

Abstract
Cannabidiol (CBD) exhibits neuroprotective properties in many experimental systems. However, development of CBD as a drug has been confounded by the following: 1) low potency; 2) a large number of molecular targets; 3) marginal pharmacokinetic properties; and 4) designation as a schedule 1 controlled substance. The present work compared the properties of CBD with a novel molecule (KLS-13019) that has structural similarities to CBD. The design strategy for KLS-13019 was to increase hydrophilicity while optimizing neuroprotective potency against oxidative stress toxicity relevant to hepatic encephalopathy. The protective responses of CBD and KLS-13019 were compared in dissociated rat hippocampal cultures co-treated with toxic levels of ethanol and ammonium acetate. This comparison revealed that KLS-13019 was 31-fold more potent than CBD in preventing neuronal toxicity from the combined toxin treatment, while both compounds exhibited complete protective efficacy back to control values. In addition, treatment with KLS-13019 alone was 5-fold less toxic (TC50) than CBD. Previous studies suggested that CBD targeted the Na+ Ca2+ exchanger in mitochondria (mNCX) to regulate intracellular calcium levels, an important determinant of neuronal survival. After treatment with an inhibitor of mNCX (CGP-37157), no detectable neuroprotection from ethanol toxicity was observed for either CBD or KLS-13019. Furthermore, AM630 (CB2 antagonist) significantly attenuated CBD-mediated neuroprotection, while having no detectable effect on neuroprotection from KLS-13019. Our studies indicated KLS-13019 was more potent and less toxic than CBD. Both compounds can act through mNCX. KLS-13019 may provide an alternative to CBD as a therapeutic candidate to treat diseases associated with oxidative stress.
Keywords: Cannabidiol, oxidative stress, mitochondrial Na+ Ca2+ exchanger, neuroprotection, cannabis, hippocampal cultures, ethanol
Introduction
Therapeutic interest in cannabis has broadened significantly with compassionate use for diseases that are refractory to current therapies and by increased legalization. Although legal accessibility to cannabis has increased, the paucity of clinical trial-based evidence for reliableefficacy remains an ongoing therapeutic dilemma. Included among the diseases that cannabis has been reported to provide symptomatic relief are the following: pain management, epilepsy, anxiety, psychosis, neurodegenerative disease, post-traumatic stress disorder and movement disorders (Darkovska-Serafimovska et al, 2018; Lim et al., 2017; Stockings et al., 2018). Although the putative uses for cannabis have been unusually broad, significant concerns limit the acceptance of cannabis as a therapeutic agent by the medical community. Driven by evidence from adult users, concerns have arisen with the regular exposure to cannabis and the occurrences of mental impairments for memory, attention, decision-making processes, emotional processing and social cognition (Weinstein et al., 2016). Compounding the recognition of emerging cognitive impairments, a list of scientific and regulatory concerns includes: psychotropic action, developmental impairment, schedule 1 designation, poor standardization of doses/purity and inadequate study of formulations.
The view on the cannabis alternative to the treatment of seizures has changed recently. The U.S. Food and Drug Administration has approved the epilepsy drug Epidiolex, an agent based on cannabidiol. The approved uses for the new drug were for two rare childhood-onset forms of epilepsy: Lennox-Gastaut syndrome and Dravet syndrome. The phase III clinical trial demonstrated a reduction in the number of seizures; however, the approval of Epidiolex by the U.S. Drug Enforcement Agency is still pending. This milestone in the medical use of cannabis/cannabidiol may facilitate significantly other clinical studies; however, both cannabis and CBD remain schedule 1 drugs. While the recent FDA approval is important, significant concerns and uncertainties remain for cannabis use. Meanwhile, many patients will be driven to this alternative out of desperation due to unmet medical need after trying conventional treatments.
Despite unresolved clinical issues associated with broad therapeutic uses of cannabis, significant interest has emerged in cannabidiol, a major component of cannabis that captures apparent therapeutic value without producing psychotropic action (Iffland and Grotenhermen, 2017). Because of cannabidiol’s remarkable breath of possible uses and a putative good safety profile, this compound was identified as the starting point for our drug discovery program focused on neuroprotection from oxidative stress (Kinney et al 2016). The need for a small molecule alternative to cannabidiol was based on the following drug-related issues: low potency, a complex pharmacology with many known targets, marginal pharmacokinetic properties, poor oral bioavailability and significant gaps in the safety profile pertaining particularly to cognitive functions.
The goal of the present study was to provide detailed comparisons of CBD with our novel small molecule (KLS-13019). The aim was to address the poor drug-like properties for CBD in the treatment of the oxidative stress relevant to hepatic encephalopathy. The strategy for the design of KLS-13019 was to introduce heteroatoms on the pentyl group of CBD to increase aqueous solubility and to ensure novelty of structure. KLS-13019 was selected from a panel of new compounds based only on increasing neuroprotective potency while maintaining full efficacy against oxidative stress in dissociated rat hippocampal cultures (Kinney et al, 2016). The choice of utilizing primary hippocampal cultures to optimize this chemical series was based on the recognized function of the hippocampus as a brain area involved in learning and memory and its recognized susceptibility to oxidative stress (Hidalgo and Arias-Cavieres, 2016). Pertinent to hepatic encephalopathy, hippocampal impairment secondary to loss of hepatic function associated with oxidative stress is thought to be fundamental to the etiology of this disease (Avraham et al., 2011).
Utilizing this strategy, significant pharmacological improvement and target focus was evident with KLS-13019 in comparison to CBD. Among the enhanced properties of KLS-13019 to be reported are: 1) increased neuroprotective potency against toxicity associated with combined treatment with ethanol and ammonium acetate; 2) decreased neurotoxicity and cell death in developing hippocampal cultures; 3) an improvement in the pharmacokinetic properties in mice; 4) marked increase in oral bioavailability; and 5) a demonstration of a more focal mechanism of action that emphasized effects on the mitochondrial Na+ Ca2+ exchanger, while decreasing CB2 interactions.
Materials and Methods
Materials.
The following reagents were obtained from Sigma-Aldrich (St Louis, Mo): propidium iodide solution (1.0 mg/ml in water; P4864); 6-carboxyfluoresceine diacetate (CFDA, C5041); ethanol (459844); ammonium acetate (431311); cannabidiol solution (1.0 mg/ml in methanol; C6395); dimethyl sulfoxide (472301); 7-Chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1- benzothiazepin-2(3H)-one (CGP-37157; C8874); cyclosporin A (30024); and dantrolene (D9175). AM 630 (1120) was obtained from Tocris Bio-techne (Minneapolis, MN). The synthesis of KLS-13019 has been described previously in detail (Kinney et al., 2016). Verification of the chemical identity for KLS-13019 was determined by 1H NMR, LC/UV, and LC/MS. The purity of the new compound used in the present study was 98.6% as determined by LC/MS.
Culture model.
Dissociated hippocampal cultures derived from embryonic day 18 rats were employed as the primary screening system to test for toxicity as well as neuroprotection (Brenneman et al., 2012). In brief, hippocampal tissue was obtained commercially through Brain Bits (Springfield, IL) and cultures prepared with slight modifications to methods previously described (Brewer et al., 1993). Tissue was dissociated with a papain-based kit from Worthington Biochemical Corporation (Lakewood, NJ). The hippocampal cells were platted at low density (10,000 cell/well) in a 96-well format and maintained in serum-free medium consisting of Neurobasal Medium supplemented with B27 and GlutaMAX (Life Technologies, Carlsbad, CA). Poly-L-lysine-coated plates (BD Biosciences, Franklin Lakes, NJ) were used because of the preferential adherence and survival of neurons on this matrix support. Prior to the initiation of all experiments between days 12 and 19 in vitro, a complete change of medium was performed in a working volume of 100 μL.
Neuroprotection from oxidative stress
To study various pharmacological and toxicological properties of CBD and KLS-13019, several formulations were utilized. The choice of formulation alternatives was based on both convenience (in the case of dimethyl sulfoxide) and the need to maximize the amount of dissolved compound in solution with a clinically approved excipient: Captisol. For the studies on efficacy and potency, the compounds were dissolved to 10 mM stock solutions with dimethyl sulfoxide (DMSO) and then serially diluted with sterile Dulbecco’s phosphate buffered saline (DPBS; Sigma:D-5780) prior to testing. The concentration of DMSO never exceeded 0.1% in culture treatments. Two reagents were used to model the effect of oxidative stress-related toxicity observed in hepatic encephalopathy: ethanol and ammonium acetate. The toxic concentrations of these molecules were determined in previous experiments (Kinney et al., 2016). Previous reports have indicated that ethanol (Tomasini et al., 2016) and ammonia (Niknahad et al., 2017) produced increases in reactive oxygen species (ROS). These findings have been confrimed in our laboratory with ROS- sensitive dyes in hippocampal cultures for both ethanol and ammonium acetate. Based on our in vitro data and published clinical data, the present studies utilized a combined treatment with both 30 mM ethanol and 300 μΜ ammonium acetate to produce robust and relevant toxicities that could be prevented by the test molecules for both of the assays employed. Furthermore, the amounts of these toxic substances were relevant to hepatic encephalopathy. The amount of ammonia observed in the plasma of pre-coma hepatic encephalopathy patients have been reported to range from 150 to 750 μΜ (Ong et al., 2003). Further, the blood alcohol legal limit is 0.08% (20 mM) and the amount to produced coma 0.4% (~100 mM), within the range of concentrations utilized for the present studies.
To evaluate the compounds for neuroprotection from combined ethanol and ammonium acetate, day 14 hippocampal cultures were given a complete change of medium containing 100 μL of Neurobasal Medium with B27. The CBD was added to the hippocampal cultures for a 5 hour test period in concentrations that ranged from 0.1 to 10 μΜ with 5 replications. For KLS- 13019, concentrations ranging from 1 nM to 10 μΜ were added to the cultures. This range of concentrations was selected on the basis of previous studies which indicated that these concentrations resulted in both a no-effect concentration and a response that produced a full efficacy response that was not different from the response of control cultures. Five minutes after treatment with the test compound, 30 mM ethanol plus 300 μΜ ammonium acetate was added to the cultures for the 5 hour test period.
Fluorescence-based assays.
At the conclusion of the test period, the cultures were evaluated with fluorescent dye-based assays for cell death (propidium iodide) and for neuronal viability (6-carboxyfluorescein diacetate, CFDA). These two standard assays were chosen because they could be measured as multiplexed determinations within a single well, thereby monitoring both an increasing (cell death) parameter and a decreasing parameter (neuronal viability) after a toxic treatment of the hippocampal cultures (Brenneman et al., 2012). For the cell death assessment with the propidium iodide, slight modification to a method previously described was used (Sarafian et al., 2002). Propidium iodide (PI) stock solution of 1 mg/mL (1.5 mM) was diluted 1:30 in DPBS for a final working concentration of 50 μΜ. After removal of the growth medium, 50 μL of the 50 μΜ PI solution was added to cultures and allowed to incubate in the dark at room temperature for 15 min. On every plate, wells without cells were used to provide a blank reading that was used to subtract background fluorescence. The cultures were assessed for fluorescence intensity at Ex536/Em590 nm in a CytoFluor fluorimeter (Perceptive Biosystems). Results were expressed in relative fluorescent units (RFU) and EC50s or IC50s calculated from the dose responses of the test compound and compared to values obtained from controls and wells treated with toxin alone. Concentrations that produced half- maximal responses were determined from the curve-fitting values computed by the 4-parameter logistic analysis within SigmaPlot 11.
After the assessment of cell death, cultures were then further assayed for neuronal viability by the CFDA method (Petroski and Geller, 1994). For the neuronal viability assay, 2 mg of 6-carboxyfluorescein diacetate (CFDA) dye was dissolved in 100 mL of warmed (37° C) DPBS (Gibco: D-5780) and kept in the dark until added to the hippocampal cultures. After a complete change of medium, 100 μL CFDA dye solution was added for 15 min of incubation at 37°C in the dark. At the conclusion of the incubation period, the dye was removed from the cultures and washed once with 100 μL of DPBS. After removal of the first wash, a second wash of DPBS was added to the culture and then incubated for 30 min to allow the efflux of dye out of non-neuronal cells in the cultures. At the conclusion of the 30 min efflux period, the culture efflux medium was removed and 100 μL of 0.1% triton-X100 in water was added to the cultures before reading at Ex490/Em517 in a CytoFluor fluorimeter. Results were expressed in relative fluorescent units (RFU) and EC50’s calculated from the dose response of the test compound as described in the Statistical Analysis section.
Toxicity in hippocampal cultures
For the in vitro toxicity studies, each compound was dissolved in 2.3 mM Captisol prepared in water to a final stock concentration of 1 mM. Each compound was sonicated and agitated until clarity was achieved. Serial dilutions were made with DPBS to achieve final treatment concentrations. Captisol is polyanionic beta-cyclodextrin derivative (Ligand Pharmaceutical Co., San Diego, CA) that has been approved for preclinical and clinical studies. Preliminary studies were conducted to confirm that 2.3 mM Captisol produced no detectible toxicity in the hippocampal cultures. Because all the toxicity studies were conducted between days 12 and 19 in culture, this test system was in a neurodevelopmental stage characterized by naturally occurring cell death, extensive neurite growth, maturing neurotransmitter systems and emerging spontaneous synaptic activity. Cultures were assessed using the same fluorescent assays as utilized to study protective properties. Two types of toxicity studies were conducted: 1) concentration-effect studies to determine the TC50; and 2) time courses to describe toxic responses to neuronal viability and cell death over a period of 30 hours. The time course studies were conducted at concentrations shown to produce maximal amount of toxicity at the lowest dose for each of the two test compounds, as determined by the CFDA assay.
Statistical Analysis
All statistical comparisons were made by ANOVA, with normality of values tested by the Shapiro-Wilk test followed by a multiple comparison of means test with the Holm-Sidak method as performed through SigmaPlot 11. In general, the number of replications was five for each value, with a minimum of two replicate experiments. All EC50 and IC50 values were generated with the curve-fitting procedure provided by the four-parameter logistic analysis with in SigmaPlot 11.
Aqueous solubility determination
Aqueous solubility was determined using the shaken flask method as previously described (Alsenz and Kansy, 2007) at desired pHs of 1.2, 7.4 and 9.2 to mimic the stomach, plasma, and intestinal environments, respectively. Concentrations were determined using HPLC where a standard curve was used for reference.
Metabolic stability assay.
The stability of test compounds were determined by incubation with liver microsomes derived from rat, mouse or human. All assays were conducted by Absorption Systems (Exton, PA) by methods previously described (Clarke and Jeffrey, 2001. Test compounds at 1 μΜ were assayed at various times of incubation up to 60 min. Samples were analyzed using a LTQ-Orbitrap XL mass spectrometer. The assay was a single determination at each time point. As a metabolically labile control, 1 μΜ testosterone was tested.
Plasma protein binding
The test compounds were tested for their association with plasma proteins. Compounds were tested in an equilibrium dialysis system for their partitioning characteristics (Liu et al., 2017). Compounds were tested at 1 μΜ after 5 hours at 37 °C. Warfarin and quinidine were used as control reference compounds. These studies were completed by Shanghai ChemPartner (Shanghai, China).
Ligand binding studies.
Based on reports that CBD has been associated with neuroprotection, a series of experiments screened the responses of 10 μΜ KLS-13019 on these targets. These studies were conducted by Eurofins Cerep (Celle l’Evescault, France). PPARy agonist binding studies were conducted with 5 nM 3H-rosiglitazone in 120 min incubations at 4 degrees C in human recombinant E coli. Non-specific binding was determined in the presence of 10 μΜ rosiglitazone. The reference compound was rosiglitazone (Kd: 6 nM). For CB2 binding studies, 0.8 nM 3H-WIN-55212–2 was incubated for 120 min at 37 degrees C in human recombinant CHO cells. The reference compound was WIN-55212–2 (KD: 1.5 nM). Non-specific binding was determined with 10 μΜ WIN-55212–2. For 5HTla agonist binding, 0.5 nM 3H-8-hydroxy-2-(di-n-propylamino) tetralin (8-OH-DPAT) was incubated in human recombinant HEK-293 cells for 60 min at room temperature. Non-specific binding was measured in the presence of 10 μΜ 8-OH-DPAT. The reference compound for this assay was 8-OH-DPAT (IC50: 0.8 nM).
In addition to the target-related binding studies, hERG channel binding assay was also screened using 3 nM [3H]-dofetillide with human recombinant HEK-293 cells. For these studies, the compound was tested at 10 μΜ at room temperature for 60 min. Non-specific binding was determined with 25 μΜ terfenadine. Results were expressed as % inhibition of control specific binding. New compounds are always tested for interactions with the hERG channel because of the importance of this potassium channel in cardiac rhythm. The goal is to identify compounds that may cause QT prolongation which can cause cardiac arrhythmia and sudden death. Positive results with the binding assays are followed by more definitive tests for hERG channel interactions and QT prolongation.
CB2 pharmacology in hippocampal cultures
Because CBD may have neuroprotective effects that are mediated in part by CB2 (Castillo et al., 2010), studies were conducted with the hippocampal cultures to explore interactions with a CB2 antagonist and either CBD or KLS- 13019. In addition, a binding study screen with a CB2 agonist suggested a low potency interaction with 10 μΜ KLS-13019. Therefore, the selective CB2 antagonist AM 630 (Pertwee, 2005) was utilized to investigate the potential interactions between CB2 and the test compounds using a neuroprotection assay. Concentrations of AM 630 ranging from 0.1 to 100 nM were incubated for 5 min prior to incubating the test compounds with the cultures. After an additional 5 min, 30 mM ethanol was added to the cultures to produce toxicity. The duration of the test period was for 5 hour followed by the CFDA neuronal viability assay.
Pharmacokinetic studies.
These studies were completed by Shanghai ChemPartner (Shanghai, China). Male CD1 mice aged 6–8 weeks and weighing 18 to 22 g were used for experimentation after a minimum of 5 days of acclimation. Fasted animals were administered a single dose of test substance by oral gavage with a dose of 10 mg/kg body weight in 5% N,N- dimethylacetamide, 5% polyoxyl 15 hydroxystearate (Kolliphor HS 15) and 90 % saline at a dose volume 10 mL/kg body weight or intravenously (via tail vein) with a dose of 2 mg/kg body weight at a dose volume of 10 mL/kg body weight.
Under mild anesthesia, blood samples were collected the retro-orbital route into labeled centrifuge tubes containing anticoagulant (K2EDTA) during the next 24 hours post dose. Collected blood samples were centrifuged at 3500 rpm for 10 minutes and plasma were separated and stored at −80°C until analyzed. Homogenized brain samples were stored at −80°C until analysis by LC-MS\MS. Drug concentration in plasma was quantified with an API 6500 triple quadrupole LC-MS/MS-021 system.
Results
Comparison of Structure and Drug-like Properties
Cannabidiol (CBD) and the structurally related compound KLS-13019 have been shown previously to be neuroprotective from oxidative stress-mediated toxicity (Kinney et al., 2016). In the present studies, further research has been conducted on the drug-like properties, cellular toxicity, mechanisms of action and additional neuroprotective characteristics for each of these two compounds against a combined toxin treatment. The structure and pertinent drug-like properties of CBD and KLS-13019 are summarized in Table 1. As shown, the structural difference between the two compounds was confined to the pentyl side chain of CBD. Increasing the hydrophilicity of CBD with the introduction of an azetidine amide on the side chain produced a > five-fold increase in the aqueous solubility of KLS-13019 in comparison to CBD. Further, the Log P of the new compound was significantly reduced to a more drug-like value as described in Lipinski’s rule of five, although otherwise CBD does not violate these rules (Lipinski et al., 2001).
Table 1:
Comparison of Structure and Properties of Cannabidiol and KLS-13019
| Cannabidiol | KLS-13019 | |
|---|---|---|
| Structure | 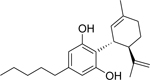 |
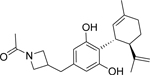 |
| Aqueous solubility | 17 μM | 87 μM |
| Log P | 7.11 | 3.42 |
| Mouse Plasma PK Parameters a |
||
| F% Cmax at 10 mg/kg p.o. Tmax p.o. |
8% 51 ng/ml 0.5 hr |
67% 1080 ng/ml 0.25 hr |
| T 1/2 p.o. | 3.45 hour | 5.31 hours |
| AUC (hr*ng/mL) p.o. | 268 | 617 |
| CL (L/h/kg) IV 2 mg/kg Vss (L/kg) IV 2 mg/kg |
7.2 7.5 |
4.6 3.2 |
| Microsomal stability | ||
| Human T 1/2 min | 3.75 | 5.69 |
| Mouse T 1/2 min | <5 | 5.67 |
| hERG Potassium channel |
||
| % Inhibition of specific binding | 73 % | 12% |
| Plasma protein binding | ||
| Human % bound | 100% | 99.2% |
| Mouse % bound | 100 % | 99.2 % |
Oral dose of 10 mg/kg in CD1 mice and IV dose of 2 mg/kg
Another important consequence produced with the strategy of increasing the hydrophilicity of CBD was improved early ADME (Absorption, Distribution, Metabolism and Elimination) properties. Pharmacokinetic (PK) properties were determined after administering the two compounds both orally (p.o.) and intravenously (i.v.) to mice at 10 and 2 mg/kg, respectively. Importantly, based on the comparative results of blood concentrations, the oral bioavailability (% F) was calculated to be substantially improved for KLS-13019 (67%) in comparison to that of CBD (8%). Both compounds exhibited rapid absorption into plasma, with the time to maximum concentration (Tmax) for KLS-13019 displaying a more rapid appearance (0.25 hour) in comparison to CBD. The concentration at Tmax (Cmax) for KLS-13019 was more than 20-fold greater than that observed for CBD. Furthermore, the AUC (Area-Under-the- Curve) or total exposure of the compounds revealed a 2.3-fold increase in KLS-13019 compared to CBD. The terminal half-life after p.o. administration was determined to be 3.5 hours for CBD and 5.3 hours for KLS-13019. As measured with the i.v. arm of the experiment, clearance (CL) was shown to be slow for both compounds: 7.2 and 4.6 mL/min/kg, respectively for CBD and KLS-13019. The apparent volume of distribution at steady state (Vss) was also determined after i.v. administration; with relatively high values for both compounds being observed, suggesting a high distribution to the tissues.
An significant screen of any new drugs is for interactions with the hERG potassium channel that may predict a cardiac liability for QT prolongation. In the present study, CBD was found to displace specific binding of [3H]-dofetilide by 75% at 10 μΜ. In contrast, KLS-13019 exhibited 12% displacement in this assay.
Microsomal Stability
Microsomal stability is commonly used to model susceptibility to metabolism. During one hour incubations, both compounds exhibited similarities, with both human and mouse microsomes metabolizing > 95% of the compounds as monitored by mass spectrometry.
Plasma protein binding
The test compounds were tested for their association with plasma proteins after equilibrium dialysis. Both test compounds were found to be highly bound (99–100%) to plasma proteins for both human and mouse plasma.
Neuroprotection in Hippocampal Cultures
Neuroprotection from toxicity associated with combined treatment with 30 mM ethanol and 300 μΜ ammonium acetate of hippocampal cultures was used as a model to evaluate CBD and KLS-13019 as potential therapeutics for hepatic encephalopathy. Two multiplexed assays were used to assess cell culture viability: a propidium iodide (PI) assay to estimate changes in the amount of cell death and a CFDA assay to estimate changes in neuronal viability (Brenneman et al., 2012). Neuroprotection was observed as an increase in fluorescence for CFDA and a decrease in fluorescence for the PI assay. For this experiment conducted between days 12–14 in culture, the combined toxin treatment produced a decrease in the CFDA fluorescence to 76 ± 4 % of that for control cultures. With the PI assay, the increase in fluorescence associated with cell death after treatment with the toxins was 142 ± 10 % of that observed with control cultures. The comparative protective effects of the test compounds on combined treatment with ethanol and ammonium acetate toxicities in hippocampal cultures are summarized in Table 2. With the CFDA neuronal viability assay, a 31-fold increase in potency was observed with KLS-13019 in comparison to CBD as determined by their relative EC50s. In the propidium iodide assay, a 15-fold increase in potency was observed with KLS-13019 in comparison to CBD, as determined by their relative IC50s. Although the potencies of CBD and KLS-13019 varied significantly between the two assays, full efficacy in providing neuroprotection back to control levels was observed with both compounds and with both assays.
Table 2.
Comparison of potencies of cannabidiol and KLS-13019 for neuroprotection from ethanol and ammonium acetate toxicity in hippocampal cultures1
| Compound2 | Toxins | Assay3 | EC50 for CFDA IC50 for PI nM |
|---|---|---|---|
| Cannabidiol | 30 mM Ethanol + 300 μM Ammonium Acetate |
CFDA PI |
4000 ± 500 3200 ± 584 |
| KLS-13019 | 30 mM Ethanol + 300 μM Ammonium Acetate |
CFDA PI |
127 ± 27 203 ± 76 |
All incubations were for 5 hours at 37° C in E18 hippocampal cultures between days 12–14.
Compounds were tested at 5 concentrations with an N = 5 for each of two replicate plates.
CFDA is a dye-related assay for neuronal viability and PI is dye-related test for cell death.
Toxicity in Hippocampal Cultures
An essential goal of the comparative analysis of the two compounds was the evaluation of toxicity that occurred with the treatment of each compound alone in the hippocampal cultures. As shown in figure 1A, toxicity was observed for both compounds by the CFDA viability assay after 5 hours of treatment on day 14 in culture. With the viability assay, the TC50 was 17 μΜ for CBD. In the cell death assay, a similar TC50 (15 μΜ) was observed for CBD. In comparison, toxicity for KLS-13019 was also observed under the same experimental conditions, with a TC50 of 81 μΜ for the neuronal viability assay. For the cell death assay, the estimated TC50 was 100 μΜ for KLS-13019. These data indicated that the TC50 for KLS-13019 was 5 to 6-fold greater than that of CBD as assessed by the viability assay and cell death assays. In contrast to their differences in TC50, these studies conducted with day 14 cultures indicated that CBD produced toxicity to 71 ± 3% of control values while KLS-13019 exhibited 73 ± 3 % of control; thus, the maximal amount of neuronal toxicity or cell death was not different between the two compounds, only their TC50s.
Fig. 1.
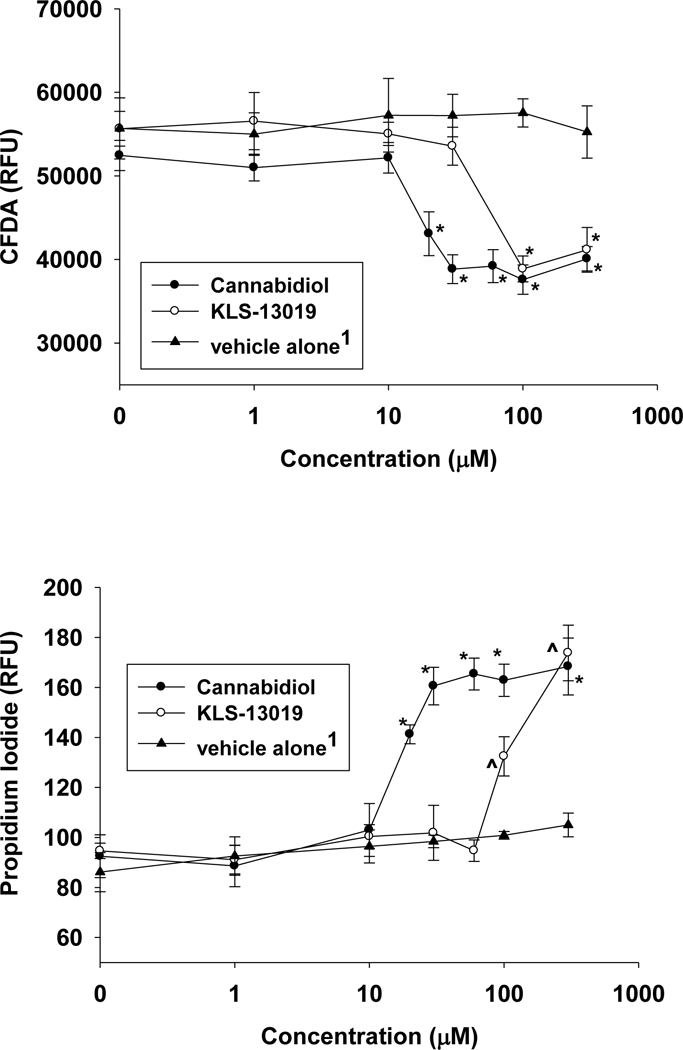
The comparison of toxicity between cannabidiol and KLS-13019 in day 14 hippocampal cultures. Both compounds were dissolved in Captisol and various concentrations of the two test compounds were incubated for 5 hours. Each point is the mean ± the standard error of 10 determinations from two experiments. 1The vehicle alone values were shown for the corresponding amounts of Captisol as that used to solubilize the test compounds, not the actual concentration of Captisol. la Toxic responses were measured by decreases in relative fluorescent units (RFU) from the CFDA neuronal viability assay. For CBD treatment (closed circles), significant decreases (P < 0.001) from control values were designated with an asterisk. Responses for the treatment with KLS-13019 are shown as open circles, with significant decreases (P< 0.001) from control designated with Λ. Treatment with the vehicle alone was shown with closed triangles, and no significant differences from control were observed, lb Cell death was estimated by the increase in relative fluorescence units from propidium iodide assay. The cell death assay was conducted in the same wells as that for the neuronal viability assay. The symbols for the treatment groups and statistical comparisons were the same as those shown in la. Significant increases (P < 0.02) in propidium iodide relative fluorescence were observed at all concentrations from 20 to 300 μΜ after CBD treatment. With KLS-13019 treatment, significant increases (P< 0.001) were observed at 100 and 300 μΜ.
Based on the experiments presented in Figure 1A that were conducted after 5 hours treatment, follow-up studies addressed the possibility that increasing the duration of treatment period might produce greater levels of toxicity. As shown in Figure 2AB, increasing the duration of the treatment period to 30 hours did not produce greater toxicity for either compound in comparison to that observed after 5 hours treatment. These observations of similar toxicities after 5 and 30 hours of treatment were apparent from both assays. In addition, comparisons of toxicity at shorter durations of treatment were also tested: 30 min, 60 min and 3 hours. With both compounds, significant decreases (P < 0.05) in the neuronal viability assay were observed within 30 min, with progressive increases in toxicity being observed to the maximum at 5 hours. In contrast, the cell death assay revealed no significant differences from control after either 30 min or 1 hour treatments for both compounds. Thus, longer treatment durations were required to produce cell death than to elicit deficits in neuronal viability. However, significant increases in propidium iodide accumulation were observed for both compounds after 3 hours treatment. These data indicate that the there was significantly more toxicity with CBD treatment than with KLS-130109 in regard to both TC50 in the neuronal viability assay and the time required to produce similar toxic effects from control with the cell death assay. However, the maximal achieved toxicities (66 ± 3 % and 62 ± 3% of control for CBD and KLS-13019, respectively) in the day 19 hippocampal cultures were not significantly different between the two compounds as assessed by the neuronal viability assay.
Fig. 2.
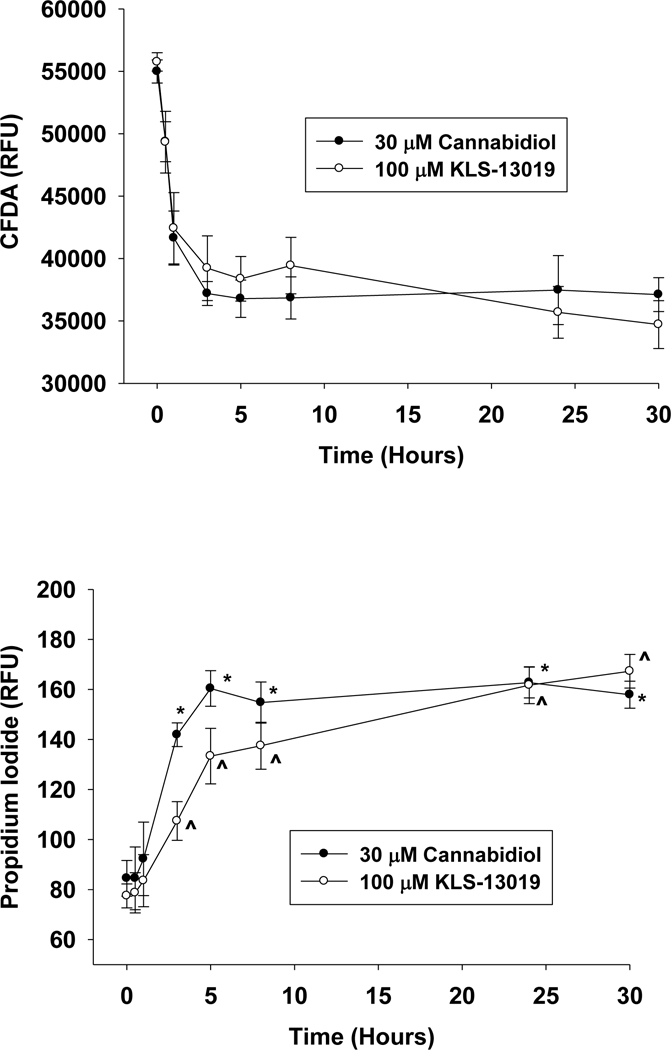
The time course of toxic responses to 30 μΜ cannabidiol and 100 μΜ KLS-13019 are shown for day 18–19 hippocampal cultures. Each point is the mean ± the standard error of 10 determinations from two replicate experiments. 2a Toxic responses were measured by decreases in the relative fluorescence from the CFDA neuronal viability assay. Responses after treatment with 30 μΜ cannabidiol are shown with closed circles and those treated with 100 μΜ KLS-13019 are shown with open circles. The concentrations of the test compounds were chosen on the basis of the neuronal viability results shown in Figure 1a, utilizing the lowest concentration which elicited the maximum toxicity after a 5 hour treatment. Significant differences (P< 0.05) from control were observed at 30 min for both compounds, with all other time points also showing significant differences (P < 0.02) for both compounds. 2b Cell death responses were measured with the propidium iodide assay after treatment with 30 μΜ cannabidiol (closed circles) or 100 μΜ KLS-13019 (open circles). For both compounds, no significant changes from controls were observed at 30 min or 60 min of incubation. Significant differences (P < 0.02) from control were observed at all time points ≥ 3 hours for both CBD and KLS-130139.
Mechanism of action
A feature of CBD pharmacology is the large (>12) number of molecular targets that have been reported to mediate the many responses of this component of cannabis. Because of the focus on neuroprotection from oxidative stress, preference in the selection of CBD-related targets in this study was given to those that have been associated with protective properties. Furthermore, because of the reported role of the mitochondrial sodium- calcium exchanger (mNCX) in regulating intracellular calcium of hippocampal neurons (Ryan et al., 2009) and the recognized importance of intracellular calcium as a determinant of neuronal survival (Randall and Thayer, 1992), our primary experimental focus was on comparing the properties of CBD and KLS-13019 on mNCX. A pharmacological approach was taken for the mNCX target with a widely used inhibitor: CGP-37157 (Cox et al., 1993; Ryan et al., 2009). KLS-13019-mediated protection from ethanol toxicity was shown to be blocked with CGP- 37157 treatment during a five hour test period. Concentration-dependent effects of this inhibition are presented for the neuronal viability assay (Figure 3A) and for the cell death assay (Figure 3B). These data indicate that in cultures treated with 30 mM ethanol, the protection provided by KLS-13019 treatment was completely prevented by the mNCX inhibitor. Similar observations were observed for 10 μΜ CBD as a protecting compound and ammonium acetate as a toxic agent. The half maximal potencies for CGP-37157-mediated inhibition of the various protective activities are summarized in Table 3. This summary of all the CGP-37157 data indicated the concentrations producing half-maximal decrease in protection were very similar between 10 μΜ CBD and 100 nM KLS-13019, for any given specific toxicity/assay comparison.
Fig. 3.
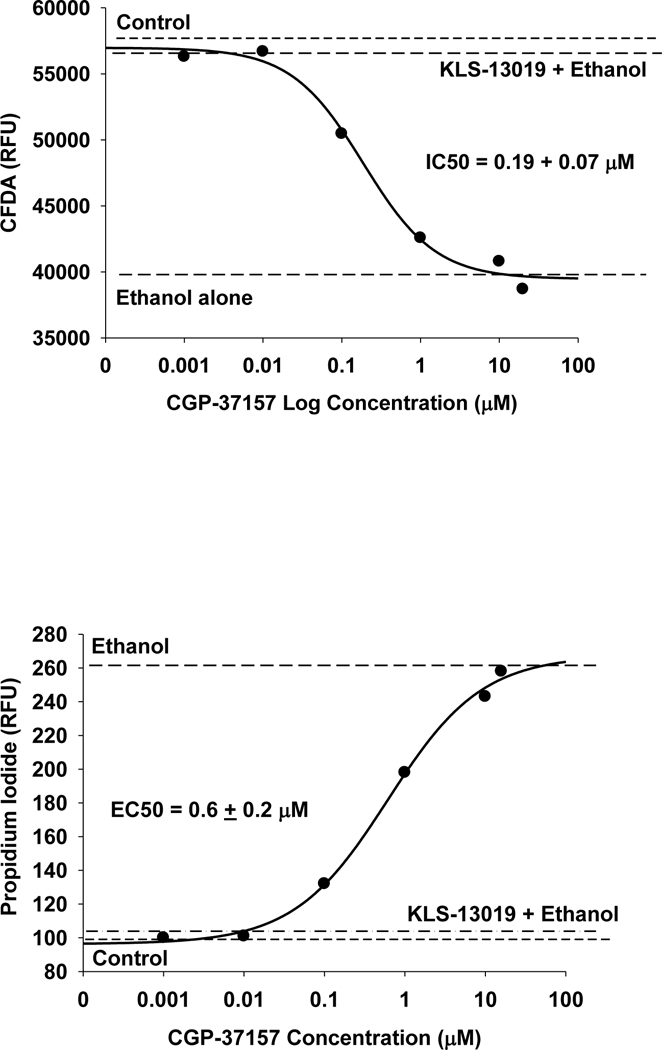
The determination of the efficacy and potency of the mNCX inhibitor (CGP-37157) on KLS-13019 mediated protection from 30 mM ethanol toxicity produced in hippocampal cultures. Representative data from two replicate experiments are shown. Each point is the mean of 5 determinations. Various concentrations of the CGP-371157 were tested for their effect in blocking neuroprotection produced by 100 nM KLS-13019 during a 5 hour test period. The EC50 and IC50 ± the standard error for the inhibitor were calculated from the four-parameter logistic analysis. The various dashed lines show the effects of various controls including 30 mM ethanol alone, non-treated control, and 100 nM KLS-13019 plus 30 mM ethanol. 3a The CFDA neuronal viability assay was used to assess the response of the hippocampal cultures. 3b The propidium iodide assay was used to measure relative changes in cell death produced by the various treatments.
Table 3.
Comparison of CGP37157 IC50/EC50s for Inhibition of Protection from Ammonium Acetate and/or Ethanol Produced by Cannabidiol or KLS-13019 Treatment
| Compound Concentration |
CG P-37157-mediated inhibition of protection IC50 for PI and EC50 for CFDA assays (μΜ)a |
||||||
|---|---|---|---|---|---|---|---|
| Assay | PI | CFDA | PI | CFDA | PI | CFDA | |
| Toxin(s) | Ethanolb | Ethanol | AmAcc | AmAc | E + AmAc | E + AmAc | |
| CBD (10 μΜ) |
1.0 ±0.6 | 0.4 ±0.1 | 0.9 ± 0.7 | 0.8 ± 0.4 | 4 ± 0.8 | 0.7 ± 0.2 | |
| KLS-13019 (0.1 μΜ) |
0.6 ±0.2 | 0.19 ±.07 | 0.9 ± 0.2 | 0.8 ± 0.5 | 3 ± 1 | 0.7 ± 0.4 | |
CFDA is a neuronal viability assay and PI is a cell death assay. All assays were multiplexed and the duration of treatment was 5 hours, with a 5 min pre-incubation consisting of a various concentrations (0.001 to 20 μM of CGP-37157 to determine half maximal responses (N =5 with + standard error).
ethanol (30 mM)
ammonium acetate (300 μM) c ammonium acetate (300 μM)
Because the specificity of CGP-37157 occurs over a narrow range of concentrations (1– 20 μΜ), it was important to demonstrate that all of the IC50s and EC50s resided within this concentration range and that toxicity was not a confounding effect of this inhibitor. Therefore, it was essential to demonstrate if CGP-37157 had any toxic effects in the hippocampal test system. As shown in Figures 4AB, the toxic effects of CGP-37157 were limited to concentrations greater than or equal to 33 μΜ, a concentration that was not attained in the present mechanism studies. Thus, the observed blocking action of CGP-37157 on neuroprotection was not confounded by toxicity. Together, these data indicate that all of the detectible neuroprotection provided by either 10 μΜ CBD or 100 nM KLS-13019 were effectively prevented by non-toxic concentrations of CGP-37157, To screen other regulatory sources of intracellular calcium that may contribute to the action of the two test compounds, two other pharmacological approaches were tested utilizing the same experimental conditions as described for CGP-37157. For these studies, only the CFDA assay was used. Cyclosporin A, an inhibitor of the mitochondrial permeability transition pore, when tested from 0.1 to 30 μΜ had no effect on the protective activity produced by either test compound (data not shown). Likewise, dantrolene, an inhibitor of ryanodine receptor channels that are linked to endoplasmic release of calcium, when tested from 0.1 to 10 μΜ had no effect on protective activity produced by either test compound (data not shown).
Fig. 4.
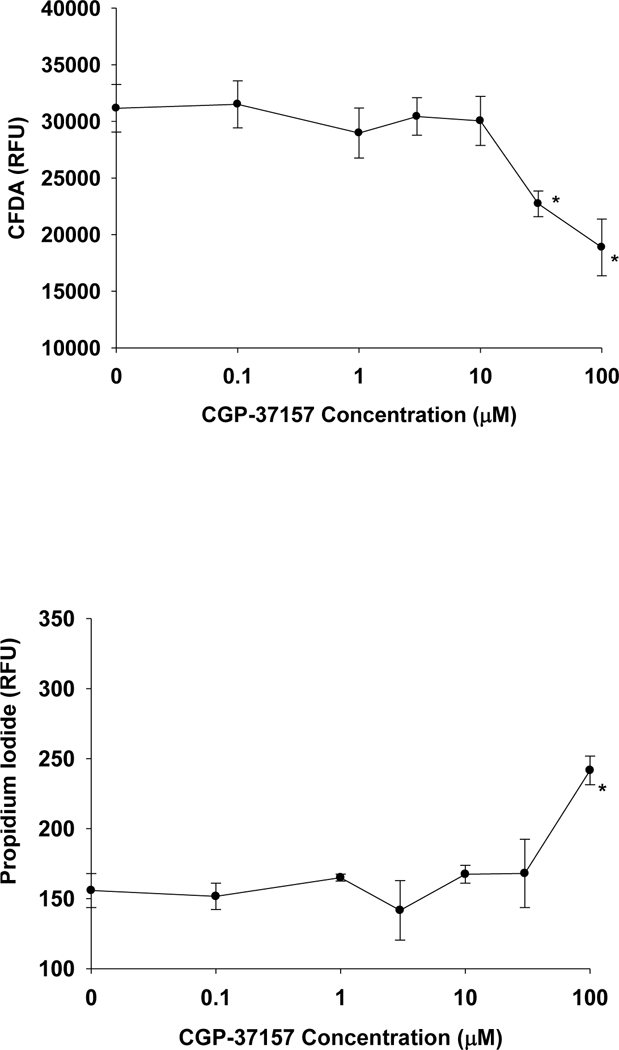
Determination of toxic concentrations of the mNCX inhibitor CGP-37137 in hippocampal cultures. A representative experiment from two replicate studies is shown, with each data point representing the mean ± the standard error of five determinations. The time of incubation was 5 hours with day 14 hippocampal cultures. 4a The CFDA neuronal viability assay indicated that significant toxicity (P < 0.001) was observed at 30 and 100 μΜ of the mNCX inhibitor. 4b As determined by the cell death assay, significant increases in the relative fluorescence of propidium iodide was observed only at 100 μΜ.
While mNCX was the major focus of these studies, other neuroprotective targets were also screened that have a reported relationship to CBD-mediated protection: CB2, 5HT-la, TRPV1 and PPAR γ (Bih et al., 2015). For binding interactions with CB2, CHO cells with human recombinant CB2 were incubated with 0.8 nM 3H-WIN-55212–2 and 10 μΜ of KLS- 13019. This study revealed a 55% displacement of radioactivity with 10 μΜ of the novel compound. Because this value suggested a possible low potency interaction with CB2, followup studies that measured the effect of AM 630 (a specific CB2 antagonist) on KLS-13019- mediated neuroprotection against ethanol toxicity were conducted. As shown in Figure 5, concentrations of the CB2 antagonist AM 630 ranging from 0.1 to 100 nM had no effect of KLS- 13019 (100 nM) mediated protection from 30 mM ethanol in the neuronal viability assay. In contrast, significant (P< 0.02) attenuation of the CBD-mediated neuroprotection was observed at 1, 10 and 100 μΜ. Significantly, none of these concentrations of AM 630 produced complete blockade of the CBD-mediated neuroprotection. Based on the observed protective signal in the neuronal viability assay, an estimated 70 ± 7 % of the CBD-mediated protection from ethanol toxicity was blocked by the CB2 antagonist. Together, these data indicate that the two compounds have significantly different properties in regard to the CB2 interactions relative to neuroprotection from ethanol toxicity, with KLS-13019 exhibiting none in this neuronal viability assay.
Fig. 5.
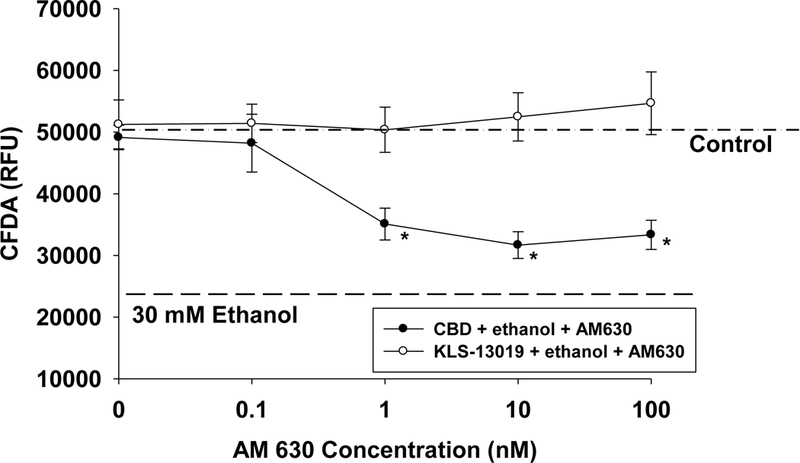
The effects of a CB2 antagonist (AM630) on neuroprotection provided by either cannabidiol or KLS-13019 were compared in hippocampal cultures co-treated with 30 mM ethanol. The responses were assessed with the CFDA neuronal viability assay. Each value is the mean of 5 determinations ± the standard error of a representative experiment. Various concentrations of AM630 were added to cultures co-treated with either 10 μΜ CB (closed circles) or 100 nM KLS-13019 (open circles). In cultures treated with 10 μΜ CBD, the concentration-effect study of AM630 revealed significant (P < 0.02) decreases in neuroprotection at concentrations ranging from 1 to 100 nM. Reference lines are used to show the mean values for control cultures (dot-dashed) and those cultures treated only with 30 mM ethanol (dashed line). At all of the concentrations tested for AM630, no significant differences from controls values were observed in cultures treated with 100 nM KLS-13019 plus 30 mM ethanol.
Other screening studies were conducted with 10 μΜ KLS-13019 and receptor binding to a 5HT-la agonist, TRPV1 (agonist and antagonist) and PPARγ (agonist). These four receptor binding studies indicated no detectable interactions with KSL-13019 (data not shown). Together, these data indicated a marked sensitivity to the mNCX inhibitor, while no potent interactions were observed with other known targets of CBD that have been linked to protective actions.
Discussion
Efficacy and Potency of Neuroprotection
Two structurally related compounds have been compared for neuroprotection from toxicity associated with combined ethanol and ammonium acetate treatment. Against the combined toxins, there was a 31-fold increase in potency with KLS-13019 in comparison to CBD as determined in the neuronal viability assay. Of interest in conducting the experiments with the combined toxins was the comparison of this potency to that observed for individual toxic agents reported in previous studies that were conducted under the same experimental conditions (Kinney et al, 2016). Whereas the half- maximal response for KLS-13019 was 127 nM against the combined toxins, the individual EC50 values were 23 nM and 50 nM for ethanol and ammonium acetate, respectively. Thus, the EC50 of KLS-13019 (127 nM) for the combined toxin treatment was more than the amount of the sum (73 nM) of the EC50s for individual toxins. In general, 10 μΜ CBD was required for full efficacy in the hippocampal cultures against oxidative stress toxicity, a potency that was similar to that reported for sensory neurons (Ward et al, 2017) and cortical neurons (Hampson et al., 1998). In comparison, 100–700 nM KLS-13019 was required to produce full efficacy to individual and combined toxins.
While we have confirmed that CBD is effective in preventing oxidative stress-related toxicity in a primary neuronal cultures, previous studies have indicated that CBD was also effective in preventing ROS-associated toxicity in immune cells that are relevant to many diseases (Booz et al., 2011). Thus, the protective effects of CBD are clearly not confined to either neurons or immune cells.
While the present studies have focused on a hippocampal culture system as a means of assessing responses to oxidative stress, previous in vivo studies have shown that CBD produced an improvement in both brain and liver function in a mouse model of hepatic encephalopathy, one of our two current therapeutic priorities. These studies suggested that behavioral abnormalities produced by liver failure can be attenuated with 5 mg/kg cannabidiol administered one day after eliciting liver toxicity with thioacetamide (Avraham et al., 2011). These studies provide a valuable perspective on the properties of CBD in this severe test of protective efficacy. In other studies, CBD has been shown to protect the liver in binge alcohol-induced steatosis through mechanisms involving inhibition of oxidative stress and increased autophagy (Yang et al., 2014. Further in vivo studies are in progress for KLS-13019 to demonstrate its efficacy in animal models of hepatic encephalopathy and chemotherapy-induced neuropathic pain (Ward et al., 2017).
Toxicity
Equally imperative to the determination of comparative potency was the evaluation of toxic responses between the two test compounds in hippocampal cultures. While there is scant evidence for CBD toxicity in other systems, the hippocampal cultures present asignificantly challenging test system that combines both high susceptibility to oxidative stress and the vulnerability of increased sensitivity to toxic substances that resides in a developing neural system. Our studies indicate that both compounds elicited a toxic response in hippocampal cultures; however, there was a 5-fold increase in the TC50 in both assays with KLS- 13019, indicating a statistically significant decrease in toxicity in comparison to CBD. The propidium iodide assay confirmed that there was significantly more toxicity with CBD than with KLS-13019. Despite these apparent different toxicological responses in TC50s, there was one clear similarity between the two compounds: there were no statistically different responses in the maximal toxic responses produced for the two compounds. Although it is not known what neuronal phenotypes may be responding to these compounds, it is very clear that not all neurons were vulnerable to these agents and that the maximal toxic signal in the neuronal viability assay was not different between the two compounds.
In addition to concerns of CBD-related toxicity on hippocampal function, there is additional evidence that CBD interferes with contextual fear memory consolidation by reducing prefrontal cortex influence of cortico-limbic structures (Rossignoli et al., 2017). Several reports have suggested that CBD may disrupt the fear memory consolidation indirectly through anandamide -mediated activation of dorsal hippocampal CB1 and CB2 receptors (Stern et al., 2017). In addition, CBD has been shown to block the formation of fear-related memory through a 5-HTlA-dependent mechanism in the ventral tegmental area (Norris et al., 2016). While these studies suggest multiple ways through which CBD may alter learning and memory, further investigations will be required to establish the long-term clinical consequences of these compounds along with an exploration of the pathways which may be modified or disrupted by
CBD and KLS-13019.
An important perspective on the toxicity of these compounds is the relevancy of the concentrations used in the cell culture experiments: are these toxic levels ever achieved in vivo ? In previous studies in both animal models and humans, there is a suggestion that these levels may not be attained with acute high doses (Iffland and Grotenhermen, 2017). However, there is the additional variable that needs to be addressed experimentally: do accumulating amounts of the lipid-soluble CBD after an extended duration of treatment ever reach toxic levels? Before assurance can be given as to their safety, long term studies in both animal models and humans are obligatory to establish the dosage and treatment duration limits for both compounds and their potential effects on cognitive functions.
Drug -like properties
Although efficacy and toxicity are often the first criteria of evaluation for any emerging drug candidate, an analysis of drug-like properties remains an obligatory and practical step that requires evaluation and potentially optimization. In the comparative analysis for drug-like properties, KLS-13019 clearly was superior in all measured parameters to CBD, particularly in the oral bioavailability and pharmacokinetic parameters (T1/2 and Cmax). Producing an increase in the aqueous solubility of CBD by the introduction of heteroatoms into the pentyl group resulted in the desired changes in the pharmacokinetic properties that indicated a better drug profile. KLS-13019 had an > 8-fold increase if F% in comparison to CBD, making it a clear choice for this important property of oral bioavailability. Furthermore, the mouse PK data and the in vitro screening tests of cellular permeability predicted that KLS-13019 would have better intestinal and blood - brain barrier properties in comparison to CBD. Together, these data indicate the marked improvement of the bioavailability of KLS13019 in comparison to CBD. The T 1/2 of KLS-113019 was also increased by more than 50% in comparison to CBD, suggesting the duration of action of the new compound may be significantly improved in vivo. The marked increase in Cmax for KLS- 13019 indicted a substantial (21-fold) increase in blood levels in comparison to CBD after oral administration of 10 mg/kg.
Mechanism of Action
The literature regarding the mechanisms of action for CBD portends these pharmacological interactions to be arguably the most confounding aspects of this cannabis-related compound (Bih et al., 2015). The broad array of molecular targets reported for CBD could be viewed as a positive, with the purported diversity of therapeutic uses being potentially predictive from the array of 65 candidate targets. Even with more limiting considerations given to more relevant targets that are activated by concentrations at < 10 μΜ that have been associated with neuroprotection or suppression of neuropathic pain, there are still at least six credible targets that mediate CBD-related actions. These protective targets include CB2 (Castillo et al., 2019), PPARγ (Aghaei et al., 2014), mNCX (Ryan et al., 2009), 5HT-la (Russo et al, 2005; Pazos et al., 2013), alpha 1 glycine receptor (Ahrens et al., 2010) and the adenosine A2A receptor (Mecha et al., 2013). While these targets were all of interest in our evaluation for protective actions, our experimental choice of the mitochondrial sodium calcium exchanger (mNCX) was highly influenced by the recognized importance of the regulation of intracellular calcium as major determinant of neuronal damage and death during oxidative stress (Randall and Thayer, 1992). Of critical importance was the previous work by Ryan et al (2009) which indicated that blocking the mNCX pharmacologically with a specific inhibitor resulted in the loss of CBD-mediated calcium homeostasis in hippocampal cultures. As with any pharmacological approach to drug mechanism, a cautionary perspective should be taken, as specificity is often equivocal and target-associated interaction should always be viewed as a concentration-dependent process. Although the mNCX is clearly an interacting target with CGP-37157, other targets may be involved with this inhibitor (Neumann et al, 2011). Molecular approaches are in progress to further test mNCX as a neuroprotective mediator of CBD and KLS-13019. Other regulators of intracellular calcium were without effect on KLS-13019 neuroprotection after treatment with specific blockers to these targets. In addition, our pharmacological screens suggested that only two of these targets were likely to interact with KLS-13019 to mediate neuroprotection: CB2 and mNCX. The findings with the mNCX inhibitor suggested that this target was relevant to the neuroprotection elicited by KLS-13019. The absence of an effect on KLS-13019-mediated neuroprotection after treatment with a potent and specific inhibitor of CB2 suggests that this target was not involved in protection from ethanol toxicity
Summary
The comparative studies described herein support the concept that both cannabidiol and KLS-13019 have potential therapeutic value. Because CBD is currently in patients through the expanded legal use of the medical cannabis, CBD already has achieved at least an anecdotal level of therapeutic acceptance by many caregivers. With the recent FDA approval for Epidiolex, a new era in the treatment of rare childhood-onset forms of seizures has begun. The purpose of our studies has been to perform side-by-side comparisons of the two compounds for neuroprotection, toxicity, drug-like properties and shared mechanisms of action. Our studies have raised concerns on the potential problems with acute treatment of these compounds that can produce toxicity in hippocampal neurons, important cellular mediators of learning and memory. Until high dose and long-term studies are adequately conducted, the final conclusion on CBD toxicity and its effects on learning and memory will remain unresolved. Our view is that although KLS-13019 has significantly improved drug-like properties in comparison to CBD, albeit likely to have a more limited therapeutic scope than CBD. Our best evidence of this shift in therapeutic use is the lack of anticonvulsant activity with KLS-13019 observed in animal models that have established highly predictive value for anti-seizure efficacy in humans. Based on our studies, it appears that KLS-13019 is not just a “super-CBD”; rather, the new compound is a non-schedule one agent that has been optimized for drug-like and neuroprotective properties for the treatment of neurological diseases involving oxidative stress.
Finally, while our belief is that CBD has significant challenges in regard to both drug-like properties and scientifically designed formulations, its use should be regarded as one of immense potential benefit due to its remarkable breadth of pharmacological actions. However, widespread use of CBD and medical cannabis currently should be tempered because of the paucity of both scientific rigor and established medical benefit. Clearly, more clinical evaluations are urgently needed for placebo-controlled, double blind studies to establish efficacy and toxicity parameters for dosage and frequency of treatment for both CBD and strains of CBD-rich cannabis. While compounds, like KLS-13019, may emerge to provide high quality, small molecule alternatives for specific therapeutic applications, the more immediate need is for a standardization of CBD and CBD-rich cannabis formulations that can provide predictable outcomes for patients desperate for treatments to intractable diseases.
References
- Aghaei I, Shabani M, Doustar N, Nazeri M, Dehpour A (2014) Peroxisome proliferator- activated receptor-γ-acti vation attenuates motor and cognition impairments inducted by bile duct ligation in a rat model of hepatic cirrhosis. Pharmacol Biochem Behav 120: 133–139. [DOI] [PubMed] [Google Scholar]
- Ahrens J, Demir R, Leuwer M, de la Roche J, Krampfl K, Foadi N, Karst M, Haeseler G (2009) The nonpsychotropic cannabinoid cannabidiol modulates and directly activates alpha-1 and alpha-1-Beta glycine receptor function. Pharmacol 83: 217–222. [DOI] [PubMed] [Google Scholar]
- Alsenz J, Kansy M (2007) High throughput solubility measurement in drug discovery and development. Adv Drug Delivery Rev 59:546–567. [DOI] [PubMed] [Google Scholar]
- Avraham Y, Grigoriadis NC, Poutahidis T, Vorobiev L, Magen I, Ilan Y, Mechoulam R, Berry EM (2011) Cannabidiol improves brain and liver function in a fulminant hepatic failure-induced model of hepatic encephalopathy in mice. Brit J Pharmacol 162: 1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bih CI, Chen T, Nunn AVW, Bazelot M, Dallas M, Whalley BJ (2015) Molecular targets of cannabidiol in neurological disorders. Neurotherap 12: 699–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman DE, Smith GR, Zhang Y, Du Y, Kondaveeti SK, Zdilla MJ, Reitz AB (2012) Small- molecule anticonvulsant agents with potent in vitro neuroprotection. J Mol Neurosci 47: 368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booz GW (2011) Cannabidiol as an emergent therapeutic strategy for lessening the impact of inflammation on oxidative stress. Free Radical Biol Med 51: 1054–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993) Optimized survival of hippocampal neurons in B27 supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res 35: 567–576. [DOI] [PubMed] [Google Scholar]
- Castillo A, Tolon MR, Fernandez-Ruiz J, Romero J, Martinez-Orgado J (2010) The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol Dis 37: 434–440. [DOI] [PubMed] [Google Scholar]
- Clarke SW, Jeffrey P (2001) Utility of metabolic stability screening: comparison of in vitro and in vivo clearance. Xenobiotica 31: 591–598. [DOI] [PubMed] [Google Scholar]
- Cox DA, Conforti L, Sperelakis N, Matlib MA (1993) Selectivity of inhibition of Na+ - Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J Cardiovasc Pharmacol 21: 595–599. [DOI] [PubMed] [Google Scholar]
- Darkovska-Serafimovska M, Serafimovska T, Arsova-Sarafinovska Z, Stefanoski S, Keskovski Z, Balkanov T. (2018) Pharmacotherapeutic considerations for use of cannabinoids to relieve pain in patients with malignant diseases. J Pain Res 11: 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlayson K, Turnbull L, January CT, Sharkey J, Kelley JS. (2001) [3H]dofetilide binding to HERG transfected membranes: a potential high throughput preclinical screen. Eur J Pharmacol 430: 147–148. [DOI] [PubMed] [Google Scholar]
- Hampson AJ, Grimaldi M, Axelrod J, Wink D (1998) Cannabidiol and (−)Δ9- tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA 95: 8268–8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo C and Arias-Cavieres A. (2016) Calcium, reactive oxygen species and synaptic plasticity. Physiology (Bethesda) 31: 201–205. [DOI] [PubMed] [Google Scholar]
- Iffland K, Grotenhermen F (2017) An update on safety and side effects of cannabidiol: a review of clinical data and relevant animal studies. Cannabis and Cannabinoid Res. 2.1: 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney WA, McDonnell ME, Zhong HM, Liu C, Yang L, Ling W, Qian T, Chen Y, Cai Z, Petkanas D, Brenneman DE (2016) Discovery of KLS-13019, a cannabidiol-derived nneuroprotective agent, with improved potency, safety, and permeability. ACS Med. Chem. Lett. 7: 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K, See YM, Lee J. (2017) A systematic review of the effectiveness of medical cannabis for psychiatric, movement and neurodegenerative disorders. Clin Psychopharm Neurosci 15:301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46: 3–26. [DOI] [PubMed] [Google Scholar]
- Liu H, Wu PP, Yang MJ, Men L, Lin HL, Zhao YL, Tang X, Yu ZG (2016) Application of a UPLC-MS/MS method to the protein binding study of TM-2 in rat, human and beagle dog plasma. J Pharm Anal 6: 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecha M, Feliu A, Inigo PM, Mestre L, Carrillo-Salinas FJ, Guaza C. (2013) Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: a role for A2A receptors. Neurobiol Dis 59: 141–150. [DOI] [PubMed] [Google Scholar]
- Neumann JT, Diaz-Sylvester PL, Fleischer S, Copello JA (2011) CGP-37157 inhibits the sarcoplasmic reticulum Ca2+ ATPase and activates ryanodine receptor channels in striated muscle. Mol Pharmacol 79: 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niknahad H, Jamshidzadeh A, Heidari R, Zarei M, Ommati MM (2017) Ammonia-induced mitochondrial dysfunction and energy metabolism disturbances in isolated brain and liver mitochondria, and the effect of taurine administration: relevance to hepatic encephalopathy treatment. Clin Exp Hepatol 3: 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris C, Loureiro M, Kramar C, Zunder J, Renard J, Rushlow W, Laviolette SR (2016) Cannabidiol modulates fear memory formation through interactions with serotonergic transmission in the mesolimbic system. Neuropsychopharmacol 41: 2839–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong JP, Aggarwal A, Krieger D, Easley KA, Karafa MT, Van Lente F, Arroliga AC, Mullen KD (2003) Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med 114: 188–193. [DOI] [PubMed] [Google Scholar]
- Pazos MR, Mohammed N, Lafuente H, Santos M, Martinez-Pinilla E, Moreno E, Valdizan E, Romero J, Pazos A, Franco R, Hillard CJ, Alvarez FJ, Martinez-Orgado J (2013) Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: role of 5HT(1A) and CB2 receptors. Neuropharm 71: 282–291. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (2005). Pharmacological actions of cannabinoids. Handbook Exp Pharmacol 168: 1–51. [DOI] [PubMed] [Google Scholar]
- Petroski RE, Geller HM (1994) Selective labeling of embryonic neurons cultures on astrocyte monolayers with 5(6)-carboxyfluorescein diacetate (CFDA). J Neurosci Methods 52: 23–32. [DOI] [PubMed] [Google Scholar]
- Randall RD, Thayer SA (1992) Glutamate-induced calcium transient triggers calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci 12: 1882–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignoli MT, Lopes-Aguiar C, Ruggiero RN, Do Val da Silva RA, Bueno-Junior LS, Kandratavicius L, Peixoto-Santos JE, Crippa JA, Cecilio-Hallak JE, Zuardi AW, Szawka RE, Anselmo-Franci J, Leite JP, Romcy-Pereira RN (2017) Selective post-training time window for memory consolidation interference of cannabidiol into the prefrontal cortex: reduced dopaminergic modulation and immediate gene expression in limbic circuits. Neurosci 350: 85–90. [DOI] [PubMed] [Google Scholar]
- Russo EB, Burnett A, Hall B, Parker KK. (2005) Agonistic properties of cannabidiol at 5HT1a receptors. Neurochem. Res 30: 1037–1043. [DOI] [PubMed] [Google Scholar]
- Ryan D, Drysdale AJ, Lafourcade C, Pertwee RG, Platt B. (2009) Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J Neurosci 29: 2053–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafian TA, Kouyoumjian S, Tashkin D, Roth MD. (2002) Synergistic cytotoxicity of 9- tetrahydrocannabianol and butylated hydroxyanisole. Tox Letters 133: 171–179. [DOI] [PubMed] [Google Scholar]
- Stern CAJ, da Silva TR, Raymundi AM, de Souza CP, Hiroaki-Sato VA, Kato L, Guimaraes FS, Andreatini R, Takahashi RN, Bertoglio LJ. (2017) Cannabidiol disrupts the consolidation of specific and generalized fear memories via dorsal hippocampal CB1 and CB2 receptors. Neuropharm. 125: 220–230. [DOI] [PubMed] [Google Scholar]
- Stockings E, Zagie D, Campbell G, Weier M, Hall WD, Nielsen S, Herkes GK, Farrell M, Degenhardt L. (2018) Evidence for cannabis and cannabinoids for epilepsy: a systematic review of controlled and observational evidence. J Neurol Neurosurg Psychiatry 89: 741–753. [DOI] [PubMed] [Google Scholar]
- Tomasini MC, Borelli AC, Beggiato S, Tanganelli S, Loche A, Cacciaglia R, Ferraro L, Antonelli T. (2016) GET3 prevents ethanol-induced neurotoxicity in primary cultures of rat hippocampal neurons. Alcohol Alcohol 51: 128–135. [DOI] [PubMed] [Google Scholar]
- Ward SJ, Riggs D, Tuma R, Kinney WA, Petkanas D. Brenneman DE (2017) Neuroprotective and anti-inflammatory effects of KLS-13019 and cannabidiol in in vitro and in vivo models of chemotherapy-induced neuropathic pain. International Cannabinoid Research Society Symposium, June 22, 2017, Montreal, Canada, 27: P1–40. [Google Scholar]
- Weinstein A, Livny A, Weizman A. (2016) Brain imaging studies on the cognitive, pharmacological and neurobiological effects of cannabis in humans: evidence from studies of adult users. Curr Pharm Des 22: 6366–6379. [DOI] [PubMed] [Google Scholar]
- Yang L, Rozenfeld R, Wu D, Devi LA, Zhang Z, Cederbaum A. (2014) Cannabidiol protects liver from binge alcohol-induced steatosis by mechanisms including inhibition of oxidative stress and increase in autophagy. Free Radical Biol Med 68: 260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]