized in alkaline conditions shows only broad featureless signals. A potential cause for the observed signal broadening is an opening of the dextrin ring in the alkaline oxidation reaction. After breaking of the CD ring (deuterated), water becomes less favorable solvent for it, leading to aggregation of the molecules. On the other hand, the spectrum of TO-CD from neutral reaction conditions still showed signals with narrow line widths, albeit overlapped with increased complexity, suggesting that the ring structure remained intact during the oxidation. However, some evidence of degradation of the CD molecule can be observed, for example, the relatively weak unresolved signals at 4.2–4.0 ppm. According to diffusion ordered spectrum (DOSY, data not shown), these belong to a species with a slightly larger diffusion coefficient, which could indicate breaking of the small portion from CD into smaller fragments. Earlier, alkaline TEMPO-mediated oxidation has been utilized in the selectively oxidation of C6 hydroxyls of a CD molecule.27 In that study, the alkaline TEMPO-oxidation was carried out in an ice bath that caused slower reaction (while also preventing unwanted side reactions), which can explain the difference with our results conducted in room temperature. Moreover, it has been shown that compared with the alkaline TEMPO-oxidation, the neutral conditions favors a more gentle carboxylation of the cellulosic materials.34
Surface Plasmon Resonance (SPR) To Verify Conjugation of TO-CD to Cellulose and EE2 Capture
CNF films were used as models for the surface of wood fiber. The SPR study showed that the chitosan adsorbed irreversibly onto the CNF surface (Figure 4a). The average thickness of the adsorbed chitosan layer was estimated to be approximately 0.53 nm (surface coverage of approximately 93 ng/cm2). In an acidic solution, the amine group of chitosan are positively charged, producing a favorable electrostatic interaction between slightly anionic CNF and positively charged chitosan.29,53 Moreover, chitosan and cellulose have identical backbones that cause a natural affinity for adhesion.54 These two factors cause the chitosan to adsorb strongly onto cellulose with a flat adsorption configuration.29 When TO-CD with EDC and NHS was injected onto the chitosan-modified surface, a large raise in the SPR signal was observed. This is explained by the liquid effect since the SPR method is also sensitive to the changes in the refractivity of the bulk liquid.55 After rinsing, the signal did not return to the starting level, which indicates that the CD was bound onto the chitosan modified cellulose surface. The average thickness of the CD layer was approximately 1 nm, which correlates rather well with the height of a β-CD molecule (0.78 nm).56 This result suggests that the adhered CD layer was likely in the form of a monolayer. The SPR estimation were carried out with pure CNF surfaces, whereas the fiber yarn contain 10% of PAA. Its effect on the chitosan adsorption and subsequent CD coupling was not studied with the SPR. We can assume that anionically charged PAA increases chitosan adsorption on the fiber surfaces via electrostatic interaction. Moreover, small part of carboxyls of PAA can be coupled with amines of chitosan within the EDC/NHS activation.
Figure 4.
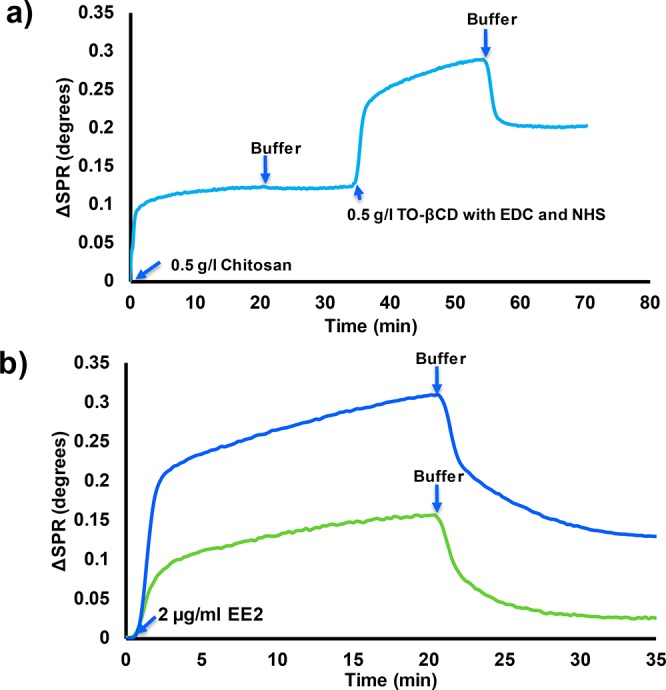
(a) Surface plasmon resonance (SPR) spectrum on the conjugation of TEMPO-oxidized β-cyclodextrin on the CNF surface with EDC/NHS chemistry by using chitosan as an anchor. (b) SPR spectra on the adsorption of 17α-ethinyl estradiol (EE2) on the chitosan modified CNF with (blue curve) and without (green curve) conjugated β-cyclodextrin.
The binding of 17α-ethinyl estradiol (EE2) on the CD modified CNF surface was tested also by SPR. It was observed that the adsorption of the EE2 was five-fold higher when conjugated CD was present on the chitosan-modified CNF surface (Figure 4b). The thicknesses of the adsorbed EE2 layers on the chitosan modified CNF without and with conjugated CD were approximately 0.095 and 0.5 nm, respectively. The small EE2 adsorption on chitosan is mostly caused by the hydrophobicity of EE2 (solubility in water 9.2 μg/mL).57 The development of biosensors to detect steroid hormones from sewage waters by using the SPR method has already been discussed in detail.20
The topological changes on the CNF surface after chitosan adsorption and subsequent CD conjugation were imaged by AFM. The pure CNF surface composed of cellulose fibrils that were evenly spread on the SPR sensors (Figure 5a). When chitosan was adsorbed onto the CNF surface, no drastic changes in topology of the fibrils were observed (Figure 5b). This is accordance with the SPR measurement where the adsorbed chitosan layer was observed to be thin. The RMS roughness values of the CNF surface with and without adsorbed chitosan were 4.53 ± 0.3 and 3.75 ± 0.25 μm, respectively. This result suggests that chitosan forms an even adsorption layer on the CNF surface. Also, changes on the surface topography after CD conjugation were limited (Figure 5c). The dimensions of a CD molecule have been reported to be 0.78 × 15.4 nm2.56 Therefore, when these molecules are packed densely on a surface, no significant changes in topography should be observed by AFM (the radius of the used AFM tip was 8 nm). The AFM measurements were carried out with pure CNF films, whereas fiber yarn contain 10% of PAA, the effect of which on the topography of CNF surface was not studied with AFM. In the manufacture of fiber yarn, PAA attachment on the fiber surfaces takes place through precipitation with ethanol and subsequent evaporation, which are difficult to analyze with CNF thin films.
Figure 5.

AFM height images of (a) pure CNF surface, (b) chitosan modified CNF surface, and (c) β-cyclodextrin functionalized CNF surface by using chitosan as an anchor. The Z-scale of the images is 10 nm.
Preparation of CD Functionalized Fiber Yarn
The conjugation of TO-CD onto the fiber yarn was carried out similarly as in the SPR studies. The fiber yarn was kept in a chitosan solution followed by the purification with the buffer to release loosely adhered chitosan. Then the chitosan-modified fiber yarn was placed into the TO-CD solution with EDC and NHS for overnight. When the reaction was carried out, the CD-fiber yarn was purified with buffer washing and dried in laboratory conditions. The conjugation process was followed indirectly by using the FTIR-ATR technique. The fiber yarn contains 10% of PAA as a wet strength additive that is seen as two new peaks at 1710 and 1560 1/cm (Figure 6), compared to the spectrum of the neat fibers, which correlate with the carboxylic acid and carboxylic salt.51 The fingerprint region (900–1200 1/cm) of cellulose was identical to that reported for native cellulose.58 When the fiber yarn was treated with chitosan, a small decrease in the peak at 1560 1/cm (carboxylate) was observed. Moreover, a small elevation at 1640 1/cm was observed that correlates with the NH-bending of primary amines.51 When TO-CD was conjugated onto the chitosan modified fiber yarn, a clear elevation at 1640 1/cm was observed again that correlates with the amide bond formation (peak range for amide bond is 1680–1630).51 It is important to note that from the FTIR signal it is not possible to isolate the small effect of possible amine bond formation in the interface between chitosan and PAA. However, taking the SPR and FTIR measurements together, the success in the conjugation of TO-CD onto the fiber yarn can be concluded. The fiber yarn with and without chitosan adsorption and subsequent TO-CD conjugation were also characterized by utilizing the SEM imaging (Figure S1, Supporting Information). However, no significant changes in the topography of the yarns were observed. This further proves that the investigated conjugation method was gentle for cellulose materials.
Figure 6.

FTIR spectra of raw pulp used to prepare the fiber yarn, fiber yarn containing 10% PAA, chitosan modified fiber yarn, and CD decorated chitosan modified fiber yarn.
Capture of EE2 from Water with CD-Fiber Yarn
The EE2 capture of the prepared CD-fiber yarn was evaluated by using indirect UV–vis absorbance measurements. The investigation was performed by comparing the change in the EE2 concentration when a piece of CD-fiber yarn was kept in the solution for 30 min. Before the measurements, the CD-fiber yarn was kept in the given buffer to prevent any effect from bulk liquid absorption that could drive hormone transport. First, a calibration curve was measured for EE2 solutions (EE2 concentrations from 0 to 1.25 μg/mL in 50 mM phosphate buffer at pH 7.4, Figure 7). When pieces of wet CD-fiber yarn were kept in the measurement cuvettes, clear changes in the absorbances of the EE2 solutions were observed. However, it is important to note that the UV–vis method is not capable of detecting EE2 concentrations < 0.1 μg/mL, the limit of UV–vis for samples of real sewage water. When the EE2 concentration was below 0.4 μg/mL, the prepared CD-fiber yarn removed almost all of the EE2 from the solution. Above 0.4 μg/mL, EE2 removal was limited by the capacity of the CD-fiber yarn to capture EE2 molecules. The effect of CD molecules on EE2 capture is evident since the chitosan modified fiber yarn without CD did not cause any change in the absorbance of the EE2 solutions. The EE2 capture capacity of the CD-fiber yarn was estimated based on the UV-measurements and gravimetry. The CD-fiber yarn was estimated to bind up to 2.5 mg/g of EE2 (Figure 7) since the equilibrium of the EE2 capture was not achieved within studied conditions. The shape of the EE2 capture isotherm was following rather well (in low concentrations, the measurement accuracy of the UV-absorbance may vary the curve profile) the model of Langmuir I isotherm that is used to express monolayer type adsorption behavior on surfaces.59 The use of carbon materials to bind EE2 from water have been studied in literature. Solid carbon fibers have reported to bind 1.8 mg/g of EE2 from aqueous solutions.60 Moreover, fullerene and carbon nanotubes have been reported to bind EE2 0.23 and 0.1 mg/g, respectively,61 whereas activated carbon absorbent takes 10.4–27.6 mg/g ethinyl estradiol due to the extremely high surface area.62 Regenerated cellulose has been reported to be a relatively poor absorbent material for EE2.63 Thus, the observed capture properties of the CD-fiber yarn were competitive when compared to the literature values. However, the EE2 capture with CD is based on nonselective hydrophobic interactions. The specific capture can be achieved by utilizing antibodies as we have shown earlier for different target molecules with CNF filaments.64 Compared to active carbon and other materials used for EE2 capture, the CD-fiber yarn allows to produce filters enabling size exclusion and affinity separation. The CD-fiber yarn based membranes and nonwovens can be designed to reduce the trans-membrane pressure and conveniently be modified to induce selectivity as well as used in multiple capturing cycles since CD moieties can be regenerated.21 Moreover, the CD-fiber yarn can be utilized in disposable applications and incinerated to destroy the captured hormones. However, it is important to note that the capture ability of the developed CD-fiber yarn need to be tested later with real estrogen containing hormone water samples to further proof the concept. Overall, the study shows that the investigated fiber yarn material exhibits interesting swelling properties that can be combined with specific functionalities for affinity filtration applications.
Figure 7.
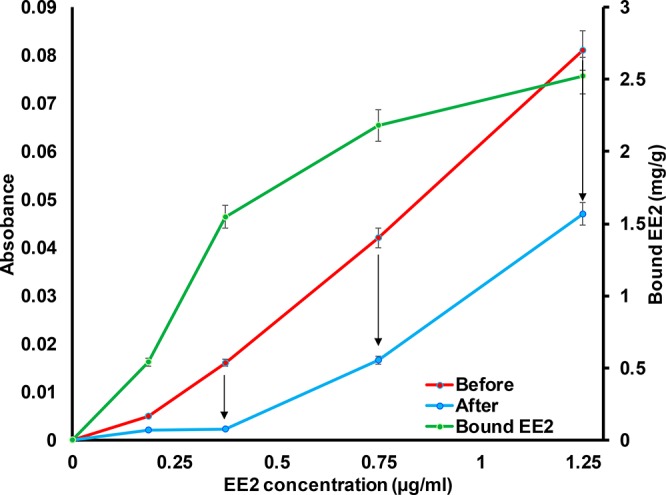
UV–vis absorbance at 280 nm of 17α-ethinyl estradiol (EE2) in 50 mM phosphate buffer at pH 7.4 before and after contacting 2.5 cm long β-cyclodextrin modified fiber. The β-cyclodextrin modified fiber yarns were swollen in the given buffer solution before being placed in the measurement cuvette (liquid volume 3 mL) for 30 min. Before light absorbance measurement, the yarns were removed from the cuvettes. The bound EE2 (mg/g CD-fiber yarn) was calculated based on the concentration of EE2 change and weight-length of the yarn.
Conclusions
Cellulose fiber yarns carrying cyclodextrin were used for 17α-ethinyl estradiol (EE2) capture. The prepared fiber yarn presented an open inner structure that swelled significantly in water. The produced yarn material resists compression while also exhibiting good water absorption properties under tension forces. The conjugation of cyclodextrin onto the fiber yarn was carried out by utilizing aqueous EDC/NHS chemistry using chitosan as an anchor molecule. The carboxylation of cyclodextrin was carried out by using neutral TEMPO-mediated oxidation, and the successful insertion was verified using FTIR and NMR spectroscopies. The conjugation of carboxylated cyclodextrin onto the surfaces of the fiber yarn and capture of EE2 was estimated by surface plasmon resonance (SPR) with cellulose nanofibril (CNF) thin films. The SPR studies revealed that the conjugation of carboxylated cyclodextrin onto chitosan modified CNF was successful and the prepared CD surface captured five-fold more EE2 than the reference surface. Finally, the preparation of the cyclodextrin functionalized fiber yarn was manufactured and the capture of EE2 was confirmed with UV–vis measurements. The EE2 capture potential of the prepared yarn was 2.5 mg/g.
Acknowledgments
Hille Rautkoski and Vuokko Liukkonen are thanked for preparing of cellulose fibre yarn. Ville Klar is thanked for helping with the development of suitable spinning machinery. This work was carried out under the funding of the Design Driven Value Chains in the World of Cellulose project (DWOC) funded by Tekes (Finnish Funding Agency for Innovation).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.7b01765.
SEM images of the pure, chitosan modified, and cyclodextrin functionalized fiber yarn (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Schlamadinger B.; Marland G. The role of forest and bioenergy strategies in the global carbon cycle. Biomass Bioenergy 1996, 10 (5–6), 275–300. 10.1016/0961-9534(95)00113-1. [DOI] [Google Scholar]
- Sathre R.; Gustavsson L. Using wood products to mitigate climate change: External costs and structural change. Appl. Energy 2009, 86 (2), 251–257. 10.1016/j.apenergy.2008.04.007. [DOI] [Google Scholar]
- Sjöström E.Wood Chemistry: Fundamentals and Applications; Academic Press: San Diego, 1993. [Google Scholar]
- Klemm D.Comprehensive Cellulose Chemistry. Vol. 1, Fundamentals and Analytical Methods; Wiley-VCH: Weinheim, 1998. [Google Scholar]
- O’Sullivan A. Cellulose: the structure slowly unravels. Cellulose 1997, 4 (3), 173–207. 10.1023/A:1018431705579. [DOI] [Google Scholar]
- Howsmon J. A. Water Sorption and the Poly-Phase Structure of Cellulose Fibers. Text. Res. J. 1949, 19 (3), 152–162. 10.1177/004051754901900303. [DOI] [Google Scholar]
- Ciolacu D.; Ciolacu F.; Popa V. I. Amorphous cellulose--structure and characterization. Cellul. Chem. Technol. 2011, 45 (1), 13. [Google Scholar]
- Cook J. G.Handbook of Textile Fibres: Man-Made Fibres; Elsevier Science, 1984. [Google Scholar]
- Tenhunen T.-M.; Hakalahti M.; Kouko J.; Salminen A.; Härkäsalmi T.; Pere J.; Harlin A.; Hänninen T. Method for Forming Pulp Fibre Yarns Developed by a Design-driven Process. BioResources 2015, 11 (1), 2492–2503. 10.15376/biores.11.1.2492-2503. [DOI] [Google Scholar]
- Adeel M.; Song X.; Wang Y.; Francis D.; Yang Y. Environmental impact of estrogens on human, animal and plant life: A critical review. Environ. Int. 2017, 99, 107–119. 10.1016/j.envint.2016.12.010. [DOI] [PubMed] [Google Scholar]
- Bartelt-Hunt S.; Snow D. D.; Damon-Powell T.; Miesbach D. Occurrence of steroid hormones and antibiotics in shallow groundwater impacted by livestock waste control facilities. J. Contam. Hydrol. 2011, 123 (3), 94–103. 10.1016/j.jconhyd.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Ying G.-G.; Kookana R. S.; Ru Y.-J. Occurrence and fate of hormone steroids in the environment. Environ. Int. 2002, 28 (6), 545–551. 10.1016/S0160-4120(02)00075-2. [DOI] [PubMed] [Google Scholar]
- Fine D. D.; Breidenbach G. P.; Price T. L.; Hutchins S. R. Quantitation of estrogens in ground water and swine lagoon samples using solid-phase extraction, pentafluorobenzyl/trimethylsilyl derivatizations and gas chromatography–negative ion chemical ionization tandem mass spectrometry. J. Chromatogr. A 2003, 1017 (1), 167–185. 10.1016/j.chroma.2003.08.021. [DOI] [PubMed] [Google Scholar]
- Johnson A. C.; Dumont E.; Williams R. J.; Oldenkamp R.; Cisowska I.; Sumpter J. P. Do Concentrations of Ethinylestradiol, Estradiol, and Diclofenac in European Rivers Exceed Proposed EU Environmental Quality Standards?. Environ. Sci. Technol. 2013, 47 (21), 12297–12304. 10.1021/es4030035. [DOI] [PubMed] [Google Scholar]
- Beaumont J. L.; Lemort N. Oral contraceptive, pulmonary artery thrombosis and anti-ethinyl-oestradiol monoclonal IgG. Clin. Exp. Immunol. 1976, 24 (3), 455–463. [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jiang L.; Xiang X.; Xu S.; Wen R.; Liu X. Competitive sorption between 17α-ethinyl estradiol and bisphenol A/4-n-nonylphenol by soils. J. Environ. Sci. 2013, 25 (6), 1154–1163. 10.1016/S1001-0742(12)60165-X. [DOI] [PubMed] [Google Scholar]
- Martínez N. A.; Pereira S. V.; Bertolino F. A.; Schneider R. J.; Messina G. A.; Raba J. Electrochemical detection of a powerful estrogenic endocrine disruptor: Ethinylestradiol in water samples through bioseparation procedure. Anal. Chim. Acta 2012, 723, 27–32. 10.1016/j.aca.2012.02.033. [DOI] [PubMed] [Google Scholar]
- Yañez C.; Basualdo J.; Jara-Ulloa P.; Squella J. A. Inclusion complexes of estrone and estradiol with β-cyclodextrin: Voltammetric and HPLC studies. J. Phys. Org. Chem. 2007, 20 (7), 499–505. 10.1002/poc.1186. [DOI] [Google Scholar]
- Buijs J.; Hlady V. Adsorption Kinetics, Conformation, and Mobility of the Growth Hormone and Lysozyme on Solid Surfaces, Studied with TIRF. J. Colloid Interface Sci. 1997, 190 (1), 171–181. 10.1006/jcis.1997.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habauzit D.; Chopineau J.; Roig B. SPR-based biosensors: a tool for biodetection of hormonal compounds. Anal. Bioanal. Chem. 2007, 387 (4), 1215–1223. 10.1007/s00216-006-0958-4. [DOI] [PubMed] [Google Scholar]
- Alsbaiee A.; Smith B. J.; Xiao L.; Ling Y.; Helbling D. E.; Dichtel W. R. Rapid removal of organic micropollutants from water by a porous β-cyclodextrin polymer. Nature 2016, 529 (7585), 190–194. 10.1038/nature16185. [DOI] [PubMed] [Google Scholar]
- Sobrinho J. L. S.; Soares M. F.; Labandeira J. J. T.; Alves L. D. S.; Rolim Neto P. J. Improving the solubility of the antichagasic drug benznidazole through formation of inclusion complexes with cyclodextrins. Quim. Nova 2011, 34, 1534–1538. 10.1590/S0100-40422011000900010. [DOI] [Google Scholar]
- Pavlov G. M.; Korneeva E. V.; Smolina N. A.; Schubert U. S. Hydrodynamic properties of cyclodextrin molecules in dilute solutions. Eur. Biophys. J. 2010, 39, 371–379. 10.1007/s00249-008-0394-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crini G. Review: A History of Cyclodextrins. Chem. Rev. 2014, 114 (21), 10940–10975. 10.1021/cr500081p. [DOI] [PubMed] [Google Scholar]
- Hermanson G. T.Bioconjugate Techniques; Academic Press: San Diego, CA, 2008; Vol. 2. [Google Scholar]
- Bhaskara-Amrit U. R.; Agrawal P. B.; Warmoeskerken M. M. C. G. Applications of β-cyclodextrins in textiles. Autex Res. J. 2011, 11 (4), 94–101. [Google Scholar]
- Fraschini C.; Vignon M. R. Selective oxidation of primary alcohol groups of β-cyclodextrin mediated by 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO). Carbohydr. Res. 2000, 328 (4), 585–589. 10.1016/S0008-6215(00)00129-4. [DOI] [PubMed] [Google Scholar]
- Fras Zemljič L.; Strnad S.; Šauperl O.; Stana-Kleinschek K. Characterization of Amino Groups for Cotton Fibers Coated with Chitosan. Text. Res. J. 2009, 79 (3), 219–226. 10.1177/0040517508093592. [DOI] [Google Scholar]
- Orelma H.; Filpponen I.; Johansson L.-S.; Laine J.; Rojas O. J. Modification of Cellulose Films by Adsorption of CMC and Chitosan for Controlled Attachment of Biomolecules. Biomacromolecules 2011, 12 (12), 4311–4318. 10.1021/bm201236a. [DOI] [PubMed] [Google Scholar]
- Da Roz A. L.; Leite F. L.; Pereiro L. V.; Nascente P. A. P.; Zucolotto V.; Oliveira O. N. Jr.; Carvalho A. J. F. Adsorption of chitosan on spin-coated cellulose films. Carbohydr. Polym. 2010, 80 (1), 65–70. 10.1016/j.carbpol.2009.10.062. [DOI] [Google Scholar]
- Ravi Kumar M. N. V. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 46 (1), 1–27. 10.1016/S1381-5148(00)00038-9. [DOI] [Google Scholar]
- Abbott A. P.; Capper G.; Davies D. L.; Rasheed R. K.; Tambyrajah V. Novel solvent properties of choline chloride/urea mixtures. Chem. Commun. 2003, (1), 70–71. 10.1039/b210714g. [DOI] [PubMed] [Google Scholar]
- Spoljaric S.; Salminen A.; Luong N.; Seppälä J. Crosslinked nanofibrillated cellulose: poly(acrylic acid) nanocomposite films; enhanced mechanical performance in aqueous environments. Cellulose 2013, 20 (6), 2991–3005. 10.1007/s10570-013-0061-x. [DOI] [Google Scholar]
- Saito T.; Hirota M.; Tamura N.; Kimura S.; Fukuzumi H.; Heux L.; Isogai A. Individualization of Nano-Sized Plant Cellulose Fibrils by Direct Surface Carboxylation Using TEMPO Catalyst under Neutral Conditions. Biomacromolecules 2009, 10 (7), 1992–1996. 10.1021/bm900414t. [DOI] [PubMed] [Google Scholar]
- Isogai A.; Kato Y. Preparation of polyuronic acid from cellulose by TEMPO-mediated oxidation. Cellulose 1998, 5 (5), 153–164. 10.1023/A:1009208603673. [DOI] [Google Scholar]
- Staros J. V.; Wright R. W.; Swingle D. M. Enhancement by N-hydroxysulfosuccinimide of water-soluble carbodiimide-mediated coupling reactions. Anal. Biochem. 1986, 156 (1), 220–222. 10.1016/0003-2697(86)90176-4. [DOI] [PubMed] [Google Scholar]
- Orelma H.; Filpponen I.; Johansson L.-S.; Österberg M.; Rojas O.; Laine J. Surface Functionalized Nanofibrillar Cellulose (NFC) Film as a Platform for Immunoassays and Diagnostics. Biointerphases 2012, 7 (1–4), 1–12. 10.1007/s13758-012-0061-7. [DOI] [PubMed] [Google Scholar]
- Jung L. S.; Campbell C. T.; Chinowsky T. M.; Mar M. N.; Yee S. S. Quantitative Interpretation of the Response of Surface Plasmon Resonance Sensors to Adsorbed Films. Langmuir 1998, 14 (19), 5636–5648. 10.1021/la971228b. [DOI] [Google Scholar]
- Iwamoto S.; Isogai A.; Iwata T. Structure and Mechanical Properties of Wet-Spun Fibers Made from Natural Cellulose Nanofibers. Biomacromolecules 2011, 12 (3), 831–836. 10.1021/bm101510r. [DOI] [PubMed] [Google Scholar]
- Fink H.-P.; Weigel P.; Purz H.; Ganster J. Structure formation of regenerated cellulose materials from NMMO-solutions. Prog. Polym. Sci. 2001, 26 (9), 1473–1524. 10.1016/S0079-6700(01)00025-9. [DOI] [Google Scholar]
- Lindström T.; Wågberg L.; Larsson T.. On the nature of joint strength in paper-A review of dry and wet strength resins used in paper manufacturing. In 13th Fundamental Research Symposium; The Pulp and Paper Fundamental Research Society: Cambridge, UK, 2005; Vol. 1, pp 457–562. [Google Scholar]
- Hauru L. K. J.; Hummel M.; Michud A.; Sixta H. Dry jet-wet spinning of strong cellulose filaments from ionic liquid solution. Cellulose 2014, 21 (6), 4471–4481. 10.1007/s10570-014-0414-0. [DOI] [Google Scholar]
- Walther A.; Timonen J. V. I.; Díez I.; Laukkanen A.; Ikkala O. Multifunctional High-Performance Biofibers Based on Wet-Extrusion of Renewable Native Cellulose Nanofibrils. Adv. Mater. 2011, 23 (26), 2924–2928. 10.1002/adma.201100580. [DOI] [PubMed] [Google Scholar]
- Lundahl M. J.; Cunha A. G.; Rojo E.; Papageorgiou A. C.; Rautkari L.; Arboleda J. C.; Rojas O. J. Strength and Water Interactions of Cellulose I Filaments Wet-Spun from Cellulose Nanofibril Hydrogels. Sci. Rep. 2016, 6, 30695. 10.1038/srep30695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundahl M. J.; Klar V.; Wang L.; Ago M.; Rojas O. J. Spinning of Cellulose Nanofibrils into Filaments: A Review. Ind. Eng. Chem. Res. 2017, 56 (1), 8–19. 10.1021/acs.iecr.6b04010. [DOI] [Google Scholar]
- Stamm A. J.Wood and Cellulose Science; Ronald Press Co.: New York, 1964. [Google Scholar]
- Moore A. T.; Scott L. W.; deGruy I. V.; Rollins M. L. The Swelling of Cotton in Water. Text. Res. J. 1950, 20 (9), 620–630. 10.1177/004051755002000904. [DOI] [Google Scholar]
- Welo L. A.; Ziifle H. M.; Loeb L. Swelling Capacities of Fibers in Water. Text. Res. J. 1952, 22 (4), 254–261. 10.1177/004051755202200403. [DOI] [Google Scholar]
- Kreze T.; Stana-Kleinschek K.; Ribitsch V. The sorption behaviour of cellulose fibres. Lenzinger Berichte 2001, 80, 28–33. [Google Scholar]
- Ibbett R. N.; Hsieh Y.-L. Effect of fiber swelling on the structure of lyocell fabrics. Text. Res. J. 2001, 71 (2), 164–173. 10.1177/004051750107100212. [DOI] [Google Scholar]
- Coates J. Interpretation of infrared spectra, a practical approach. Encycl. Anal. Chem. 2006, 1. 10.1002/9780470027318.a5606. [DOI] [Google Scholar]
- Salvatierra D.; Jaime C.; Virgili A.; Sánchez-Ferrando F. Determination of the Inclusion Geometry for the β-Cyclodextrin/Benzoic Acid Complex by NMR and Molecular Modeling. J. Org. Chem. 1996, 61 (26), 9578–9581. 10.1021/jo9612032. [DOI] [Google Scholar]
- Myllytie P.; Salmi J.; Laine J. The influence of pH on the adsorption and interaction of chitosan with cellulose. Bioresources 2009, 4 (4), 1647–1662. [Google Scholar]
- Mishima T.; Hisamatsu M.; York W. S.; Teranishi K.; Yamada T. Adhesion of [beta]–glucans to cellulose. Carbohydr. Res. 1998, 308 (3–4), 389–395. 10.1016/S0008-6215(98)00099-8. [DOI] [Google Scholar]
- Schasfoort R. B. M.; Tudos A. J.; Gedig E. T.. Handbook of Surface Plasmon Resonance; The Royal Society of Chemistry: Cambridge, UK, 2008. [Google Scholar]
- Chierotti M. R.; Gobetto R. Solid-state NMR studies of weak interactions in supramolecular systems. Chem. Commun. 2008, (14), 1621–1634. 10.1039/b711551b. [DOI] [PubMed] [Google Scholar]
- Shareef A.; Angove M. J.; Wells J. D.; Johnson B. B. Aqueous Solubilities of Estrone, 17β-Estradiol, 17α-Ethynylestradiol, and Bisphenol A. J. Chem. Eng. Data 2006, 51 (3), 879–881. 10.1021/je050318c. [DOI] [Google Scholar]
- Oh S. Y.; Yoo D. Il; Shin Y.; Seo G. FTIR analysis of cellulose treated with sodium hydroxide and carbon dioxide. Carbohydr. Res. 2005, 340 (3), 417–428. 10.1016/j.carres.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Adamson A.; Gast A.. Physical Chemistry of Surfaces; Wiley-Interscience, 1997. [Google Scholar]
- Hristovski K. D.; Nguyen H.; Westerhoff P. K. Removal of arsenate and 17α-ethinyl estradiol (EE2) by iron (hydr)oxide modified activated carbon fibers. J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng. 2009, 44 (4), 354–361. 10.1080/10934520802659695. [DOI] [PubMed] [Google Scholar]
- Pan B.; Lin D.; Mashayekhi H.; Xing B. Adsorption and Hysteresis of Bisphenol A and 17α-Ethinyl Estradiol on Carbon Nanomaterials. Environ. Sci. Technol. 2008, 42 (15), 5480–5485. 10.1021/es8001184. [DOI] [PubMed] [Google Scholar]
- Han J.; Qiu W.; Cao Z.; Hu J.; Gao W. Adsorption of ethinylestradiol (EE2) on polyamide 612: Molecular modeling and effects of water chemistry. Water Res. 2013, 47 (7), 2273–2284. 10.1016/j.watres.2013.01.046. [DOI] [PubMed] [Google Scholar]
- Han J.; Meng S.; Dong Y.; Hu J.; Gao W. Capturing hormones and bisphenol A from water via sustained hydrogen bond driven sorption in polyamide microfiltration membranes. Water Res. 2013, 47 (1), 197–208. 10.1016/j.watres.2012.09.055. [DOI] [PubMed] [Google Scholar]
- Vuoriluoto M.; Orelma H.; Lundahl M.; Borghei M.; Rojas O. J. Filaments with Affinity Binding and Wet Strength Can Be Achieved by Spinning Bifunctional Cellulose Nanofibrils. Biomacromolecules 2017, 18 (6), 1803–1813. 10.1021/acs.biomac.7b00256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.