

Abstract
Nanobubble nucleation is a problem that affects efficiency in electrocatalytic reactions since those bubbles can block the surface of the catalytic sites. In this article, we focus on the nucleation rate of O2 nanobubbles resulting from the electrooxidation of H2O2 at Pt disk nanoelectrodes. Bubbles form almost instantaneously when a critical peak current, inbp, is applied, but for lower currents, bubble nucleation is a stochastic process in which the nucleation (induction) time, tind, dramatically decreases as the applied current approaches inb, a consequence of the local supersaturation level, ζ, increasing at high currents. Here, by applying different currents below inbp, nanobubbles take some time to nucleate and block the surface of the Pt electrode at which the reaction occurs, providing a means to measure the stochastic tind. We study in detail the different conditions in which nanobubbles appear, concluding that the electrode surface needs to be preconditioned to achieve reproducible results. We also measure the activation energy for bubble nucleation, Ea, which varies in the range from (6 to 30)kT, and assuming a spherically cap-shaped nanobubble nucleus, we determine the footprint diameter L = 8–15 nm, the contact angle to the electrode surface θ = 135–155°, and the number of O2 molecules contained in the nucleus (50 to 900 molecules).
Introduction
The generation of bubbles in chemical reactions is a process that has been well known by scientists throughout history. When these bubbles are extremely small (on the order of nanometers), the problem becomes much more challenging to analyze.1−3 However, the technology used to visualize such surface nanobubbles has been very recently developed.2,4−6 The biggest concern that surface nanobubbles cause is their generation in chemical reactions, such as electrolysis7 and catalysis.8 Nanobubbles nucleating on top of reacting surfaces or electrodes influence the efficiency of chemical reactions since they partially block the reactive surface and consequently impede the reaction of interest.9 A similar situation occurs in the case of nanodroplet and nanocrystal nucleation.10 In other scenarios, such as redox reactions in cells, the nanobubbles can form within the nanoprobes and induce current amplification.11 The high internal pressures of nanobubbles make their behavior rather different from that of micro- or macrobubbles.2,12 Nanobubbles often adhere to the surface at which they originate, forming a spherical cap that strongly attaches to the active surface.2,13 Without pinning, i.e., when the nanobubbles are not attached to a specific location on the surface, due to the high pressure inside them,14,15 nanobubbles would dissolve extremely rapidly once the reaction stops. However, if there are pinning sites and constant gas supersaturation is provided at the surface, then nanobubbles on reacting surfaces are very stable16,17 and do not dissolve.2 Molecular dynamics simulations18 support this view.
In this article, we measure the nucleation rate of single O2 nanobubbles generated at Pt nanodisk electrodes by the electro-oxidation of H2O2. When the local dissolved O2 concentration at the nanoelectrode is sufficiently high,19 a nanobubble nucleates and blocks the reacting surface, as depicted in Figure 1. We study the factors affecting the nucleation rate of O2 nanobubbles under different applied currents.
Figure 1.
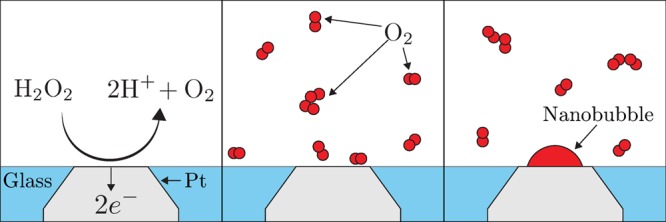
O2 nanobubble generation by electro-oxidation of H2O2. When the O2 concentration at the nanoelectrode is sufficiently high, a nanobubble nucleates after some time and partially blocks the electrode surface.
Experimental Methods
The fabrication and measurement of the size of Pt nanoelectrodes are described in detail in the Supporting Information (section 1). All experiments were performed in an aqueous solution of 1 M H2O2 and 1 M HClO4, prepared using purified deionized water (18.2 MΩ·cm).
A HEKA EPC10 patch clamp amplifier was used to collect current, i, voltage, E, and time, t, data, which were filtered with a 4-pole Bessel low-pass filter at 10 kHz and sampled at 50 kHz. A LabVIEW program employing a FPGA card (National Instruments, PCIe-7852) was used to monitor the voltage and to control current steps for galvanostatic experiments. The program was capable of lowering the current to 0 nA within 80 μs after the detection of nanobubble nucleation. A mercury sulfate electrode (BASi) was employed (E = +0.64 V relative to a normal hydrogen electrode, NHE) as a reference/counter electrode in a two-electrode configuration. For convenience, all potentials are presented vs NHE.
Results and Discussion
Cyclic Voltammogram in a Solution of 1 M H2O2 and 1 M HClO4
The generation of a single nanobubble at a Pt nanoelectrode can be observed in cyclic voltammetric measurements, as reported in previous work.20,21Figure 2 shows the cyclic voltammogram of a 6-nm-radius electrode in an aqueous solution of 1 M H2O2 and 1 M HClO4.
Figure 2.
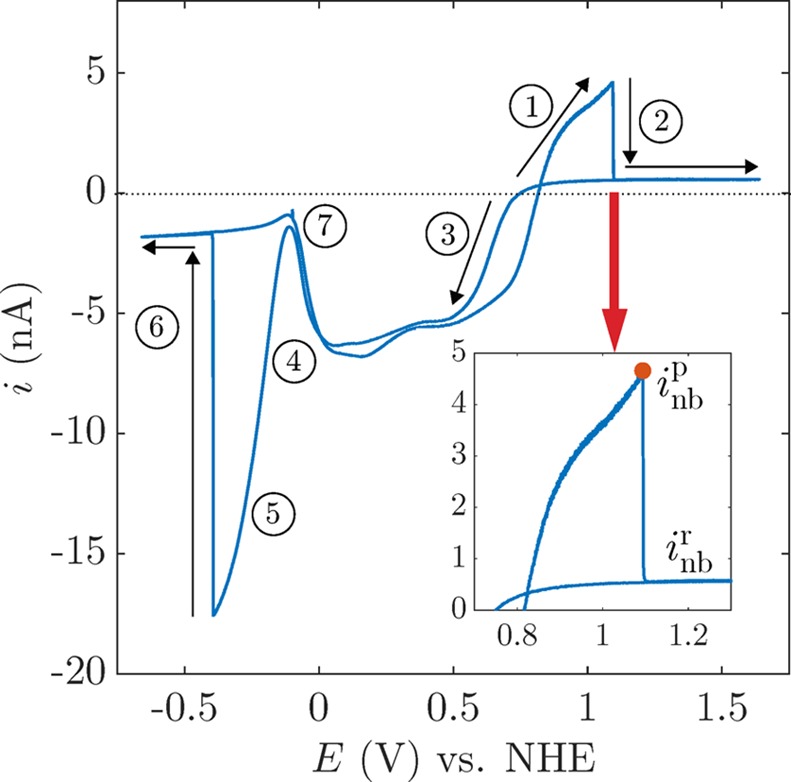
Cyclic voltammogram of a 6-nm-radius Pt nanoelectrode in a solution of 1 M H2O2 and 1 M HClO4. The red dot on the close-up area corresponds to the peak current inbp at which a nanobubble nucleates and then grows to block the electrode surface. The scan rate is 200 mV/s.
The i–E response reflects several potential-dependent electrochemical reactions,21 as labeled in Figure 2:
-
1.
H2O2 → 2H+ + 2e– + O2: above 0.8 V, H2O2 is electrochemically oxidized to produce dissolved O2. The higher the current, the faster the rate of O2 production and the higher the local supersaturation.19
-
2.
O2 nanobubble formation: when the current reaches the peak value, inbp, the concentration of O2 is sufficiently high that a nanobubble nucleates at the nanoelectrode, grows, and blocks it, as depicted in Figure 1. Consequently, the current rapidly drops to a residual current, inb, which corresponds to the balance of the steady-state O2 dissolution from the bubble to the bulk with the O2 production at the circumference of the nanoelectrode (which is not fully covered by the nanobubble).20,22 Increasing the voltage from that point on causes no change in the current, which remains constant at inbr.20 This current also stays constant when we subsequently reduce the voltage until O2 electrogeneration ceases and the nanobubble dissolves.
-
3.
H2O2 + 2e– + 2H+ → 2H2O: from 0.8 to 0 V, H2O2 is reduced to form H2O, resulting in a cathodic current.
-
4.
H+ + e– → Pt–H: a monolayer of H· is reductively adsorbed at the Pt surface,23 reducing the rate of H2O2 reduction, resulting in a decrease in the cathodic current.
-
5.
2H+ + 2e– → H2: from −0.1 to −0.4 V, protons are reduced to produce H2.
-
6.
H2 nanobubble formation: when enough H2 molecules cluster together, a H2 nanobubble nucleates at the nanoelectrode, as indicated by a sudden decrease in current to a potential-independent residual current.
-
7.
As the electrode potential is scanned toward positive voltages (from 0 to 0.8 V), H desorbs from the Pt and the rate of H2O2 reduction increases.
This cyclic voltammogram may be scanned hundreds of times, with repeated formation and dissolution of the O2 and H2 nanobubbles on each scan.24 However, large variability in inbp is observed after an extensive repetition of voltammetric cycles. This is likely due to the restructuring of the surface in the repetitive scans, as the voltammetric responses involve H· absorption and desorption (from 0 to 0.8 V, steps 3 and 7 in Figure 2) and the oxidation and reduction of H2O2 involve the generation of PtOx as well as the reduction of PtOx to Pt.23 However, the self-decomposition of H2O2 to O2 caused by the Pt surface25,26 does not play a significant role in the case of eventual bubble nucleation since the O2 generation rate is negligible compared to the gas production rate once a certain current is applied.21 The electrode apparent radius a is also affected during the application of the conditioning cycles (section 1 in the Supporting Information); consequently, inb may be affected.21,22,27Figure 3 shows several cyclic voltammograms recorded at the same electrode, displaying a different inbp in every cycle. However, by reducing the scan range or applying a “conditioning cycle”, a reproducible electrode surface and a constant apparent radius, and therefore a consistent inb, can be achieved. We can thus influence the surface chemistry of the Pt electrode, achieving a more reproducible inbp.28Figure 4 presents several voltammograms of the same electrode covering different scan ranges, with the resulting variability of inb shown in Figure 4d. It is important to notice the different mean values of inbp for different scan ranges, which emphasizes the importance of the surface chemistry on bubble nucleation.
Figure 3.
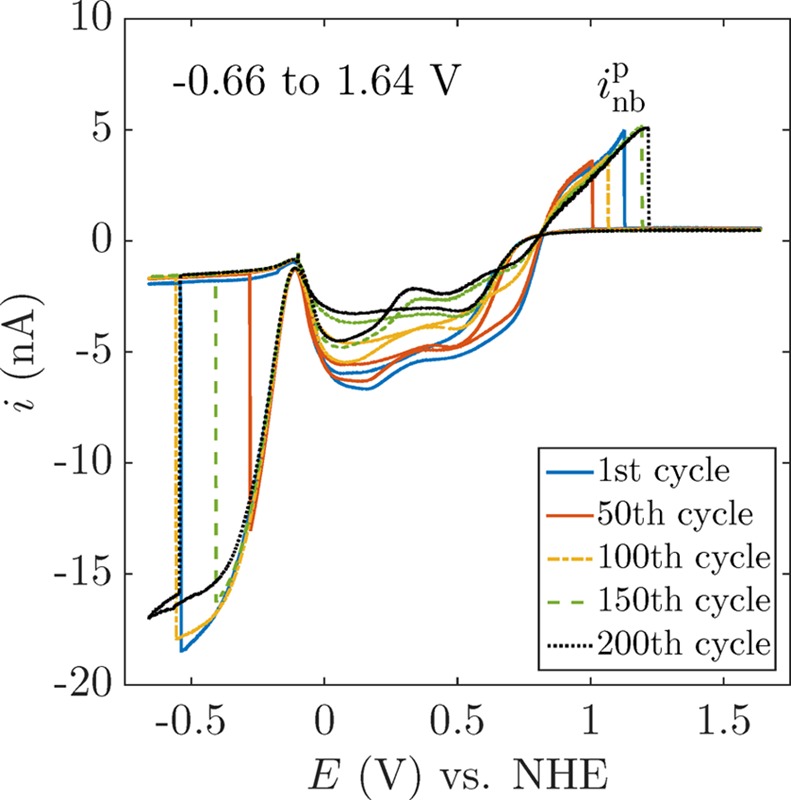
Multiple voltammetric cycles demonstrating the generation of O2 and H2 nanobubbles. Note that the curves show a large variation. More specifically, the current inbp at which an O2 nanobubble forms varies significantly from cycle to cycle. The voltammograms were recorded at a 6-nm-radius electrode at a scan rate of 200 mV/s.
Figure 4.

(a–c) Multiple cycles of voltammograms performed over different potential scan ranges for the determination of experimental conditions giving rise to reproducible inbp for O2 nanobubble formation on a 6-nm-radius electrode. (d) inb vs cycle number for O2 nanobubble formation for different scan ranges corresponding to voltammograms in panels a–c and Figure 3. The voltammetric cycle shown in panel (c) is the one that involves the least surface chemistry and thus, results in the most reproducible results (green diamonds in panel (d)). Note that the mean value of inbp changes for different scan ranges, again highlighting the importance of the surface chemistry.
Once we have secured a reproducible inbp, we can turn to the objective of our story: the measurement of the different rates at which nanobubbles nucleate if a certain current below inb is applied.
Measurement of the Nucleation Rate
Our experiments aim at measuring the time to nucleate a nanobubble as a function of the applied current iapp. In the experiments described above, the nanobubbles nucleate “instantaneously” on the voltammetric time scale when the current reaches inbp (which is approximately linearly related to the electrode apparent measurable radius a(21,22)). However, if iapp is lower than inb, then a measurable stochastic induction time is required for nucleation.21
As shown in Figure 4d, at least 40 cycles need to be performed before a good inbp reproducibility is reached. Therefore, we designed the conditioning cycle shown in Figure 5a to obtain reproducible results during the nucleation rate experiments. First, the voltage is swept positively until an O2 nanobubble nucleates and blocks the electrode. The voltage is then stepped to E = 0.64 V for a defined time, tdis, to allow the nanobubble to dissolve; this dissolution is followed by a stabilization process in which the electrode surface is conditioned by holding it at a voltage, Estab, for a time, tstab. Finally, we let the electrode rest at E = 0.64 V for a time trest before repeating the cycle. Conditioning waveforms using different combinations of tdis, tstab, Estab, and trest have been experimentally tried and are presented in the table in Figure 5c. The optimized conditions correspond to tdis = 1 s, tstab = 2 s, Estab = 0.89 V, and trest = 1 s, corresponding to conditioning protocol #4 (green diamonds) in Figure 5b, where we represent inb vs the cycle number corresponding to the most representative data among 20 different conditioning configurations. The different i–E vs t plots of these conditioning cycles have been included in the Supporting Information (Figure S4).
Figure 5.

(a) Voltammetric electrode conditioning cycle used to rapidly achieve a state where the O2 nanobubble forms at a consistent current. (b) inbp for the most representative combinations of tdis, tstab, Estab, and trest as indicated in the table in panel (c). The configuration with the smallest standard deviation used throughout this article corresponds to configuration 4, with tdis = 1 s, tstab = 2 s, Estab = 0.89 V, and trest = 1 s. (c) Table with the different configurations for conditioning the electrode to obtain reproducible results. The configuration which achieves the lowest standard deviation is highlighted in blue (diamond symbol) and is used in the remainder of the article and is referred to as “the electrode conditioning cycle”.
Now we can measure the nucleation rate at different supersaturation levels, which are directly controlled by the applied current.29 We chose to control current rather than voltage because the experiment is very sensitive to any drift in the voltage. For example, in the region very close to inbp, a change of 20 mV generated a change of 8% in current, as can be seen in Figure 6a, in which we plot a voltammogram for O2 nanobubble nucleation. Such an apparent “small variation” in current causes a significant variation in the nucleation rate of nanobubbles. Control of the voltage is difficult to achieve with the desired precision. Therefore, we decided to control the current, which can be precisely adjusted to the level of pA.
Figure 6.

(a) Forward scan of a voltammogram for bubble formation at a 41-nm-radius Pt electrode. Inset: the range of iapp where bubble nucleation times are measured. (b) Experimental sequence used to determine bubble nucleation time as a function of iapp. (c) Applied current cycle (top) and measured potential (bottom) for determining the bubble nucleation time at the same electrode as in panel (a). The different times are defined similarly to the intervals in Figure 5a, whereas istab is the measured value of the current obtained during the application of Estab in the same figure.
A LabVIEW script was used to control the current iapp applied to the electrode. By subsequent step increases from ∼0.7inbp to ∼1.0inb, we can measure different nucleation times, which become smaller the closer iapp is to inbp. The applied current loop is represented in Figure 6c, in which istab corresponds to the current response obtained during the application of Estab in the course of electrode conditioning (Figure 5a). Once a nanobubble forms, according to the voltammogram presented in Figure 6a, if iapp is maintained fixed at a certain value, then the voltage will dramatically increase to values that correspond to water oxidation, damaging the nanoelectrode. Therefore, a threshold voltage Ethres = 1.5 V is established, so when a nanobubble nucleates and the voltage spikes, iapp will automatically return to zero.
We design a full experiment as shown in the flowchart of Figure 6b. First, the electrode surface is conditioned by applying the cycle in Figure 5a over a hundred times with voltage control. Afterward, we switched to current control, applying a fixed iapp until a nanobubble nucleates and blocks the electrode, while measuring the corresponding nucleation time tind. For the purpose of avoiding electrode surface reconditioning, if no bubble nucleated after 30 s, the current was manually set to zero and the process continued as designed. We repeated this loop for different currents 10 times before conditioning the electrode surface again. Several voltage responses are shown for a 41-nm-radius electrode corresponding to different iapp values in Figure 7 (see more results for different currents and different electrodes in the Supporting Information, Figures S5–S7). It can be perfectly appreciated that the closer iapp is to inbp, the shorter tind becomes, e.g., tind ≈ 15 s at iapp = 11.3 nA (Figure 7a), whereas when iapp is increased by 0.8 nA, tind drastically decreases to ∼7 ms (Figure 7d). An increase of 7% in iapp causes a decrease in the nucleation time of 3 orders of magnitude. We report the different nucleation times tind for different iapp values on an a = 41 nm electrode in Figure 8a (refer to the Supporting Information, Figures S8 and S9, for the results corresponding to the different electrodes used in Figures S6 and S7). The stochasticity of the process can be appreciated in the different nucleation times measured at the same iapp. Notably, the closer iapp is to inb (inbp = 12.2 nA for this case), the lower the variability of tind, i.e., the curves for different repetitions lie closer together. This effect originates from the shorter exposure time to a certain iapp, thus avoiding any reconditioning of the electrode surface. The shorter the exposure, the less the surface chemistry and consequently the more reproducible the results. For values of inb of approximately 0.9inbp, the stochastic variability in tind ranges over 2 to 3 orders of magnitude,29 moving toward values with variability within 1 order of magnitude for 0.99inb.
Figure 7.

Potential–time measurements to determine the time needed to nucleate an O2 nanobubble. Each panel shows a different iapp < inbp = 12.2 nA and is presented on a different time scale. The electrode was 41 nm in radius. In some repetitions, for the lowest iapp, bubble nucleation did not occur within 30 s; therefore, the process was manually stopped. Panel (c) illustrates the interval in which the nucleation time, tind, is measured, from the moment in which the current is applied until the moment in which the nanobubble nucleates (indicated by a sudden increase in the voltage as the bubble blocks the surface of the nanoelectrode). The arrows indicate the cycle number.
Figure 8.

(a) Experimentally measured tind for different iapp values on a 41-nm-radius Pt electrode. (b) Corresponding cumulative probability P of a nanobubble nucleation event for iapp = 11.4, 11.6, 11.7, 11.8, 11.9, and 12 nA (other currents have similar results but are not shown, aiming for clarity in the figure). The theoretical curves correspond to the best fit of eq 1 to the data. The curves become increasingly vertical with increasing iapp: the higher the current, the higher the nucleation rate J.
The cumulative probability of a nanobubble nucleating at a nanoelectrode can be expressed as P = N(t)/NT, where N(t) represents the number of nanobubbles whose nucleation occurs before a specific time t and NT is the total number of nucleation events recorded at a certain iapp.30 This cumulative probability can be theoretically expressed by an exponential relationship:30,31
| 1 |
where J refers to the nucleation rate at a specific current and tlimit is the shortest accessible experimental time, which can be physically associated with the response time of the electronic circuit coupled to the capacitance of the electrochemical cell29 and determines the minimum experimentally achievable tind. By fitting eq 1 to the data in Figure 8a, the nucleation rate, J, for each value of iapp can be measured as a best-fitting parameter. An estimate can be calculated as J ≈ 1/(t̅ind – tlimit), where t̅ind is the mean value of the nucleation time at a certain iapp. A single value of tlimit was fit to all measurements with each electrode. The probability distribution for the different iapp values in Figure 8a is represented in Figure 8b. The best match between the fits and experiments occurs for intermediate levels of iapp since for the lower values, nanobubbles take a longer time to nucleate and therefore the nanoelectrode surface can be reconditioned to a different state, whereas for iapp very close to inbp the process is so fast that any uncertainty in tlimit may result in a very significant uncertainty in J.
The nucleation rate, J, depends on iapp, increasing its value as iapp increases, as can be depicted from the more vertical sigmoidal curves in Figure 8b for higher values of iapp. From Classical Nucleation Theory (CNT), the nucleation rate of a nanobubble at the surface of the electrode can be expressed as9,12,16,32,33
| 2 |
where J0 is the pre-exponential factor which describes the statistical molecule-by-molecule process of nucleus growth (which can be considered to be constant with respect to i), γ is the surface tension of the gas–liquid interface (variations in the surface tension at molecular length scale, which have been shown to be minimal down to 10 nm,20 are not considered), ϕ(θ) = (1 + cos(θ))2(2 – cos(θ))/4 is a geometric function which depends on the nanobubble contact angle θ to the electrode surface24,34,35 (which implicitly accounts for the minute effect of the surface tension between the electrode and the bubble36), kT = 4.11 × 10–21 J is the product of Boltzmann constant k and thermodynamics temperature T = 298 K, and Ps and P0 refer to the pressure in the bubble and the ambient pressure in the bulk liquid, respectively. Note again that in the derivation of eq 2 a spherical cap shape of the bubble nucleus has been assumed. Once van der Waals forces and thus the disjoining pressure play a role, this is no longer the case and the nanobubble shape may differ from that of a perfect spherical cap.37 Given the nanometric size of the bubbles, these considerations may apply. These deviations from a perfect spherical-cap shape are nevertheless known to be very small,37 so our assumption may still be valid.
Assuming Fickian diffusion on a planar disk electrode, we can relate the steady-state current i to the local concentration of O2 being produced at the electrode surface, CO2surface:21,22,35
| 3 |
In eq 3, n = 2 is the number of exchanged electrons per molecule of O2 generated, F = 96 485 C/mol is Faraday’s constant, DO2 = 1.67 × 10–9 m2/s is the diffusivity of O2,38 and CO2surface and CO2 are the O2 concentrations locally at the surface of the electrode and in the bulk, respectively. CO2bulk is approximately zero compared to CO2 in our experiments. Applying Henry’s law, we can relate this supersaturated concentration at the electrode surface to Ps and therefore35
| 4 |
where KH = 1.283 × 10–5 mol/m3Pa is Henry’s constant for O2.39Equation 4 can be rewritten as
| 5 |
where B = 16πγ3ϕ(θ)/3kT and J0 are calculated as best-fit parameters. Figure 9 shows experimentally measured nucleation rates vs the bracketed expression in eq 5 for three different electrodes, indeed displaying the linear behavior suggested by this equation. From the slope B, we can extract the contact angle θ. See below.
Figure 9.

Logarithmic linear relation between nucleation rate J and the inverse of the squared supersaturated pressure difference for (a) a = 3 nm, (b) a = 41 nm, and (c) a = 51 nm. An outlier in (a) has been indicated by a black circle.
The nucleation rate is related to the activation energy Ea of a nanobubble nucleation by Arrhenius’ law.32 Therefore, we can achieve a one-to-one relation between both physical magnitudes:29
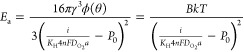 |
6 |
Ea thus decreases with increasing current; i.e., Ea decreases with increasing supersaturation ζ = (cs – c0)/c0, where cs and c0 refer to the O2 concentration at supersaturation and under ambient conditions. These concentrations are related to pressure by Henry’s law at constant temperature, thus ζ can be rewritten as the pressure difference, ζ = (Ps – P0)/P0. Associating this definition with eqs 2 and 4, we obtain an expression for the supersaturation level at the nucleation time for a bubble:
| 7 |
The different Ea values for different electrodes are represented in Figure 10a vs the corresponding levels of ζ achieved at different iapp values and tabulated in Table 1. As expected, the increase in supersaturation causes a decrease in the activation energy for every case.
Figure 10.

(a) Activation energy, Ea, as a function of the supersaturation, ζ, at different electrodes. Ea decreases with increasing i, i.e., increasing ζ. (b) Sketch of a surface nanobubble nucleus under the assumption of a spherical cap shape. The contact angle, θ, is defined on the water side, where hnb corresponds to the nucleus height and L determines the nucleus footprint. Inset: geometric relation ϕ(θ) = (1 + cos(θ))2(2 – cos(θ))/4 needed for the calculation of the nucleus volume assuming a spherical cap shape. The framed region indicates the domain in which all of the nanobubbles studied in this article are situated.
Table 1. Fitting Parameters B and J0 in Equation 5, Activation Energy Ea from Equation 6, and Supersaturation ζ at Bubble Nucleation from Equation 7 for Different Electrodesa.
| a(nm) | B(Pa2) | ln(J0s) | Ea/kT | ζ |
|---|---|---|---|---|
| 3 | 9.0 ± 1.0 × 1016 | 14.3 ± 1.1 | 12.5 – 7.9 ± 1.2 | 835–1050 |
| 41 | 8.3 ± 1.3 × 1015 | 32.2 ± 4.2 | 30.3 – 26.4 ± 4.2 | 160–175 |
| 51 | 1.6 ± 0.5 × 1016 | 12.1 ± 2.1 | 9.5 – 5.7 ± 2.1 | 410–525 |
From Figure 10a, there is no apparent relation between the different values of the activation energy Ea and the apparent radius of the electrode a nor is there a clear relation between a and fitting parameters B and J0, as indicated in Table 1.
As mentioned above, the contact angle θ can be calculated from the slope B, namely, by the implicit equation:
| 8 |
For all the different electrodes, θ ranges from 135 to 155° (inset in Figure 10b), similar to the case of H2 nanobubbles.29 The radius of curvature of the nucleus for an O2 nanobubble has been reported to be rnb = 10 nm,21,22,40 which implies a Laplace pressure of Pnb = 2γ/rnb = 14.2 MPa. The extremely high pressure within the nanobubble justifies the surprisingly high supersaturation levels achieved locally around it at nucleation. Knowing rnb and θ, the nanobubble nucleus geometry is fully determined, assuming a spherical cap shape22,33,40 (Figure 11 and Table 2).
Figure 11.

(a) Scale drawing41 of a nanobubble nucleus at an a = 41 nm electrode. The white circles (best seen in panel (b)) represent Pt atoms, whereas the red ones represent O atoms. The molecular structure is defined in detail in panel (c). The close-up view of the area covered by the nanobubble nucleus is depicted from the top and the side in panel (b). The nucleus (as shown) initially covers a very small portion of the electrode surface before growing and blocking the majority of the electrode. The volume occupied by the O2 molecules contained in the nucleus is small compared to the total nucleus volume, which may suggest that the nucleus shape may be deformed by the disjoining pressure.37
Table 2. Geometrical Dimensions of Different Nanobubble Nucleia.
| a(nm) | inbp(nA) | θ(deg) | L(nm) | hmb(nm) | nnb |
|---|---|---|---|---|---|
| 3 | 5.3 | 144 ± 12 | 11.4 ± 3.2 | 2.0 ± 1.2 | 550–900 |
| 41 | 12.1 | 156 ± 1 | 4.1 ± 1 | 0.9 ± 0.0.1 | 50–85 |
| 51 | 45 | 151 ± 2 | 4.8 ± 0.3 | 1.2 ± 0.0.1 | 100–170 |
Apparent electrode radius, a; peak current, inbp; contact angle, θ; nucleus footprint, L; nucleus height, hnb; and number of O2 molecules in the nucleus, nnb. The tolerances are the standard deviations which are derived from the calculation of the contact angle, θ, from fitting parameter B.
From the small size of the bubble nucleus footprint, L, when compared to the elctrode radius, a (Table 2), we conclude that the nanobubble nucleus covers a very small portion of the electrode. There is one exception to this case: a = 3 nm. In this particular case, the nucleus of the nanobubble is larger than the electrode. This result has several possible origins: either the nucleus is attached to some irregularities on the nanoelectrode surface, i.e., the electrode has a very nonflat surface, or the Pt disk is recessed within the glass seal,42 providing an apparent radius determined volumetrically to be much smaller than the actual electrode size, or the disjoining pressure within the nanobubble deforms its shape in a high manner such that the assumption of a spherical cap is no longer applicable. On the other hand, the total volume occupied by the number of molecules contained in the nuclei (∼50–900) is small compared to the presumed initial bubble volume. From the drawing to scale in Figure 11, we can appreciate this fact: within the nanobubble nucleus volume, there is empty space which cannot be occupied by the number of molecules present. This may result in a deformed shape of the nanobubble nucleus due to the disjoining pressure.37 From our results in Tables 1 and 2, we can conclude that there is a direct relationship between the supersaturation level ζ and the number of molecules in the nucleus nnb. However, there is no apparent relation between nnb and inbp nor a. This issue can be due to the electrode surface properties and especially the surface chemistry that applies to the electrode during its conditioning to achieve reproducible results.
Conclusions
The nucleation of single O2 nanobubbles at a Pt nanoelectrode has been studied in detail. The importance of surface chemistry has been exposed throughout all of this research. The necessary treatment of the Pt surface to generate a reproducible peak current inbp at which nanobubbles are formed is of extreme importance if good stochastic results are to be obtained concerning the nucleation rate of nanobubbles (the corresponding analysis for a non-preconditioned electrode can be found in section 4 in the Supporting Information). In a region very close to inb, the nucleation time tind rapidly changes with a small variation on the order of tenths of a nA in the applied current iapp. The nucleation rate J(iapp) can be calculated from the different induction times for bubbles to nucleate when iapp is fixed at a certain level below inbp. The Classical Nucleation Theory (eq 2) provides an accurate mathematical expression for J. Because of stochasticity, the nucleation time can vary for the same iapp within 2 orders of magnitude. The higher the iapp, the higher the supersaturation ζ and consequently the higher the nucleation rate. ζ values are large in an area local to where the nanobubble nucleates, which results from the high Laplace pressure of the nanobubble nucleus due to its small radius. From the different measured J at different ζ, the activation energies Ea have been derived along with the contact angle θ between the bubble and the electrode surface if the geometry of the nucleus of the nanobubble is approximated as a spherical cap. We can estimate the number of O2 molecules contained in the critical bubble nucleus, which is higher for higher ζ; i.e., the more molecules are locally produced, the more molecules will form the nanobubble nucleus. Though disjoining pressure may affect the nanobubble final shape,37 for the scope of this research the assumption of a spherical cap is more than justified since the disjoining pressure barely cause the bubble shape to deviate from a spherical cap.
Acknowledgments
This work was supported by The Netherlands Centre for Multiscale Catalytic Energy Conversion (MCEC), an NWO Gravitation program funded by the Ministry of Education, Culture and Science of the government of The Netherlands. Nanobubble research in the White Research Group is funded by the Office of Naval Research (N00014-16-1-2541).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.langmuir.8b01372.
Fabrication of nanoelectrodes. Nanoelectrode conditioning. Nucleation rate measurements for additional values of iapp and electrodes. Bubble nucleation measurements without electrode surface conditioning. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Peng H.; Birkett G. R.; Nguyen A. V. Progress on the surface nanobubble story: What is in the bubble? Why does it exist?. Adv. Colloid Interface Sci. 2015, 222, 573–580. 10.1016/j.cis.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Lohse D.; Zhang X. Surface nanobubbles and nanodroplets. Rev. Mod. Phys. 2015, 87, 981–1035. 10.1103/RevModPhys.87.981. [DOI] [Google Scholar]
- Craig V. S. J. Very small bubbles at surfaces – the nanobubble puzzle. Soft Matter 2011, 7, 40–48. 10.1039/C0SM00558D. [DOI] [Google Scholar]
- Li M.; Tonggu L.; Zhan X.; Mega T. L.; Wang L. Cryo-EM visualization of nanobubbles in aqueous solutions. Langmuir 2016, 32, 11111–11115. 10.1021/acs.langmuir.6b00261. [DOI] [PubMed] [Google Scholar]
- Wang L.; Wang X.; Wang L.; Hun J.; Wang C. L.; Zhao B.; Zhang X.; Tai R.; He M.; Chen L.; Zhang L. Formation of surface nanobubbles on nanostructured substrates. Nanoscale 2017, 9, 1078–1086. 10.1039/C6NR06844H. [DOI] [PubMed] [Google Scholar]
- Ma W.; Hu K.; Chen Q.; Zhou M.; Mirkin M. V.; Bard A. J. Electrochemical size measurement and characterization of electrodeposited platinum nanoparticles at nanometer resolution with Scanning Electrochemical Microscopy. Nano Lett. 2017, 17, 4354–4358. 10.1021/acs.nanolett.7b01437. [DOI] [PubMed] [Google Scholar]
- Verhaart H. F. A.; de Jonge R. M.; van Stralen S. J. D. Growth rate of a gas bubble during electrolysis in supersaturated liquid. Int. J. Heat Mass Transfer 1980, 23, 293–299. 10.1016/0017-9310(80)90117-9. [DOI] [Google Scholar]
- Somorjai G. A.; Li Y.. Introduction to Surface Chemistry and Catalysis, 2nd ed.; John Wiley and Sons: Hoboken, NJ, 2010. [Google Scholar]
- Dapkus K. V.; Sides P. J. Nucleation of electrolytically evolved hydrogen at an ideally smooth electrode. J. Colloid Interface Sci. 1986, 111, 133–151. 10.1016/0021-9797(86)90014-7. [DOI] [Google Scholar]
- Velmurugan J.; Noël J.-M.; Nogala W.; Mirkin M. V. Nucleation and growth of metal on nanoelectrodes. Chem. Sci. 2012, 3, 3307–3314. 10.1039/c2sc21005c. [DOI] [Google Scholar]
- Ying Y.-L.; Hu Y.-X.; Gao R.; Yu R.-J.; Gu Z.; Lee L. P.; Long Y.-T. Asymmetric nanopore electrode-based amplification for electron transfer imaging in live cells. J. Am. Chem. Soc. 2018, 140, 5385–5392. 10.1021/jacs.7b12106. [DOI] [PubMed] [Google Scholar]
- Lubetkin S.; Blackwell M. The nucleation of bubbles in supersaturated solutions. J. Colloid Interface Sci. 1988, 126, 610–615. 10.1016/0021-9797(88)90161-0. [DOI] [Google Scholar]
- Hampton M. A.; Nguyen A. V. Nanobubbles and the nanobubble bridging capillary force. Adv. Colloid Interface Sci. 2010, 154, 30–55. 10.1016/j.cis.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Forest T. W.; Ward C. A. Effect of a dissolved gas on the homogeneous nucleation pressure of a liquid. J. Chem. Phys. 1977, 66, 2322–2330. 10.1063/1.434267. [DOI] [Google Scholar]
- Hemmingsen E. A. Cavitation in gas-supersaturated solutions. J. Appl. Phys. 1975, 46, 213–218. 10.1063/1.321323. [DOI] [Google Scholar]
- Wilt P. M. Nucleation rates and bubble stability in water-carbon dioxide solutions. J. Colloid Interface Sci. 1986, 112, 530–538. 10.1016/0021-9797(86)90122-0. [DOI] [Google Scholar]
- Pan G.; Yang B. Effect of surface hydrophobicity on the formation and stability of oxygen nanobubbles. ChemPhysChem 2012, 13, 2205–2212. 10.1002/cphc.201100714. [DOI] [PubMed] [Google Scholar]
- Maheshwari S.; van der Hoef M.; Zhang X.; Lohse D. Stability of surface nanobubbles: a molecular dynamics study. Langmuir 2016, 32, 11116–11122. 10.1021/acs.langmuir.6b00963. [DOI] [PubMed] [Google Scholar]
- Rubin M. B.; Noyes R. M. Thresholds for nucleation of bubbles of N2 in various solvents. J. Phys. Chem. 1992, 96, 993–1000. 10.1021/j100181a082. [DOI] [Google Scholar]
- German S. R.; Edwards M. A.; Chen Q.; White H. S. Laplace pressure of individual H2 nanobubbles from pressure-addition electrochemistry. Nano Lett. 2016, 16, 6691–6694. 10.1021/acs.nanolett.6b03590. [DOI] [PubMed] [Google Scholar]
- Ren H.; German S. R.; Edwards M. A.; Chen Q.; White H. S. Electrochemical generation of individual O2 nanobubbles via H2O2 oxidation. J. Phys. Chem. Lett. 2017, 8, 2450–2454. 10.1021/acs.jpclett.7b00882. [DOI] [PubMed] [Google Scholar]
- German S. R.; Edwards M. A.; Chen Q.; Liu Y.; Luo L.; White H. S. Electrochemistry of single nanobubbles. Estimating the critical size of bubble-forming nuclei for gas-evolving electrode reactions. Faraday Discuss. 2016, 193, 223–240. 10.1039/C6FD00099A. [DOI] [PubMed] [Google Scholar]
- Katsounaros I.; Schneider W. B.; Meier J. C.; Benedikt U.; Biedermann P. U.; Auer A. A.; Mayrhofer K. J. J. Hydrogen peroxide electrochemistry on platinum: towards understanding the oxygen reduction reaction mechanism. Phys. Chem. Chem. Phys. 2012, 14, 7384–7391. 10.1039/c2cp40616k. [DOI] [PubMed] [Google Scholar]
- Lubetkin S. D. Why is it much easier to nucleate gas bubbles than theory predicts?. Langmuir 2003, 19, 2575–2587. 10.1021/la0266381. [DOI] [Google Scholar]
- MacInnes D. A. The mechanism of the catalysis of the decomposition of hydrogen peroxide by colloidal platinum. J. Am. Chem. Soc. 1914, 36, 878–881. 10.1021/ja02182a011. [DOI] [Google Scholar]
- Garwig P. L.Heterogeneous Decomposition of Hydrogen Peroxide by Inorganic Catalysis; PN: 1966. [Google Scholar]
- Chen Q.; Wiedenroth H. S.; German S. R.; White H. S. Electrochemical nucleation of stable N2 nanobubbles at Pt nanoelectrodes. J. Am. Chem. Soc. 2015, 137, 12064–12069. 10.1021/jacs.5b07147. [DOI] [PubMed] [Google Scholar]
- Svetovoy V. B.; Sanders R. G. P.; Elwenspoek M. C. Transient nanobubbles in short-time electrolysis. J. Phys.: Condens. Matter 2013, 25, 184002. 10.1088/0953-8984/25/18/184002. [DOI] [PubMed] [Google Scholar]
- German S. R.; Edwards M. A.; Ren H.; White H. S. Critical nuclei size, rate, and activation energy of H2 gas nucleation. J. Am. Chem. Soc. 2018, 140, 4047–4053. 10.1021/jacs.7b13457. [DOI] [PubMed] [Google Scholar]
- Brandel C.; ter Horst J. H. Measuring induction times and crystal nucleation rates. Faraday Discuss. 2015, 179, 199–214. 10.1039/C4FD00230J. [DOI] [PubMed] [Google Scholar]
- Hyde M. E.; Compton R. G. A review of the analysis of multiple nucleation with diffusion controlled growth. J. Electroanal. Chem. 2003, 549, 1–12. 10.1016/S0022-0728(03)00250-X. [DOI] [Google Scholar]
- Blander M.; Katz J. L. Bubble nucleation in liquids. AIChE J. 1975, 21, 833–848. 10.1002/aic.690210502. [DOI] [Google Scholar]
- Kashchiev D.; van Rosmalen G. M. Review: nucleation in solutions revisited. Cryst. Res. Technol. 2003, 38, 555–574. 10.1002/crat.200310070. [DOI] [Google Scholar]
- Butt H.-J.; Kappl M. Normal capillary forces. Adv. Colloid Interface Sci. 2009, 146, 48–60. 10.1016/j.cis.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Luo L.; Faraji H.; Feldberg S. W.; White H. S. Electrochemical measurements of single H2 nanobubble nucleation and stability at Pt nanoelectrodes. J. Phys. Chem. Lett. 2014, 5, 3539–3544. 10.1021/jz501898r. [DOI] [PubMed] [Google Scholar]
- Fletcher N. H. Size effect in heterogeneous nucleation. J. Chem. Phys. 1958, 29, 572–576. 10.1063/1.1744540. [DOI] [Google Scholar]
- Svetovoy V. B.; Dević I.; Snoeijer J. H.; Lohse D. Effect of disjoining pressure on surface nanobubbles. Langmuir 2016, 32, 11188–11196. 10.1021/acs.langmuir.6b01812. [DOI] [PubMed] [Google Scholar]
- Wakabayashi N.; Takeichi M.; Itagaki M.; Uchida H.; Watanabe M. Temperature-dependence of oxygen reduction activity at a platinum electrode in an acidic electrolyte solution investigated with a channel flow double electrode. J. Electroanal. Chem. 2005, 574, 339–346. 10.1016/j.jelechem.2004.08.013. [DOI] [Google Scholar]
- Tham M. K.; Walker R. D.; Gubbins K. E. Diffusion of oxygen and hydrogen in aqueous potassium hydroxide solutions. J. Phys. Chem. 1970, 74, 1747–1751. 10.1021/j100703a015. [DOI] [Google Scholar]
- Kikuchi K.; Ioka A.; Oku A.; Tanaka Y.; Saihara Y.; Ogumi Z. Concentration determination of oxygen nanobubbles in electrolyzed water. J. Colloid Interface Sci. 2009, 329, 306–309. 10.1016/j.jcis.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Batsanov S. S. Van der Waals radii of elements. Inorg. Mater. 2001, 37, 871–885. 10.1023/A:1011625728803. [DOI] [Google Scholar]
- Chen Q.; Luo L.; White H. S. Electrochemical generation of a hydrogen bubble at a recessed platinum nanopore electrode. Langmuir 2015, 31, 4573–4581. 10.1021/acs.langmuir.5b00234. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.