

Abstract

We present a general thermodynamic top-down analysis of the effects of oxidants and pH on dopamine oxidation and cyclization, supplemented with UV–vis and electrochemical studies. The model is applicable to other catecholamines and various experimental conditions. The results show that the decisive physicochemical parameters in autoxidation are the pK values of the semiquinone and the amino group in the oxidized quinone. Addition of Ce(IV) or Fe(III) enhances dopamine oxidation in acidic media in aerobic and anaerobic conditions by the direct oxidation of dopamine and, in the presence of oxygen, also by the autoxidation of the formed semiquinone. At pH 4.5, the enhancement of the one-electron oxidation of dopamine explains the overall reaction enhancement, but at a lower pH, cyclization becomes rate-determining. Oxidation by Cu(II) at reasonable rates requires the presence of oxygen or chloride ions.
Introduction
Melanins are a group of ubiquitous natural pigments with diverse structures and biological functions, widely distributed in all kinds of organisms.1 The most studied of them are the dark eumelanins found in the skin, hair, and eyes of animals, including humans. They are heterogeneous polymeric materials that are formed by the enzymatic or chemical oxidation of various nitrogen-containing natural phenols, mainly the amino acid tyrosine and the neurotransmitters dopamine (DA) and l-3,4-dihydroxyphenylalanine (l-dopa). In biological systems, all the roles of melanins are not known, but they act, for example, as pigments, photoprotectors, and antioxidants.2,3 Eumelanins have received attention because of their versatile physical and chemical properties.4,5 They are nontoxic, biodegradable, semiconducting materials obtained from natural sources or easily synthesized and chemically modified. They effectively bind many metals and scavenge free radicals and can turn practically all absorbed light into heat. A wide range of applications have been suggested for melanin-type materials, for example, drug release,6 heavy-metal capture,7 radical scavenging,8 irradiation protection,9 energy storage10,11 and harvesting,12 structural coloration,13 biosensors and bioanalysis,14,15 bioimaging,16,17 and photothermal therapy.18,19 A synthetic material called polydopamine (polyDOPA), possessing practically identical properties to those of eumelanin, can be obtained via the chemical oxidation of dopamine by dissolved oxygen in basic aqueous solutions.20 Both polydopamine and eumelanin have complex and still debated structures, in which the major structural moieties are assumed to be catecholic or quinonoid 5,6-dihydroxyindole (DHI) units, their derivatives, and oligomers.11,21−29 The structural components are bound together via covalent or noncovalent interactions; neither eumelanin nor polydopamine can be considered true polymers but represent a mixture of various oligomeric species with structural and redox disorder.30
Although the structure of the final material is not known for certain, the initial steps leading to polydopamine formation are well established (Scheme 1). The autoxidation of dopamine (DA) by oxygen in a basic medium proceeds via dopamine semiquinone (DSQ) to dopaminequinone (DQ), which undergoes intramolecular cyclization to leucodopaminechrome (DAL). Further oxidation yields dopaminechrome (DAC), which can rearrange to 5,6-dihydroxyindole (DHI). Subsequent oxidation and coupling reactions then lead to the final insoluble product.
Scheme 1. Initial Steps in Dopamine Autoxidation.

The synthesis conditions may influence the properties of the melanin-type materials. In cells, melanogenesis takes place enzymatically within a biological matrix, and a similar biomatrix has been used as a template for the spontaneous oxidation of DHI, too.31 The autoxidation of dopamine by dissolved oxygen produces an aqueous suspension of nanoparticles and a thin globular film on immersed surfaces.20 However, many applications would require the material mainly in the form of a thin film, and smoother melanin-type films have been prepared by dissolving nanoparticles in concentrated ammonia or by using oxidative layer-by-layer multilayers.32,33 Oxygen produces highly reactive radicals, which can attack the material, and decomposition due to the splitting of the catechol moiety has been observed in natural and synthetic melanin.34,35 The autoxidation of dopamine is carried out in basic solutions, which ensures the deprotonation of the amino group required for the intramolecular Michael addition.36 On the other hand, autoxidative polydopamine formation has recently been reported even at pH 1.5 under hydrothermal conditions.37 Oxidizing salts, like ammonium persulfate, sodium periodate, and sodium chlorate, can also be used for polydopamine synthesis.38,39 In addition, several metal ions have been shown to catalyze or initiate dopamine oxidation. Redox-active transition metals, such as Ce(IV),33 Cu(II),40,41 Fe(III),42 and Mn(III),43 especially can be used for dopamine oxidation, in either the presence or absence of oxygen. This is a kind of biomimetic approach because many enzymes that catalyze the oxidation of phenols (e.g., tyrosinase and catechol oxidases) contain copper in their active sites.44 It is noteworthy that oxidizing salts and transition-metal ions can also induce dopamine oxidation in weakly acidic solutions.33,38,40−42 The effect of the oxidant on the properties of the polydopamine nanoparticles and thin films has been studied, especially with Cu(II) and oxidizing salts as the oxidants, and those products have been compared with the autoxidized products.39−41
A lot of work on dopamine (or dopa) autoxidation or metal-induced oxidation can be found in literature, including thorough kinetic analysis, but most of it is within a rather limited neutral or mildly alkaline pH range.29,35,45−49 Our previous work was based on using Ce(IV) as an oxidant in mildly acidic conditions (pH 4.5).33 We were also interested in the reported unexpected formation of polydopamine at very low pHs.37 Aiming at the production of good-quality melanin-type films with oxidative multilayers, we are interested in using more benign biocompatible metals, such as iron or copper. Therefore, in line with the suggestions in the literature, we feel that a more thorough investigation of the effects of pH and oxidants on the dopamine oxidation, ring closure, and polydopamine formation is needed.23,36 As the processes leading to polydopamine are extremely complicated, we limit our analysis only to the details of the initial steps in the overall pathway (Scheme 1) but in a wide pH range of 1–10 (in more alkaline solutions, quinones are not stable and participate in side reactions). Our approach is based on a thermodynamic analysis of the initial processes, upon which general models are built. The goal is a pH profile of the triggering events in the autoxidation reaction and a firmer understanding of the role of transition metals. The models devised can also be applied to other catecholamines under a variety of conditions. We supplement the theoretical modeling with experimental studies using UV–vis spectroscopy and electrochemistry for characterizing the initial steps in the process.
Experimental Section
Materials and Methods
Dopamine hydrochloride, cerium(IV) ammonium nitrate, and iron(III) chloride (all from Sigma-Aldrich); copper(II) sulfate pentahydrate (Merck); and catechol (Acros Organics) were used as received. All solutions were prepared in water distilled twice in quartz vessels. Numerical calculations were done using the Mathcad software (PTC).
pH Dependence of Dopamine Oxidation
Acidic dopamine solutions (0.1 mM) containing dissolved oxygen with or without metal ions (metal/dopamine ratio of 10:1) were titrated with 0.5 M NaOH. After the addition of each titrant, the UV–vis spectrum was recorded when the pH was stabilized (after a few minutes). For the long-duration oxidation studies, 0.1 mM dopamine solutions with or without metal ions (metal/dopamine ratio of 4:1) at different pHs were allowed to react in open vessels for 20 h at room temperature and then analyzed spectrophotometrically. Before the spectral measurements, the sample solutions were dispersed in an ultrasonic bath (USC500THD ultrasonic cleaner) when necessary. The pHs of the solutions were adjusted using 10 mM sulfuric acid, formate, acetate, phosphate, or citrate phosphate buffers (autoxidation).
Catechol and Dopamine Oxidation
Dopamine (0.1 mM) oxidation with dissolved oxygen was carried out in a stirred solution in a spectrophotometric cuvette at 50 °C and pH 8.5 (45 mM Tris buffer), and the oxidation in the presence of metal ions was carried out at pH 4.5 (10 mM acetate buffer). The metal/dopamine ratios were 1:1 for Ce(IV) and Fe(III) and 6:1 for Cu(II). For the anaerobic oxidation experiments, the dopamine and metal-ion solutions were deaerated with nitrogen and added to cuvettes through septum caps. With Cu(II), oxidation was followed also in the presence and absence of 0.1 M NaCl. The oxidation of catechol was carried out similarly in 0.1 M acetate buffer, but the concentrations of catechol and the metal ions were 0.2 mM. All measurements were performed at room temperature (except those for the autoxidation of dopamine).
Characterization
UV–vis spectra were measured using an HP 8453 Diode Array UV–vis spectrophotometer equipped with a Peltier temperature controller. Electrochemical studies were carried out with a computer-controlled potentiostat (Iviumstat, Ivium Technologies), a conventional three-electrode cell at room temperature with a 1 mm diameter gold working electrode (Cypress Systems), and a platinum-wire auxiliary electrode. The gold electrode was electrochemically cleaned by cycling in ultrapure 0.5 M sulfuric acid until a stable voltammogram of pure gold was obtained.50 The reference electrode was a miniature Ag/AgCl electrode, calibrated using a Ag/AgCl (3.5 M KCl) reference electrode, against which all experimental results are given. Its standard potential is +0.205 V above that of the normal hydrogen electrode (NHE), which is used as a reference in the theoretical results. All measurements were done at pH 4.5 (0.1 M acetate buffer containing 0.1 M NaNO3). The concentrations of catechol and metal ions were 1 mM, and the catechol and metal-ion solutions were deaerated with nitrogen for 30 min and added to the cells under a constant nitrogen atmosphere. Cyclic voltammograms were recorded using a sweep rate of 10 mV/s. In the alternating-current (ac) voltammograms, the potential scan rate was 5 mV/s, upon which an 11 Hz sinusoidal excitation with a 10 mV rms amplitude was added, and the rms ac-current magnitude was recorded.
Results and Discussion
Dissolved Dioxygen as Oxidant
The spontaneous autoxidation of dopamine by dissolved oxygen, the most common path to polydopamine, serves as a reference system for our discussion. The autoxidation is carried out in basic aqueous media, often at slightly elevated temperatures to speed up the relatively slow process. The spectral evolution during the autoxidation of a 0.1 mM dopamine solution in a Tris pH 8.5 buffer at 50 °C is shown in Figure S1 in the Supporting Information. The spectra show the formation of a transient species with an absorption maximum at ca. 480 nm, identified as dopaminechrome (DAC), which is formed from the cyclic leuco form (dopamineleucochrome, DAL) produced by intramolecular cyclization.35 The further reactions of dopaminechrome to 5,6-dihydroxyindole (DHI) and indolequinone (IQ) are slow, which leads to its accumulation in the solution.35,51 Dopaminechrome has another absorption band at 300 nm, which hides the possible signal due to the rapidly decaying dopamine semiquinone with a band at 305 nm.51 From the beginning, absorption increases also at 300–350 nm. Dopaminequinone has an absorbance maximum at longer wavelength (395 nm),35 we attribute these changes to the formation of the cyclized products, dopaminechrome and, especially, leucodopaminechrome. The leuco form has its absorption maximum at ca. 285 nm and a tail of higher absorptivity than that of dopamine.52 Later in the reaction, the absorbance increases over the whole observed spectral range, and the spectra begin to resemble the characteristic featureless polydopamine–melanin spectrum, masking further detailed evolution. In this discussion, we concentrate only on those initial triggering steps leading to the formation of the first cyclized products over a large pH range.
There are two separate critical steps in the formation of dopaminechrome and its further-rearrangement products. The first is the initial oxidation of dopamine to dopaminesemiquinone and its subsequent oxidation to dopaminequinone. These reactions are basically reversible. The second step is the practically irreversible intramolecular cyclization to form leucodopaminechrome, which transforms to dopaminechrome. This irreversible step can drive the overall reaction even under unfavorable conditions.
It is known that hydrogen peroxide is produced upon the oxidation of dopamine to dopaminequinone.53 This suggests that in the beginning, the two-electron redox systems O2(aq)/H2O2 and dopaminequinone/dopamine are crucial to the overall redox reaction leading to dopaminequinone. Protons are involved in these redox reactions, and the speciation of the compounds depends on pH. The effect of solution pH can be taken into account by calculating the formal standard redox potentials using the reported redox potentials and the pK values for the species involved.48,54,55 Although the formal two-electron redox potentials of the redox pairs O2(aq)/H2O2 and DQ/DQH2 (DQ denotes the catechol/quinone moiety) would suggest that a two-electron oxidation of dopamine by the oxygen/hydrogen peroxide redox pair is thermodynamically favorable over the whole pH range (Figure 1), molecular oxygen is actually a poor oxidant.56,57 The ground state of oxygen is a triplet state (3Σg–), and its direct two-electron redox reactions with organic species, which are usually in singlet states, are spin-forbidden.55,58 This creates a large activation barrier and makes the reactions slow. Therefore, the peroxide pathway must proceed through two one-electron steps with a superoxide radical anion, O2·–, or a perhydroxyl radical, HO2· (pKHO2· = 4.8),55 as an intermediate, depending on the pH. However, the standard redox potential of the aqueous O2·–/O2 couple is very low (EO2·–/O2 = −0.18 V vs NHE), which also makes the one-electron reduction of oxygen thermodynamically unfavorable in most cases. However, the superoxide radical anion, O2·–, and perhydroxyl radical, HO2·, are much stronger oxidants.42 On the other hand, dopamine has an inverted order of the one-electron redox potentials, which is typical for quinones in aqueous media.48 It should be noted here that care has to be taken when comparing the redox-potential values given in the literature as many of them refer to the biochemical standard state (pH 7). In addition, the strictly thermodynamic standard state for oxygen is an ideal gaseous substance at 1 bar, whereas a more appropriate standard state for reactions of dissolved oxygen refers to 1 M aqueous solutions (as here), leading to a value that is 0.15 V higher.
Figure 1.
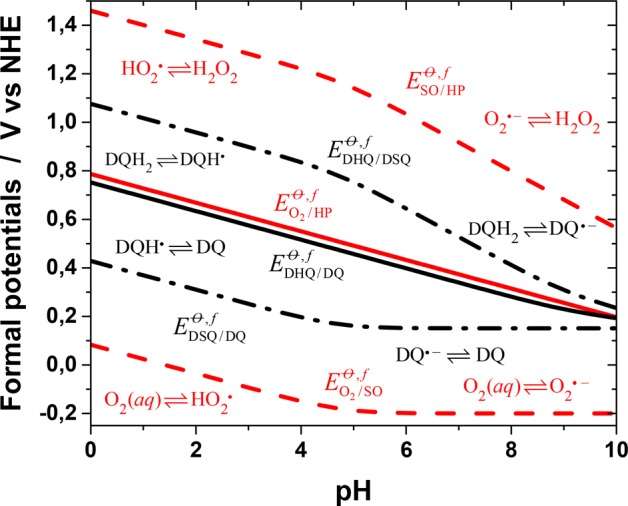
Formal standard two-electron (solid lines) and one-electron (dashed lines) redox potentials of Scheme 2 as a function of pH for the dopamine/dopaminesemiquinone/dopaminequinone (black lines) and oxygen/superoxide/hydrogen peroxide (red lines) redox systems. The dominant species involved at the different pH ranges are shown in the figure. All data refer to 25 °C.
On the basis of kinetic modeling, it has been suggested that the rate-determining step in dopamine oxidation is the formation of monodeprotonated dopamine.42 However, proton transfer to and from oxygen and nitrogen is very fast, and in the discussion of proton-coupled electron transfer, the protonation reactions are generally assumed to be in equilibrium. In accordance with this assumption, we discuss the two one-electron redox processes required to oxidize dopamine to dopaminequinone and reduce oxygen to hydrogen peroxide, using the square-scheme model suggested by Laviron,59 which is the standard model used to describe proton-coupled redox processes.60 Hydrogen peroxide is a weak acid (pKH2O2 = 11.7);55 dopamine and its semiquinone have several protonation states in the normal pH range, and both redox systems can be presented by incomplete 3 × 3 square schemes (Scheme 2). The total two-electron two-proton (2e–, 2H+) redox processes of both systems can be described by two one-electron redox reactions, and their formal standard potentials are drawn in the form of a Pourbaix diagram in Figure 1 as a function of pH (the calculation details are in the Supporting Information). The curves have been calculated using the value pKDQH· = 4.7 for dopamine semiquinone48,54 and the first macroscopic pK of dopamine for the deprotonation of the first catecholic hydroxyl (vide infra). The effect of the protonation state of the amino group is neglected.
Scheme 2. Incomplete Square Schemes Used to Calculate the Apparent One-Electron Redox Potentials EO2/SOθ,f, ESO/HP, EDQ/DSQθ,f, and EDSQ/DHQ (vs NHE at 25 °C) of (A) Oxygen Species and (B) Dopamine Species.

pH-independent general acronyms: SO = superoxide, HP = hydrogen peroxide, DQ = dopaminequinone, DSQ = dopaminesemiquinone, and DHQ = dopaminehydroquinone.
In the following, the generic acronyms SO, HP, DHQ, and DSQ (Scheme 2) will be used for superoxide, hydrogen peroxide, dopaminehydroquinone, and dopaminesemiquinone species, respectively, irrespective of their protonation state (dopaminequinone, DQ, is not protonated in the normal pH range). It is evident from Figure 1 that the first one-electron step in the autoxidation of dopamine
| 1 |
is always thermodynamically highly unfavorable, although it becomes less so at pH > 5.
In polydopamine formation, the second critical reaction step involves the cyclization of the nonprotonated amino side chain to form a 5-ring. This Michael N-addition requires that the catechol moiety be in the quinone form. Therefore, the reaction must produce dopaminequinone from the semiquinone form. After the initial oxidation to form superoxide and the semiquinone radicals, the reaction can be thought to proceed in several possible reversible paths, for example:
| 1′ |
| 2 |
| 3 |
| 4 |
| 5 |
The conditional equilibrium constants for each reaction can be calculated as a function of pH using the one-electron formal standard potentials in Figure 1 and are presented in Figure 2. The first reaction is the same as the initial oxidation by molecular dioxygen, but when the reaction products accumulate, the backward reaction has to be considered too. The first and second reactions are thermodynamically unfavorable over the whole pH range. Reaction 3 is favorable because superoxide radicals are much better oxidizing agents than dioxygen, and the reaction between superoxide and semiquinone radicals (reaction 4) is highly favorable over the whole pH range. The last reaction (reaction 5) is the semiquinone disproportionation equilibrium, which always lies on the side of the products. Dopaminequinone is formed in reactions 2, 4, and 5, which probably all contribute to its formation during the reaction. However, initially, the concentrations of superoxide and dopaminesemiquinone radicals are very low because of the slowness of the initial oxidation reaction. Therefore, reaction 4, which is between SO and DSQ and is thermodynamically highly favorable, should be the most important one because the reactants, born in the initial oxidation reaction, are very close to each other, possibly still within the encounter complex. Later, when more DSQ and SO have been generated in the solution, the significance of dopamine oxidation by SO and the disproportionation increase. The disproportionation reaction can, in effect, produce an effect known as self-acceleration of hydroquinone oxidation because of the chemical feedback it provides.53
Figure 2.
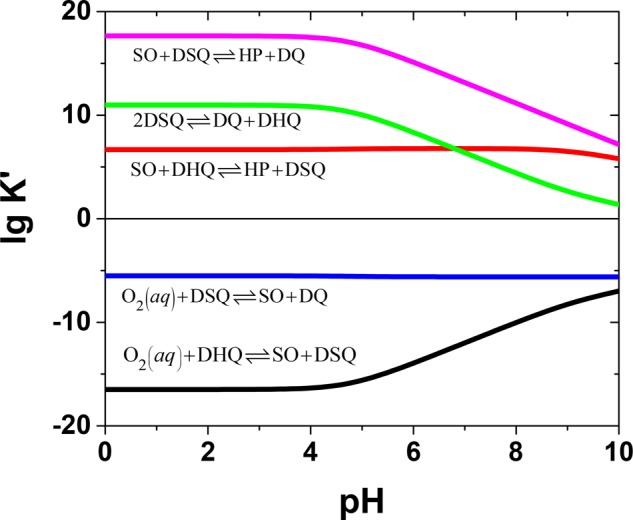
Conditional equilibrium constants, K′, as a function of pH for various redox reactions between oxygen and dopamine species.
The pH dependence of dopamine autoxidation and cyclization was studied experimentally using two approaches (Figure 3). First, 0.1 mM dopamine solutions at different pHs were allowed to react in open vessels for 20 h and analyzed spectrophotometrically. A long reaction time was used in order to also see possible changes in acidic solutions, in which the reactions are slow. However, the long time scale means that the intermediate species may already have disappeared. Second, an acidic dopamine solution was titrated with concentrated NaOH, and the spectrum was recorded after each addition when the pH was stabilized. It should be noted that this titration method is not well-defined in a thermodynamic or kinetic sense, but we have previously used it successfully to study the Ce(IV)-induced oxidation of dopamine.33 The time scale of titration is short (minutes), helping the observation of intermediate species, but each addition causes a local, transient pH rise, which can affect the reaction, especially at low pHs.
Figure 3.
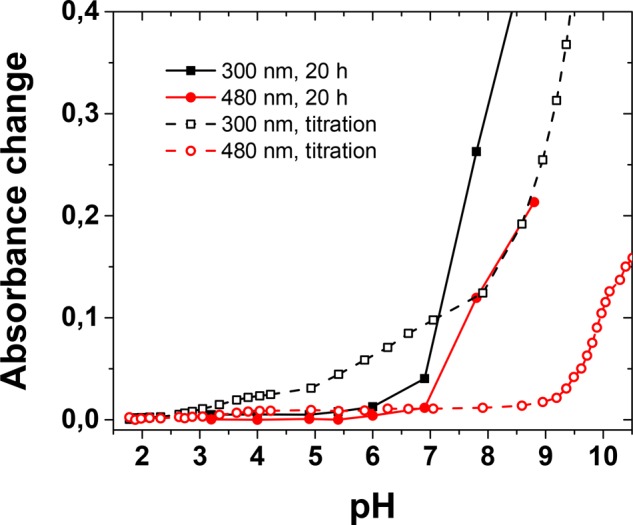
Evolution of absorbance at 300 nm (black lines and symbols) and 480 nm (red lines and symbols) in the autoxidation of a 0.1 mM dopamine solution as a function of pH during titration with 0.5 M NaOH or after 20 h of reaction time.
In the 20 h spectra, absorbance only slightly increases above pH 5 at ca. 300 nm, and after that, an increase is observed over a large wavelength range, with no clear band attributable to dopaminechrome. In the titration spectra, the first change below ca. pH 3 takes place also around 300 nm, and a separate weak band assigned to dopaminechrome forms after pH 3.5 at 480 nm.35 On the other hand, dopaminequinone should have a band at 395 nm, which is not observed.35 The absorption increase at ca. 300 nm can be assigned mostly to the formation of cyclic leucodopaminechrome, on the basis of the UV-spectrum of the corresponding form of dopa.47,52 However, we attribute these first observed changes in the titration spectra to local pH transients. Clear changes take place only above ca. pH 5, and the onset of spectral changes at 480 nm is shifted to pH 6–7, and rapid growth takes place at ca. pH 9. On the other hand, in the case of the long reaction time, all dopaminechrome has reacted further, and the spectra are obscured by other products (DHI and polydopamine), as is the case in the titration spectra at high pH, too. However, both results show that the effective onset pH for autoxidation is ca. pH 5, but the reaction rate increases markedly only at higher pHs. This is well in accordance with the calculated pH profiles for the process (vide infra).
In order to quantitatively discuss the pH dependence of dopamine autoxidation and intramolecular cyclization during the initial stages of the reaction, we describe the system by the following simple model (DAL = leucodopaminechrome, DAC = dopaminechrome):
| 6 |
This model describes the triggering events in dopamine autoxidation, and we ignore the generation of dopaminequinone via disproportionation and other reactions that are important at later stages in the process. Here, k1f, k1b, k2f, and k2b are the average apparent one-electron-forward- and backward-charge-transfer rate constants for the first and second redox steps, respectively, and kc is the apparent first-order rate constant for the ring closure (vide infra). Considering the highly unfavorable thermodynamics of the reaction between oxygen and dopamine, the first one-electron transfer should initially determine the rate of dopamine autoxidation. In order to describe the pH dependence of the redox rates, we use the classical Marcus theory to estimate the redox rate constants (see the Supporting Information).61 The electron-transfer rate constants are given by
| 7 |
where ΔG1,2θ′ = −FΔE1,2. Here, ΔG1,2θ′ and ΔE1,2 are the pH-dependent conditional Gibbs-free-energy change and the formal potential difference, respectively, for redox steps 1 or 2; λ = λis + λos is the reorganization energy, given as the sum of the inner- and outer-sphere energies; and kapp0 is the maximum apparent rate constant including the equilibrium constant for encounter-complex formation. The backward-electron-transfer rates are calculated using the conditional equilibrium constants for the steps, as k1,2;b = K1,2′/k1,2;f. Unfortunately, no reorganization energies for the relevant reactions were found in the literature, and we used the values reported for the self-exchange reactions of O2·–/O2 and the p-benzoquinone species as rough approximations.62,63 These yield a value of λ ∼ 1.5 eV,61 which is really a crude estimate. However, it turns out that the value of the reorganization energy does not have a marked influence on the form of the pH profiles calculated for the process.
The next step in the initial pathway to polydopamine is intramolecular cyclization. It takes place as a Michael N-addition of the unprotonated amino group to the oxidized catechol ring, which is subsequently reduced to hydroquinone. Nitrogen with its lone-pair electrons attacks the positively charged C5 atom in dopaminequinone (Scheme 1). The C–N-bond formation is followed by the loss of two protons and the rearomatization of the catechol ring. The nitrogen electrons act as reducing agents in this process. The macroscopic-pK values for dopamine are pK1 = 8.87 or 8.89, pK2 = 10.63 or 10.41, and pK3 = 13.1, from two independent sources.64,65 However, because of the zwitterionic nature of dopamine, the protonation and deprotonation process should be described using the microscopic-protonation constants (Scheme S1 in the Supporting Information). The distribution diagram (Figure S19) shows that the first macroscopic constant corresponds mainly to the deprotonation of the first hydroxyl group, and pK3 clearly represents the deprotonation of the second hydroxyl group. Therefore, the macroscopic-pK1 value has been used to draw the Pourbaix diagram for dopamine above. However, the macroscopic-pK2 value does not correspond to the acidity constant, pKNH3, of the protonated amino group in the oxidized dopaminequinone, for which a value of pKNH3 = 9.58 has been obtained using the dimethoxy derivative.45
The dopaminequinone-intramolecular-cyclization reaction is relatively rapid (the first-order rate constant is kc0 = 25.6 s–1 at 25 °C),45 which can be attributed to the facile formation of 5-membered rings via an exo-trig reaction.66 In all-carbon systems, this is due to favorable ring strain and entropy contributions at the transition-state geometry, and with catecholamines, the cyclization reaction is strongly favored by entropic factors, too.45,67 The cyclization rate of catecholamines increases strongly with pH. The further oxidation of the cyclic leuco form to the aminochrome form is very fast with dopa (3,4-dihydroxyphenylalanine), and the cyclization rate is identical to the observed rate of dopachrome formation. The pKNH3 value in dopa is ca. 2 units lower than in dopamine, which we suggest to be due to intramolecular hydrogen bonding, producing a ca. 10-fold difference in the cyclization rates. The oxidation of the leuco form to the aminochrome form (dopachrome or dopaminechrome) by dopa(mine)quinone is very fast, but the oxidation by dissolved oxygen is spin-forbidden and rather slow.49,51 However, because the cyclization reaction is practically irreversible, the further steps do not affect the modeling of the initial oxidation and ring-closure steps. The further rearrangement of dopaminechrome (DAC) to 5,6-dihydroxyindole (DHI) and its oxidation are relatively slow at physiological pH, leading to the accumulation of dopaminechrome under those conditions.35,68
Several kinetic studies carried out in mildly basic solutions suggest that the rate of dopamine oxidation is inversely proportional to the hydrogen-ion concentration, which has been explained by assuming the deprotonation of the hydroxyl groups or the charged amino group to be the rate-determining step.45 However, the proton-transfer kinetics of aliphatic amines is close to diffusion control in aqueous media.69 For ethylamine, the protonation and deprotonation rates have been determined to be 1.4 × 107 s–1 and 3.2 × 1010 M–1 s–1, respectively. For aliphatic amines, the rates are almost constant, although steric hindrance can decrease the protonation rate. Applying steady-state approximation to the deprotonated form and using the protonation rate of ethylamine as an approximation for the amino group in dopaminequinone (see the Supporting Information), the cyclization rate can be expressed as
| 8 |
where [DQNH2] is the concentration of dopaminequinone with a deprotonated amino group, CDQ is the total concentration of dopaminequinone, and kc0 is the first-order rate constant (estimated from the literature).45
The general kinetic model above can be discussed using different levels of approximation. In this work, we focus on the pH dependence of the initial reactions that are required to trigger the process. Therefore, we make the simplifying assumption that the concentrations of dopamine and dissolved oxygen are constant; that is, [DHQ] = CQ and [O2(aq)] = CO2 (≈ 0.25 mM at 25 °C in air). This also approximates the real reaction conditions in an open vessel at a low pH (slow reaction) and with a high dopamine concentration. Under this assumption, the kinetic scheme allows an analytic solution (for details, see the Supporting Information). The unknown pre-exponential factors, k1f0 and k2f, in the redox rate constants were estimated using the reported electron-transfer rates at pH 7.4.49 This is an approximation, too, because in the pH range considered here, differently protonated species participate in the reactions, and the rate coefficients may vary accordingly. The exact expressions are quite complicated, but they show that the DQ concentration remains low and is inversely proportional to the cyclization rate, decreasing with a pH-dependent time constant of τ = 1/kc. The DQ formation rate is given by
| 9 |
and that of leucodopaminechrome by
| 10 |
The rate of DAL formation increases with time, because the DQ concentration grows, and is zero in the beginning as no DQ is present. In order to compare the oxidation and cyclization kinetics in the early stages of the process, we looked at the initial DQ-formation rate (ξDQ) and the pH dependence of the DAL-formation rate at t ∼ 0 (ξDAL). The results, which can be understood as relative probabilities for triggering the processes at different pHs, are plotted in Figure 4 and calculated using a wide range of reorganization energies.
Figure 4.
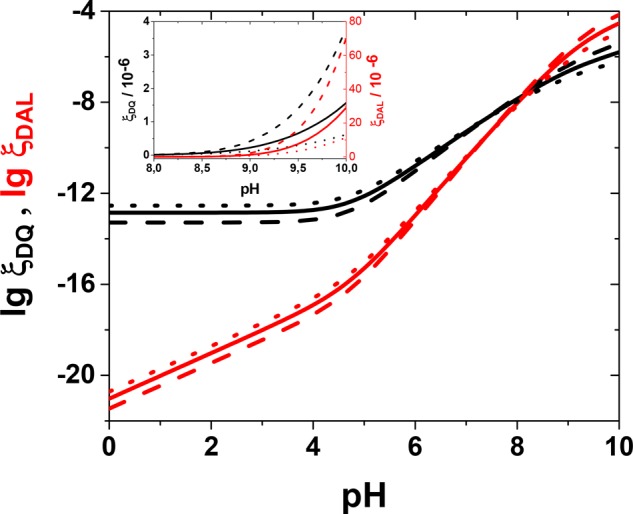
Calculated pH profiles for the initial formation (ξ, eqs 9 and 10) of dopaminequinone (ξDQ, black lines; initial rate in units of dm3 mol–1 s–1) and leucodopaminechrome (ξDAL, red lines; in units of dm3 mol–1 s–2, actual rate obtained by multiplying by time as long as t ≪ 1/kc), based on the kinetic model in the text. Solid lines, λ = 1.5 eV; dashed lines, λ = 1.8 eV; dotted lines, λ = 1.2 eV. Calculated for CQ = 1 mM, CO2 = 0.25 mM, and 25 °C. The inset shows the same curves on a linear scale.
The value of the reorganization energy does not markedly affect the rates and, more importantly, the form of their pH profiles. The calculations predict that the DQ-formation rate will be constant but small below ca. pH 5 but will increase above it. Similarly, the DAL-formation rate accelerates above pH 5 because of the higher DQ concentration, but a significant increase is observed only at high pHs (ca. 8–9). Both results are in accordance with experimental data. There are, therefore, two physicochemical parameters that control the autoxidation process of dopamine and, evidently, other catecholamines, too. They are the pK value of the hydroxyl group in the semiquinone radical (here 4.7) and the pK of the protonated amino group (9.6) in the quinone form.
The model allows us to make some major predictions and interpretations of the autoxidation process. The cyclization rate increases with time but is very low below ca. pH 5. It levels off at high pHs as all amino groups become deprotonated. The dopaminequinone-formation rate depends exponentially on the cyclization rate, and at low pHs, where the cyclization can be neglected, a steady increase of dopaminequinone is observed. Under these conditions, the reaction slowly tends toward an equilibrium determined by the first two redox steps. Therefore, at low pHs, cyclization is the rate-determining step in the process, not dopaminequinone formation. However, if there existed a mechanism for the enhancement of cyclization, then polydopamine could be formed also in highly acidic media. We think that this explains the recently reported synthesis of polydopamine at very low pHs under hydrothermal conditions.37 In aqueous media, acid dissociation is entropically favored, which lowers the pK value at higher temperatures. In addition, at high temperatures and pressures, the dielectric constant of water decreases, which profoundly affects all processes involving charged species, including protonation reactions.70 Specifically, the formation of a dopamine zwitterion becomes less favorable. Assuming little effect on dopamine oxidation, dopaminequinone with an increased fraction of neutral amino groups is formed under these conditions, allowing cyclization to take place. In addition, the cyclization reaction is enhanced at high temperatures also because it is entropically favored.45
The autoxidation model presented here is, of course, an oversimplification and should be applied only to the initial phases triggering the process. Later, disproportionation reactions and the role of superoxide radicals become important. In addition, the rate of the initial one-electron autoxidation of dopamine is very low; therefore, if semiquinone or superoxide radicals can be formed by some other pathway, the reaction can proceed more effectively, also at a low pH. This will be considered in the next section when discussing catalysis by metal ions.
Transition-Metal-Assisted Oxidation
Transition metals are known to catalyze autoxidation reactions and, in fact, metals bound to biomolecules or as traces in buffers have been suggested to be responsible for the observed autoxidation reactions in biological and experimental systems, respectively.48,53,58 Detailed kinetic analysis of iron- and copper-induced oxidation of dopa and dopamine can be found in the literature.29,42,48,49
In this work, we have studied dopamine oxidation and dopaminechrome formation under aerobic and anaerobic conditions in a pH range of 1–10 using three redox-active transition-metal ions, Ce(IV), Fe(III), and Cu(II). Under aerobic conditions, the general pH dependence of the reactions with different metal ions was tested using long reaction times and titration with NaOH, similar to that described above for the autoxidation reaction (Figure 5). The previous results with Ce(IV) are similar to those obtained with Fe(III).33 The early absorbance increase at 300 and 480 nm is attributed to the formation of leucodopaminechrome and dopaminechrome, respectively, whereas no direct evidence for dopaminequinone can be observed in the spectra. The observed pH shifts in the reaction onset, referring to the autoxidation reaction, are so large (2–6 pH units) that they require explanation in spite of the uncertainties involved.
Figure 5.
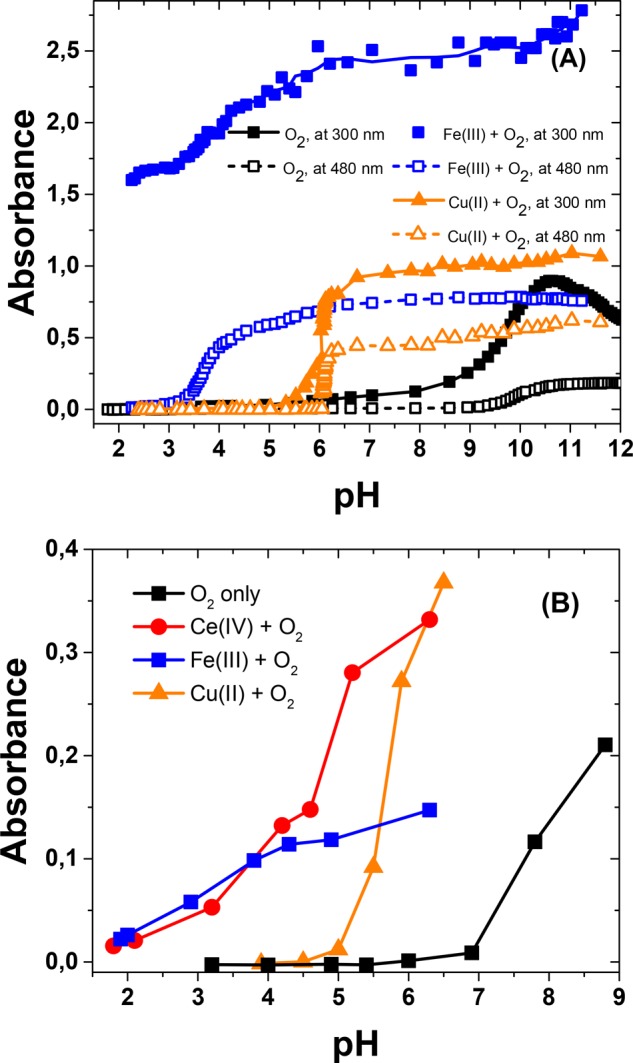
(A) Absorbance change during the titration of 0.1 mM dopamine with 0.5 M NaOH at 300 nm (solid lines, filled symbols) and 480 nm (dashed lines, open symbols) in the presence of dissolved oxygen only or with Fe(III) or Cu(II). (B) Absorbance changes at 480 nm after a reaction time of 20 h in the presence of O2 only or with 0.4 mM Fe(III), Ce(IV), or Cu(II).
The cyclization process is limited by the deprotonation of the amino group, and Figure 5 seems to imply shifts in its apparent pK value. The influence of a nearby charge on the pK value of an ionizable group is a well-known phenomenon in proteins.71 Similarly, the positive metal ion bound to the catecholate moiety electrostatically affects the protonated amino group in the molecule. This interaction thermodynamically favors its deprotonation and decreases its apparent pK value. Simple thermodynamic arguments (see the Supporting Information) suggest that this would lead to a shift of only ΔpK ∼ −0.45 per unit positive charge, which is, in fact, well in accordance with the difference between the microscopic-protonation constants, pk2 and pk12. However, metal complexation takes place with a catecholate dianion, which lowers the effective positive charge, and the effect on the amino group in dopaminequinone is questionable as a result of the poor complexing capacity of the quinone form. Therefore, electrostatic interactions must be ruled out as the cause of the shifts observed.
We can envision several possible reasons for the enhanced rate at low pHs. First, transition-metal ions may oxidize dopamine to semiquinone. Second, in the presence of oxygen, metal ions may induce the formation of reactive oxygen species. Third, complex formation with the catechol moiety may enhance either the oxidation kinetics, by alleviating the spin restrictions,53 or the thermodynamics, by forming a noninnocent complex with a lower redox potential than that of dopamine. In addition, dopamine–metal–oxygen complexes can enhance the reaction.58 The first alternative depends on the oxidation power of the metals. Dopamine is a strongly complexing ligand, and metals are predominantly attached to the catechol moiety, forming mono-, bis-, or tris-complexes. If the catechol moiety (Cat) is an innocent ligand, the redox properties of the ligand and the metal center can be treated separately. This should be the case at least with catecholates of the first-row transition metals (e.g., Fe, Cu), which can display valence tautomerism (intracomplex charge transfer) with localized charges because of reversible metal–ligand electron transfer.72,73
| 11 |
Interestingly, the oxidation of catechol-terminated self-assembled monolayers has been reported to shift cathodically in solutions containing transition metals.74
In order to understand the effects of the metal ions, we first studied the metal-assisted oxidation of catechol, in which case further reactions do not interfere, using low equimolar concentrations in order to slow down the process. The two-electron redox potential of catechol (+0.32 V vs Ag/AgCl at pH 4.5) is very close to that of dopamine in the whole pH range, but the one-electron potentials (+0.12 and +0.77 V vs Ag/AgCl at pH 4.5) are cathodically and anodically shifted with respect to those of dopamine (Figure S20). In both the presence and absence of oxygen, mixing equimolar amounts of catechol and Ce(IV) or Fe(III) solutions (at pH 4.5) results in the rapid formation of a band due to o-quinone (at ca. 390 nm), together with a decrease of absorbance of catechol and the metal. Using the reported molar absorbance of the quinone form at 390 nm (1370 M–1 cm–1),75 we can estimate that approximately half of the catechol is oxidized to quinone almost immediately by Ce(IV), and practically all the metal oxidant is consumed. In principle, this can take place either via the one-electron oxidation of catechol molecules and the consumption of all the metal, followed by semiquinone disproportionation, or via successive one-electron oxidations of catechol by the metal. In our previous work with cerium-based oxidative multilayers, the experimental evidence supported the first pathway with the complete loss of the oxidant.33 With Ce(IV), the o-quinone band decreases with time, and an isosbestic point can be seen at ca. 427 nm before the absorbance increases further above 400 nm, and a rather featureless spectrum is obtained. This further reaction can be attributed to polycatechol formation.76 Qualitatively similar behavior is seen with Fe(III), but no isosbestic point is observed, and the formation of o-quinone is slower. The band due to the 1:1 Fe(III)–catechol complex forms immediately at 700 nm as approximately one-half of the catechol is complexed.77 Concomitant with o-quinone formation, the Fe–catechol complex is broken down. In this case, the mechanism probably resembles a combination of the pathways discussed above. Cu(II) does not produce any reaction in the presence or absence of oxygen, and the final spectrum of the solution (after ca. 1 h) is identical to the sum of the component spectra.
Electrochemical experiments on the metal-catechol mixtures yielded interesting results (Table 1 and Figure 6). In the cyclic voltammograms the two-electron redox wave of catechol dominates in every case. The formal standard potential of the catechol/o-quinone pair obtained is +0.34 V vs Ag/AgCl, very close to the calculated value. The cyclic voltammograms display some smaller humps on both sides of the main peak, which cannot be accurately characterized (with Fe, there is also a small peak at ca. 0 V, which can be attributed to the Fe(III) ↔ Fe(II) transformation). Therefore, we have applied ac-voltammetry, which is a much more sensitive method, to study these systems. In ac-voltammograms, a clearly defined redox wave can be seen on both sides of the central catechol/o-quinone two-electron redox process (Figure 6B). The formal standard potential of the first of these peaks does not depend on the metal (Fe or Ce; with Cu the possible peak is masked by the Cu(II)/Cu(I)/Cu(0) redox system) in solution, indicating that it is related to the catechol moiety. The spectral measurements showed the generation of o-quinone in the reaction mixture, which must be preceded by semiquinone formation. Therefore, we attribute this more cathodic peak to the one-electron redox process of the semiquinone/o-quinone pair because the observed potentials are very close to the calculated one. The other peaks on the anodic side of the two-electron catechol/o-quinone wave are more problematic because their potential depends on the metal and is clearly more cathodic than expected for the catechol/semiquinone one-electron process. We tentatively suggest that these peaks correspond to the two-electron oxidation of metal-bound catechol. Spectra show that, in the case of Fe(III), approximately half of the catechol is complexed with metal under these conditions. The positive charge of the metal ion increases the oxidation potential, and its dependence on the metal may also imply some redox-state mixing (ligand noninnocence), especially with the Ce complex.
Table 1. Observed Formal Standard Potentials (V vs Ag/AgCl) from the ac-Voltammetry of Equimolar Mixtures of Metals and Catechol at pH 4.5a.
| catechol | Fe(III) + catechol | Ce(IV) + catechol | Cu(II) + catechol |
|---|---|---|---|
| (+0.12)b | +0.15 | +0.15 | nac |
| +0.34 | +0.35 | +0.34 | +0.34 |
| (+0.77)b | +0.60 | +0.51d | +0.50 |
Maximum errors less than ±0.01 V unless otherwise stated.
Calculated values for the one-electron potentials at pH 4.5 (SQ ↔ Q and HQ ↔ SQ, respectively).
Not available.
±0.05 V (a shoulder).
Figure 6.
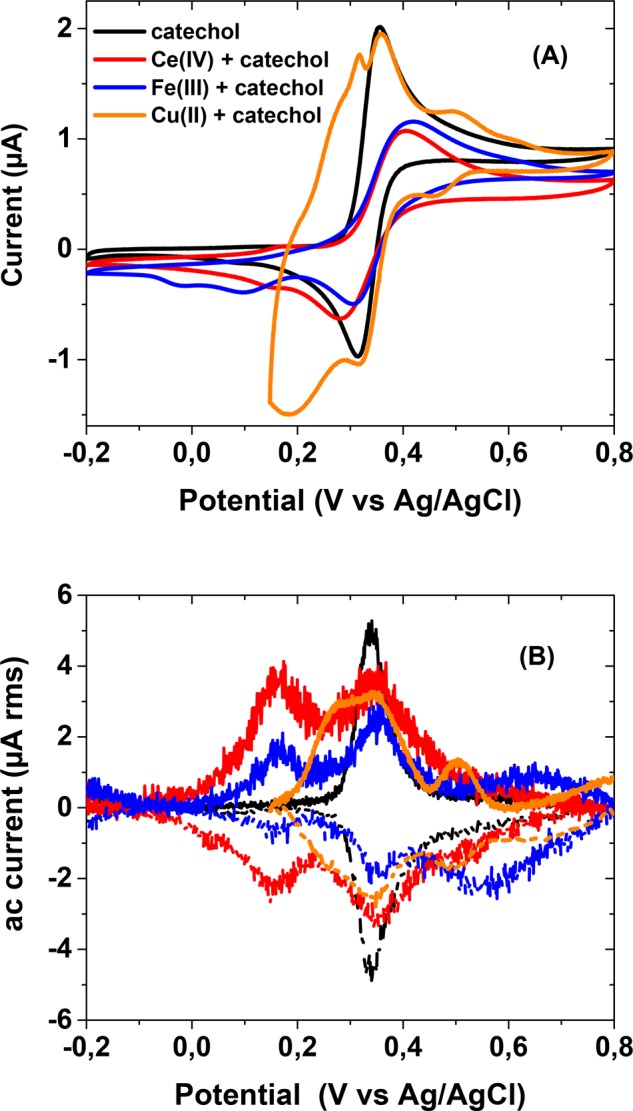
(A) Cyclic voltammograms and (B) ac-voltammograms of catechol and catechol with Ce(IV), Fe(III), or Cu(II) in the absence of dissolved oxygen. The line colors are the same in A and B. For the cyclic voltammetry, the sweep rate was 10 mV/s in the range −0.2 V ↔ +0.8 V (with Cu, +0.15 V ↔ +0.8 V). For the ac-voltammetry, the solid lines indicate anodic scans, and the dashed lines indicate cathodic scans; it is corrected for a linear background. The ac-voltammograms (B) with the metal ions have been multiplied by a factor of 7 for better comparison.
Spectroscopic studies with dopamine at pH 4.5 were done similarly using low equimolar amounts of reactants to limit the contribution from follow-up reactions (see the Supporting Information for the spectra). We have focused on this mildly acidic pH value because it was also used in previous studies by us and others.33,39−41 In addition, the pH-series study shows that at much lower pHs, no reaction takes place at a noticeable rate, whereas in solutions closer to neutral, autoxidation starts to influence the results. When dopamine is used instead of catechol, a rapid increase of a clear band at 480 nm is observed when equimolar amounts of dopamine and Ce(IV) or Fe(III) are mixed, under both aerobic and anaerobic conditions, followed by a slower decrease, forming a trace typical for an intermediate (Figure 7). No clear spectral band assignable to dopaminequinone could be observed, implying a rapid formation of the cyclized (and tautomerized) form dopaminechrome, which then reacts further. Absorbance slowly increases above ca. 550 nm, for example, at 620 nm, which corresponds to the absorbance maximum of indolequinone.33 The reported molar-absorptivity value of dopaminechrome (3280 M–1 cm–1)48 allows us to estimate that with Ce(IV), most of the oxidant is consumed. With Cu(II), no reaction took place at this pH without oxygen.
Figure 7.

Evolution of absorbance at 480 nm (corresponding to the maximum of dopaminechrome) during the oxidation of 0.1 mM dopamine at pH 4.5. The metal concentrations were 0.1 mM with Ce(IV) and Fe(III) and 0.6 mM with Cu.
In order to justify the observations, we have constructed the Pourbaix diagrams of the metals, together with dopamine (and oxygen), as shown in Figure 8 (see the Supporting Information for details). The redox properties of transition metals in aqueous solutions are quite complicated because the metal speciation strongly depends on the pH and complexing ligands. In addition, poorly soluble species can be formed, and a different diagram has to be drawn for each metal concentration separately. All side reactions of the oxidized or reduced forms of the metals can be accounted for by using the side-reaction coefficient, αox(red), defined as [ox(red)]tot = αox(red)[ox(red)]free.78 The formal standard redox potential of the redox pair ox + ze– ⇌ red is then given by
| 12 |
where Eox/redθ is the standard redox potential of the ox/red pair. In the UV–vis spectra, the metal concentrations are quite low, and the diagram here corresponds to a total nominal concentration of 0.1 mM for the oxidized and reduced forms of each metal (the methods detailed in the Supporting Information allow the diagram to be drawn for other concentrations and complexing agents). The formal potential varies with pH because of the formation of hydroxo complexes and solid hydroxides or oxides, and the appearance of solid precipitates causes sharp bends in the curves. Dopamine, and catechols in general, are strongly complexing ligands themselves, but assuming reversible complex formation and intracomplex charge transfer, the complexation with an innocent redox-active ligand is thermodynamically identical to the case with no metal complexation (see the Supporting Information). Therefore, the presence of dopamine is not assumed to influence the oxidizing power of the metals in Figure 8.
Figure 8.
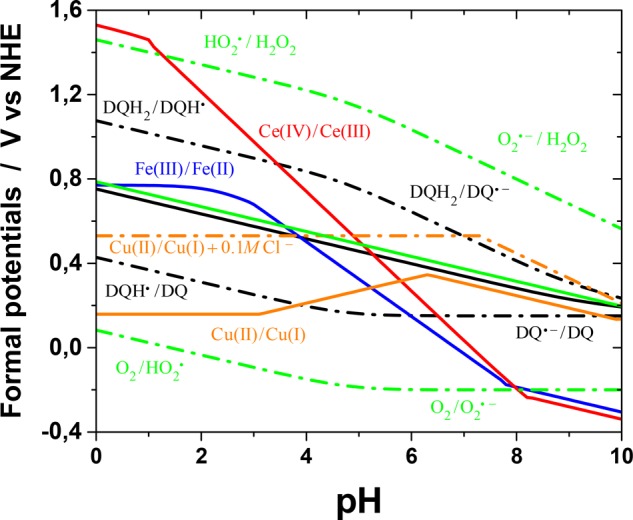
Comparison of the calculated formal one-electron potentials of the transition-metal redox systems and other relevant redox pairs (at 25 °C) as a function of pH. For each metal, the calculations refer to the potential of the solution containing both oxidation states at the nominal total concentration 0.1 mM. With Ce and Fe, only the soluble hydroxo complexes, hydroxides, and oxides are considered. With Cu, the chloro complexes are also considered. The solid black and green lines are the two-electron formal potentials of the dopamine/dopaminequinone and oxygen/hydrogen peroxide pairs, respectively.
Figure 8 shows that Ce(IV) is highly oxidizing only in acidic conditions. At low pHs, Fe(III) is also a rather good oxidizing agent, but both metals rapidly lose their oxidizing power as the pH increases. The one-electron oxidation of dopamine to semiquinone is favorable only by Ce at low pHs. In the absence of oxygen, this is the first reaction in the dopamine-oxidation process, and thermodynamics suggests rapid semiquinone formation in acidic solutions (and reasonable rates also at pH 4.5, studied here). Semiquinones tend to disproportionate at all pHs, forming dopaminequinone. On the other hand, if not all Ce(IV) is consumed in the initial oxidation of dopamine to semiquinone, the remaining oxidant can rapidly oxidize semiquinone to dopaminequinone. In both cases, half of the original dopamine will be converted to the quinone form. The same conclusions apply to the reaction of Ce with catechol, too. The formal potential of the Fe(III)/Fe(II) pair is much lower in acidic media, and assuming comparable reorganization energies, the oxidation of dopamine to semiquinone should be slower, in accordance with the experimental results. However, disproportionation and semiquinone oxidation can take place easily, as with cerium, leading to the same overall conversion, although at a lower apparent rate. The differences in the rate of the initial oxidation step explain the different dopaminechrome transients in Figure 7. No Fe–dopamine complex is observed, contrary to the oxidation of catechol, which implies fast intracomplex charge transfer. If semiquinone radicals are formed via some other route, both Ce(IV) and Fe(III) can readily oxidize them to dopaminequinone below ca. pH 6–7. In addition, if oxygen is present in the solution, reaction 2 becomes important. Its equilibrium constant is on the order of 10–5 in the whole pH range (Figure 2), but both metals produce DSQ in acidic and neutral solutions. On the other hand, the products are removed from the solution as DQ quickly cyclizes, and SO easily reacts further because of its high oxidizing potential. Therefore, reaction 2 is driven to the right, which enhances dopaminechrome formation. These factors contribute to the large onset pH shifts in Figure 5. However, at physiological and higher pHs, both Ce(IV) and Fe(III) actually become rather poor oxidizing agents for dopamine.
The behavior of Cu(II) reserves further analysis. In nature, enzymes catalyzing the oxidation of phenols and catechols usually contain copper in their active sites.44 Tyrosinase and catechol oxidase, enzymes important in melanogenesis, have a dicopper active site that can reversibly bind dioxygen as a bridge between the copper atoms. Catechol also binds to this dicopper core, although the exact bonding mode is unknown. Metal–dioxygen complexes have been suggested as being involved in the other metal-enhanced, nonenzymatic, aerobic oxidation processes of dopamine and catechol, too.58 Actually, in such complexes, dioxygen is assumed to be reduced to a superoxide radical.79 Mechanisms involving redox reactions between iron or copper and oxygen species have also been devised for kinetic studies of the metal-assisted aerobic oxidation of dopamine.48,49 Especially, as Fe(II) or Cu(I) accumulate during the reaction, oxygen may provide a chemical feedback route by regenerating the oxidized transition metals. It is evident from Figure 8 that thermodynamically, under anaerobic conditions, Cu(II) is only a slightly better oxidant than oxygen. However, chloride ions have an enhancing effect, which has been attributed to strong Cu(I)–chloro complexes.48 In this work, the experimental and theoretical results confirm that chloride ions enhance the rate of dopamine oxidation by Cu(II) and allow us to attribute the effect to the increased oxidizing power of the Cu(II)/Cu(I) pair, in accordance with previous studies. Calculations further show that under the conditions studied, this is due to the stabilization of Cu(I) by soluble chloro complexes above a chloride concentration of ca. 1 mM and not to the formation of insoluble CuCl (see the Supporting Information, Figure S22). In mildly acidic and neutral solutions containing chloride, Cu(II) can drive reaction 2 to the right, similar to Ce(IV) and Fe(III), as discussed above. Interestingly, calculations indicate that in alkaline and neutral solutions (ca. pH > 6), including at physiological pH, Cu(II) is actually a better oxidizing agent than Ce(IV) or Fe(III) (however, the redox potentials are dependent on all complexing species in the medium). The clear threshold pH of 5–6 in Figure 5 coincides with the increase of the oxidizing power of Cu(II). Furthermore, thermodynamics shows that the stable potential range of Cu(I) is very narrow,80 which suggests that Cu(II) could actually act as a two-electron oxidant, contrary to what Fe(III) and Ce(IV) can do, especially as the second one-electron oxidation potential of dopamine is low under these conditions. It is important to notice that, in this study, the observed ineffectiveness of Cu(II) alone in the oxidation of catechol or dopamine is actually a matter of the time scale. The half-life of intramolecular electron transfer in a Cu(II)–catechol complex is reported to be approximately 10 h at room temperature.81 Previous work on Cu(II)-assisted dopamine oxidation and polymerization have also demonstrated the very slow rate of the process.39−41
As a summary, the spectral and electrochemical results show that Ce(IV), Fe(III), and Cu(II) in chloride-containing solutions, can oxidize dopamine (Ce and Fe can oxidize catechol, too) to semiquinone, increasing its formation at mildly acidic pHs. When dissolved oxygen is present, the metals enhance reaction 2, which further accelerates the whole process. Semiquinone can disproportionate or be oxidized by oxygen/superoxide to o-quinone, as in autoxidation, but all the metals can also directly oxidize semiquinone. With catechol, in the time scale of the experiments, the main product is o-quinone (which slowly produces polycatechol), whereas only dopaminechrome and further products are seen with dopamine (at pH 4.5). This suggests that the ring-closure reaction is fast enough to consume practically all the dopaminequinone formed. The metal-assisted pathway suggested by the analysis is shown in Scheme 3, in which the steps shown with dashed lines can take place under aerobic conditions (possible metal–oxygen–dopamine complexes are omitted).
Scheme 3. Pathways of Dopamine Oxidation and Ring Closure in the Presence of a Transition Metal.

The steps possible in the presence of oxygen are shown with dashed lines.
An important question is whether transition metals enhance the cyclization reaction, too. Although the catalysis of the autoxidation reaction by transition metals has been much studied, mostly close to physiological pH, the effects of metals on this subsequent reaction have received much less attention. In organic solvents, metal-catalyzed Michael aza-addition to α,β-unsaturated ketones has been shown to be due to Brönsted acid catalysis by formed protons.82 The spectra obtained during the anaerobic oxidation of dopamine in the presence of metals at pH 4.5 allow us to estimate the initial rate of dopaminechrome formation. Initially, absorbance at 480 nm increases linearly in the mixture of 0.1 mM Ce(IV) and 0.1 mM dopamine (Figure 7), yielding an initial rate of ca. 1.4 × 10–8 M s–1. Under similar conditions, half of catechol is immediately oxidized to o-quinone. The cyclization is a unimolecular process and must be proportional to the quinone concentration. With these assumptions, the value of 2.3 × 10–4 s–1 is obtained for the observed dopaminechrome-formation-rate coefficient. On the other hand, we obtain a value of 2.0 × 10–4 s–1 for the cyclization rate at pH 4.5 from eq 8. Considering the uncertainties involved, these two values are in remarkably good accordance with each other and suggest that the unassisted rate of ring closure is enough to cyclize practically all dopaminequinone formed in the Ce(IV)-enhanced oxidation. In case of Fe(III) and Cu(II) with chloride, the oxidation reaction is slower than with Ce(IV). Therefore, at pH 4.5, the enhancement of dopamine oxidation due to thermodynamic factors is enough to explain the positive effects of all the studied redox-active transition-metal ions on dopaminechrome formation. However, this pH lies within an intermediate range between two possibilities. At low pHs, the rate of dopamine oxidation by metals increases, and the cyclization will become rate-determining. On the other hand, at high pHs, the thermodynamic driving force for the oxidation to semiquinone decreases, but the rate constant of cyclization increases.
Conclusions
A general thermodynamic model was developed to explain the pH dependence of the oxidation and cyclization of dopamine both in autoxidation and in the presence of transition-metal ions. Combined with a simple model mechanism, pH-dependent relative probabilities could be obtained for the initial triggering steps in autoxidation. This model is applicable to the reactions of other catecholamines, too. In autoxidation, the most important physicochemical parameters are the pK values of the corresponding semiquinone radical and the pendent amino group in the oxidized quinone form.
The addition of oxidizing transition-metal ions (Ce4+, Fe3+) to a dopamine solution greatly accelerates the reaction at mildly acidic pHs and shifts the effective dopamine oxidation and cyclization range down by several pH units. A thermodynamic model easily applied to many different cases was created to explain the enhancement. The model and experimental studies show that these two metals can accelerate the oxidation of dopamine to the semiquinone form in mildly acidic solutions in the absence of oxygen. The semiquinone formed is transformed to dopaminequinone, which rapidly cyclizes and tautomerizes to dopaminechrome. In acidic media, the model predicts that the initial oxidation rate is higher with Ce(IV) than with Fe(III), which is also experimentally observed. In mildly acidic conditions, this enhancement of the dopamine-to-semiquinone-oxidation rate can explain the overall rate enhancement in the process leading to dopaminechrome (and further products in the polydopamine pathway). In acidic media, Cu(II) alone is not a good enough oxidizing agent to initiate the process (at a reasonable rate) but requires the presence of chloride ions or dissolved oxygen. Thermodynamic considerations show that chloride ions increase the formal Cu(II)/Cu(I) redox potential because of the formation of strong Cu(I)–chloro complexes. Interestingly, in neutral (physiological) and basic media, Cu(II) becomes a better oxidant than Fe(III) or Ce(IV).
We are well aware that the reaction is much more complicated than it is discussed in this work, which focuses on the early stages of the process where many complications due to the further reactions of the formed products can be neglected. However, it shows that the thermodynamic top-down approach can clarify the first steps in polydopamine formation over a wide variety of experimental conditions.
Acknowledgments
L.M. gratefully acknowledges the Emil Aaltonen Foundation for research grants (grant numbers 160165 and 170166 N).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.8b02304.
Compilation of relevant spectral data; UV–vis spectra of dopamine and catechol oxidation and cyclization; discussion of dopamine-protonation equilibria; discussion of the effects of Coulombic interactions on protonation constants; discussion on dopamine cyclization; calculation of the formal redox potentials of dopamine, catechol, Ce(IV)/Ce(III), Fe(III)/Fe(II), and Cu(II)/Cu(I); solution of the kinetic model of dopamine oxidation and cyclization; and author contributions (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Solano F. Melanins: Skin Pigments and Much More—Types, Structural Models, Biological Functions, and Formation Routes. New J. Sci. 2014, 2014, 1–28. 10.1155/2014/498276. [DOI] [Google Scholar]
- Simon J. D.; Peles D. N. The Red and the Black. Acc. Chem. Res. 2010, 43, 1452–1460. 10.1021/ar100079y. [DOI] [PubMed] [Google Scholar]
- Bustamante J.; Bredeston L.; Malanga G.; Mordoh J. Role of Melanin as a Scavenger of Active Oxygen Species. Pigm. Cell Res. 1993, 6, 348–353. 10.1111/j.1600-0749.1993.tb00612.x. [DOI] [PubMed] [Google Scholar]
- Meredith P.; Sarna T. The Physical and Chemical Properties of Eumelanin. Pigm. Cell Res. 2006, 19, 572–594. 10.1111/j.1600-0749.2006.00345.x. [DOI] [PubMed] [Google Scholar]
- Cicoira F.; Santato C.; Pezzella A.; Wünsche J.. Eumelanin: An Old Natural Pigment and a New Material for Organic Electronics – Chemical, Physical, and Structural Properties in Relation to Potential Applications. In Organic Electronics: Emerging Concepts and Technologies; Cicoira F., Santato C., Eds.; Wiley-VCH: Weinheim, Germany, 2013; pp 113–137. [Google Scholar]
- Araújo M.; Viveiros R.; Correia T. R.; Correia I. J.; Bonifácio V. D.; Casimiro T.; Aguiar-Ricardo A. Natural Melanin: A Potential pH-Responsive Drug Release Device. Int. J. Pharm. 2014, 469, 140–145. 10.1016/j.ijpharm.2014.04.051. [DOI] [PubMed] [Google Scholar]
- Kim D. J.; Ju K.-Y.; Lee J.-K. The Synthetic Melanin Nanoparticles Having an Excellent Binding Capacity of Heavy Metal Ions. Bull. Korean Chem. Soc. 2012, 33, 3788–3792. 10.5012/bkcs.2012.33.11.3788. [DOI] [Google Scholar]
- Ju K.-Y.; Lee Y.; Lee S.; Park S. B.; Lee J.-K. Bioinspired Polymerization of Dopamine to Generate Melanin-Like Nanoparticles Having an Excellent Free-Radical-Scavenging Property. Biomacromolecules 2011, 12, 625–632. 10.1021/bm101281b. [DOI] [PubMed] [Google Scholar]
- Rageh M. M.; El-Gebaly R. H.; Abou-Shady H.; Amin D. G. Melanin Nanoparticles (MNPs) Provide Protection against Whole-Body E -Irradiation Pound in Mice via Restoration of Hematopoietic Tissues. Mol. Cell. Biochem. 2015, 399, 59–69. 10.1007/s11010-014-2232-y. [DOI] [PubMed] [Google Scholar]
- Kim Y. J.; Wu W.; Chun S.-E.; Whitacre J. F.; Bettinger C. J. Biologically Derived Melanin Electrodes in Aqueous Sodium-Ion Energy Storage Devices. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20912–20917. 10.1073/pnas.1314345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. J.; Khetan A.; Wu W.; Chun S.-E.; Viswanathan V.; Whitacre J. F.; Bettinger C. J. Evidence of Porphyrin-Like Structures in Natural Melanin Pigments Using Electrochemical Fingerprinting. Adv. Mater. 2016, 28, 3173–3180. 10.1002/adma.201504650. [DOI] [PubMed] [Google Scholar]
- Lee J.-W.; Cho H.-B.; Nakayama T.; Sekino T.; Tanaka S.-I.; Minato K.; Ueno T.; Suzuki T.; Suematsu H.; Tokoi Y.; et al. Dye-Sensitized Solar Cells Using Purified Squid Ink Nanoparticles Coated on TiO2 Nanotubes/nanoparticles. J. Ceram. Soc. Jpn. 2013, 121, 123–127. 10.2109/jcersj2.121.123. [DOI] [Google Scholar]
- Wu T.-F.; Hong J.-D. Dopamine-Melanin Nanofilms for Biomimetic Structural Coloration. Biomacromolecules 2015, 16, 660–666. 10.1021/bm501773c. [DOI] [PubMed] [Google Scholar]
- Martín M.; González Orive A.; Lorenzo-Luis P.; Hernández Creus A.; González-Mora J. L.; Salazar P. Quinone-Rich Poly (dopamine) Magnetic Nanoparticles for Biosensor Applications. ChemPhysChem 2014, 15, 3742–3752. 10.1002/cphc.201402417. [DOI] [PubMed] [Google Scholar]
- Wang D.; Chen C.; Ke X.; Kang N.; Shen Y.; Liu Y.; Zhou X.; Wang H.; Chen C.; Ren L. Bioinspired Near-Infrared-Excited Sensing Platform for in Vitro Antioxidant Capacity Assay Based on Upconversion Nanoparticles and a Dopamine–Melanin Hybrid System. ACS Appl. Mater. Interfaces 2015, 7, 3030–3040. 10.1021/am5086269. [DOI] [PubMed] [Google Scholar]
- Ju K.-Y.; Lee J. W.; Im G. H.; Lee S.; Pyo J.; Park S. B.; Lee J. H.; Lee J.-K. Bio-Inspired, Melanin-like Nanoparticles as a Highly Efficient Contrast Agent for T 1-Weighted Magnetic Resonance Imaging. Biomacromolecules 2013, 14, 3491–3497. 10.1021/bm4008138. [DOI] [PubMed] [Google Scholar]
- Fan Q.; Cheng K.; Hu X.; Ma X.; Zhang R.; Yang M.; Lu X.; Xing L.; Huang W.; Gambhir S. S.; et al. Transferring Biomarker into Molecular Probe: Melanin Nanoparticle as a Naturally Active Platform for Multimodality Imaging. J. Am. Chem. Soc. 2014, 136, 15185–15194. 10.1021/ja505412p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L.-S.; Cong Z.-X.; Cao J.-B.; Ke K.-M.; Peng Q.-L.; Gao J.; Yang H.-H.; Liu G.; Chen X. Multifunctional Fe3O4@ Polydopamine Core–shell Nanocomposites for Intracellular mRNA Detection and Imaging-Guided Photothermal Therapy. ACS Nano 2014, 8, 3876–3883. 10.1021/nn500722y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J.; Park W.; Park S.; Na K. Photosensitizer-Conjugated Hyaluronic Acid-Shielded Polydopamine Nanoparticles for Targeted Photomediated Tumor Therapy. ACS Appl. Mater. Interfaces 2016, 8, 7739–7747. 10.1021/acsami.6b01664. [DOI] [PubMed] [Google Scholar]
- Lee H.; Dellatore S. M.; Miller W. M.; Messersmith P. B. Mussel-Inspired Surface Chemistry for Multifunctional Coatings. Science 2007, 318, 426–430. 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebscher J.; Mrowczynski R.; Scheidt H. A.; Filip C.; Hadade N. D.; Turcu R.; Bende A.; Beck S. Structure of Polydopamine: A Never-Ending Story?. Langmuir 2013, 29, 10539–10548. 10.1021/la4020288. [DOI] [PubMed] [Google Scholar]
- D’ Ischia M.; Napolitano A.; Ball V.; Chen C.-T.; Buehler M. J. Polydopamine and Eumelanin: From Structure–Property Relationships to a Unified Tailoring Strategy. Acc. Chem. Res. 2014, 47, 3541–3550. 10.1021/ar500273y. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Ai K.; Lu L. Polydopamine and Its Derivative Materials: Synthesis and Promising Applications in Energy, Environmental, and Biomedical Fields. Chem. Rev. 2014, 114, 5057–5115. 10.1021/cr400407a. [DOI] [PubMed] [Google Scholar]
- Hong S.; Na Y. S.; Choi S.; Song I. T.; Kim W. Y.; Lee H. Non-Covalent Self-Assembly and Covalent Polymerization Co-Contribute to Polydopamine Formation. Adv. Funct. Mater. 2012, 22, 4711–4717. 10.1002/adfm.201201156. [DOI] [Google Scholar]
- Chen C.-T.; Ball V.; de Almeida Gracio J. J.; Singh M. K.; Toniazzo V.; Ruch D.; Buehler M. J. Self-Assembly of Tetramers of 5, 6-Dihydroxyindole Explains the Primary Physical Properties of Eumelanin: Experiment, Simulation, and Design. ACS Nano 2013, 7, 1524–1532. 10.1021/nn305305d. [DOI] [PubMed] [Google Scholar]
- Alfieri M. L.; Micillo R.; Panzella L.; Crescenzi O.; Oscurato S. L.; Maddalena P.; Napolitano A.; Ball V.; d’Ischia M. Structural Basis of Polydopamine Film Formation: Probing 5,6-Dihydroxyindole-Based Eumelanin Type Units and the Porphyrin Issue. ACS Appl. Mater. Interfaces 2018, 10, 7670. 10.1021/acsami.7b09662. [DOI] [PubMed] [Google Scholar]
- Okuda H.; Yoshino K.; Wakamatsu K.; Ito S.; Sota T. Degree of Polymerization of 5,6-dihydroxyindole-derived Eumelanin from Chemical Degradation Study. Pigm. Cell Melanoma Res. 2014, 27, 664–667. 10.1111/pcmr.12254. [DOI] [PubMed] [Google Scholar]
- Della Vecchia N. F.; Avolio R.; Alfè M.; Errico M. E.; Napolitano A.; d’ Ischia M. Building-Block Diversity in Polydopamine Underpins a Multifunctional Eumelanin-Type Platform Tunable Through a Quinone Control Point. Adv. Funct. Mater. 2013, 23, 1331–1340. 10.1002/adfm.201202127. [DOI] [Google Scholar]
- Mondal S.; Thampi A.; Puranik M. Kinetics of Melanin Polymerization during Enzymatic and Nonenzymatic Oxidation. J. Phys. Chem. B 2018, 122, 2047–2063. 10.1021/acs.jpcb.7b07941. [DOI] [PubMed] [Google Scholar]
- Micillo R.; Panzella L.; Iacomino M.; Prampolini G.; Cacelli I.; Ferretti A.; Crescenzi O.; Koike K.; Napolitano A.; d’Ischia M. Eumelanin Broadband Absorption Develops from Aggregation-Modulated Chromophore Interactions under Structural and Redox Control. Sci. Sci. Rep. 2017, 7, 41532. 10.1038/srep41532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Vecchia N. F.; Cerruti P.; Gentile G.; Errico M. E.; Ambrogi V.; d’ Errico G.; Longobardi S.; Napolitano A.; Paduano L.; Carfagna C.; et al. Artificial Biomelanin: Highly Light-Absorbing Nano-Sized Eumelanin by Biomimetic Synthesis in Chicken Egg White. Biomacromolecules 2014, 15, 3811–3816. 10.1021/bm501139h. [DOI] [PubMed] [Google Scholar]
- Bothma J. P.; de Boor J.; Divakar U.; Schwenn P. E.; Meredith P. Device-Quality Electrically Conducting Melanin Thin Films. Adv. Mater. 2008, 20, 3539–3542. 10.1002/adma.200703141. [DOI] [Google Scholar]
- Salomäki M.; Tupala M.; Parviainen T.; Leiro J.; Karonen M.; Lukkari J. Preparation of Thin Melanin-Type Films by Surface-Controlled Oxidation. Langmuir 2016, 32, 4103–4112. 10.1021/acs.langmuir.6b00402. [DOI] [PubMed] [Google Scholar]
- Szpoganicz B.; Gidanian S.; Kong P.; Farmer P. Metal Binding by Melanins: Studies of Colloidal Dihydroxyindole-Melanin, and Its Complexation by Cu (II) and Zn (II) Ions. J. Inorg. Biochem. 2002, 89, 45–53. 10.1016/S0162-0134(01)00406-8. [DOI] [PubMed] [Google Scholar]
- Bisaglia M.; Mammi S.; Bubacco L. Kinetic and Structural Analysis of the Early Oxidation Products of Dopamine: Analysis of the Interactions with -Synuclein. J. Biol. Chem. 2007, 282, 15597–15605. 10.1074/jbc.M610893200. [DOI] [PubMed] [Google Scholar]
- Yang J.; Stuart M. A. C.; Kamperman M. Jack of All Trades: Versatile Catechol Crosslinking Mechanisms. Chem. Soc. Rev. 2014, 43, 8271–8298. 10.1039/C4CS00185K. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Fan H.; Wang L.; Jin Z. Oxidative Self-Polymerization of Dopamine in an Acidic Environment. Langmuir 2015, 31, 11671–11677. 10.1021/acs.langmuir.5b02757. [DOI] [PubMed] [Google Scholar]
- Wei Q.; Zhang F.; Li J.; Li B.; Zhao C. Oxidant-Induced Dopamine Polymerization for Multifunctional Coatings. Polym. Chem. 2010, 1, 1430–1433. 10.1039/c0py00215a. [DOI] [Google Scholar]
- Ponzio F.; Barthès J.; Bour J.; Michel M.; Bertani P.; Hemmerlé J.; d’ Ischia M.; Ball V. Oxidant Control of Polydopamine Surface Chemistry in Acids: A Mechanism-Based Entry to Superhydrophilic-Superoleophobic Coatings. Chem. Mater. 2016, 28, 4697–4705. 10.1021/acs.chemmater.6b01587. [DOI] [Google Scholar]
- Bernsmann F.; Ball V.; Addiego F.; Ponche A.; Michel M.; Gracio J. J. d. A.; Toniazzo V.; Ruch D. Dopamine– Melanin Film Deposition Depends on the Used Oxidant and Buffer Solution. Langmuir 2011, 27, 2819–2825. 10.1021/la104981s. [DOI] [PubMed] [Google Scholar]
- Ball V.; Gracio J.; Vila M.; Singh M. K.; Metz-Boutigue M.-H.; Michel M.; Bour J.; Toniazzo V.; Ruch D.; Buehler M. J. Comparison of Synthetic Dopamine?Eumelanin Formed in the Presence of Oxygen and Cu2+ Cations as Oxidants. Langmuir 2013, 29, 12754–12761. 10.1021/la4029782. [DOI] [PubMed] [Google Scholar]
- Linert W.; Herlinger E.; Jameson R. F.; Kienzl E.; Jellinger K.; Youdim M. B. H. Dopamine, 6-Hydroxydopamine, Iron, and Dioxygen — Their Mutual Interactions and Possible Implication in the Development of Parkinson’s Disease. Biochim. Biophys. Acta, Mol. Basis Dis. 1996, 1316, 160–168. 10.1016/0925-4439(96)00020-8. [DOI] [PubMed] [Google Scholar]
- Barreto W. J.; Ponzoni S.; Sassi P. A Raman and UV-Vis Study of Catecholamines Oxidized with Mn(III). Spectrochim. Acta, Part A 1998, 55, 65–72. 10.1016/S1386-1425(98)00164-4. [DOI] [Google Scholar]
- Koval I. A.; Gamez P.; Belle C.; Selmeczi K.; Reedijk J. Synthetic Models of the Active Site of Catechol Oxidase: Mechanistic Studies. Chem. Soc. Rev. 2006, 35, 814–840. 10.1039/b516250p. [DOI] [PubMed] [Google Scholar]
- Young T. E.; Babbitt B. W. Electrochemical Study of the Oxidation of Alpha-Methyldopamine, Alpha-Methylnoradrenaline, and Dopamine. J. Org. Chem. 1983, 48, 562–566. 10.1021/jo00152a029. [DOI] [Google Scholar]
- Herlinger E.; Jameson R. F.; Linert W. Spontaneous Autoxidation of Dopamine. J. Chem. Soc., Perkin Trans. 2 1995, 2, 259–263. 10.1039/p29950000259. [DOI] [Google Scholar]
- Land E. J.; Ito S.; Wakamatsu K.; Riley P. A. Rate Constants for the First Two Chemical Steps of Eumelanogenesis. Pigm. Cell Res. 2003, 16, 487–493. 10.1034/j.1600-0749.2003.00082.x. [DOI] [PubMed] [Google Scholar]
- Pham A. N.; Waite T. D. Cu(II)-Catalyzed Oxidation of Dopamine in Aqueous Solutions: Mechanism and Kinetics. J. Inorg. Biochem. 2014, 137, 74–84. 10.1016/j.jinorgbio.2014.03.018. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Pham A. N.; Waite T. D. Elucidation of the Interplay between Fe(II), Fe(III), and Dopamine with Relevance to Iron Solubilization and Reactive Oxygen Species Generation by Catecholamines. J. Neurochem. 2016, 137, 955–968. 10.1111/jnc.13615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerstein-Kozlowska H.Surfaces, Cells and Solutions for Kinetic Studies. In Comprehensive Treatise of Electrochemistry; Plenum Press: New York, 1984; Vol. 9, pp 15–60. [Google Scholar]
- Thompson A.; Land E. J.; Chedekel M. R.; Subbarao K. V.; Truscott T. G. A Pulse Radiolysis Investigation of the Oxidation of the Melanin Precursors 3,4-Dihydroxyphenylalanine (dopa) and the Cysteinyldopas. Biochim. Biophys. Acta, Gen. Subj. 1985, 843, 49–57. 10.1016/0304-4165(85)90048-0. [DOI] [PubMed] [Google Scholar]
- Wyler H.; Chiovini J. Die Synthese von Cyclodopa (Leukodopachrom). Helv. Chim. Acta 1968, 51, 1476–1494. 10.1002/hlca.19680510636. [DOI] [PubMed] [Google Scholar]
- Song Y.; Buettner G. R. Thermodynamic and Kinetic Considerations for the Reaction of Semiquinone Radicals to Form Superoxide and Hydrogen Peroxide. Free Radical Biol. Med. 2010, 49, 919–962. 10.1016/j.freeradbiomed.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Rivera A. E.; Corona-Avendaño S.; Alarcón-Angeles G.; Rojas-Hernández A.; Ramírez-Silva M. T.; Romero-Romo M. A. Spectrophotometric Study on the Stability of Dopamine and the Determination of Its Acidity Constants. Spectrochim. Acta, Part A 2003, 59, 3193–3203. 10.1016/S1386-1425(03)00138-0. [DOI] [PubMed] [Google Scholar]
- Koppenol W. H.; Stanbury D. M.; Bounds P. L. Electrode Potentials of Partially Reduced Oxygen Species, from Dioxygen to Water. Free Radical Biol. Med. 2010, 49, 317–322. 10.1016/j.freeradbiomed.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Krumova K.; Cosa G.. Overview of Reactive Oxygen Species. In Singlet Oxygen: Applications in Biosciences and Nanosciences; Nonell S., Flors C., Eds.; RSC Publishing: Cambridge, 2016; Vol. 1, pp 1–21. [Google Scholar]
- Valentine J. S.Dioxygen Reactions. In Bioinorganic Chemistry; Bertini I., Gray H. B., Lippard S. J., Valentine J. S., Eds.; University Science Books: Herndon, VA, 1994; pp 253–313. [Google Scholar]
- Miller D. M.; Buettner G. R.; Aust S. D. Transition Metals as Catalysts of “autoxidation” Reactions. Free Radical Biol. Med. 1990, 8, 95–108. 10.1016/0891-5849(90)90148-C. [DOI] [PubMed] [Google Scholar]
- Laviron E. Electrochemical Reactions with Protonations at Equilibrium 0.8. the 2 E, 2 H+ Reaction (9-Member Square Scheme) for a Surface Or for a Heterogeneous Reaction in the Absence of Disproportionation and Dimerization Reactions. J. Electroanal. Chem. Interfacial Electrochem. 1983, 146, 15–36. 10.1016/S0022-0728(83)80110-7. [DOI] [Google Scholar]
- Costentin C.; Robert M.; Savéant J.-M. Update 1 of: Electrochemical Approach to the Mechanistic Study of Proton-Coupled Electron Transfer. Chem. Rev. 2010, 110, PR1–PR40. 10.1021/cr100038y. [DOI] [PubMed] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Methods. Fundamentals and Applications, 2nd ed.; John Wiley & Sons: New York, 2001. [Google Scholar]
- German E. D.; Kuznetsov A. M.; Efremenko I.; Sheintuch M. Theory of the Self-Exchange Electron Transfer in the Dioxygen/Superoxide System in Water. J. Phys. Chem. A 1999, 103, 10699–10707. 10.1021/jp9920141. [DOI] [Google Scholar]
- Stack A. G.; Rosso K. M.; Smith D. M. A.; Eggleston C. M. Reaction of Hydroquinone with Hematite II. Calculated Electron-Transfer Rates and Comparison to the Reductive Dissolution Rate. J. Colloid Interface Sci. 2004, 274, 442–450. 10.1016/j.jcis.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Martin R. B. Zwitterion Formation Upton Deprotonation in L-3,4-Dihydroxyphenylalanine and Other Phenolic Amines. J. Phys. Chem. 1971, 75, 2657–2661. 10.1021/j100686a021. [DOI] [PubMed] [Google Scholar]
- Kiss T.; Gergely A. Complexes of 3,4-Dhydroxyphnyl Derivatives, III. Equilibrium Study of Parent and Some Mixed Ligand Complexes of Dopamine, Alanine and Pyrocatechol with nickel(II), copper(II) and zinc(II) Ions. Inorg. Chim. Acta 1979, 36, 31–36. 10.1016/S0020-1693(00)89367-6. [DOI] [Google Scholar]
- Baldwin J. E. Rules for Ring Closure. J. Chem. Soc., Chem. Commun. 1976, 734–736. 10.1039/c39760000734. [DOI] [Google Scholar]
- Casadei M. A.; Galli C.; Mandolini L. Ring-Closure Reactions. 22. Kinetics of Cyclization of Diethyl (ω-Bromoalkyl)malonates in the Range of 4- to 21-Membered Rings. Role of Ring Strain. J. Am. Chem. Soc. 1984, 106, 1051–1056. 10.1021/ja00316a039. [DOI] [Google Scholar]
- Segura-Aguilar J.; Paris I.; Muñoz P.; Ferrari E.; Zecca L.; Zucca F. A. Protective and Toxic Roles of Dopamine in Parkinson’s Disease. J. Neurochem. 2014, 129, 898–915. 10.1111/jnc.12686. [DOI] [PubMed] [Google Scholar]
- Eigen M.; Maass G.; Schwarz G. Schallabsorptionsmessungen zum Studium des Einflusses sterischer Faktoren und hydrophober Wechselwirkungen auf die Geschwindigkeit protolytischer Reaktionen. Z. Phys. Chem. 1971, 74, 319–330. 10.1524/zpch.1971.74.3_6.319. [DOI] [Google Scholar]
- Weingärtner H.; Franck E. U. Supercritical Water as a Solvent. Angew. Chem., Int. Ed. 2005, 44, 2672–2692. 10.1002/anie.200462468. [DOI] [PubMed] [Google Scholar]
- Pace C. N.; Grimsley G. R.; Scholtz J. M. Protein Ionizable Groups: pK Values and Their Contribution to Protein Stability and Solubility. J. Biol. Chem. 2009, 284, 13285–13289. 10.1074/jbc.R800080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skara G.; Gimferrer M.; De Proft F.; Salvador P.; Pinter B. Scrutinizing the Noninnocence of Quinone Ligands in Ruthenium Complexes: Insights from Structural, Electronic, Energy, and Effective Oxidation State Analyses. Inorg. Chem. 2016, 55, 2185–2199. 10.1021/acs.inorgchem.5b02543. [DOI] [PubMed] [Google Scholar]
- Kaim W.; Schwederski B. Non-Innocent Ligands in Bioinorganic Chemistry-An Overview. Coord. Chem. Rev. 2010, 254, 1580–1588. 10.1016/j.ccr.2010.01.009. [DOI] [Google Scholar]
- Brooksby P. A.; Schiel D. R.; Abell A. D. Electrochemistry of Catechol Terminated Monolayers with Cu(II), Ni(II) and Fe(III) Cations: A Model for the Marine Adhesive Interface. Langmuir 2008, 24, 9074–9081. 10.1021/la8007816. [DOI] [PubMed] [Google Scholar]
- Albarran G.; Boggess W.; Rassolov V.; Schuler R. H. Absorption Spectrum, Mass Spectrometric Properties, and Electronic Structure of 1,2-Benzoquinone. J. Phys. Chem. A 2010, 114, 7470–7478. 10.1021/jp101723s. [DOI] [PubMed] [Google Scholar]
- Jabbari M.; Gharib F. Kinetics and Mechanism of the Reaction of Catechol with Ceric Ion in the Presence and Absence of iridium(III) Catalyst in Acidic Media. Monatsh. Chem. 2012, 143, 997. 10.1007/s00706-011-0676-2. [DOI] [Google Scholar]
- Sever M. J.; Wilker J. J. Visible Absorption Spectra of Metal–catecholate and Metal–tironate Complexes. Dalton Trans. 2004, 1061–1072. 10.1039/B315811J. [DOI] [PubMed] [Google Scholar]
- Sucha L.; Kotrlý S.. Solution Equilibria in Analytical Chemistry; Van Nostrand Reinhold: London, 1972. [Google Scholar]
- Holland P. L. Metal–dioxygen and Metal–dinitrogen Complexes: Where Are the Electrons?. Dalton Trans. 2010, 39, 5415. 10.1039/c001397h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer G. K.; Pesterfield L. L.. The Aqueous Chemistry of the Elements; Oxford University Press: Oxford, 2010. [Google Scholar]
- Kamau P.; Jordan R. B. Kinetic Study of the Oxidation of Catechol by Aqueous Copper(II). Inorg. Chem. 2002, 41, 3076–3083. 10.1021/ic010978c. [DOI] [PubMed] [Google Scholar]
- Wabnitz T. C.; Yu J.; Spencer J. B. Evidence That Protons Can Be the Active Catalysts in Lewis Acid Mediated Hetero-Michael Addition Reactions. Chem. - Eur. J. 2004, 10, 484–493. 10.1002/chem.200305407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.