

Abstract
Flexible polypeptide linkers composed of glycine and serine are important components of engineered multidomain proteins. We have previously shown that the conformational properties of Gly-Gly-Ser repeat linkers can be quantitatively understood by comparing experimentally determined Förster resonance energy transfer (FRET) efficiencies of ECFP-linker-EYFP proteins to theoretical FRET efficiencies calculated using wormlike chain and Gaussian chain models. Here we extend this analysis to include linkers with different glycine contents. We determined the FRET efficiencies of ECFP-linker-EYFP proteins with linkers ranging in length from 25 to 73 amino acids and with glycine contents of 33.3% (GSSGSS), 16.7% (GSSSSSS), and 0% (SSSSSSS). The FRET efficiency decreased with an increasing linker length and was overall lower for linkers with less glycine. Modeling the linkers using the WLC model revealed that the experimentally observed FRET efficiencies were consistent with persistence lengths of 4.5, 4.8, and 6.2 Å for the GSSGSS, GSSSSS, and SSSSSS linkers, respectively. The observed increase in linker stiffness with reduced glycine content is much less pronounced than that predicted by a classical model developed by Flory and co-workers. We discuss possible reasons for this discrepancy as well as implications for using the stiffer linkers to control the effective concentrations of connected domains in engineered multidomain proteins.
The generation of fusion proteins consisting of multiple protein domains is a popular and highly successful approach to engineering new protein functions. While in some applications the linker between the domains merely separates two protein domains and allows their independent folding, in many cases linker properties directly affect the functional properties of the fusion proteins.1 An important example is that of genetically encoded fluorescent sensor proteins based on Förster resonance energy transfer (FRET). These sensors consist of donor and acceptor fluorescent domains (FPs) fused to ligand binding domains, in such a way that ligand binding changes the distance and orientation of the FPs, resulting in a change in emission color. The dynamic range of these sensors (i.e., the relative difference in FRET between the free and ligand-bound form of the sensor) is determined by the distance between the FPs in the on and off states. Optimal design of these sensor proteins thus requires quantitative understanding of the effect of linker length and linker flexibility on the conformational behavior of these fusion proteins.2−4 The conformational behavior of peptide linkers also affects the local effective concentrations of protein domains, which is an important parameter that determines the oligomeric state of fusion proteins [e.g., single-chain variable antibody fragments (scFv)],5,6 the affinities of multivalent interactions,7,8 the binding properties of fusions to Fc domains,9 transferrin,10 and albumin,11 and the catalytic activity of bifunctional enzymes.12,13
The linkers used in the construction of multidomain proteins typically consist of repeats of glycine and serine residues. The combination of flexible and hydrophilic residues in these linkers prevents the formation of secondary structures and reduces the likelihood that the linkers will interfere with the folding and function of the protein domains. In a previous study, we showed that the amount of energy transferred between cyan and yellow fluorescent domains connected by linkers consisting of GlyGlySer repeats could be quantitatively understood by random coil models that describe the peptide linker as a wormlike chain (WLC) with a persistence length of 4.5 Å or a Gaussian chain (GC) with a characteristic ratio of 2.3.14 These models also allowed the calculation of effective concentrations as a function of linker length and distance, providing quantitative understanding of intramolecular domain–domain interactions.15 Both models, originally developed in polymer physics, have been extensively used to describe polypeptides. The GC model describes the chain as a series of rigid segments (residues) with a completely random orientation. The WLC model describes the polypeptide as a continuous semiflexible tube with a persistent memory of its direction. For short or very stiff chains, the WLC model describes the properties of real polymers more accurately than the GC model does, while the two models are identical in the limit of long, flexible chains. Our previous analysis shows that while flexible linkers are attractive for applications in which relatively short distances are covered (<60 Å), they are much less effective in spanning longer distances, such as the distance between the two antigen binding sites present in a single antibody.2,16 Similarly, the relatively compact nature of the random coil structures formed by these linkers gives rise to a substantial amount of FRET for the so-called low-FRET state in many FRET sensors, which is detrimental for their dynamic range.
In this work, we explore whether the stiffness of polypeptide linkers can be increased by systematically decreasing the glycine content of Gly/Ser linkers from the value of 67% in GGS linkers to 33, 17, and 0%. Our approach was inspired by early work of Flory and co-workers, who applied models originally developed for polymer chemistry to describe the random coil distribution of polypeptide chains.17,18 In Flory’s GC model, the polypeptide chain is modeled as a freely jointed chain of Cα atoms in which the stiffness of the chain is described by characteristic ratio C∞. The Flory model established a relationship between glycine content and characteristic ratio, with C∞ ranging from 2 for polyglycine to 9 for polyalanine (or any other residue except proline).18 To experimentally test this hypothesis, we constructed fusion proteins consisting of ECFP and EYFP connected by linkers containing various numbers of (GSSGSS)n, (GSSSSS)n, or (S6)n repeats. The amount of energy transfer observed for each of these fusion proteins was subsequently used to determine the effect of glycine content on linker stiffness, using the same model previously applied to ECFP–EYFP fusion proteins containing a GlyGlySer linker. While an increase in linker stiffness was observed with a decrease in glycine content, the magnitude of the effect is more subtle than that predicted by the Flory model. The implications of these findings for the design of FRET sensors and other multidomain proteins are discussed.
Materials and Methods
Molecular Cloning
The fusion proteins were designed on the basis of the ECFP-linker-EYFP 9 (CLY-9) series described previously,14 with the minor modifications of incorporating a Strep tag at the C-termini of the proteins and changing the linker sequences (see Figure S1 for sequence details). pET-28a(+)-CLY-(G2S4)9, pET-28a(+)-CLY-(GS5)9, and pUC-57-(S6)9 were ordered from Genscript (Piscataway, NJ). To obtain pET-28a(+)-CLY-(S6)9, the DNA encoding the (S6)9 linker was amplified via polymerase chain reaction (PCR) from pUC-57-(S6)9 using primers 5′-caagtccggaattcgttcgag-3′ and 5′-caccatccgcggtgtgga-3′ and Phusion High Fidelity DNA polymerase [New England Biolabs (NEB)]. The pET-28a(+)-CLY-(G2S4)9 plasmid was linearized by PCR using primers 5′-tccacaccgcggatggtg-3′ and 5′-ctcgaacgaattccggacttg-3′, and the product was isolated from impurities by gel extraction. The two PCR products were combined in a CPEC reaction, essentially as described previously.19 The correct sequence was confirmed by Sanger sequencing. The shorter CLY genes were obtained by partial digestion and religation as previously described.14 For the CLY-(S6) series, partial digestion was performed with SacI, while the other series used BamHI. For all reactions, 1 μg of DNA was incubated with either 0.5, 1, 1.5, or 2 units of enzyme at 37 °C for 1 h. Samples were then ligated as described. Colony PCR of the linker region, using primers shown above, was performed to check for altered linker lengths, and the coding regions were verified by Sanger sequencing.
Protein Expression and Purification
Proteins were expressed in Escherichia coli BL21(DE3) (Merck Novagen). Bacteria were grown overnight at 37 °C in 5 mL of LB medium with 50 μg mL–1 kanamycin, then diluted into 250 mL of fresh LB with kanamycin, and grown at 37 °C, while being shaken at 250 rpm until OD600 reached 0.6. Induction was performed by adding isopropyl β-d-1-thiogalactopyranoside to a final concentration of 0.1 mM and decreasing the temperature to 22 °C. Following expression for 16 h [except for CLY-(S6)8 and CLY-(S6)9, which were expressed for 4 h to prevent aggregation], cultures were centrifuged at 20000g for 20 min, and the pellets were frozen in liquid nitrogen and stored at −80 °C. Cells were resuspended in 10 mL of BugBuster (Merck) and 10 μL of Benzonase (Merck) and lysed at room temperature for 30 min. Lysates were centrifuged at 40000g for 40 min, and the supernatants were filtered through 0.2 μm syringe filters prior to chromatography. Proteins were first purified using nickel affinity chromatography using gravity-flow HisBind Ni-NTA-agarose (Merck) columns with a bed volume of 2 mL, according to the manufacturer’s instructions, except that the wash buffer contained 30 mM imidazole and the elution buffer 200 mM imidazole. Eluates were further purified using gravity-flow Strep-Tactin Sepharose columns (IBA Life Sciences) according to the manufacturer’s instructions. CLY-(S6)8 was further purified using size exclusion chromatography (SEC) over a 26/60 HiLoad Superdex column in 20 mM Tris-HCl (pH 8) and 100 mM NaCl.
Fluorescence Spectroscopy
Unless stated otherwise, fluorescence spectroscopy was performed at a protein concentration of 200 nM in fluorescence buffer [20 mM Tris-HCl (pH 8.0) containing 100 mM NaCl, 20 μM EDTA, and 10% (v/v) glycerol]. Fluorescence emission spectra of the CLY proteins were recorded on a Varian Cary Eclipse spectrophotometer, using 420 nm excitation light with a slit width of 5 nm and an emission slit width of 5 nm. Spectra were smoothed by averaging over a sliding 5 nm interval and normalized at the intensity at 475 nm. Fluorescence anisotropy measurements were taken with excitation at 512 nm (slit width of 5 nm) and emission ranging from 527 to 529 nm with a 0.2 nm interval. The G factor was calculated before each measurement. The average anisotropy from these wavelengths was used. FRET efficiencies were determined by measuring three quantities for each CLY protein. (1) Donor intensity FAD(λDex,λDem) was measured by exciting at 420 nm and recording emission from 470 to 480 nm. (2) Acceptor intensity FAD(λDex,λA) was measured by exciting at 420 nm and recording emission from 523 to 533 nm. (3) Direct acceptor excitation intensity FAD(λAex,λA) was measured by exciting at 514 nm with a slit width of 2.5 nm and recording emission from 523 to 533 nm. From these values, the FD(λDex,λA) and FA(λDex,λA) parameters were obtained using eqs 1 and 2, respectively.
| 1 |
| 2 |
Equation 1 was derived by comparing the intensities at 475 and 528 nm in the emission spectrum of 200 nM ECFP in fluorescence buffer, excited at 420 nm. For eq 2, FA(λDex,λA) and FA(λAex,λA) were measured for a series of free EYFP concentrations in fluorescence buffer, and a straight line was fit through the FA(λDex,λA)/FA(λAex,λA) plot (Figure S2). These equations can be used because the two following conditions are met: (1) the acceptor does not significantly emit at the donor emission wavelength, and (2) the donor is not significantly excited at the acceptor excitation wavelength. FD(λDex,λA) and FA(λDex,λA) were used to calculate FRET efficiency E using eq 3 (adapted from ref (14)). The εA(λDex)/εD(λD) extinction coefficient ratio of 0.0366 was taken from ref (14).
| 3 |
Wormlike Chain and Gaussian Chain Models
The end-to-end distance probability distributions of a linker according to the WLC model are given by eqs 4 and 5.
| 4 |
where PWLC(re) is the probability of a given end-to-end distance re; lc is the contour length of the chain, which is equal to the number of residues n times the length of one residue b0, which is 3.8 Å; lp is the persistence length; and w is a nonclosed form of expression:
| 5 |
The end-to-end distance probability distribution PG(re) according to the GC model is given by eqs 6 and 7.
| 6 |
| 7 |
where C∞ is the characteristic ratio, defined by Brant and Flory.17 These probability distributions were normalized by dividing them by their integral from 0 to infinity (approximated by 600 Å, at which the probability was on the order of 10–60) to satisfy the criterion that the probability of a linker having any end-to-end distance must be 1.
To calculate the average FRET efficiency ⟨E⟩ for each re, a slightly adapted form of the previously described method was used.14 From the ends of the linker, two vectors, vc and vy, were drawn to the centers of the fluorescent proteins as shown in Figure 1 (vc points from the start of the linker to the center of ECFP, and vy points from the end of the linker to the center of EYFP). For each end-to-end distance, many orientations of vc and vy are possible. Both vectors were placed at a distance re and rotated over two spheres, approximated by polyhedra. These were obtained starting from octahedra by bisecting each edge. The midpoints of edges on the same face were connected, and the edges of the created triangles were bisected again. The 66 resultant points were moved outward onto the surface of spheres with radii |vc| and |vy| (20 and 24 Å, respectively). The average FRET efficiency ⟨E⟩ for a single re was calculated by placing the centers of the spheres at a distance re and calculating rc and E [using the Förster equation (eq 8)] for each of the 66 × 66 orientations. The values of E that satisfied the rc ≥ 22 Å condition were then averaged. This 22 Å represents the distance between the chromophores in the GFP dimer and was introduced to account for the fact that the fluorescent proteins cannot occupy the same space.
| 8 |
Figure 1.
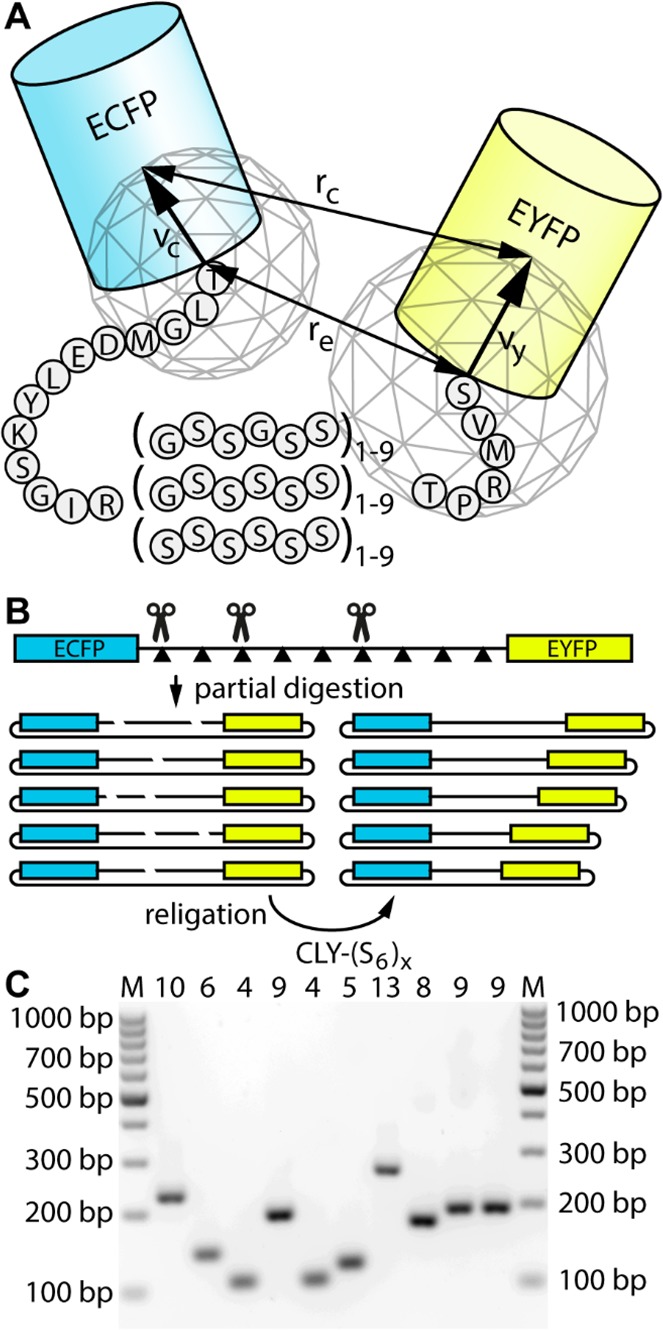
Cyan-linker-yellow (CLY) proteins for studying the behavior of flexible linkers. (A) Schematic representation of the CLY fusion protein. The region between parentheses was varied in length and glycine content. re is the distance between the ends of the linker. rc is the distance between the two chromophores. vc is the vector connecting the start of the linker to the ECFP chromophore. vy is the vector connecting the end of the linker to the EYFP chromophore. The two wireframe polyhedra depict the different orientations over which both ECFP and EYFP were averaged for calculation of interchromophore distances and FRET efficiencies. (B) Schematic representation of the partial digestion and religation method used to obtain multiple linker length variants. (C) Gel image of colony PCR products encompassing the linker regions of partially digested (with 0.1–1 unit of SacI) and religated (S6)9 constructs.
Förster distance R0 of the ECFP–EYFP pair has previously been determined to be 48 Å, and this value was used throughout this work.14 The ensemble average ⟨E⟩ensemble for a given CLY protein was obtained by multiplying the probability of each re, either PG(re) or PWLC(re), by the associated ⟨E⟩ and integrating the resultant curve over all re.
Calculation of the Effective Concentration
The effective concentration for the formation of an intramolecular complex is proportional to probability density p(re) for the distance re that the linker needs to bridge in the complex, which can be obtained from the end-to-end probability [PWLC(re)] according to eq 9.43
| 9 |
PWLC(re) was obtained using eqs 4 and 5. When re, lc, and lp are given in decimeters, the effective concentration Ceff is obtained by dividing p(re) by Avogadro’s constant:
| 10 |
Results
Three series of ECFP-linker-EYFP fusion proteins were constructed in which the linker contained between one and nine GSSGSS, GSSSSSS, or SSSSSSS repeats (Figure 1A). These proteins are termed CLY-(G2S4)n, CLY-(GS5)n, and CLY-(S6)n, in which n indicates the number of repeats. Except for the introduction of different linkers and an additional C-terminal Strep tag for easy purification, these fusion proteins are identical to the previously reported CLYx proteins, which contained one to nine GGSGGS repeats and for the sake of consistency are now termed CLY-(G4S2)n. In all constructs, a 13-amino acid sequence is present between the last residues in the ECFP domain core and the first residue of the repeat sequence while six amino acids are present between the C-terminus of the repeat sequence and the first N-terminal amino acid of the EYFP core. In our analysis, we assume these residues to be part of the flexible linker (Figure 1A). To efficiently generate expression constructs with many different linker lengths, we used a strategy in which a construct with nine repeats was partially digested with restriction enzymes, followed by religation to generate shorter linker lengths. BamHI sites were introduced into the G2S4 and GS5 linkers, whereas SacI was used in the polyserine linker (Figure 1B and Figure S1 for exact protein sequences). By digesting with small amounts of the enzyme, we cleaved only a fraction of the restriction sites. Upon religation, this resulted in a mixture of products of different lengths that can be distinguished by colony PCR (Figure 1C) and by sequencing. In this way, a diverse library of CLY proteins was obtained with linkers (G2S4)1,2,3,5,6,7,9, (GS5)1,2,3,5,6,9, and (S6)1,2,4,5,6,8,9, spanning lengths from 25 (6 + 19) to 73 (54 + 19) residues and having glycine contents of 0, 16.7, and 33.3%. All CLY proteins were expressed in E. coli and purified by nickel affinity chromatography and subsequently by Streptactin affinity chromatography. Almost all proteins were obtained in good yield, except for CLY-(S6)8 and CLY-(S6)9, which were produced as mostly insoluble protein following overnight expression. Decreasing the time of protein expression to 4 h resulted in most of the proteins being in the soluble fraction, however. Because of the stronger aggregation tendency of proteins with long polyserine linkers, CLY-(S6)8 and CLY-(S6)9 were also purified using SEC to remove residual protein oligomers, which could otherwise have interfered with the determination of FRET efficiencies. The monomeric state of all proteins was verified by measuring the fluorescence anisotropy using direct excitation of the EYFP domain (Table 1). Oligomerization of YFP fusion proteins typically results in a large decrease in fluorescence anisotropy because of homo-FRET between EYFP domains. Almost all fusion proteins showed anisotropies slightly above the value for free EYFP (0.33), providing clear evidence of their monomeric state. The anisotropy of CLY-(S6)9 was 0.32, which is slightly below that of EYFP, but still far above the value of a soluble aggregate, which is typically observed below 0.2.20
Table 1. Experimental and Calculated Properties of CLY Proteins.
| CLY | no. of amino acidsa | Gly (%)a | Eobsb | ⟨E⟩ensemblec | ⟨re⟩ensemble (Å)c | anisotropy |
|---|---|---|---|---|---|---|
| (G4S2)1 | 23 | 26.1 | 0.71d | 0.67/0.68 | 23.8/23.1 | 0.370d |
| (G4S2)2 | 29 | 34.5 | 0.65d | 0.64/0.65 | 26.6/26.0 | 0.352d |
| (G4S2)3 | 35 | 40.0 | 0.63d | 0.61/0.62 | 29.2/28.5 | 0.351d |
| (G4S2)4 | 41 | 43.9 | 0.59d | 0.58/0.59 | 31.5/30.9 | 0.350d |
| (G4S2)5 | 47 | 46.8 | 0.56d | 0.56/0.57 | 33.7/33.1 | 0.350d |
| (G4S2)6 | 53 | 49.1 | 0.52d | 0.54/0.54 | 35.8/35.1 | 0.343d |
| (G4S2)7 | 59 | 50.8 | 0.51d | 0.51/0.52 | 37.7/37.1 | 0.348d |
| (G4S2)8 | 65 | 52.3 | 0.48d | 0.49/0.50 | 39.6/38.9 | 0.341d |
| (G4S2)9 | 71 | 53.5 | 0.43d | 0.47/0.48 | 41.4/40.7 | 0.332d |
| (G2S4)1 | 25 | 16.0 | 0.64 | 0.63/0.64 | 27.4/27.1 | 0.363 |
| (G2S4)2 | 31 | 19.4 | 0.62 | 0.60/0.60 | 30.4/30.2 | 0.368 |
| (G2S4)3 | 37 | 21.6 | 0.58 | 0.57/0.57 | 33.1/33.0 | 0.365 |
| (G2S4)5 | 49 | 24.5 | 0.51 | 0.51/0.51 | 38.0/38.0 | 0.363 |
| (G2S4)6 | 55 | 25.5 | 0.49 | 0.49/0.49 | 40.2/40.2 | 0.362 |
| (G2S4)7 | 61 | 26.2 | 0.45 | 0.46/0.46 | 42.4/42.4 | 0.360 |
| (G2S4)9 | 73 | 27.4 | 0.40 | 0.42/0.42 | 46.3/46.3 | 0.361 |
| (GS5)1 | 25 | 12.0 | 0.64 | 0.62/0.62 | 28.6/28.2 | 0.362 |
| (GS5)2 | 31 | 12.9 | 0.61 | 0.58/0.69 | 31.8/31.4 | 0.363 |
| (GS5)3 | 37 | 13.5 | 0.49 | 0.55/0.55 | 34.6/34.3 | 0.363 |
| (GS5)5 | 49 | 14.3 | 0.50 | 0.49/0.49 | 39.7/39.5 | 0.358 |
| (GS5)6 | 55 | 14.5 | 0.48 | 0.47/0.47 | 42.0/41.9 | 0.354 |
| (GS5)9 | 73 | 15.1 | 0.39 | 0.40/0.40 | 48.3/48.2 | 0.352 |
| (S6)1 | 25 | 8.0 | 0.61 | 0.57/0.58 | 32.4/32.3 | 0.362 |
| (S6)2 | 31 | 6.5 | 0.60 | 0.53/0.53 | 35.9/35.9 | 0.366 |
| (S6)4 | 43 | 4.7 | 0.44 | 0.47/0.46 | 42.0/42.3 | 0.362 |
| (S6)5 | 49 | 4.1 | 0.42 | 0.44/0.43 | 44.8/45.2 | 0.360 |
| (S6)6 | 55 | 3.6 | 0.39 | 0.41/0.41 | 47.4/47.9 | 0.358 |
| (S6)8 | 67 | 3.0 | 0.37 | 0.37/0.37 | 52.2/52.8 | 0.355 |
| (S6)9 | 73 | 2.7 | 0.32 | 0.35/0.35 | 54.4/55.2 | 0.321 |
The linker length and glycine content were calculated from the entire linker, including the flexible C- and N-termini of ECFP and EYFP.
Eobs is the experimentally determined FRET efficiency.
⟨re⟩ensemble and ⟨E⟩ensemble are calculated using WLC/GC with (lp = 3.7 Å)/(C∞ = 1.9), (lp = 4.5 Å)/(C∞ = 2.4), (lp = 4.9 Å)/(C∞ = 2.6), and (lp = 6.2 Å)/(C∞ = 3.4) for G4S2, G2S4, G1S5, and S6, respectively.
Values reported in ref (14).
Figure 2 shows the fluorescence emission spectra for the CLY proteins with three different linker types as a function of linker length. When the emission spectra are normalized at the ECFP emission at 475 nm, a clear decrease in EYFP emission (527 nm) is observed with an increase in linker length for each series of sensor proteins, consistent with a decrease in FRET efficiency. In addition, although the effect is quite subtle, a consistent trend can be observed of decreasing FRET with decreasing glycine content. To correlate these spectral properties with linker stiffness, we first calculated the energy transfer efficiency (Eobs) for each sensor protein based on the enhanced acceptor fluorescence emission according to eq 3. This method is essentially the same as that used previously, except that we did not use an external EYFP reference sample to correct for direct EFYP excitation at 420 nm. Instead, this contribution was determined by direct excitation of the EYFP domain in the CLY protein at 512 nm, using a calibration curve to correlate the EYFP intensity observed at 512 nm to the EYFP intensity obtained using 420 nm excitation (eq 2; Figure S2). Table 1 shows the obtained values of Eobs for the three series of CLY proteins examined here and compares them to the values of the CLY proteins with the (G4S2)n linkers. Overall, proteins with G2S4 and GS5 linkers of the same length showed similar values of Eobs, ranging from 0.64 for the shortest linkers (25 amino acids) to 0.39–0.40 for the longest linkers containing nine repeats (73 amino acids). Although the differences are modest, these FRET efficiencies are consistently lower than those obtained previously for the G4S2 linkers but higher than those of CLY proteins with linkers containing only serine.
Figure 2.
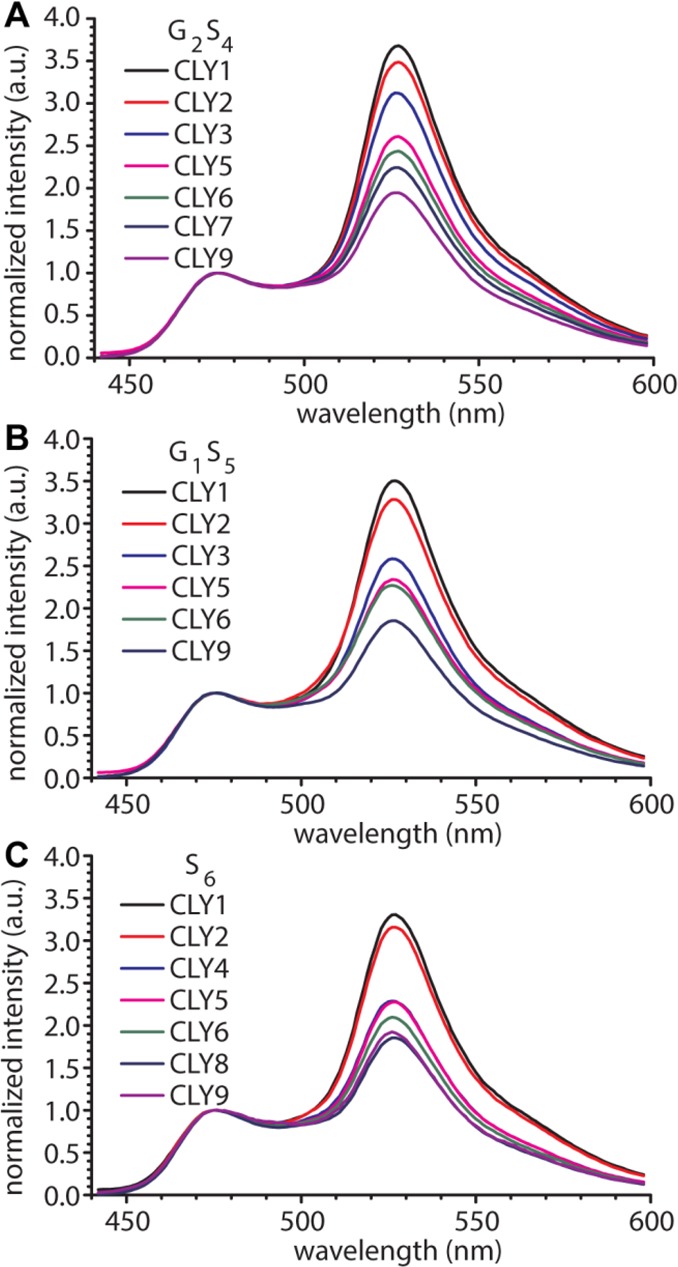
Spectra of CLY proteins with linkers of varying lengths and glycine contents. Normalized fluorescence emission spectra of 200 nM CLY proteins with different lengths of (A) (G2S4) linkers, (B) (GS5) linkers, and (C) (S6) linkers.
While these results show that a decrease in glycine content indeed results in an increase in linker stiffness, the influence does not appear to be very strong. To directly correlate the observed energy transfer efficiencies with linker stiffness, two models were used to describe the conformational behavior of the peptide linker. The GC model describes the polymer chain as a series of n rigid segments (residues) with constant length b0 (equal to the length of one amino acid, 3.8 Å), joined by completely flexible hinges. To account for restrictions in dihedral angles of real polypeptides, a “characteristic ratio” Cn is incorporated. This parameter depends on the chain length, but at lengths of >30 residues, it approaches a constant value, C∞. This parameter thus describes the stiffness of the chain. The GC model was used by Flory and co-workers to establish the initial relationship between the glycine content and random coil stiffness.17,18 The WLC model describes the peptide chain as a continuous semiflexible tube with a contour length lc and a persistent memory of its direction. This correlation of the direction of the tube at points i and i + Δl decays exponentially with Δl. The “persistence length”, lp, is inversely related to the rate of this decay (the length where the correlation is 1/e) and is therefore a measure of the stiffness of the chain. Short, stiff linkers (lc < lp) effectively behave like rods, while at long contour lengths, the chain behaves in a Gaussian fashion where lp = C∞ × b0/2. The advantage of the WLC model is that it can also be used to describe the conformational properties of relatively short and/or stiff polymers, whereas application of the GC model using the single C∞ is valid for only long flexible linkers. Because the linkers used here are relatively long and flexible (vide infra), both models yielded very similar results. The results obtained using the WLC model will be described here, while results obtained using the GC model are shown in the Supporting Information.
The WLC model can be used to calculate the probability distribution of the end-to-end distance (re) of the linker as a function of linker length and persistence length. Figure 3A shows the calculated probability distributions for linkers of the same length (73 amino acids) with values of lp ranging from the predicted value for polyglycine (3.7 Å) to that of polyalanine (17.1 Å). To theoretically predict FRET efficiencies for a given linker length and stiffness and enable a comparison with experimental results, the interchromophore distances (rc) must be calculated from the end-to-end distances. The chromophores of fluorescent proteins are located at the centers of the β-barrel structure, a considerable distance from the point of attachment of the linker. Furthermore, each end-to-end distance is sampled by many linker conformations with the fluorescent proteins oriented in different directions, resulting in many different possible values of rc for each re. To account for these different orientations, we used a numerical model in which the two vectors that connect the end of each linker with the chromophores are independently rotated over a sphere to sample all possible conformations. The interchromophore distances and FRET efficiencies were calculated for each of these orientations and then averaged. In contrast to our previous study, conformations with interchromophore distances of <22 Å were excluded from the analysis, because these conformations are sterically prohibited. Twenty-two angstroms is the hydrodynamic radius of GFP,21 and the interchromophore distance in the GFP dimer.22 Taking the excluded volume of the fluorescent proteins into account leads to larger values for ⟨rc⟩ and smaller values for ⟨E⟩ensemble. The effect is most pronounced at low re values and gradually decreases until 66 Å (the 22 Å cutoff plus the combined lengths of vc and vy). By repeating this procedure for every re, we obtained relationships between re and the average interchromophore distance ⟨rc⟩, and average FRET efficiency ⟨E⟩ (Figure 3B). Multiplying ⟨E⟩ at every re by the probability distribution and integrating over all values of re yield the ensemble average FRET efficiency (⟨E⟩ensemble) for a CLY protein that can be compared to observed FRET efficiency Eobs (Figure 3C).
Figure 3.
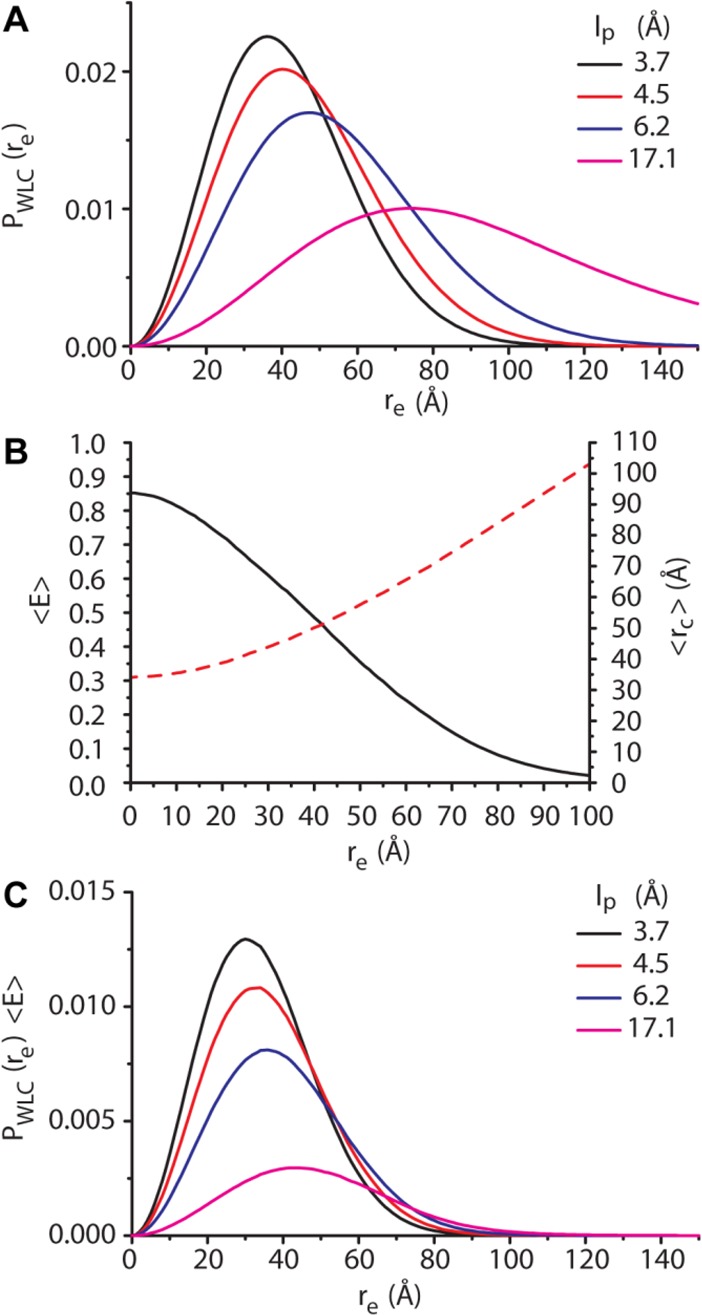
Predicting FRET efficiencies of CLY proteins with the wormlike chain model. (A) End-to-end distance probability distributions of WLCs with a contour length of 73 amino acids and different persistence lengths. (B) Numerical relationship between the end-to-end distance and the average interchromophore distance (red dashed line) and the average FRET efficiency (black solid line). (C) Curves obtained by multiplying the probability density distributions in panel A by the average FRET efficiency at each end-to-end distance. The area under these curves is the ensemble average FRET efficiency for a CLY protein.
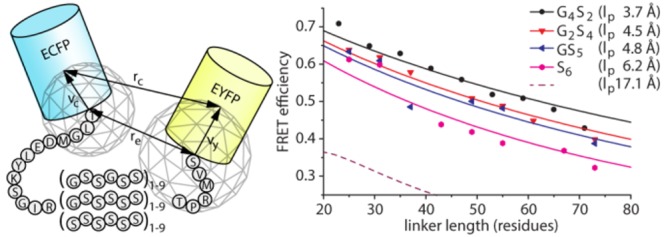
The observed energy transfer efficiencies (Eobs) of the three series of CLY proteins were compared to curves of ⟨E⟩ensemble versus linker length as predicted by the WLC model for different persistence lengths (Figure 4 and Table 1). The WLC models that best describe the experimental data had persistence lengths of 3.7, 4.5, 4.8, and 6.2 Å for G4S2, G2S4, GS5, and S6 linkers, respectively. The lp of 3.7 Å for the G4S2 linker reported here is slightly lower than that reported previously (4.5 Å), which results from the exclusion of conformations in our analysis that are sterically impossible (rc < 22 Å). These persistence lengths correspond to characteristic ratios C∞ of 1.9 (G4S2), 2.4 (G2S4), 2.6 (GS5), and 3.4 (S6) (Figure S3). While the C∞ value for the G4S2 linker is close to that predicted for an all-glycine linker (C∞ = 2.1), the dependence of C∞ on glycine content is significantly smaller than that predicted by the Flory model (lp = 17.1 Å and C∞ = 9.3 for the polyserine linker).
Figure 4.
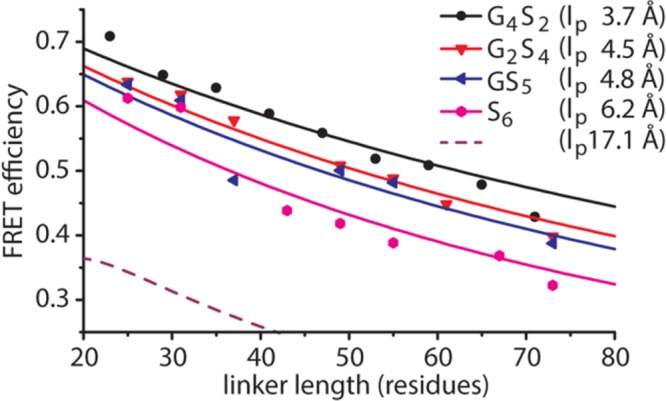
Comparison of experimental FRET efficiencies with those predicted by a wormlike chain model. Symbols show Eobs values for all CLY proteins. Solid lines represent best fit modeled ⟨E⟩ensemble vs linker length curves with indicated values of lp. Best fits were determined by minimizing the average root-mean-square deviation (RMSD) between the model and experimental data. The purple dashed line (lp = 17.1 Å) is the modeled FRET efficiency vs linker length curve for polyserine linkers according to theoretical predictions for which lp = C∞ × b0/2.18
Discussion
Flexible linkers consisting of repeats of serine and glycine are widely used in the construction of multidomain proteins, but thus far, the relationship between the relative glycine content and the flexibility of these linkers had not been systematically studied, at least not in the context of multidomain proteins. Using a previously developed approach to provide a quantitative understanding of the amount of energy transferred in FRET sensor proteins containing flexible GlyGlySer linkers, we here determined the stiffness of linkers containing 33, 16.7, and 0% glycine. We found that the amount of energy transfer observed in these fusion proteins can be satisfactorily described by modeling the peptide linker as a random coil, using a single value to describe the linker stiffness (persistence length and characteristic ratio for WLC and GC models, respectively) over the entire range of linker lengths. The model was improved to exclude conformations that are sterically impossible (rc < 22 Å), which yielded a slightly lower value of the persistence length for the SerGlyGly linkers (lp = 3.7 Å vs lp = 4.5 Å). All of the other variables used in this model were obtained independently and were not adjusted to obtain a better fit of the model with the experimental data. When calculating E for each conformation, we used a single orientation factor κ2 of 2/3, an assumption that was recently confirmed to be valid for systems with unrestricted motions.23 Even in situations with incomplete dynamic orientational averaging on the time scale of donor excitation, the error introduced by assuming κ2 = 2/3 is typically smaller than the experimental error, as long as some reorientation occurs and the system is not fully static.44 In our calculations, we also assumed that the 13 residues at the C-terminus of ECFP and four residues at the N-terminus of EYFP are part of the linker, as these are not part of the β-barrel structure of the fluorescent domains. Three of these 17 residues are glycines (18%), which may explain why proteins with short polyserine linkers show a FRET efficiency slightly higher than that predicted by the model.
While an increase in linker stiffness was observed with a decrease in glycine content, the effect is less pronounced than that predicted by the classical GC model of Flory and co-workers.17,18 Glycine can adopt dihedral angles that are inaccessible to the other amino acids, but recent work has shown that dihedral angles for Cα-substituted amino acids are not merely “allowed” or “forbidden”, as assumed in the Flory model, but have intrinsic preferences for different angles.24−28 Another important assumption in Flory’s random coil theory is that the dihedral angles for a given residue are not affected by the identity or conformation of nearby residues (isolated pair hypothesis). Experimental studies of various polypeptides have shown that this hypothesis is not correct.27,29,30 A third explanation for the relatively short end-to-end distances we find may be found in solvent conditions. The WLC and GC models assume ideal chain statistics, relevant under θ-solvent conditions, under which attractive chain–chain interactions exactly compensate for excluded volume effects. The question of whether physiological environments constitute θ-solvents for unfolded polypeptide chains is a topic of ongoing debate. Physiological buffers were found to be θ-solvents for some intrinsically disordered proteins,45−48 but other evidence suggests that water may constitute a poor solvent for other unstructured polypeptide chains, which form collapsed globules rather than coils in the absence of a denaturant49−53 (reviewed in refs (54) and (55)). Importantly, chain collapse in water was also observed for polyglycine in MD simulations56,57 as well as in experimental studies.58 Backbone–backbone interactions are thought to be responsible for this compaction, as fully N-methylated polyglycine does not collapse.59 The presence of side chains was proposed to lower the effective concentrations of backbone amides, thereby expanding the chains.60 This mechanism may explain the increase in average end-to-end distance going from glycine-rich to glycine-poor linkers even if physiological conditions would constitute a poor solvent for the linkers. Although our primary objective was to gain a quantitative understanding of linker behavior in fusion proteins, the sets of CLY proteins that we generated here may be of interest to those studying the biophysics of natively unfolded polypeptides.
While our work is the first to study the effect of glycine content in linkers connecting two protein domains, other studies have also systematically studied the relation between glycine content and peptide flexibility. Triplet–triplet energy transfer has been used to investigate the rate of first contact formation between two residues at the ends of a chain as a function of chain length, comparing Gly-Ser repeats to polyserine repeats and investigating lengths of 3–60 amino acids.31 The rate of first contact formation in polyserine linkers was found to be only 2-fold slower than in linkers containing equal amounts of serine and glycine. The Bowler group studied the stability of an intramolecular loop formed between a histidine residue on one end and a coordinated Fe3+ ion in a covalently attached heme group at the other end under denaturing conditions, comparing different lengths of glycine-rich and alanine-rich loops.32,33 The stabilities were related to the chain stiffness, and although no absolute characteristic ratios were reported for glycine-rich and alanine-rich loops, a 1.6-fold increase in C∞ was observed with a decrease in glycine content from 60 to 4%. This modest increase in C∞ is not consistent with Flory’s predictions but agrees well with the 1.8-fold increase in C∞ observed in our work when comparing C∞ values for G4S2 and S6 linkers. Thus, while Flory’s model correctly predicts a correlation between glycine content and chain flexibility, it clearly underestimates the effective flexibility of non-glycine residues. The proteins reported here may provide a useful experimental system for benchmarking Monte Carlo-based methods and molecular dynamics force fields to more realistically describe the conformational ensembles of random coil structures observed in flexible peptide loops, intrinsically disordered proteins, or proteins under denaturing conditions.31,34−37
Our motivation for studying the effect of glycine content on linker stiffness was to allow a more rational design of the conformational behavior of multidomain proteins. On the basis of the predictions of the Flory model, we anticipated a pronounced effect of polyserine linkers on the energy transfer efficiency between fluorescent domains connected by these flexible linkers. Many FRET sensors undergo a transition from a low-FRET state in which the donor and acceptor domains do not interact to a high-FRET state in which ligand binding or protein phosphorylation induces an intramolecular domain interaction that brings the two fluorescent domains into the proximity of each other.3,38−40 Komatsu et al. showed that very long flexible linkers of up to 244 residues are therefore required to obtain kinase FRET sensors with large dynamic ranges.3 Our results show that replacement of SerGlyGly linkers in FRET sensors with polyserine linkers will decrease the energy transfer efficiency of the off state by approximately 10% when using the same linker length. The relatively small decrease in average energy transfer efficiency is due to not only the modest change in persistence length (from 3.7 to 6.2 Å) but also an intrinsic feature of the relative compactness of random coil structures.14 Because of the nonlinear dependence of energy transfer efficiency on the distance and the broad distribution of end-to-end distances for a given linker, changes in average end-to-end distance do not efficiently translate into changes in the ensemble energy transfer efficiency. Alternative strategies for attenuating energy transfer efficiency in the off state of these sensors are the introduction of rigid peptide blocks4 and the use of polyproline linkers, a strategy that has been successfully applied in FRET- and BRET-based sensors developed by the Johnsson group.41,42 While the use of these linkers represents a viable strategy for sensor improvement, the conformational behavior of these linkers is more complex and cannot be modeled using the analytical models employed here.
The persistence length of flexible linkers also determines the effective concentrations of intramolecular interaction partners that they connect and in this way affects the strength of their interaction. The probability of finding a given end-to-end distance (i.e., the distance the linker must span to allow an interaction to occur) can be used to calculate the effective concentration (Ceff) of the interaction partners at this distance. Figure 5 compares the linker length dependence of Ceff for linkers with persistence lengths of 3.7 Å (GlyGlySer) and 6.2 Å (all Ser) for distances ranging from 0 to 100 Å. When the distance that the linker needs to bridge is small (0–20 Å), flexible linkers provide slightly higher effective concentrations, but the differences are not large. However, for distances of ≥40 Å, polyserine linkers become more effective. In general, polyserine linkers provide relatively high effective concentrations over a broad range of linker lengths, whereas the values of Ceff for flexible GlyGlySer linkers are lower and rapidly decrease with a decrease in linker length. The reason that larger distances of stiffer linkers result in higher effective concentrations is that the number of conformations that can span this distance is higher for a stiffer linker than for a more flexible linker of the same length (see Figure 3A). The effects on Ceff are particularly substantial for a distance of 100 Å, where, e.g., an all-serine linker of 60 amino acids shows a Ceff 30-fold higher than that of a GGS linker of the same length. The analysis shown in Figure 5 thus represents a useful reference for designing optimal linkers in multivalent binding proteins or protein switches based on intramolecular domain interactions.
Figure 5.
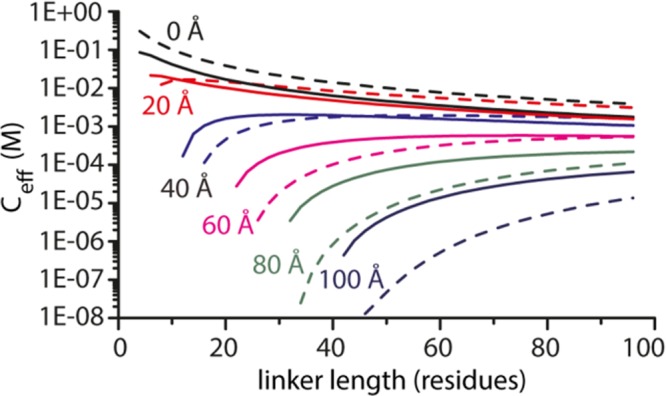
Effect of linker stiffness on effective concentration. Effective concentrations (Ceff) were calculated using the wormlike chain model as described by Zhou43 for distances ranging from 0 to 100 Å. Solid lines represent Ceff as a function of linker length for polyserine linkers (lp = 6.2 Å). Dashed lines represent Ceff as a function of linker length for GlyGlySer linkers (lp = 3.7 Å).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00902.
Protein sequences, correlation between EYFP excitation at 514 and 420 nm, and fitting of experimental FRET efficiencies using the GC model (PDF)
Author Contributions
M.v.R. and M.K. contributed equally to this work.
This work was supported by an ERC starting grant (ERC-2011-Stg 28055).
The authors declare no competing financial interest.
Supplementary Material
References
- Chen X.; Zaro J. L.; Shen W.-C. (2013) Fusion protein linkers: property, design and functionality. Adv. Drug Delivery Rev. 65, 1357–1369. 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golynskiy M. V.; Rurup W. F.; Merkx M. (2010) Antibody detection by using a FRET-based protein conformational switch. ChemBioChem 11, 2264–2267. 10.1002/cbic.201000143. [DOI] [PubMed] [Google Scholar]
- Komatsu N.; Aoki K.; Yamada M.; Yukinaga H.; Fujita Y.; Kamioka Y.; Matsuda M. (2011) Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 22, 4647–4656. 10.1091/mbc.E11-01-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Huang Z.; Zhang C.; Dong B.-J.; Guo R.-H.; Yue H.-W.; Yan L.-T.; Xing X.-H. (2016) Construction of a linker library with widely controllable flexibility for fusion protein design. Appl. Microbiol. Biotechnol. 100, 215–225. 10.1007/s00253-015-6985-3. [DOI] [PubMed] [Google Scholar]
- Atwell J. L.; Breheney K. A.; Lawrence L. J.; McCoy A. J.; Kortt A. A.; Hudson P. J. (1999) ScFv multimers of the anti-neuraminidase antibody NC10: length of the linker between V(H) and V(L) domains dictates precisely the transition between diabodies and triabodies. Protein Eng., Des. Sel. 12, 597–604. 10.1093/protein/12.7.597. [DOI] [PubMed] [Google Scholar]
- Dolezal O.; Pearce L. A.; Lawrence L. J.; McCoy A. J.; Hudson P. J.; Kortt A. A. (2000) ScFv multimers of the anti-neuraminidase antibody NC10: shortening of the linker in single-chain Fv fragment assembled in V(L) to V(H) orientation drives the formation of dimers, trimers, tetramers and higher molecular mass multimers. Protein Eng., Des. Sel. 13, 565–574. 10.1093/protein/13.8.565. [DOI] [PubMed] [Google Scholar]
- Dong J.; Kojima T.; Ohashi H.; Ueda H. (2015) Optimal fusion of antibody binding domains resulted in higher affinity and wider specificity. J. Biosci. Bioeng. 120, 504–509. 10.1016/j.jbiosc.2015.03.014. [DOI] [PubMed] [Google Scholar]
- Lee M.; Bang K.; Kwon H.; Cho S. (2013) Enhanced antibacterial activity of an attacin-coleoptericin hybrid protein fused with a helical linker. Mol. Biol. Rep. 40, 3953–3960. 10.1007/s11033-012-2472-4. [DOI] [PubMed] [Google Scholar]
- Silacci M.; Baenziger-Tobler N.; Lembke W.; Zha W.; Batey S.; Bertschinger J.; Grabulovski D. (2014) Linker length matters, Fynomer-Fc fusion with an optimized linker displaying picomolar IL-17A inhibition potency. J. Biol. Chem. 289, 14392–14398. 10.1074/jbc.M113.534578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amet N.; Lee H.-F.; Shen W.-C. (2009) Insertion of the designed helical linker led to increased expression of Tf-based fusion proteins. Pharm. Res. 26, 523–528. 10.1007/s11095-008-9767-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H. L.; Yao X. Q.; Xue C.; Wang Y.; Xiong X. H.; Liu Z. M. (2008) Increasing the homogeneity, stability and activity of human serum albumin and interferon-α2b fusion protein by linker engineering. Protein Expression Purif. 61, 73–77. 10.1016/j.pep.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Haga T.; Hirakawa H.; Nagamune T. (2013) Fine tuning of spatial arrangement of enzymes in a PCNA-mediated multienzyme complex using a rigid poly-L-proline linker. PLoS One 8, e75114. 10.1371/journal.pone.0075114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramlich P. A.; Westbroek W.; Feldman R. A.; Awad O.; Mello N.; Remington M. P.; Sun Y.; Zhang W.; Sidransky E.; Betenbaugh M. J.; Fishman P. S. (2016) A peptide-linked recombinant glucocerebrosidase for targeted neuronal delivery: design, production, and assessment. J. Biotechnol. 221, 1–12. 10.1016/j.jbiotec.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers T. H.; Van Dongen E. M. W. M.; Faesen A. C.; Meijer E. W.; Merkx M. (2006) Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry 45, 13183–13192. 10.1021/bi061288t. [DOI] [PubMed] [Google Scholar]
- Van Dongen E. M. W. M.; Evers T. H.; Dekkers L. M.; Meijer E. W.; Klomp L. W. J.; Merkx M. (2007) Variation of linker length in ratiometric fluorescent sensor proteins allows rational tuning of Zn(II) affinity in the picomolar to femtomolar range. J. Am. Chem. Soc. 129, 3494–3495. 10.1021/ja069105d. [DOI] [PubMed] [Google Scholar]
- Janssen B. M. G.; Lempens E. H. M.; Olijve L. L. C.; Voets I. K.; Van Dongen J. L. J.; De Greef T. F. A.; Merkx M. (2013) Reversible blocking of antibodies using bivalent peptide-DNA conjugates allows protease-activatable targeting. Chem. Sci. 4, 1442–1450. 10.1039/c3sc22033h. [DOI] [Google Scholar]
- Brant D. A.; Flory P. J. (1965) The configuration of random polypeptide chains. II. Theory. J. Am. Chem. Soc. 87, 2791–2800. 10.1021/ja01091a003. [DOI] [Google Scholar]
- Miller W. G.; Brant D. A.; Flory P. J. (1967) Random coil configurations of polypeptide copolymers. J. Mol. Biol. 23, 67–80. 10.1016/S0022-2836(67)80067-6. [DOI] [Google Scholar]
- Quan J.; Tian J. (2011) Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat. Protoc. 6, 242–251. 10.1038/nprot.2010.181. [DOI] [PubMed] [Google Scholar]
- Chan F. T. S.; Kaminski C. F.; Kaminski Schierle G. S. (2011) HomoFRET fluorescence anisotropy imaging as a tool to study molecular self-assembly in live cells. ChemPhysChem 12, 500–509. 10.1002/cphc.201000833. [DOI] [PubMed] [Google Scholar]
- Hink M. A.; Griep R. A.; Borst J. W.; van Hoek A.; Eppink M. H. M.; Schots A.; Visser A. J. W. G. (2000) Structural dynamics of green fluorescent protein alone and fused with a single chain Fv protein. J. Biol. Chem. 275, 17556–17560. 10.1074/jbc.M001348200. [DOI] [PubMed] [Google Scholar]
- Yang F.; Moss L. G.; Phillips G. N. (1996) The molecular structure of green fluorescent protein. Nat. Biotechnol. 14, 1246–1251. 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- Kyrychenko A.; Rodnin M. V.; Ghatak C.; Ladokhin A. S. (2017) Joint refinement of FRET measurements using spectroscopic and computational tools. Anal. Biochem. 522, 1–9. 10.1016/j.ab.2017.01.011. [DOI] [PubMed] [Google Scholar]
- Beck D. A. C.; Alonso D. O. V.; Inoyama D.; Daggett V. (2008) The intrinsic conformational propensities of the 20 naturally occurring amino acids and reflection of these propensities in proteins. Proc. Natl. Acad. Sci. U. S. A. 105, 12259–12264. 10.1073/pnas.0706527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childers M. C.; Towse C.-L.; Daggett V. (2016) The effect of chirality and steric hindrance on intrinsic backbone conformational propensities: Tools for protein design. Protein Eng., Des. Sel. 29, 271–280. 10.1093/protein/gzw023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzkee N. C.; Fleming P. J.; Rose G. D. (2005) The protein coil library: a structural database of nonhelix, nonstrand fragments derived from the PDB. Proteins: Struct., Funct., Genet. 58, 852–854. 10.1002/prot.20394. [DOI] [PubMed] [Google Scholar]
- Perskie L. L.; Street T. O.; Rose G. D. (2008) Structures, basins, and energies: a deconstruction of the protein coil library. Protein Sci. 17, 1151–1161. 10.1110/ps.035055.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A. K.; Colubri A.; Zaman M. H.; Koide S.; Sosnick T. R.; Freed K. F. (2005) Helix, sheet, and polyproline II frequencies and strong nearest neighbor effects in a restricted coil library. Biochemistry 44, 9691–9702. 10.1021/bi0474822. [DOI] [PubMed] [Google Scholar]
- Pappu R. V.; Srinivasan R.; Rose G. D. (2000) The Flory isolated-pair hypothesis is not valid for polypeptide chains: implications for protein folding. Proc. Natl. Acad. Sci. U. S. A. 97, 12565–12570. 10.1073/pnas.97.23.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth S. A.; Lewis M. C.; Karplus P. A. (2016) Beyond basins: φ,ψ preferences of a residue depend heavily on the φ,ψ values of its neighbors. Protein Sci. 25, 1757–1762. 10.1002/pro.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger F.; Fierz B.; Bieri O.; Drewello M.; Kiefhaber T. (2003) Dynamics of unfolded polypeptide chains as model for the earliest steps in protein folding. J. Mol. Biol. 332, 265–274. 10.1016/S0022-2836(03)00892-1. [DOI] [PubMed] [Google Scholar]
- Tzul F. O.; Kurchan E.; Bowler B. E. (2007) Sequence composition effects on denatured state loop formation in iso-1-cytochrome c variants: polyalanine versus polyglycine Inserts. J. Mol. Biol. 371, 577–584. 10.1016/j.jmb.2007.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan M. L.; Bowler B. E. (2012) Scaling properties of glycine-rich sequences in guanidine hydrochloride solutions. Biophys. J. 102, 1969–1978. 10.1016/j.bpj.2012.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H.-P.; Paul W.; Binder K. (2010) Polymer chain stiffness vs. excluded volume: a Monte Carlo study of the crossover towards the worm-like chain model. EPL 92, 28003. 10.1209/0295-5075/92/28003. [DOI] [Google Scholar]
- Mittal A.; Lyle N.; Harmon T. S.; Pappu R. V. (2014) Hamiltonian switch metropolis Monte Carlo simulations for improved conformational sampling of intrinsically disordered regions tethered to ordered domains of proteins. J. Chem. Theory Comput. 10, 3550–3562. 10.1021/ct5002297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal S., Mackernan D., and Coker D. F. (2015) Multiscale modelling of unimolecular FRET probes using Monte Carlo simulations. XXVI IUPAP Conference on Computer Physics, CCP2014 (Coker D. F., Tang Y., Sandvik A. W., and Campbell D. K., Eds.) IOP Publishing, Philadelphia, Vol. 640. [Google Scholar]
- Maupetit J.; Tuffery P.; Derreumaux P. (2007) A coarse-grained protein force field for folding and structure prediction. Proteins: Struct., Funct., Genet. 69, 394–408. 10.1002/prot.21505. [DOI] [PubMed] [Google Scholar]
- Miyawaki A.; Llopis J.; Heim R.; McCaffery J. M.; Adams J. A.; Ikura M.; Tsien R. Y. (1997) Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388, 882–887. 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Evers T. H.; Appelhof M. A. M.; Meijer E. W.; Merkx M. (2008) His-tags as Zn(II) binding motifs in a protein-based fluorescent sensor. Protein Eng., Des. Sel. 21, 529–536. 10.1093/protein/gzn029. [DOI] [PubMed] [Google Scholar]
- Thestrup T.; Litzlbauer J.; Bartholomäus I.; Mues M.; Russo L.; Dana H.; Kovalchuk Y.; Liang Y.; Kalamakis G.; Laukat Y.; Becker S.; Witte G.; Geiger A.; Allen T.; Rome L. C.; Chen T.-W.; Kim D. S.; Garaschuk O.; Griesinger C.; Griesbeck O. (2014) Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat. Methods 11, 175–182. 10.1038/nmeth.2773. [DOI] [PubMed] [Google Scholar]
- Griss R.; Schena A.; Reymond L.; Patiny L.; Werner D.; Tinberg C. E.; Baker D.; Johnsson K. (2014) Bioluminescent sensor proteins for point-of-care therapeutic drug monitoring. Nat. Chem. Biol. 10, 598–603. 10.1038/nchembio.1554. [DOI] [PubMed] [Google Scholar]
- Xue L.; Yu Q.; Griss R.; Schena A.; Johnsson K. (2017) Bioluminescent antibodies for point-of-care diagnostics. Angew. Chem., Int. Ed. 56, 7112–7116. 10.1002/anie.201702403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.-X. (2004) Polymer models of protein stability, folding, and interactions. Biochemistry 43, 2141–2154. 10.1021/bi036269n. [DOI] [PubMed] [Google Scholar]
- Allen L. R.; Paci E. (2009) Orientational averaging of dye molecules attached to proteins in Förster resonance energy transfer measurements: Insights from a simulation study. J. Chem. Phys. 131, 065101. 10.1063/1.3193724. [DOI] [PubMed] [Google Scholar]
- Hofmann H.; Soranno A.; Borgia A.; Gast K.; Nettels D.; Schuler B. (2012) Polymer scaling laws of unfolded and intrinsically disordered proteins quantified with single-molecule spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 109, 16155–16160. 10.1073/pnas.1207719109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Trewhella J.; Goldenberg D. P. (2008) Small-angle X-ray scattering of reduced ribonuclease A: Effects of solution conditions and comparisons with a computational model of unfolded proteins. J. Mol. Biol. 377, 1576–1592. 10.1016/j.jmb.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgia A.; Zheng W.; Buholzer K.; Borgia M. B.; Schüler A.; Hofmann H.; Soranno A.; Nettels D.; Gast K.; Grishaev A.; Best R. B.; Schuler B. (2016) Consistent view of polypeptide chain expansion in chemical denaturants from multiple experimental methods. J. Am. Chem. Soc. 138, 11714–11726. 10.1021/jacs.6b05917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes G.; Banterle N.; Ruff K. M.; Chowdhury A.; Mercadante D.; Koehler C.; Kachala M.; Estrada Girona G.; Milles S.; Mishra A.; Onck P. R.; Gräter F.; Esteban-Martín S.; Pappu R. V.; Svergun D. I.; Lemke E. A. (2017) Decoupling of size and shape fluctuations in heteropolymeric sequences reconciles discrepancies in SAXS vs. FRET measurements. Proc. Natl. Acad. Sci. U. S. A. 114, E6342–E6351. 10.1073/pnas.1704692114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möglich A.; Joder K.; Kiefhaber T. (2006) End-to-end distance distributions and intrachain diffusion constants in unfolded polypeptide chains indicate intramolecular hydrogen bond formation. Proc. Natl. Acad. Sci. U. S. A. 103, 12394–12399. 10.1073/pnas.0604748103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters R. H.; Murphy R. M. (2009) Examining polyglutamine peptide length: a connection between collapsed conformations and increased aggregation. J. Mol. Biol. 393, 978–992. 10.1016/j.jmb.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick S. L.; Jayaraman M.; Frieden C.; Wetzel R.; Pappu R. V. (2006) Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. U. S. A. 103, 16764–16769. 10.1073/pnas.0608175103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman E.; Haran G. (2006) Coil-globule transition in the denatured state of a small protein. Proc. Natl. Acad. Sci. U. S. A. 103, 11539–11543. 10.1073/pnas.0601395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F.; Lerner E.; Sato S.; Amir D.; Haas E.; Fersht A. R. (2009) Time-resolved fluorescence resonance energy transfer study shows a compact denatured state of the b domain of protein A. Biochemistry 48, 3468–3476. 10.1021/bi801890w. [DOI] [PubMed] [Google Scholar]
- Haran G. (2012) How, when and why proteins collapse: The relation to folding. Curr. Opin. Struct. Biol. 22, 14–20. 10.1016/j.sbi.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler B.; Soranno A.; Hofmann H.; Nettels D. (2016) Single-molecule FRET spectroscopy and the polymer physics of unfolded and intrinsically disordered proteins. Annu. Rev. Biophys. 45, 207–231. 10.1146/annurev-biophys-062215-010915. [DOI] [PubMed] [Google Scholar]
- Drake J. A.; Harris R. C.; Pettitt B. M. (2016) Solvation thermodynamics of oligoglycine with respect to chain Length and flexibility. Biophys. J. 111, 756–767. 10.1016/j.bpj.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doose S. (2008) Importance of backbone and solvent properties for conformational dynamics in polypeptides. ChemPhysChem 9, 2687–2689. 10.1002/cphc.200800545. [DOI] [PubMed] [Google Scholar]
- Tran H. T.; Mao A.; Pappu R. V. (2008) Role of backbone-solvent interactions in determining conformational equilibria of intrinsically disordered proteins. J. Am. Chem. Soc. 130, 7380–7392. 10.1021/ja710446s. [DOI] [PubMed] [Google Scholar]
- Teufel D. P.; Johnson C. M.; Lum J. K.; Neuweiler H. (2011) Backbone-driven collapse in unfolded protein chains. J. Mol. Biol. 409, 250–262. 10.1016/j.jmb.2011.03.066. [DOI] [PubMed] [Google Scholar]
- Holehouse A. S.; Garai K.; Lyle N.; Vitalis A.; Pappu R. V. (2015) Quantitative assessments of the distinct contributions of polypeptide backbone amides versus side chain groups to chain expansion via chemical denaturation. J. Am. Chem. Soc. 137, 2984–2995. 10.1021/ja512062h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.