

Abstract

A continuous and scalable method for the wet spinning of cellulose nanofibrils (CNFs) is introduced in a core/shell configuration. Control on the interfacial interactions was possible by the choice of the shell material and coagulant, as demonstrated here with guar gum (GG) and cellulose acetate (CA). Upon coagulation in acetone, ethanol, or water, GG and CA formed supporting polymer shells that interacted to different degrees with the CNF core. Coagulation rate was shown to markedly influence the CNF orientation in the filament and, as a result, its mechanical strength. The fastest coagulation noted for the CNF/GG core/shell system in acetone led to an orientation index of ∼0.55 (Herman’s orientation parameter of 0.40), Young’s modulus of ∼2.1 GPa, a tensile strength of ∼70 MPa, and a tenacity of ∼8 cN/tex. The system that underwent the slowest coagulation rate (CNF/GG in ethanol) displayed a limited CNF orientation but achieved an intermediate level of mechanical resistance, owing to the strong core/shell interfacial affinity. By using CA as the supporting shell, it was possible to spin CNF into filaments with high water absorption capacity (43 g water/g dry filament). This was explained by the fact that water (used as the coagulant for CA) limited the densification of the CNF core structure, yielding filaments with high accessible area and pore density.
Keywords: wet spinning, core/shell, coaxial filaments, continuous spinning, nanocellulose, water absorption, absorbent filaments
Introduction
Fibers form the basis of a multitude of materials that capitalize on their high aspect ratio and anisotropy (different properties in the axial and radial directions). With the renewed interest to shift from synthetic fibers to renewable ones, new technologies have been introduced for their development. As an example, fibers spun from cellulose nanofibrils (CNFs) have been shown to be suitable as reinforcement and for given functional properties1,2 as well as for moisture sorption.3 The latter use, however, has been largely overlooked if compared to the traditional efforts to optimize the mechanical strength.
Interest toward renewable absorbents is growing because of the need for a sustainable, low-cost response to the growing demand for consumables, from personal care products, diapers, and towels to advanced wound dressings. Typically, absorbents are produced in the form of three-dimensional, cross-linked hydrogels,4−6 aerogels,7,8 fibers,9−11 or their combinations,12,13 often using synthetic polymers, such as acrylic acid, acrylamide, and acrylonitrile. Recently, biodegradable, biocompatible, and inexpensive alternatives have been actively searched from cellulose and its derivatives.14,15
Related to the present study, functionalized cellulose fibers have been spun into yarns with ability to both absorb water and capture pharmaceutical molecules.16 This kind of one-dimensional form of cellulose is beneficial when developing woven or nonwoven structures, which further enhance the absorption capacity beyond the limits of single filaments. When using CNFs, absorbent materials have been mostly developed in the form of aerogels,17,18 which are prepared through an expensive freeze-drying process. In this study, we produced absorbent one-dimensional filaments from CNFs through low-cost high-throughput core/shell wet spinning using supporting biopolymer shells. In addition, we demonstrate how the water absorption can be tailored through the choice of the components and the coagulation system.
In addition, we demonstrate how the shell material, which is spun with CNF, as well as the coagulant influence the alignment of cellulose fibrils in the ensuing filament. So far, the CNF orientation in spun filaments has been mainly addressed considering the extrusion speed, nozzle size, and solids content of the CNF dope,3,19−21 as well as drawing.22−26 Even though it has been shown that the coagulation conditions play an important role on the orientation and structure development of man-made fibers, such as Lyocell,27 more attention is needed to understand their effect on CNFs.
Moreover, the methods reported so far for CNF wet spinning involve rather small processing rates (Table S5, Supporting Information). For instance, the only examples available for continuous wet spinning of CNFs involve spinning rates of 2–13 m/min.24,28 Only an alternative method, such as dry spinning, has surpassed these limits (660 m/min)29 and allowed for CNF drawing in an integrated process. Herein, we demonstrate a prototype wet-spinning line with production rates of 33 m/min, which takes advantage of the role of a supporting polymer (used as a shell) to facilitate the core/shell spinning of CNF.
The selection of the shell component allows the control of the properties of the bicomponent filament. Here, two biopolymers were used, such as cellulose acetate (CA) and guar gum (GG). CA is a rather hydrophobic cellulose derivative with well-established spinnability.30,31 It can support fast spinning and even drawing of CNF, resulting in a bicomponent, core/shell filament. Furthermore, CA makes it suitable to coagulate the filament in water, which, as will be demonstrated later, maximizes the water absorption capacity of the CNF core. CA has already been blended with cellulose nanocrystals to produce fibers by electrospinning32 and dry spinning.33 On one hand, cellulose nanocrystals increased the viscosity when added to a CA solution, which was taken as evidence of the existence of cellulose–CA interactions.32 On the other hand, CA developed remarkable hydrogen bonding with cellulose nanocrystals in electrospun fibers only upon the deacetylation of CA into regenerated cellulose,34 which signifies poor compatibility between cellulose and CA. Similarly, CNF and CA are used here with the expectation that they have a limited compatibility, which can facilitate the formation of a sufficiently loose filament structure to improve the water absorbance capacity.
Compared to CA, the hydrophilic GG shell has a higher affinity with the CNF core. GG consists of galactose residues substituted on a linear mannan backbone with a mannose–galactose ratio of 1.6–1.8.35 Such composition enables abundant hydroxyl groups in GG to engage in hydrogen bonding with cellulose. Earlier, hydrophilic poly(vinyl alcohol) (PVA) has been shown to effectively (hydrogen) bond with cellulose nanocrystals in electrospun fibers, thus even decreasing the susceptibility of the fiber to humidity.36 Similarly, a strong connection can be expected between the core and the shell of a CNF/GG filament. In addition, acetone is a suitable antisolvent for both GG and CNF, enabling their fast coagulation.
A CNF filament with a shell comprising silica nanoparticles was recently proposed for the purpose of flame retardancy, which points to the possibility of core/shell filaments with multiple functions.28 In electrospinning, core/shell geometries have been employed to prepare hollow fibers by selective removal of the core component.37,38 Herein, we selectively remove the CA shell, resulting in a continuous CNF filament. Furthermore, the method proposed here can be adjusted to core/shell combinations for multifunction systems.
Experimental Section
Materials
CNF preparation is described in detail in ref (3). In brief, bleached hardwood fibers were refined and fluidized for six passes through a high-pressure microfluidizer operating at a solids content of 1.5 or 2 wt % in deionized water. The batch with a solids content of 1.5 wt % was used for preparing most of the samples. Before use, the CNF suspension was stirred for approximately 30 min to break flocs and consecutively mixed in vacuum for 10 min to remove air bubbles. For the absorbent filaments spun into water with varying drawing ratio (DR), the batch at a solids content of 2 wt % was diluted to 1 wt %.
To compare the water absorption properties of the filaments to other forms of cellulose, films were prepared from CNF hydrogels (solids content 2 wt %) and from the refined wood fibers used as the precursor for CNF preparation. These samples were first diluted with an excess of acetone to simulate the acetone exposure that CNF experiences during filament preparation. The obtained dispersions were cast on a dish and dried in air at room temperature overnight or in a fume hood for a minimum of 3 h.
CA with a degree of substitution of ∼2.5 and an average Mn of 30 000 was purchased from Sigma-Aldrich and used as a 15 wt % solution in acetone. GG (typical molecular weight in the range of 1–3 millions),39−42 also from Sigma-Aldrich, was used as a 1 wt % dispersion in deionized water. Before use, the GG dispersion was stirred for an appropriate time for the sample to foam and warm up to ∼40 °C (10–20 min). After this, the introduced air bubbles were removed by mixing in vacuum for 10 min. The GG dispersion was prepared fresh before each experiment.
Wet Spinning
The spinning conditions are specified in Table S1 of the Supporting Information. In brief, core and shell dopes were loaded in syringes connected to the core and shell sections of a coaxial needle (Figure 1a). Both components were extruded at a controlled speed into the coagulation bath and incubated in the coagulant (acetone or ethanol) for ∼5 min. Subsequently, the filament was collected on a heated winder for drying (Figure 1a). Ensuing bicomponent filaments are referred to with the names including first the core and then the shell component; for example, CNF/CA refers to a filament with a CNF core and a CA shell. The control sample with CNF only was prepared by dissolving the CA shell of the CNF/CA filament with acetone (>1 h immersion). The process was repeated twice more, using fresh acetone to ensure a CA-free, neat CNF filament. The control sample with GG only was spun to acetone using a single needle (core section only, Table S1). The control sample with CA only was spun according to the conditions used in the drawing experiments, as will be explained below.
Figure 1.
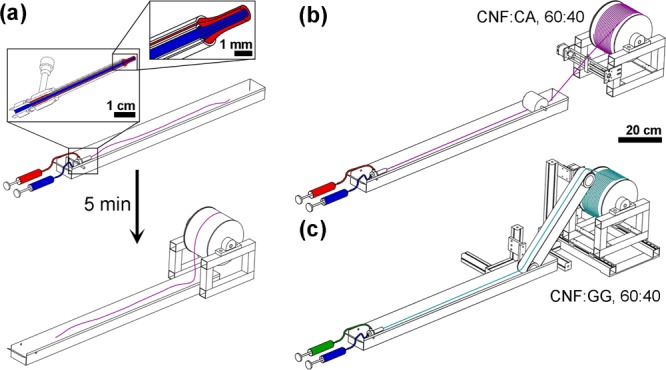
Schematic illustrations of the used spinning systems. (a) Spinning of standard samples in batches. The inset shows the structure of the core/shell needle along with the extruded filament. (b) Continuous spinning of CNF/CA core/shell filaments; CNF in blue and CA in red. (c) Continuous spinning of the CNF/GG core/shell filament, including a conveyor belt for additional support for the wet filament; CNF in blue and GG in green. Ratios of the components in the continuous filaments are noted in the image.
In order to determine the maximum production rate, CNF (1 wt %) and CA (15 wt %) were spun to a 1:1 mixture of deionized water and ethanol. The obtained filament was collected on the heated winder directly without additional incubation time (Figure 1b). In the case of CNF (1.5 wt %) with GG (1 wt %), a conveyor belt was added to the spinning line to facilitate winding because the fresh CNF/GG filament had insufficient wet strength for collection through air (Figure 1c). The spinning conditions applied in the continuous systems are presented in detail in Table S2 of the Supporting Information.
Wet Spinning of Absorbent Filaments with Drawing
A smaller version of the continuous CNF/CA system (Figure 1b), with a shorter coagulation bath (<40 cm) and a smaller winder (diameter 6 cm), was used to prepare absorbent filaments and study the effect of drawing. CNF and CA were spun into a deionized water bath and collected on a winder under direct heating with hot air. The draw was created by winding the filament faster than the extrusion speed, and the DR was calculated as the quotient between the winding and extrusion speeds. In the case of coaxial filaments, DR refers to the DR of CNF, unless specified otherwise.
The applied DR and extension rate were varied, as specified in the Supporting Information along with the spinning conditions (Tables S3 and S4). A single needle (with no shell compartment) was used for spinning a control sample of CA without CNF (Table S3). To obtain a control sample with (drawn) CNF, in the absence of CA, a portion of the CNF/CA filaments was immersed three times in acetone as explained above for ethanol-coagulated CNF/CA (Wet Spinning subsection). The success of CA removal was confirmed by Fourier transform infrared (FTIR) spectroscopy, using a Mattson 3000 FTIR spectrometer (Unicam). Sixteen scans were averaged with a resolution of 32 cm–1. Water-coagulated and drawn CNF/CA (DR = 5) was used for this measurement.
Coagulation Rate
The rate of the coagulation for CNF in given antisolvents can be estimated by calculating a parameter T for the diffusion of the water outward, from the filament to the coagulation bath, according to43
| 1 |
where Din is the diffusion coefficient of the coagulant into water (into the filament) and Dout is the diffusion coefficient of water into the coagulant (out of the filament).44 The calculation suggested that the CNF coagulates in acetone faster (T = 5580 s/cm2), compared to coagulation in ethanol (T = 54 630 s/cm2).
Two-Dimensional Wide-Angle X-ray Diffraction (2D-WAXD)
To study the orientation of the fibrils along the filament axis, 2D-WAXD patterns were captured with a PANalytical Empyrean multipurpose diffractometer (Almelo, The Netherlands) using Cu radiation with a wavelength of 0.15406 nm. The system was equipped with a focusing, graded multilayer X-ray mirror (Goebel type) for Cu radiation, a dedicated 2D-WAXD kit creating an X-ray beam with a diameter of 100 μm at the detector position, and a PIXcel3D 2 × 2 position-sensitive 2D detector imaging an angular range of ±31° with a static exposure. This detector has a pixel size of 55 μm and a detection efficiency of >95% for Cu radiation. The measurements were performed on single filaments, using a measurement time of 60 min per filament because of the low scattering efficiencies of these samples. Subsequent data treatment and azimuthal integrations along the diffraction rings were performed using the XRD2DScan software package. Before calculations, the baseline of the peaks was subtracted from the azimuthal intensity distributions, and thus, obtained data were normalized between 0 and 1.
Orientation index fc was calculated based on scattered intensity I as a function of the azimuthal angle φ for the lattice plane of 200 (the most intense peak) according to
| 2 |
where βc is the full width at half-maximum of the azimuthal peak, obtained from fitting the data with a Gaussian model following
| 3 |
where a, b, and c are peak-specific parameters. fc was determined for both azimuthal peaks, and the average of these values was reported. Also, the Herman’s orientation parameter S was calculated based on the azimuthal integration according to
| 4 |
Assuming cylindrical symmetry in the filament, average cosine ⟨cos2 γ⟩ was obtained from the average cosine of the azimuthal angle φ according to45
| 5 |
where
| 6 |
where φ0 is the azimuthal angle in the beginning of the range used for the calculation of the average cosine ⟨cos2 φ⟩. S was calculated at φ0 of 0, π/2, π, and 3π/2, and the average of these values was reported.
Filament Morphology
Filament cross sections were imaged with a Zeiss Sigma VP scanning electron microscope (Carl Zeiss Microscopy Ltd, Cambridge, UK) with 425–12 150× magnifications. The operating voltage was 2–3 kV and the working distance 6.4–16 mm. Prior to imaging, the samples were sputtered with a gold–palladium coating.
All filaments flattened upon contact with the drying winder. The cross-sectional areas were estimated assuming a rectangular geometry using the width and thickness measured, both 10 times for each sample. The widths were measured based on the images acquired with an optical microscope Leica DM750 equipped with a Leica ICC50 HD camera (Leica Microsystems, Germany). Filament thicknesses were measured with a digital micrometer gauge under a controlled pressure, according to the international standard used for paper products (ISO 534) at relative humidity (RH) 50% and 25 °C after overnight equilibration. The measured cross-sectional areas were used to normalize the values of tensile strength (i.e., maximum load divided by the cross-sectional area).
Apparent coarseness (i.e., linear density) was estimated by weighing a total length of approximately 4 m of each filament sample. The measured weight was divided with the length to attain apparent coarseness in tex (i.e., g/km). Coarseness was required to calculate filament tenacity (i.e., maximum load divided by coarseness), which provides an alternative measure of the tensile strength. Tenacity is equally important to report, as the values of tensile strength are heavily influenced by irregularities in the filament cross section. In addition to tenacity normalization, the apparent coarseness was also used to estimate the apparent density by assuming a constant cross section along the total length.
Filament Properties
For tensile testing in dry conditions, the samples were equilibrated for >12 h at RH 50%, 23 °C. For wet conditioning, the samples were immersed overnight in deionized water, prior to testing. Given the difficulties of geometry evaluation in wet conditions and as an approximation, the filament cross-sectional area in the wet state was assumed to be the same as that in the dry state. We note, though, that the filaments are expected to swell, and in the case of CNG/GG, GG can be partially dissolved during immersion.
Tensile tests were performed with a Universal Testing Instrument model 33R4204 (Instron, USA). Both dry and wet measurements were performed using a load cell of 100 N, a gauge length of 10 mm, and a test speed of 2 mm/min. Tests were repeated five times for each sample, except for the filament containing only GG, which was too weak to measure more than three repetitions in the dry state. In wet conditions, most of the samples became too weak to handle when they were soaked overnight. Only the filaments with CA only, CNF/CA coagulated in ethanol, and CNF/GG coagulated in acetone were tested using five repetitions. The sample of CNF/GG coagulated in ethanol and CNF (i.e., CNF/CA coagulated in ethanol, followed by CA removal) were tested twice.
The water absorption capacity of the filaments and comparison with the precursor materials were measured with the teabag method. Sections of a dry filament or film (∼0.2 g) were placed inside a teabag and immersed for 60 min, followed by draining and reweighing after 10 min. The same procedure was repeated for three blank teabags to obtain the absorption capacity per gram of teabag. The blank absorption was subtracted from the total water absorption of the samples to obtain the effective water absorbed by the samples, following the formula
| 7 |
where wstw is the weight of the wet sample and teabag, wsd is the weight of the dry sample, wtd is the weight of the dry teabag, and wwgt is the water absorption per gram of teabag (g water/g teabag). The measurement was repeated twice for each sample, apart from neat CA filament, which was measured only once.
Water contact angles were measured on films prepared from the same materials used for filament spinning. CNF and GG dispersions in water (solids content 1 wt %) and CA solution in acetone (solids content 15 wt %) were cast on dishes and dried overnight in air at room temperature. CNF was dried for additional 1 week and GG for additional 2 weeks in a fume hood. The initial static water contact angles of the obtained films were measured with a CAM 200 optical contact angle meter (KSV Instruments, Finland) at room temperature. Drops of 6.7 μL were deposited on the films and imaged repeatedly over a period of 17 s, starting immediately after deposition. The images were used to compute the contact angles. Five repetitions of the measurement were performed for each sample.
Results and Discussion
Coagulation Rate Determines CNF Orientation
The wet-spinning process is expected to organize the fibrils contained in the filament with a preferred orientation along the filament axis. The extent of uniaxial alignment can be influenced by the spinning conditions. Here, the orientation of the CNF component in the core/shell filaments spun at different coagulation conditions was studied by 2D-WAXD, assuming that the orientation of the cellulose crystallites provides an indication of the fibril orientation in a filament. The 2D-WAXD diffractograms for CNF-containing filaments (Figure 2a) consist of rings with radii corresponding to the scattering vector originated from the cellulose crystal planes 200 (outer ring, scattering vector 15.8 nm–1) as well as 110 and 11̅0 merged (inner ring, scattering vector 12 nm–1). These rings are highlighted as peaks in the radial integrations of the diffractograms depicted in Figure S3 of the Supporting Information. According to Figure 2a, the diffraction rings are fainter in the horizontal (filament axis) direction and more intense in the vertical one (perpendicular to the filament axis). This contrast arises from the preferred orientation of the crystallites along the filament axis direction.
Figure 2.
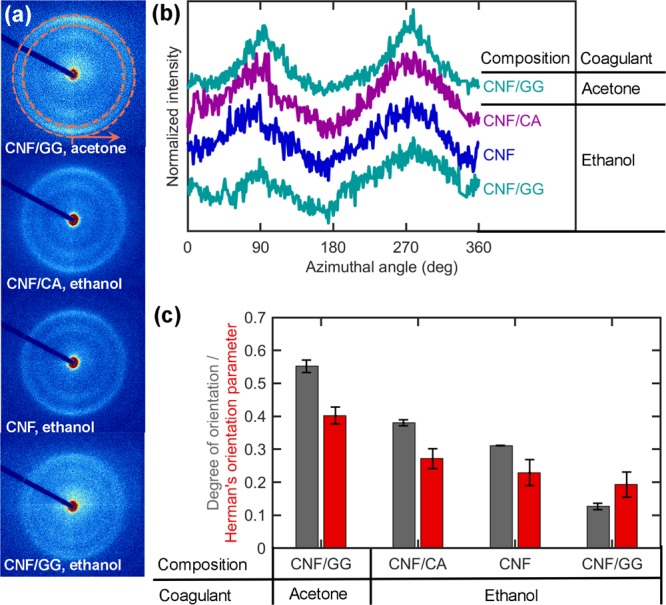
(a) 2D-WAXD diffractograms of filaments positioned horizontally. (b) Azimuthal intensity distributions of various filament samples. Orange circles in the first diffractogram in (a) indicate the position of the cellulose 200 peak and the tick and arrow the starting point and direction of the azimuthal integrations shown in (b). (c) Orientation indices (eq 2) and Herman’s orientation parameters (eq 4) of the samples calculated based on the azimuthal intensity distributions shown in (b). The error bars indicate the standard error among the parameters calculated with different peaks or values of φ0; that is, different sections in the azimuthal plot (b). Note that CNF/CA and CNF are the same filament before (CNF/CA) and after (CNF) the removal of the CA shell.
The effect of the preferred fibril alignment can be seen even more clearly when plotting the scattered intensity as a function of the azimuthal angle (Figure 2b). The intensity distributions show two peaks each, corresponding to the most intense sections of the diffraction rings in Figure 2a: one at 0 or 360° (bottom of the ring) and another one at 180° (top of the ring). These peaks appear well defined for the CNF/GG core/shell filament coagulated in acetone and the least pronounced for the same filament coagulated in ethanol.
This finding was validated by calculations (eqs 2 and 4) with the CNF/GG filament coagulated in acetone and ethanol, which indicated the highest and lowest orientation indices (0.55 ± 0.02 and 0.13 ± 0.01) as well as the highest and lowest Herman’s orientation parameters (0.40 ± 0.03 and 0.19 ± 0.04), respectively. Because these filaments are otherwise prepared similarly, the noted difference can be expected to arise from the faster CNF coagulation in acetone (T = 5580 s/cm2) than in ethanol (T = 54 630 s/cm2, eq 1). The faster the coagulation during wet spinning, the lesser the time required for the forming filament to relax back to random CNF orientation. To maximize the retained orientation, coagulation should occur faster than the relaxation time of the dope.46 Earlier, the relaxation timescale of carboxymethylated CNF from maximum alignment to maximum acceptable anisotropy (defined as broadening of the orientation distribution by 22.5°) has been estimated as ∼3.9 s. According to the present spinning experiments, coagulation of CNF was clearly not completed (filament not fully solidified) during this time. This can explain the relatively low orientation index and Herman’s orientation parameter in all the samples, compared to the maximum value (1) for a fully aligned structure.
Among the filaments that are coagulated in ethanol, the CNF alignment is enhanced by changing the shell material from GG to CA. This can be explained by the contribution of acetone present in the CA dope. Once extruded from the needle, the CNF hydrogel contacts first the shell dope, which starts to act as its primary coagulant. As ethanol diffuses through the shell into the CNF layer, it dilutes the acetone and slows down the coagulation. Nevertheless, as acetone already has had time to act on the CNF, better alignment is preserved than when using a GG shell dissolved in water, which tends to swell rather than coagulate CNF.
Most probably, effective coagulation not only preserves but also contributes to the alignment, as shown earlier by displaying CNF orientation, both during and after hydrodynamic alignment.22,47 CNF exhibited a maximum order parameter of 0.4 inside a single22 or 0.5 in a double47 flow-focusing channel. However, while traveling downstream through the channel, the initial alignment relaxed, causing its decline to values below 0.2 or 0.4, respectively. After emerging from the channel, the filament was transferred first to a water bath and then to an acetone bath and finally dried under tension. Following, the extruded filament not only regained but surpassed the initial maximum CNF alignment, achieving an order parameter of 0.522 or 0.7.47 This observation highlights the critical influence of coagulation and drying on CNF alignment. Similarly, during Lyocell spinning, cellulose molecules have been shown to first align in the spinneret and then relax in the die swell region in the beginning of the air gap.27 The lost orientation is regained and enhanced by drawing as well as by crystallization and solvent depletion during coagulation and drying.27 As such, coagulation and drying are critical steps that allow for property control of wet-spun CNF filaments.
Interestingly, the CNF/GG filament flattens less upon contact with the drying winder, after spinning in ethanol rather than in acetone (apparent thickness and width of 167 ± 5 and 353 ± 30 μm after coagulation in ethanol or 93 ± 2 and 424 ± 54 μm in acetone, respectively, Table S7 of the Supporting Information). These effects are shown in the SEM images of Figure 3e,d, respectively, and are most likely related to the spatial arrangement of CNF upon coagulation. After the coagulation in acetone or ethanol, CNF/GG contains more or less aligned fibrils, respectively (Figure 2). The more randomly oriented fibrils after ethanol coagulation may prevent the filament from flattening. Note that the coagulation rate in the respective solvents is not expected to influence the flattening resistance, at least directly, because of the 5 min coagulation time used (the filaments likely solidify approximately to their maximum extent). The effect of compression for the acetone-coagulated filament is also noted from the higher apparent density (0.89 ± 0.13 g/cm3) compared to the ethanol-coagulated one (0.60 ± 0.07 g/cm3, Table S7 of the Supporting Information). The filament densification probably occurs not only upon flattening but also already upon coagulation. Also in the case of CA, delayed coagulation has been shown to produce less dense filaments with more uniform porosity.43
Figure 3.
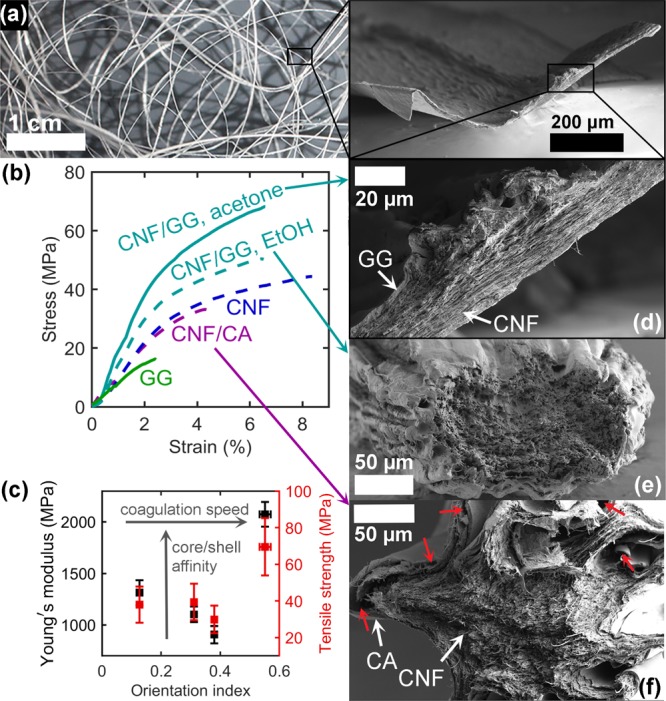
(a) Photograph of a bundle of CNF/GG filaments. The inset shows the full cross section of an acetone-coagulated filament. (b) Representative stress–strain curves of the filaments coagulated in ethanol (dashed lines) and acetone (solid lines). “CNF” refers to the CNF/CA filament after the removal of CA. (c) Young’s modulus and tensile strength as a function of the CNF orientation index along with the trends caused by increasing coagulation speed and core/shell affinity. Data points from left to right: CNF/GG, ethanol–CNF–CNF/CA–CNF–GG, acetone. (d–f) SEM images (low magnification) of the cross sections of CNF/GG from (d) acetone and (e) ethanol as well as CNF/CA (f) from ethanol. Red arrows indicate areas where the core and the shell are separated.
In CNF/CA with medium CNF orientation (Figure 2), flatter sections are found along with those that have collapsed into a starlike shape (Figure 3f). This filament had an average thickness and a width of 135 ± 9 μm and 599 ± 53 μm, respectively (Table S7, Supporting Information), suggesting a similar degree of flattening as with the acetone-coagulated CNF/GG. Flattening and star-shaped collapse can be beneficial in applications requiring filaments to be packed with a large contact area and friction, whereas a round and uniform cross section optimizes better the strength of a single filament.
Affinity between the Core and the Shell Strengthens the Filament
A bundle of CNF/GG core/shell filaments is presented in Figure 3a, and representative stress–strain curves for filaments with different core/shell combinations are shown in Figure 3b. Also, the average values for mechanical properties are shown in Table S7 of the Supporting Information. When changing the shell material from CA to GG, while keeping ethanol as the coagulant, the Young modulus of the filament (initial slope of the stress–strain curve), tensile strength (maximum stress per cross-sectional area), and tenacity (maximum stress per coarseness) increased from 907 ± 85 to 1313 ± 122 MPa, from 29.9 ± 7.5 to 38.0 ± 9.9 MPa, and from 4.01 ± 0.41 to 6.30 ± 0.90 cN/tex, respectively. This can be explained by the stronger affinity between CNF and GG, arising from the larger number of hydroxyl groups available for hydrogen bonding with cellulose. Figure 3f shows the presence of areas where the CA shell has even ripped off the CNF core, highlighting the poor interfacial affinity. In contrast, the cross section of the ethanol-coagulated CNF/GG filament does not clearly display any interface between the core and the shell.
Possibly, the slow coagulation in ethanol provides time for CNF and GG to fuse at the interface. This is supported by previous studies with electrospinning, as well-defined core and shell regions have only been attained by electrospinning of two immiscible polymers.38 Consequently, even though the CNF was poorly oriented (orientation index 0.13 ± 0.01, Herman’s orientation parameter 0.19 ± 0.04), the increased high-affinity contact area through fusing with GG led to a stronger filament than the more oriented CNF/CA system (orientation index 0.38 ± 0.01, Herman’s orientation parameter 0.27 ± 0.03, Figure 2). Furthermore, this filament benefited from the relatively uniform cross-sectional shape discussed above, which may be optimal for load bearing (Figure 3e). The strength of GG itself cannot be expected to significantly contribute to the strength of the bicomponent filament because of the weakness of the neat GG filament (Figure 3b, green).
When CNF/GG was coagulated in acetone, the core and shell sections of the filament were clearly distinguishable but closely connected with each other (Figure 3d). This observation is in line with the faster coagulation (T = 5580 s/cm2 in acetone compared to 54 630 s/cm2 in ethanol, eq 1) locking the CNF and GG in place without much time to interdiffuse. As the fast coagulation also enabled the CNF to retain more alignment (Figure 2), this filament achieved the highest Young’s modulus of 2074 ± 121 MPa, a tensile strength of 69.5 ± 15.7 MPa, and a tenacity of 7.84 ± 0.58 cN/tex (Figure 3b, cyan solid line, Table S7).
In conclusion, the stiffness and strength of coaxial filaments can be optimized by the enhanced alignment of load-bearing crystallites in the structure through fast coagulation. Nevertheless, even poor orientation caused by slow coagulation can be compensated by using chemically compatible core and shell materials, which can fuse and restructure during the prolonged coagulation to increase interparticle contact area and develop compressive strength to protect the filament from excess flattening during drying. For this kind of core/shell systems, facile spinning could be possible even if the components would have such a high affinity to aggregate in suspension and thus impair spinnability if blended into a dope prior to spinning. The concluded trends of increasing Young’s modulus with improving CNF alignment and interfacial affinity are presented in Figure 3c. In the case of tensile strength (Figure 3c, red), the experimental deviation is larger but approximately similar trend is observed as that for Young’s modulus, especially when comparing the core/shell filaments.
High-Throughput Spinnability Depends on Shell Coagulation
In order to estimate the scalability of filament production, the maximum spinning rate was determined on the laboratory scale. Spinning in the absence of a shell, the CNF filament frequently broke, making impossible the development of a high-throughput system. In contrast, CNF/GG could be continuously wet-spun into acetone at a maximum rate of 1.2 m/min (Table S2), provided that the coagulation bath was sufficiently long (24 cm filament travel in the coagulant) and a conveyor belt supported the wet filament in air (Figure 1c). Here, the reported rate refers to the winding speed, which defines the rate at which the filament is produced. The corresponding rate for CNF extrusion was 8.1 m/min (Table S2), which signifies a CNF DR of 0.15 (i.e., negative draw). At this DR and rate, uninterrupted spinning was possible until either material was exhausted from the syringe, apart from occasional breakages.
The system with CNF and CA experienced even fewer breakages and was less sensitive to drawing, when half of the ethanol used as the coagulant was replaced with water. This system could reach a maximum spinning rate of 33 m/min, when the CNF extrusion speed was 11 m/min (Table S2, CNF DR = 3), and maintain it as long as both dopes were available in the syringes. Compared to GG, the ability of CA to support CNF spinning more effectively is partly explained by the rheological behavior of these dopes, as discussed elsewhere,31 and partly owing to the fact that CA coagulates faster and thus serves as a stronger support material, immediately after extrusion from the coaxial needle.
The potential for scalable production of neat CNF filaments with this method was evaluated by verifying the selective removal of the CA shell. Successful shell removal from the core-/shell-structured filament indicated the possibility for “islands-in-the-sea” configurations by using several extruding orifices for CNF enclosed inside a larger one for CA. For this kind of process, commercial spinnerets already exist. Through dissolution of CA, individual CNF filaments could be separated from the bundle. Furthermore, the dissolved CA could be recycled to produce another batch to reduce the costs of the method.
The possibility for removing the CA shell by soaking in acetone was confirmed by FTIR. As shown in Figure 4a, the FTIR spectrum of the core/shell filament features the carbonyl and ester peaks, which were also present in the neat CA filament. These peaks essentially disappear upon the removal of the CA shell, which confirms that the remaining filament consisted almost entirely of CNF. Notably, the wide peak of the neat CNF filament assigned to hydroxyl groups (Figure 4a in blue) suggests that this filament has a greater affinity to water than the same system in the presence of the CA shell, which shows a much smaller hydroxyl peak (>80% of CA original hydroxyl moieties are substituted with acetate groups, Figure 4a, purple). Related implications on the interactions between the filaments and water will be discussed below.
Figure 4.
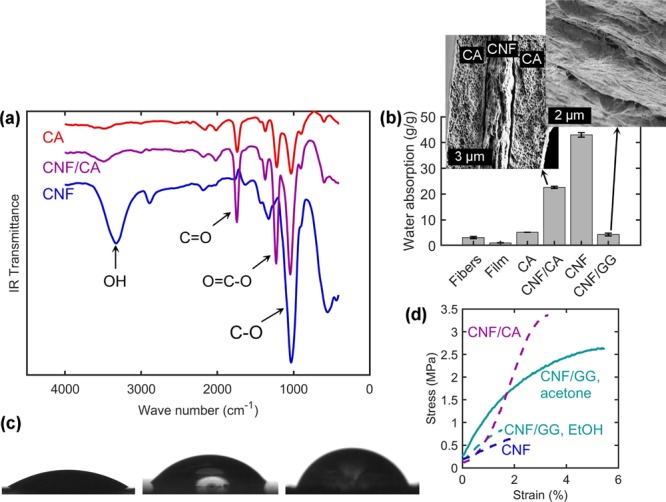
(a) FTIR spectra of CNF filaments with and without CA shell as well as a neat CA filament. (b) Water absorption capacities of core/shell filaments along with reference cellulosic materials. Error bars are based on standard deviation divided by the square root of the sample size (mostly two specimens, see Experimental Section). Insets show the SEM images (high magnification) of the cross-sectional CNF/CA and CNF/GG (only the core shown in the latter case). (c) Photographs of water drops on films cast from CNF (left), CA (middle), and GG (right). The images of CNF and CA are acquired immediately and GG 2 s after drop deposition (beginning of linearly progressing decline in Figure S4, Supporting Information). (d) Wet stress–strain curves of the filaments coagulated in ethanol (dashed lines) and acetone (solid line). “CNF” refers to the CNF/CA filament after the removal of CA.
The DRs achievable for CNF/CA were further explored by changing the coagulant to water. If enclosed in CA, CNF could be drawn in water with a DR of 9. The drawing, especially with DR > 5, did not significantly influence the mechanical properties of the CNF component (Table S6, Figure S2, Supporting Information), probably because of the lack of a coagulating effect on CNF immersed in water. As the CNFs were not forced to pack together by a coagulant, their possible structural development, upon drawing, did not manifest as an improved mechanical strength. Instead, the water coagulation system enabled filaments with enhanced porosity and thus offer potential for water absorption. The diameter of these filaments could be controlled by the extent of drawing (Table S6). For the water absorption studies discussed below, an intermediate CNF DR of 5 was selected.
Filaments for Water Absorption
The water absorption capacity of CNF/CA filaments was compared to that of CNF/GG as well as the precursor materials (Figure 4b). Remarkably, when CNFs were spun into a filament with CA instead of casting into a film from acetone, the water absorption capacity increased 20-fold (from 1.1 ± 0.1 to 22.6 ± 0.4 g/g). Apparently, the surface tension of water was able to draw the fibrils together more effectively, when drying freely in air (film), than when the CNF was covered by CA (core/shell filament). This can be possibly explained by the presence of any residual acetone, originating from the shell dope, which slightly lowers the surface tension of the water in the drying filament. However, the CNF/CA filament comprised >50 wt % CA (Figure S1, Supporting Information), which has a relatively low affinity to water because of the substituted hydroxyl groups. The full water absorption potential of CNF was exploited by removing the CA shell, almost doubling the water absorption to 43.0 ± 0.9 g/g. This is on a similar level with freeze-dried CNF structures,17,18 though the filaments have been prepared with a much more practical method relative to freeze-drying.
Furthermore, these filaments absorb significantly more water than a fiber yarn spun from macroscopic cellulose fibers (5 g/g),16 or even fluff pulp (12 g/g),48 which is a typical component in commercial absorbent products. This highlights the benefits of the increased surface area by disintegration of wood fibers to the nanoscale. In a fiber-based material, most of the water absorption can be expected to occur inside the fiber lumens as well as in the spaces between adjacent fibers. In contrast, in a CNF filament, the spaces between individual fibrils are more available for water sorption as they have been liberated from the native cellulose fiber structure. A demonstration of this effect is found with CNF materials that have been shown to sorb more moisture than the macroscopic cellulose fibers.3 It should be noted, though, that a filament form is required to capitalize on the high surface area of CNF: a CNF film cast at room temperature without heating or pressing absorbed even less (1.1 ± 0.1 g/g) than the wood fibers used as the precursor for CNF (3.1 ± 0.5 g/g, Figure 4b).
CNF filaments absorb considerably less water compared to commercial superabsorbents based on acrylic acid and acrylamide (absorption capacity 2000–3000 g/g).49,50 Nevertheless, the present results are promising, as they are on the higher end among renewable absorbents, as described above. Furthermore, the optimization of the preparation and modification, blending, and cross-linking of the CNF have not yet been explored in this context. Already, the present filaments were able to maintain their structural integrity during the absorption test and were suitable to handling in the wet condition. This is promising regarding their practical applicability in thin absorbent structures that require some level of mechanical integrity.
The absorption could be enhanced by further optimization of the filament porosity, as shown for CA filaments, which attained a higher and more uniform porosity when spun into a less aggressive coagulant.43 In the case of CNF, the same principle could be applied by changing the solvent of the shell polymer to prevent the contact with the rapidly diffusing acetone. Also, chemical modification to increase the CNF surface charge could still increase the water absorption.51 Furthermore, the filaments can be placed into a network, woven or nonwoven structure, where water droplets would be trapped between adjacent filaments in addition to the water absorbed by each single filament. Overall, CNF spun to water with a CA shell is a highly promising precursor for light and thin materials that are able to absorb large amounts of water and still be sustainable in their production and disposal.
In comparison, CNF/GG only absorbed 4.3 ± 0.6 g/g water, even though CNF and GG are both expected to have a high affinity to water owing to a large amount of available hydroxyl groups. Apparently, CNF and GG have already formed extensive hydrogen bonding, thus leaving fewer sorption sites accessible to water. Similar phenomenon has been shown previously for CNF and PVA.36 Furthermore, addition of guar derivatives has been shown to enhance the water vapor barrier properties of CNF films.52,53 The tight arrangement of CNF and GG is highlighted by the higher apparent density, 0.89 ± 0.13 g/cm3 (Table S7, Supporting Information) compared to that of water-coagulated CNF/CA (0.54 ± 0.05 g/cm3, Table S6 of the Supporting Information). The limited coagulation in the water bath favors a higher porosity in the filament, thus increasing the water absorption capacity. This is qualitatively illustrated in the high-magnification cross-sectional images of both core/shell filaments (Figure 4b, inset). Inside CA, CNF has coalesced into loosely interconnected layers, leaving void areas for water absorption, whereas inside GG, CNFs have packed into a denser arrangement.
Both the CNF film and the CNF/GG filament have been formed in acetone and thus in a nonpolar environment. These conditions have probably forced the CNF to expose its hydrophobic crystal planes. In water, an opposite phenomenon can be expected to occur, thus enhancing the hydrophilicity of the pore surfaces in CNF spun with CA. As CNF could not be wet-spun in water alone, because of its tendency to disperse in water instead of forming a recoverable filament, core/shell spinning with CA is essential to enable filaments with excellent hydrophilicity.
The hydrophilicity of CNF dried from a polar environment was corroborated by the relatively low initial water contact angle of 29 ± 6° on a surface of a CNF film cast from water. For a similar film cast from acetone, a 45% higher contact angle, 42 ± 5°, has been measured previously,3 highlighting a clear difference depending on whether the CNFs have aggregated with more exposed hydrophobic or hydrophilic crystal planes. The initial water contact angle on a CA film was even higher (51 ± 7°) as expected because of the low density of hydroxyl groups. This explains the poor water sorption capacity of a neat CA filament (5.2 g/g) and the effect of CA shell on reducing the total water sorption of CNF/CA (Figure 4b). Photographs of drops of water on the surfaces of films prepared from the filament materials are shown in Figure 4c. Counterintuitively, the water contact angle on GG is the highest. However, the steady decrease in contact angle over time (Figure S4, Supporting Information) indicates the evolution of water absorption by the GG film.
Figure 4d displays the stress–strain curves of the core/shell filaments after being soaked overnight in deionized water. The wet mechanical properties are also summarized in Table S8 of the Supporting Information. It should be noted that the Young modulus and tensile strength were calculated using the cross-sectional area measured in the dry state. The water-coagulated filaments became too weak for testing after soaking. This is due to their extremely high water sorption capacity (22.6 ± 0.4 g/g or 43.0 ± 0.9 g/g with or without CA, respectively). Evidently, if this kind of filament was used as an absorbent in an application where it also must bear load when wet, CNF cross-linking would be necessary. The filaments spun to organic solvents were stable enough for the measurement, probably owing to the nonpolar environment that promoted the formation of a slightly more hydrophobic surface, as discussed above. Still, even these filaments became significantly weaker in wet conditions (Figure 4c) compared to their dry strength (Figure 3b).
The highest wet tensile strength among the core/shell filaments was for ethanol-coagulated CNF/CA and acetone-coagulated CNF/GG (2.94 ± 0.62 and 2.70 ± 0.59 MPa, respectively, Figure 4c, Table S8 of the Supporting Information). In the case of CNF/CA, this arises from the reinforcing effect of CA, as neat CA has a significantly higher wet strength than pure CNF (Table S8, Supporting Information), owing to its lower water absorption (5.2 g/g, Figure 4b). In the case of CNF/GG, the wet strength was influenced by the coagulant, as the filaments from ethanol became more susceptible to water. Apparently, acetone coagulates CNF and GG into a structure which not only is stronger when dry but also maintains its strength when exposed to water.
Conclusions
Core/shell filaments were prepared by the wet spinning of CNF with GG or CA using different coagulants. The CNF alignment in the filament was shown to increase with accelerating coagulation time. Enhanced CNF alignment strengthened and stiffened the filament, though even poor orientation could be compensated by a strong interfacial affinity between the core and shell components. Such effects were maximized when CNF and GG were coagulated in ethanol, in which the slow coagulation allowed for the relaxation of the CNFs, which were initially aligned in the spinning needle, as well as their integration with GG at the interface.
Potential scalability of the core/shell spinning was demonstrated in a prototype spinning line that produced CNF/CA and CNF/GG filaments at rates of up to 33 and 1.2 m/min, respectively. These rates are much higher than those reported for the wet spinning of CNF previously. Furthermore, CA could be removed from CNF/CA by selective dissolution, which makes possible the recovery of the support polymer and indicates the potential of upscaling the process, for example, in an “islands-in-the-sea” configuration. Further work is needed to validate the applicability to such geometries. The removal of the CA shell from coaxial filaments, spun under conditions of limited CNF coagulation, unlocked a remarkable water absorption capacity (43 g water/g dry CNF filaments). This finding suggests an opening for this material in the area of absorbency and the design of filament-based structures for related purposes.
Acknowledgments
This work has been funded by Jenny and Antti Wihuri foundation as well as Business Finland through a strategic opening entitled Design Driven Value Chains in the World of Cellulose. This work made use of the facilities of Aalto University’s Nanomicroscopy Center. We acknowledge the Academy of Finland’s Centers of Excellence program (project 264677, HYBER) for support. Also, we are thankful to Tero Kämäräinen, Anurodh Tripathi, and Dr. Hannes Orelma for insightful discussions and Dr. Steven Spoljaric for assistance with the mechanical testing, as well as Valeria Azovskaya for the filament photographs.
Glossary
Abbreviations
- CNFs
cellulose nanofibrils
- CA
cellulose acetate
- GG
guar gum
- DR
drawing ratio
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.8b08153.
Spinning conditions for filaments prepared for different experiments; effect of drawing on absorbent filaments with CNF and CA; filament crystallinity; effect of the shell material and coagulation system on filament physical–mechanical properties; filament interactions with water; and effect of core/shell system on spinnability (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Jenny and Antti Wihuri foundation; Business Finland.
The authors declare no competing financial interest.
Supplementary Material
References
- Clemons C. Nanocellulose in Spun Continuous Fibers: A Review and Future Outlook. J. Renewable Mater. 2016, 4, 327–339. 10.7569/jrm.2016.634112. [DOI] [Google Scholar]
- Lundahl M. J.; Klar V.; Wang L.; Ago M.; Rojas O. J. Spinning of Cellulose Nanofibrils into Filaments: A Review. Ind. Eng. Chem. Res. 2016, 56, 8–19. 10.1021/acs.iecr.6b04010. [DOI] [Google Scholar]
- Lundahl M. J.; Cunha A. G.; Rojo E.; Papageorgiou A. C.; Rautkari L.; Arboleda J. C.; Rojas O. J. Strength and Water Interactions of Cellulose I Filaments Wet-Spun from Cellulose Nanofibril Hydrogels. Sci. Rep. 2016, 6, 30695. 10.1038/srep30695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Ren H.; Tong G.; Deng Y. Ultra-Lightweight Poly (Sodium Acrylate) Modified TEMPO-Oxidized Cellulose Nanofibril Aerogel Spheres and Their Superabsorbent Properties. Cellulose 2016, 23, 3665–3676. 10.1007/s10570-016-1041-8. [DOI] [Google Scholar]
- Chang C.; Duan B.; Cai J.; Zhang L. Superabsorbent Hydrogels Based on Cellulose for Smart Swelling and Controllable Delivery. Eur. Polym. J. 2010, 46, 92–100. 10.1016/j.eurpolymj.2009.04.033. [DOI] [Google Scholar]
- Trivedi P.; Schaller J.; Gustafsson J.; Fardim P. Supramolecular Design of Cellulose Hydrogel Beads. J. Renewable Mater. 2017, 5, 400–409. 10.7569/jrm.2017.634143. [DOI] [Google Scholar]
- Innerlohinger J.; Weber H. K.; Kraft G. Aerocellulose: Aerogels and Aerogel-like Materials Made from Cellulose. Macromol. Symp. 2006, 244, 126–135. 10.1002/masy.200651212. [DOI] [Google Scholar]
- Hubbe M. A.; Ayoub A.; Daystar J. S.; Venditti R. A.; Pawlak J. J. Enhanced Absorbent Products Incorporating Cellulose and Its Derivatives: A Review. BioResources 2013, 8, 6556–6629. 10.15376/biores.8.4.6556-6629. [DOI] [Google Scholar]
- Schuchardt D. R.; Berg J. C. Liquid Transport in Composite Cellulose-Superabsorbent Fiber Networks. Wood Fiber Sci. 1991, 23, 342–357. [Google Scholar]
- Weerawarna S. A.; Bing S.. Cellulose Fibers Having Superabsorbent Particles Adhered Thereto. U.S. Patent 20,080,082,067 A1, 2006.
- Foelsch L.; Rosenfeld L. G.. Absorbent Article with High Absorbency Zone. EP1023884 A2, 1998.
- Cerutti G.Absorbent Article Which Includes Superabsorbent Material Located in Discrete Pockets and Manufacturing Process. EP0875224 A1, 1997.
- Flohr A.Superabsorbent Polymers Comprising Direct Covalent Bonds between Polymer Chain Segments. EP1568385 A1, 2004.
- Bergh M.Absorbent Cellulose Based Fibers; Chalmers University of Technology, 2011. [Google Scholar]
- Dahlan N. A.; Pushpamalar J.; Veeramachineni A. K.; Muniyandy S. Smart Hydrogel of Carboxymethyl Cellulose Grafted Carboxymethyl Polyvinyl Alcohol and Properties Studied for Future Material Applications. J. Polym. Environ. 2018, 26, 2061–2071. 10.1007/s10924-017-1105-3. [DOI] [Google Scholar]
- Orelma H.; Virtanen T.; Spoljaric S.; Lehmonen J.; Seppälä J.; Rojas O. J.; Harlin A. Cyclodextrin-Functionalized Fiber Yarns Spun from Deep Eutectic Cellulose Solutions for Nonspecific Hormone Capture in Aqueous Matrices. Biomacromolecules 2018, 19, 652–661. 10.1021/acs.biomac.7b01765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin F. W.; Lund K.; Brelid H.; Theliander H. Reinforced Absorbent Material: A Cellulosic Composite of TEMPO-Oxidized MFC and CTMP Fibres. Cellulose 2012, 19, 1413–1423. 10.1007/s10570-012-9706-4. [DOI] [Google Scholar]
- Kettunen M.; Silvennoinen R. J.; Houbenov N.; Nykänen A.; Ruokolainen J.; Sainio J.; Pore V.; Kemell M.; Ankerfors M.; Lindström T.; Ritala M.; Ras R. H. A.; Ikkala O. Photoswitchable Superabsorbency Based on Nanocellulose Aerogels. Adv. Funct. Mater. 2011, 21, 510–517. 10.1002/adfm.201001431. [DOI] [Google Scholar]
- Mohammadi P.; Toivonen M. S.; Ikkala O.; Wagermaier W.; Linder M. B. Aligning Cellulose Nanofibril Dispersions for Tougher Fibers. Sci. Rep. 2017, 7, 11860. 10.1038/s41598-017-12107-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooshmand S.; Aitomäki Y.; Norberg N.; Mathew A. P.; Oksman K. Dry-Spun Single-Filament Fibers Comprising Solely Cellulose Nanofibers from Bioresidue. ACS Appl. Mater. Interfaces 2015, 7, 13022–13028. 10.1021/acsami.5b03091. [DOI] [PubMed] [Google Scholar]
- Iwamoto S.; Isogai A.; Iwata T. Structure and Mechanical Properties of Wet-Spun Fibers Made from Natural Cellulose Nanofibers. Biomacromolecules 2011, 12, 831–836. 10.1021/bm101510r. [DOI] [PubMed] [Google Scholar]
- Håkansson K. M. O.; Fall A. B.; Lundell F.; Yu S.; Krywka C.; Roth S. V.; Santoro G.; Kvick M.; Prahl Wittberg L.; Wågberg L.; Söderberg L. D. Hydrodynamic Alignment and Assembly of Nanofibrils Resulting in Strong Cellulose Filaments. Nat. Commun. 2014, 5, 4018. 10.1038/ncomms5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Rendon J. G.; Schacher F. H.; Ifuku S.; Walther A. Mechanical Performance of Macrofibers of Cellulose and Chitin Nanofibrils Aligned by Wet-Stretching: A Critical Comparison. Biomacromolecules 2014, 15, 2709–2717. 10.1021/bm500566m. [DOI] [PubMed] [Google Scholar]
- Yao J.; Chen S.; Chen Y.; Wang B.; Pei Q.; Wang H. Macrofibers with High Mechanical Performance Based on Aligned Bacterial Cellulose Nanofibers. ACS Appl. Mater. Interfaces 2017, 9, 20330–20339. 10.1021/acsami.6b14650. [DOI] [PubMed] [Google Scholar]
- Kafy A.; Kim H. C.; Zhai L.; Kim J. W.; Van Hai L.; Kang T. J.; Kim J. Cellulose Long Fibers Fabricated from Cellulose Nanofibers and Its Strong and Tough Characteristics. Sci. Rep. 2017, 7, 17683. 10.1038/s41598-017-17713-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivonen M. S.; Kurki-Suonio S.; Wagermaier W.; Hynninen V.; Hietala S.; Ikkala O. Interfacial Polyelectrolyte Complex Spinning of Cellulose Nanofibrils for Advanced Bicomponent Fibers. Biomacromolecules 2017, 18, 1293–1301. 10.1021/acs.biomac.7b00059. [DOI] [PubMed] [Google Scholar]
- Mortimer S. A.; Péguy A. A. The Formation of Structure in the Spinning and Coagulation of Lyocell Fibres. Cellul. Chem. Technol. 1996, 30, 117–132. [Google Scholar]
- Nechyporchuk O.; Bordes R.; Köhnke T. Wet Spinning of Flame-Retardant Cellulosic Fibers Supported by Interfacial Complexation of Cellulose Nanofibrils with Silica Nanoparticles. ACS Appl. Mater. Interfaces 2017, 9, 39069–39077. 10.1021/acsami.7b13466. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Orelma H.; Sneck A.; Kataja K.; Salmela J.; Qvintus P.; Suurnäkki A.; Harlin A. High Velocity Dry Spinning of Nanofibrillated Cellulose (CNF) Filaments on an Adhesion Controlled Surface with Low Friction. Cellulose 2016, 23, 3393–3398. 10.1007/s10570-016-1044-5. [DOI] [Google Scholar]
- Dickie W. A.; Sowter P. F. C.. Production of Artificial Filaments, Films, and like Materials. U.S. Patent 2,147,641 A, 1929.
- Lundahl M. J.; Berta M.; Ago M.; Stading M.; Rojas O. J.. Shear and Extensional Rheology of Aqueous Suspensions of Cellulose Nanofibrils for Biopolymer-Assisted Filament Spinning. Biomacromolecules, Manuscript Submitted to. 2018. [Google Scholar]
- Herrera N. V.; Mathew A. P.; Wang L. Y.; Oksman K. Randomly Oriented and Aligned Cellulose Fibres Reinforced with Cellulose Nanowhiskers, Prepared by Electrospinning. Plast., Rubber Compos. 2011, 40, 57–64. 10.1179/174328911x12988622800891. [DOI] [Google Scholar]
- Chen S.; Schueneman G.; Pipes R. B.; Youngblood J.; Moon R. J. Effects of Crystal Orientation on Cellulose Nanocrystals-Cellulose Acetate Nanocomposite Fibers Prepared by Dry Spinning. Biomacromolecules 2014, 15, 3827–3835. 10.1021/bm501161v. [DOI] [PubMed] [Google Scholar]
- Vallejos M. E.; Peresin M. S.; Rojas O. J. All-Cellulose Composite Fibers Obtained by Electrospinning Dispersions of Cellulose Acetate and Cellulose Nanocrystals. J. Polym. Environ. 2012, 20, 1075–1083. 10.1007/s10924-012-0499-1. [DOI] [Google Scholar]
- Cheng Y.; Prud’homme R. K.; Chik J.; Rau D. C. Measurement of Forces between Galactomannan Polymer Chains: Effect of Hydrogen Bonding. Macromolecules 2002, 35, 10155–10161. 10.1021/ma020887e. [DOI] [Google Scholar]
- Peresin M. S.; Habibi Y.; Vesterinen A.-H.; Rojas O. J.; Pawlak J. J.; Seppälä J. V. Effect of Moisture on Electrospun Nanofiber Composites of Poly(Vinyl Alcohol) and Cellulose Nanocrystals. Biomacromolecules 2010, 11, 2471–2477. 10.1021/bm1006689. [DOI] [PubMed] [Google Scholar]
- Pakravan M.; Heuzey M.-C.; Ajji A. Core-Shell Structured PEO-Chitosan Nanofibers by Coaxial Electrospinning. Biomacromolecules 2012, 13, 412–421. 10.1021/bm201444v. [DOI] [PubMed] [Google Scholar]
- Li D.; Xia Y. Electrospinning of Nanofibers: Reinventing the Wheel?. Adv. Mater. 2004, 16, 1151–1170. 10.1002/adma.200400719. [DOI] [Google Scholar]
- Scborsch C.; Gamier C.; Doublier J.-L. Viscoelastic Properties of Xanthan/Galactomannan Mixtures: Comparison of Guar Gum with Locust Bean Gum. Carbohydr. Polym. 1997, 34, 165–175. 10.1016/s0144-8617(97)00095-7. [DOI] [Google Scholar]
- Torres M. D.; Hallmark B.; Wilson D. I. Effect of Concentration on Shear and Extensional Rheology of Guar Gum Solutions. Food Hydrocolloids 2014, 40, 85–95. 10.1016/j.foodhyd.2014.02.011. [DOI] [Google Scholar]
- Frollini E.; Reed W. F.; Milas M.; Rinaudo M. Polyelectrolytes from Polysaccharides: Selective Oxidation of Guar Gum - a Revisited Reaction. Carbohydr. Polym. 1995, 27, 129–135. 10.1016/0144-8617(95)00021-x. [DOI] [Google Scholar]
- Picout D. R.; Ross-Murphy S. B.; Errington N.; Harding S. E. Pressure Cell Assisted Solution Characterization of Polysaccharides. 1. Guar Gum. Biomacromolecules 2001, 2, 1301–1309. 10.1021/bm010118n. [DOI] [PubMed] [Google Scholar]
- Tripathi A.Bioinspired Light-Weight Materials using Biopolymers: From Synthesis to Application; North Carolina State University, 2018. [Google Scholar]
- Cussler E. L.Diffusion: Mass Transfer in Fluid Systems, 3rd ed.; Cambridge University Press: Cambridge, U.K., 2009. [Google Scholar]
- Yoshiharu N.; Shigenori K.; Masahisa W.; Takeshi O. Cellulose Microcrystal Film of High Uniaxial Orientation. Macromolecules 1997, 30, 6395–6397. 10.1021/ma970503y. [DOI] [Google Scholar]
- Ferguson J.; Ibrahim K. M. The Wet Spinning of Secondary Cellulose Acetate: Solution Rheology and Filament Spinnability. Polymer 1969, 10, 135–145. 10.1016/0032-3861(69)90017-2. [DOI] [Google Scholar]
- Mittal N.; Ansari F.; Gowda K. V.; Brouzet C.; Chen P.; Larsson P. T.; Roth S. V.; Lundell F.; Wa L.; Kotov N. A. Multiscale Control of Nanocellulose Assembly: Transferring Remarkable Nanoscale Fibril Mechanics to Macroscale Fibers. ACS Nano 2018, 12, 6378–6388. 10.1021/acsnano.8b01084. [DOI] [PubMed] [Google Scholar]
- Zohuriaan-Mehr M. J.; Kabiri K. Superabsorbent Polymer Materials: A Review. Iran. Polym. J. 2008, 17, 451–477. [Google Scholar]
- Mundiyanselage T. K.; Neckers D. C. Highly Absorbing Superabsorbent Polymer. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 1357–1364. 10.1002/pola.22476. [DOI] [Google Scholar]
- Kiatkamjornwong S.; Wongwatthanasatien R. Superabsorbent polymer of poly[acrylamide-co-(acrylic acid)] by foamed polymerization. I. synthesis and water swelling properties. Macromol. Symp. 2004, 207, 229–240. 10.1002/masy.200450320. [DOI] [Google Scholar]
- Van De Ven T. G. M.; Alam N.; Antal M.; Tejado A.. Highly Charge Group-Modified Cellulose Fibers Which Can Be Made into Cellulose Nanostructures or Super-Absorbing Cellulosic Materials and Method of Making Them. WO2012119229 A1, 2011.
- Dai L.; Long Z.; Zhao Y.; Wang B.; Chen J. Comparison of Hydroxypropyl and Carboxymethyl Guar for the Preparation of Nanocellulose Composite Films. Cellulose 2016, 23, 2989–2999. 10.1007/s10570-016-0998-7. [DOI] [Google Scholar]
- Dai L.; Wang B.; Long Z.; Chen L.; Zhang D.; Guo S. Properties of Hydroxypropyl Guar/TEMPO-Oxidized Cellulose Nanofibrils Composite Films. Cellulose 2015, 22, 3117–3126. 10.1007/s10570-015-0691-2. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.