

1. Introduction
SUMOylation is an ATP-dependent post translational modification where a small ubiquitin-like modifier (SUMO) protein is covalently bound to a lysine residue in target proteins exhibiting a SUMO-interaction motif, ΨKXE/D, Ψ being a large hydrophobic amino acid [73]. Similar to ubiquitination, SUMO conjugation is achieved by different enzymes, E1-activating (SAE1/UBA2 complex), E2-conjugating (Ubc9) and various E3-ligases.
SUMOylation has been described in numerous cellular processes including nuclear-cytosolic transport, transcriptional regulation, apoptosis, protein stability, response to stress, and progression through the cell cycle [33], and is an important regulator of neuronal and synaptic function [34]. SUMO targets several cytosolic and plasma membrane proteins, including ion channels, to regulate plasma membrane depolarization or neurotransmission by modulating their stability, sub-cellular targeting, transport or interacting properties [53; 54; 72].
Collapsin response mediator protein 2 (CRMP2), is a tetrameric protein which specifies axon/dendrite fate and axonal outgrowth [37; 42], and was shown, using sequence alignment and structure analysis, to be SUMOylated on Lysine 374 [20]. We demonstrated that SUMOylation of CRMP2 at site 374 is dependent on phosphorylation at Serine 522 by cyclin-dependent kinase 5 (Cdk5) [19], which is thought to disrupt CRMP2 tetramer towards a monomeric state, thus exposing the SUMOylation site [20].
SUMOylation of CRMP2 specifically controls trafficking and surface expression of the tetrodotoxin-sensitive (TTX-S) voltage-gated sodium channel NaV1.7 [10; 19; 21]. Decreasing CRMP2 SUMOylation, using CRMP2 mutants with disrupted SUMO motifs, reduced NaV1.7 currents. As inferred from human pain disorders, NaV1.7 is a major contributor to nociception and pain signaling. Gain-of-function mutations in NaV1.7 increase neuronal excitability and underlie pain disorders whereas loss-of-function mutations render individuals insensitive to painful stimuli [14; 24; 83; 91]. The potential of targeting NaV1.7 in pain is highlighted by many patents filed and clinical trials being conducted (see [23]). As pan NaV1.x blockers have a potent analgesic effect, there is a vital need for NaV1.7-specific treatment, which is lacking in many NaV1.7 drug-development programs.
Targeting NaV1.7 indirectly, but specifically, via modulation of CRMP2 SUMOylation has been demonstrated to be a promising avenue for experimental neuropathic pain treatment [64]. In the chronic model of spared nerve injury (SNI), CRMP2 SUMOylation was increased, and reducing deSUMOylation of CRMP2, by expressing CRMP2 K374A in vivo, significantly reversed SNI mechanical allodynia [64]. Consequently, we postulated that (i) CRMP2 interacts with Ubc9 and (ii) disruption of this interaction using a CRMP2 SUMO peptide would specifically reduce NaV1.7 sodium current, resulting in a relief of neuropathic pain [10].
Here, we assessed binding between the Ubc9 E2 SUMO-conjugating enzyme and CRMP2 using microscale thermophoresis and determined that CRMP2 and Ubc9 interact directly and with low micromolar affinity. Using rational design, we identified a heptamer peptide containing the CRMP2 SUMOylation consensus site fused to the transduction domain of the HIV-1 tat protein, called t-CSM, to disrupt the CRMP2-Ubc9 interaction. This t-CSM peptide inhibited CRMP2 SUMOylation, NaV1.7 membrane trafficking, and specifically inhibited NaV1.7 sodium influx in sensory neurons. Intrathecal injection of t-CSM reversed nerve injury-induced thermal and mechanical hypersensitivity with no sedation or motor impairment in rats.
2. Methods
2.1. Animals
Adult male and female Sprague-Dawley rats (Pathogen-free male 250–275 g, female 200–225 g, Envigo, Placentia, CA) or female mice (C57BL/6NHsd, 20–24 g; Envigo) were kept in light (12-h light: 12-h dark cycle; lights on at 07:00 h) and temperature (23 ± 3°C) controlled rooms. Standard rodent chow and water were available ad libitum. University of Arizona’s College of Medicine Institutional Animal Care and Use Committee (IACUC) approved these protocols. All experiments were performed congruent with the National Institutes of Health’s Guide for Care and Use of Laboratory Animals and per the ethical guidelines reported by the International Association for the Study of Pain. Treatment and control groups consisted of randomly assigned animals and experimenters were blinded to the grouping information until the analysis was completed.
2.2. Materials
The CRMP2 SUMOylation Motif (CSM) peptide chemically linked to the cell-penetrating motif (CPP) of the HIV transactivator of transcription peptide tat (t-CSM; YGRKKRRQRRRGKMDENQ; tat sequence identified by the underlined text) was manufactured and HPLC-purified by Genscript USA Inc. (Piscataway, NJ) and was resuspended in double-distilled water or DMSO (Sigma-Aldrich, St. Louis, MO), aliquoted, and stored at −20°C. Several of our studies previously showed the lack of effect of both scramble and random peptides fused with different CPPs in both in vitro and in vivo studies [4; 26; 40; 70]. All chemicals, unless stated otherwise, were purchased from Sigma-Aldrich. Fura-2 Low Affinity and Sodium-binding Benzofuran Isophthalate Acetoxymethyl ester (SBFI-AM) was obtained from Teflabs (Austin, TX). Catecholamine A differentiated (CAD) (ECACC Cat# 08100805) cells, of mouse origin, were grown in cell culture conditions at 37°C in 5% CO2. CAD cells were grown in DMEM/F12 media supplemented with 10% FBS (Hyclone) and 1% penicillin/streptomycin sulfate from 10,000 μg/ml stock. CAD cells were chosen as a model neuron cell line due to ~80% contribution by NaV1.7 to total sodium currents as determined by isoform-specific blockade of NaV1.7 by both ProTox-II (Sigma, St. Louis, MO) and Huwentoxin-IV (Alomone Laboratories, Jerusalem, Israel) [20; 21].
2.3. Co-immunoprecipitation and immunoblotting
Co-immunoprecipitation experiments were performed on spinal cord lysates from adult Sprague-Dawley rats using lysates homogenized in RIPA buffer as described previously [26]. Samples were then processed by immunoblotting as described [4; 5]. Blots were probed with anti-Ubc9 (Catalogue# ab21193, Abcam), or anti-CRMP-2 (Catalogue# 2993, Sigma-Aldrich) (both 1:1000). At least three blots were done per experiment.
2.4. Purification of recombinant human CRMP2-His and Ubc9-GST
Recombinant human CRMP2-His was expressed and purified as described previously [26]. The pure CRMP2 protein was concentrated to 30 mg/mL, flash frozen in liquid nitrogen and stored at −80°C. Ubc9-GST fusion protein was expressed and purified as previously described in [30].
2.5. Microscale Thermophoresis (MST)
MST, an assay that monitors the thermophoretic movement of molecules in optically generated microscopic temperature gradients thereby permitting analysis of biomolecular interaction was performed as described previously [86; 87]. In MST, increasing concentrations of unlabeled ligand are mixed with the fluorescently labeled biomolecule which is kept at a constant concentration. Purified CRMP2-His was fluorescently labelled with a His-Tag labeling kit RED-Tris-NTA (Nanotemper, Germany) per manufacturer’s instructions. Two hundred nanomolar of CRMP2-His (in PBS supplemented with 0.05% Tween-20 (PBS-T buffer)) was mixed with 1M NT-647-His-labeling dye. After a 30 min incubation at room temperature, the labeled CRMP2 was pelleted by centrifugation at 15,000Xg for 10 min at 4°C. 50 nM of NT-647-CRMP2 was mixed with different concentrations of Ubc9-GST in PBS-T and incubated at room temperature for 10 min. The thermophoresis readouts were captured on a Monolith NT.115 (Nanotemper, Germany) using premium MST capillaries, at 40% LED and 40% MST power. Measurements were done in triplicates. Data analysis was performed with the MO Affinity Analysis software (Nanotemper) using the Kd model (standard fitting model derived from law of mass action).
2.6. Ubc9-CRMP2 interaction measured by amplified luminescent proximity homogeneous alpha assay (ALPHA)
ALPHA experiments were performed per instructions supplied by PerkinElmer (USA). After a first mix of 200 nM of Ubc9-GST with 800 nM CRMP2-GST, 10 μL of donor and 15 μL acceptors beads (both at 10mg/mL) were added and incubated for one hour. The reaction was read at an emission wavelength of 615 nm using an excitation wavelength of 680 nm, on a PerkinElmer EnVisionTM plate reader.
2.7. Immunoprecipitation (IP) of endogenously SUMOylated proteins and CRMP2 from cell lysate
CAD cells, treated with 20 μM t-CSM peptide (or control, i.e. 0.01% DMSO) overnight, were lysed into the immunoprecipitation buffer exactly as previously described [11]. Immunoblotting was performed with a anti-CRMP2 antibody (Catalogue #2993, Sigma) on samples immune-captured by immunoprecipitation with a SUMO1 antibody (Catalogue #S8070, Sigma, St. Louis, MO).
2.8. Patch clamp electrophysiology
Whole cell voltage clamp recordings were performed at room temperature on CAD cells using an EPC 10 Amplifier (HEKA Electronics, Germany) exactly as described previously [20; 21].
2.9. Primary dorsal root ganglion (DRG) neuronal cultures
DRGs from all levels were acutely dissociated using methods as described previously [26]. Rat DRG neurons were isolated from Sprague-Dawley rats using previously developed procedures [26; 63].
2.10. Sodium fluorescence imaging
Sodium fluorescence imaging of rat cultured DRG neurons grown for 4 days in vitro (4 DIV) was performed exactly as described before [6]. Changes in cytosolic Na+ concentration ([Na+]c) were examined using the Na+-sensitive dye SBFI-AM (Molecular Probes, Eugene, OR). Neurons were loaded with 9 μM SBFI-AM for 1 hour at 37ºC. [Na+]c was calculated using equations [32] as described previously [16], assuming a dissociation constant of 15.7 mM for SBFI.
2.11. NaV1.7 surface staining
DRG neurons in culture were treated with either 20 μM of t-CSM or 0.1% DMSO overnight. The neurons were washed in ice-cold PBS twice before incubation (40 min) with anti-NaV1.7 antibody (extracellular epitope, dilution 1/40, Cat# ASC-027, Alomone labs, Israel) for ice. Cells were washed in ice-cold PBS thrice before fixation with 4% paraformaldehyde (10 min, 4°C). The neurons were next kept in blocking buffer (BSA (3% w/v) in PBS) for 20 min at room temperature (RT) before addition of the (goat anti rabbit Alexa Fluor 488 secondary antibody (Catalogue # A-11008, Thermofisher) for 1 hour at RT. After 3 washes with PBS, the coverslips were fixed and imaged with a Nikon Eclipse Ti-U (Nikon instruments) using a Photometrics cooled CCD camera CoolSNAPES2 (Roper Scientific, Tucson, AZ) run by Nis Elements software (version 4.20, Nikon Instruments). The fluorescence intensity for each cell was quantified and normalized to the area of the region of interest using NiS elements software. To obtain the region of interest, the perimeter of each neuron was manually traced.
2.12. Spared nerve injury model of neuropathic pain
The biceps femoris muscle was dissected to bare the 3 terminal branches of the sciatic nerve to create the spared nerve injury as described previously [15]. Sham animals also underwent the same operation; however, the nerves were exposed and not ligated. Before any testing, animals were allowed to recover for 5–7 days.
2.13. Indwelling intrathecal catheter
Rats were sedated using a xylazine/ketamine/ (12/80 mg/kg intraperitoneal; Sigma-Aldrich) and placed in a stereotaxic head holder. Following the exposition and incision of the cisterna magna, an 8-cm catheter (PE-10; Stoelting) was implanted as previously reported [89].
2.14. Testing of allodynia
The assessment of tactile allodynia was done exactly as reported before using the “up and down” method described before [9]. Data were expressed as the mean withdrawal threshold.
2.15. Writhing test
The acetic acid-induced writhing test is a model of visceral pain where a chemical irritant -acetic acid- is injected intraperitoneally, inducing abdominal contractions and extension of back limbs, collectively defined as ‘writhes’ [55]. Animals were first allowed to acclimate with the clear Plexiglas chamber used for the experiment during 15 min or until explorative behavior ceased. Mice were then immediately injected in the peritoneum of the left lower quadrant of the abdomen with 300 μl of 1% acetic acid in normal saline, via a 27-gauge needle. After the acetic acid injection, mice were placed in the testing chamber and their subsequent nociceptive behavior was recorded and scored (in an investigator blinded manner): writhing behavior was assessed between 5 and 25 min after the injection. t-CSM peptide (4 μg or the vehicle 0.1% DMSO in the control group) were administered i.t. (in a volume of 10 μl), 60 min before the acetic acid injection.
2.16. Rotarod
A week after surgical placement of the intrathecal catheters, the rats were trained and then assessed (every 30 min for 3 hours) for their capability to stay on a rotating rod following intrathecal delivery of peptide (or vehicle) as described before [71]. For SNI rats training consisted of 2 training sessions on a non-rotating rotarod followed by 2 training sessions where the rotarod was set at 4 and 8 rpm respectively after intrathecal injection of t-CSM or vehicle as indicated. Testing was done on an accelerating rotarod (4 to 40 rpm, 5min) and repeated every half hour for the first hour after drug administration and every hour for 4 more hours for a total of 5 hours.
2.17. Data Analysis
Statistical significance was determined using GraphPad Software (LaJolla, CA). Student’s t-test or a one- or two-way ANOVA followed by post hoc comparisons (Dunnett’s or Tukey’s test) were used to assess the statistical significance of differences between means. Results were stated as significant when the corresponding P value was less than 0.05.
3. Results
3.1. Ubc9 interacts directly with CRMP2
We previously reported that the CRMP2 protein sequence has a canonical ψ-K-X-E/D SUMOylation motif which accommodates E2-conjugating enzyme Ubc9-mediated addition of SUMO [20; 21]. Here, we asked whether, during the course of SUMOylation, CRMP2 interacts with Ubc9. In the present study, co-immunoprecipitation experiments revealed an interaction between Ubc9 and CRMP2 in CAD cells (Fig. 1).
Figure 1. CRMP2 co-immunoprecipitates with Ubc9 in CAD cells.
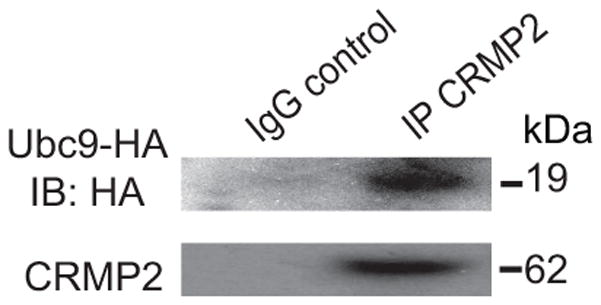
Lysates from mouse CAD cells transfected with HA-tagged Ubc9 were immunoprecipitated using an antibody against CRMP2. The samples were processed for immunoblotting with antibodies against Ubc9 and CRMP2. The blot is representative of 3 individual experiments.
Microscale thermophoresis (MST) was used to test if there is direct binding between CRMP2 and Ubc9 using purified recombinant proteins. In MST, an infrared laser generates precise microscopic heat gradients within capillaries containing a fluorescently labeled protein (i.e., CRMP2). The diffusion of labeled molecules along these temperature gradients is measured in the presence of ranging concentrations of an unlabeled ligand (Ubc9) [39; 75]. As the concentration of Ubc9 increases, it binds to CRMP2 altering its thermodiffusion out of the heated infrared spot, resulting in a change in the MST signal and providing a record of the binding between CRMP2 and Ubc9 (Fig. 2A). As a negative control, CRMP2 was also incubated with various concentrations of GST; no association could be detected between GST and CRMP2 (Fig. 2B, C). NT647-labeled CRMP2 (100 nM) was mixed with varying concentrations of Ubc9 (0.003–100 μM) and apparent Kd values were calculated by fitting curves using the standard fitting model derived from law of mass action. The MST experiments revealed that Ubc9 binds to CRMP2 with an apparent Kd of 12.2 ± 2.7 μM (Fig. 2C). ATP (1 mM), a necessary co-factor for the SUMOylation machinery, did not alter the affinity of the Ubc9/CRMP2 interaction (Fig. 2C). Divalent cations MgCl2 (200 mM) or CaCl2 (200 mM), believed to favor CRMP2 tetramerization [51], also did not alter the Kd values of the Ubc9/CRMP2 interaction (Fig. 2C). These results establish CRMP2 as a bona fide binding partner of Ubc9.
Figure 2. Ubc9-CRMP2 interaction measured by microscale thermophoresis (MST).

(A) Thermographs of Ubc9-GST (0.003 - 100 μM) binding to increasing concentrations of NT-647 labeled CRMP2 altered thermodiffusion providing well-defined curves. (B) Thermographs of GST (0.007 to 120 μM) provided no thermodiffusion of NT-647 labeled CRMP2. The grey bar (at ~2 s) represents the steady-state time point at which the MST measurements were analyzed for the graphs shown in panel C. (C) MST values were used to determine dissociation constant for binding of Ubc9 to CRMP2. The data were fitted as described in the Methods section yielding an apparent Kd of 12.2 ± 2.7 μM (r2 = 0.98). ATP (1mM) had no effect on the dissociation constant (Kd = 7.6 ± 2.9 μM, r2 = 0.91). CaCl2 or MgCl2 (200 mM) had no significant effect on the Kd values (26 ± 1.7 μM, r2 = 0.98 and 25.5 ± 3.0 μM, r2 = 0.98, respectively). No association could be detected between GST and CRMP2, thus no binding curve was extractable. Data is presented as means ± SEM (n=3). Some error bars are smaller than the symbols.
3.2. Rational design of a CRMP2 SUMOylation motif “decoy” peptide
We previously identified CRMP2 SUMOylation as a potential biomarker for neuropathic pain [64]. In rodents inflicted with a spared nerve injury (SNI) mimicking chronic pain, CRMP2 SUMOylation was found to be increased and preventing SUMOylation of CRMP2, by expressing the SUMO-null CRMP2 K374A in vivo, significantly reversed SNI-induced mechanical allodynia [64]. The sequence GKMDENQ (Fig. 3; SUMOylated lysine at position 374 underlined) in CRMP2 conforms to a canonical SUMOylation motif [84]. Reasoning that a peptide encompassing this region (CRMP2 SUMOylation motif (CSM)) could serve as a “decoy” and act as a sink for SUMOylation by Ubc9, we fused CSM to the cell penetrating transduction domain of the HIV-1 trans-activator of transcription (tat, t) creating the cell permeable peptide t-CSM. The resulting t-CSM peptide was incubated with Ubc9 to assess binding using MST (Fig. 3D), revealing an interaction with a Kd of 0.4 ± 0.1 μM.
Figure 3. Structure guided rational design of a CRMP2 SUMOylation Motif (CSM) “decoy” peptide.

(A) Surface representation of CRMP2 monomer (subunit A, PDB ID: 5uqc [20]) with K374 (spheres) within the GKMDENQ peptide shown in CPK coloring (green, carbon; red, oxygen; blue, nitrogen; yellow sulfur). Close-up view of GKMDENQ peptide in surface (B) and cartoon (C) representations. A CRMP2 heptamer peptide fused with the transduction domain of the HIV-1 tat protein was designed to disrupt Ubc9-CRMP2 interaction and designated as tat-CRMP2 SUMOylation Motif (t-CSM), it includes the residue K374 (red) and the surrounding amino acids. (D) Thermographs of NT-647 labeled Ubc9 binding to various concentrations of t-CSM were used to determine dissociation constant for binding of t-CSM peptide to labelled Ubc9, apparent Kd = 0.4 ±0.1 μM. The data was fitted using the Hill method (r2 = 0.96).
3.3. Inhibition of Ubc9-CRMP2 interaction using t-CSM peptide
Next, we tested if t-CSM peptide could inhibit the Ubc9-CRMP2 interaction. For this we initially turned to the amplified luminescent proximity homogeneous assay (ALPHA), a bead-based assay amenable to high throughput screening and miniaturization (Fig. 4A) [22]. Briefly, we incubated purified CRMP2-His protein bound to a Ni-chelate acceptor bead and Ubc9-GST protein bound by a glutathione-coated donor bead (Fig. 4B). If an acceptor bead comes into proximity with a donor bead, the acceptor bead emits a fluorescence signal between 520–620 nm. The donor bead contains a photosensitizer, phthalocyanine, that converts ambient oxygen to an excited singlet form of oxygen. This singlet oxygen will react with a thioxene derivative on the acceptor bead culminating in a chemiluminescent reaction. This reaction occurs only if the beads are within 200 nm of each other. The strength of the singlet oxygen reaction is also proportional to the amount of analyte present. In the absence of acceptor beads or any beads, no ALPHA signal was produced (Fig. 4C). Incubating various concentrations of Ubc9 (from 0.005 to 5 μM) with 1 μM CRMP2 which gave an apparent Kd of 0.8 ± 0.1 μM (Fig. 4D); this binding was stable over at least an 18 hr window. No binding could be detected between Ubc9 and CRMP2 in the presence of 10 μM t-CSM peptide; the data could not be fitted to a curve (r2 = 0.23) (Fig. 4D).
Figure 4. t-CSM peptide disrupts the Ubc9-CRMP2 interaction as determined via ALPHA.

(A) ALPHA assay schematic. The ALPHA assay signal is generated following the transfer of singlet oxygen between nickel-chelate donor beads (CRMP2-His) and a glutathione acceptor bead chelated to Ubc9-GST. (B) Black and white image of Coomassie-stained gel of 1 and 5 μg of purified CRMP2-His and Ubc9-GST (arrowheads). (C) Steady state binding of 200 nM Ubc9-GST to 800 nM CRMP2-His over 24 hr with a stable signal over ~18 hr. In presence of both donor and acceptor beads, the ALPHA signal was always at least 500 times higher than the signal generated in absence of acceptor beads or without any beads (negative controls). For this ALPHA experiment (performed in triplicate), the signal/background ratio was ~50 with a Z’ factor (index for quality control) of 0.71. (D) ALPHA signal generated by the binding of CRMP2 with ranging concentration of Ubc9. The data was fitted using the Hill method (r2 = 0.96). The apparent Kd is 0.8 ± 0.2μM. No association between CRMP2 and Ubc9 could be detected in presence of 1 μM t-CSM. Data is presented as means ± SEM (n=3). Error bars are smaller than the symbols.
To test if t-CSM could be useful in inhibiting the Ubc9-CRMP2 interaction, we performed MST analyses between CRMP2 and Ubc9 in the presence of increasing concentrations of t-CSM. Ubc9-CRMP2 binding was reduced by t-CSM in a concentration-dependent manner: 2.5 μM t-CSM reduced the binding of Ubc9-GST to CRMP2 (Kd = 80 ± 14 μM) while 5 μM t-CSM reduced it even further (Kd = 137 ± 14 μM, respectively) (Fig. 5). No association could be detected between CRMP2 and Ubc9 at 10 and 20μM of t-CSM (Fig. 5). In separate pull-down experiments with bacterially produced Ubc9-GST, CRMP2 binding to Ubc9 was also reduced by t-CSM in spinal cord lysates (data not shown). Collectively, these results show that the Ubc9-CRMP2 interaction can be recapitulated both with recombinant proteins and in native tissue and that t-CSM can interfere with this interaction.
Figure 5. t-CSM peptide disrupts the Ubc9-CRMP2 interaction.
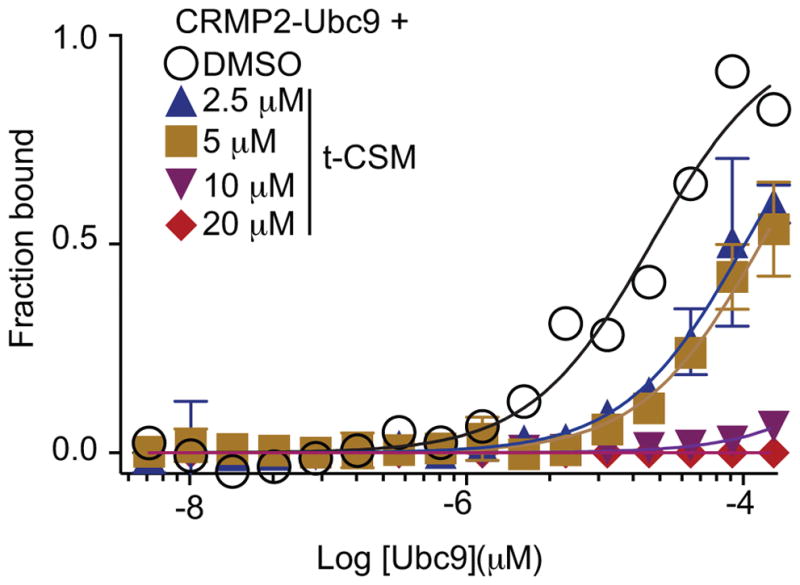
Microscale thermophoresis (MST) analyses was used to determine the binding between Ubc9-GST (0.003 – 100 μM) and NT-647 labeled CRMP2 (50 nM) in the absence (DMSO, 0.1%) or presence of increasing concentrations of t-CSM peptide. MST data were fitted as described in the Methods section. Ubc9-CRMP2 binding was reduced by t-CSM in a concentration-dependent manner: t-CSM, at 2.5 and 5μM, reduced the binding of Ubc9-GST to CRMP2 (Kd = 80 ± 14 μM and 137 ± 14 μM, respectively). No association could be detected between CRMP2 and Ubc9 at 10 μM and 20μM of t-CSM. Data is presented as means ± SEM (n=3). Error bars are smaller than the symbols.
3.4. t-CSM peptide decreases SUMOylation of CRMP2 in CAD cells
Having established that t-CSM can interfere with the Ubc9-CRMP2 interaction, we next sought to investigate whether cellular dialysis of t-CSM could prevent CRMP2 SUMOylation. Since there is no known antibody that selectively identifies SUMOylated CRMP2, we performed the following experiment. We prepared lysates from CAD cells in a denaturing lysis buffer, then heat denatured them to completely unfold and disrupt non covalent protein complexes and to inactivate SUMO isopeptidases [88]; the mouse CAD (CNS catecholaminergic cells) neuronal cell line were chosen because they expresses high levels of NaV1.7 [81]. An immunoprecipitation was then performed with a SUMO1 antibody followed by immunoblotting with a CRMP2 antibody to detect SUMOylated CRMP2. As shown in Fig. 6A–B, expression of a SUMO-null (K374A) CRMP2 or dialysis of the t-CSM “decoy” peptide decreased the amount of detectable SUMOylated CRMP2. These results demonstrate that endogenous CRMP2 SUMOylation is inhibited by the t-CSM peptide.
Figure 6. t-CSM blocks SUMOylation of CRMP2 in CAD cells.
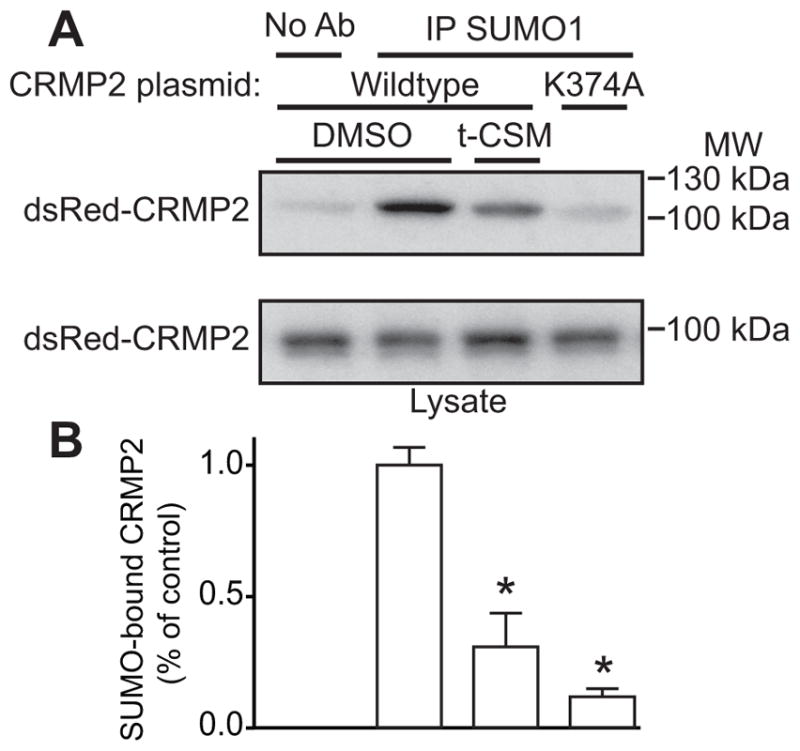
(A) Immunoprecipitates (IP) enriched with a SUMO1 antibody from CRMP2- (treated with 0.1% DMSO or 20 μM t-CSM peptide overnight) and CRMP2-K374A- (negative control) expressing catecholamine A differentiated (CAD) cells probed with an anti-CRMP2 polyclonal antibody. The leftmost lane (top panel) shows a negative control where anti-SUMO1 antibody was omitted. Lysates of cells are shown (bottom panel) to demonstrate equal amounts of total CRMP2. Representative Western blots show a significant reduction of CRMP2 SUMOylation with t-CSM peptide or in cells expressing a SUMO-incompetent CRMP2. (B) Bar graph showing the quantitative analysis of relative SUMOylated CRMP2 levels for the indicated conditions. Mean ± SEM, * p < 0.05 compared to SUMOylated CRMP2 level in DMSO treated cells, one-way ANOVA with Tukey’s post-hoc test; n=4.
3.5. t-CSM peptide decreases Na+ currents in CAD cells
We previously reported that CRMP2-K374A, a non-SUMOylated mutant of CRMP2, impairs sodium current density in CAD cells [19; 21]. If preventing CRMP2 SUMOylation were achieved by cellular dialysis of t-CSM, then we would expect NaV1.7 current density to be diminished consistent with our published data. Current-voltage relationships in transfected CAD cells were recorded by the application of 5-ms step depolarizations ranging from −70 mV to +60 mV (in +5 mV increments) from a holding potential of −80 mV (Fig. 7A). The transient inward current in CAD cells activated between −40 and −30 mV and reached its peak at 0 to +10 mV (Fig. 7B). Peak inward Na+ currents were measured and expressed as normalized peak current density to account for variations in cell size. Sodium currents were reduced to 42.5 ± 5.9% (n=6) in CAD cells treated with 20 μM of t-CSM compared to those treated with DMSO (0.1%) (Fig. 7C). There was no difference in Boltzmann parameters of voltage dependence of activation, half-activation voltage (V½), and slope factor (k) between the two treatment conditions (Fig. 7D).
Figure 7. t-CSM decreases Na+ current density in CAD cells.
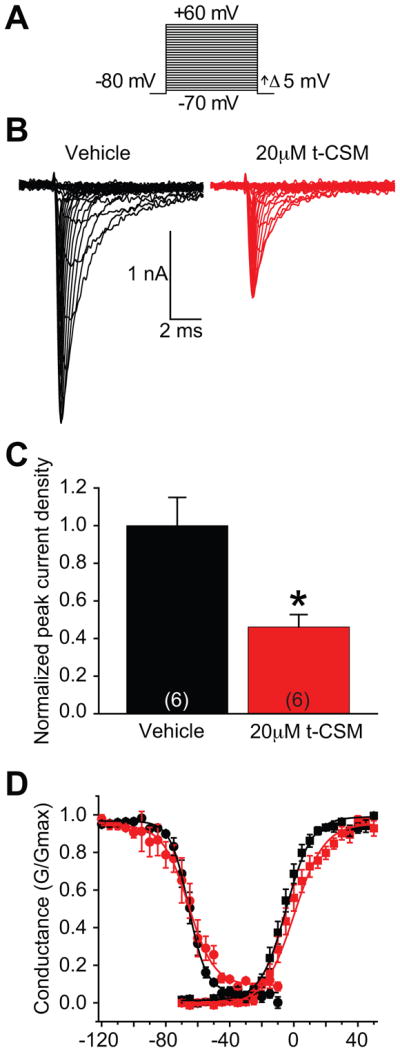
(A) Voltage protocol for current-voltage relationship: Na+ currents were evoked by 5-ms voltage steps from −70 to +60 mV in 5-mV increments from a holding potential of −80 mV. (B) Na+ current traces were measured from vehicle treated (black) or 20 μM t-CSM treated (red) neurons. (C) Summary current density in the absence or presence of t-CSM (normalized to 34.2 ± 4.7 pF and 28.6 ± 3.3 pF, respectively). Incubation of CAD cells with t-CSM produced a marked decreased in current density. (*p < 0.05, Student’s t-test, n = 6 cells). (D) Representative Boltzmann fits for activation and steady-state inactivation for NaV1.7. No significant differences were detected in half-maximal voltage or slope properties of fast inactivation or activation. Data show means ±SEM (n=6).
3.6. t-CSM attenuates ouabain-induced increase in cytosolic Na+ in primary sensory neurons
The above data showing t-CSM peptide inhibits NaV1.7 currents (Fig. 8) is consistent with our published results demonstrating diminution of NaV1.7 currents in cells lines and in sensory neurons expressing the SUMO-null CRMP2-K374A mutant [19–21]. Notwithstanding that conventional patch clamp remains the sole technique with sufficiently high time resolution and sensitivity required for precise and direct characterization of ion channel properties, it is universally accepted that patch clamp is a slow, labor-intensive, and thus expensive, technique. In anticipation of a future high throughput screening campaign for SUMOylation inhibitors of Ubc9 that we will likely identify in the ALPHA screen, we developed a low-to-medium throughput sodium fluorescence assay. Changes in cytolosolic Na+ ([Na+]c) were monitored using the Na+-sensitive fluorescent dye, SBFI-AM. Ouabain, an inhibitor of Na+/K+-ATPase, was used to increase [Na+]c, which was monitored using SBFI. Ouabain resulted in an increase in [Na+]c (Fig. 8A). Pre-treatment of neurons with t-CSM (10 μM for 12 hours) strongly attenuated ouabain-induced increase in [Na+]c (Fig. 8B). The ouabain-induced increase in [Na+]c was also inhibited by tetrodotoxin (TTX, 1 μM), a pan-blocker of NaV1.1 to NaV1.7 channels [44], and Protox (10 μM), a specific blocker of NaV1.7 [74], suggesting that this increase in [Na+]c is mediated by voltage-gated Na+ channels and, specifically, by NaV1.7 (Fig. 8C, D). Ouabain-induced changes in [Na+]c over time were quantified by calculating the area under the curve (AUC) (Fig. 8E). Taken together, the data obtained with Protox and inhibition of ouabain-induced increase in [Na+]c by t-CSM suggest that this peptide reduces NaV1.7 function as well.
Figure 8. Peptide t-CSM, tetrodotoxin (TTX), and Protox attenuate ouabain-induced increase in cytosolic Na+ ([Na+]c) in primary sensory neurons.

Neurons loaded with SBFI-AM, a Na+ sensitive fluorescent dye, were treated with 1 mM ouabain alone (A) or in combination with 1μM TTX (C) or 10μM Protox (D), as indicated. (B) Neurons were pre-incubated with 10μM t-CSM for 12 hours, and then loaded with SBFI-AM and taken into the experiment. t-CSM (10μM) was present in the medium during SBFI loading and then during the experiment. (E) Statistical analysis of ouabain-induced changes in [Na+]c. The ouabain-induced changes in [Na+]c over time were quantified by calculating the area under the curve (AUC). The AUC was calculated for 1200 seconds following application of ouabain and other drugs. Data are means ± SEM, *p<0.05, **p<0.01. n=5 independent experiments from the indicated number of total cells.
3.7. t-CSM peptide impedes membrane trafficking of NaV1.7 in DRGs
Next, we used immunofluorescent microscopy to determine if t-CSM affected the subcellular localization of NaV1.7. Sensory neurons were incubated with 20 μM t-CSM peptide and examined after live labeling of NaV1.7 with an antibody targeting an extracellular epitope of NaV1.7. Visual inspection of the micrographs (Fig. 9A) demonstrated that, in control cells treated with vehicle, the fluorescent signal for extracellular NaV1.7 appeared in the vicinity of the plasma membrane of the DRG neurons. As illustrated in Fig. 9A, prominent membranous NaV1.7 staining was observed in the absence of peptide (top panel) but significantly reduced in the presence of t-CSM (bottom panel). Quantification from 30–45 cells revealed an ~80% reduction in surface NaV1.7 (Fig. 9B). Taken together, these results suggest that, t-CSM related inhibition of CRMP2 SUMOylation prevents NaV1.7 from being targeted to the DRG neuronal membrane.
Figure 9. t-CSM restricts NaV1.7 membrane localization in DRG sensory neurons.

After an overnight treatment with either 0.1% DMSO or 20 μM t-CSM, Nav1.7 membrane localization was detected in sensory neurons. (A) Representative fluorescent micrographs of DRG neurons immunostained with an antibody against an extracellular epitope for NaV1.7. Fluorescent signal can be detected when NaV1.7 was localized at the plasma membrane in live DRG neurons. (B) Bar graph showing the relative surface NaV1.7 signal compared to the levels in the 0.1% DMSO-treated cells. Fluorescent signal from (A) was quantified and normalized to the area of the cell of interest. Treating the cells with 20 μM of t-CSM resulted in decreased membrane localization of NaV1.7. Mean ± SEM, *p<0.05, Student’s t-test (n=30 to 45 cells).
3.8. t-CSM peptide reverses neuropathic pain
Nav1.7 is upregulated and promotes ongoing pain in animal models of chronic peripheral inflammation and in peripheral nerve injury [43; 77]. Recently, we reported that (i) NaV1.7 upregulation in chronic neuropathic pain is accompanied by a concomitant increase in CRMP2 SUMOylation, and (ii) loss of CRMP2 SUMOylation, mimicked by expressing a non-SUMOylated mutant of CRMP2 (i.e., CRMP2 K374A) in vivo, reversed SNI-induced mechanical allodynia [64]. Because increased NaV1.7 following nerve injury influences the hyperexcitability of sensory neurons, which leads to neuropathic pain, here we tested whether the “decoy peptide” based on the CRMP2 SUMOylation motif would reverse SNI-induced mechanical allodynia. The spared nerve injury model resulted in significant mechanical and thermal hypersensitivity in both male and female rats (Fig. 10, post-surgery group) on day 7 after injury. Spinal delivery of t-CSM (10μg/5μL) resulted in a significant long-lasting reversal of both thermal and mechanical hypersensitivity (Fig. 10A, B) that returned to the injury baseline by 24 hrs. These results provide proof-of-concept for targeting Ubc9-CRMP2 interaction to curb NaV1.7 activity and control nociceptive behaviors.
Figure 10. t-CSM peptide reverses spared nerve injury (SNI) chronic pain.

Rats developed mechanical allodynia (A) and thermal hyperalgesia (B) 7 days after SNI but not with after a sham injury. Intrathecal injection (black arrow) of t-CSM (10μg/5μl), but not vehicle (0.01% DMSO), significantly reversed the mechanical allodynia, measured by paw withdrawal threshold (in grams), and thermal hyperalgesia, as measure by paw withdrawal latency (in sec), for at least 6 hours after injection (n=6; *p< 0.05 vs. pre-injection baseline, 2-way ANOVA, post hoc Student-Neuman-Kuels). Brown and green symbols represent female SNI rats (n=6 each) treated with vehicle or t-CSM, respectively.
3.9.t-CSM does not affect visceral pain
Although inhibition of tetrodotoxin-sensitive channels was reported to block inflammatory and visceral pain [31; 52], pharmacological block of NaV1.7 alone in the viscera may be insufficient in targeting chronic visceral pain [36]. Therefore, we tested the potential efficacy of t-CSM in the acetic acid-writhing test wherein NaV1.7 is predicted not to contribute. Intraperitoneal injection of acetic acid (1%) in female C57BL/6NHsd mice induced characteristic writhing behaviors typified by contractions of the abdomen, twisting and turning of the trunk, arching of the back and extension of the hind limbs (Fig. 11). In mice injected with t-CSM (i.t., 4μg/5μl), 1 hour prior to the acetic acid injection, the number of observed writhing events was 35 ± 7 (n=7), which was not different from mice pre-treated spinally with 0.1% DMSO (34 ±4 (n=8) (Fig. 11). Thus, t-CSM does not contribute to suppression of visceral pain.
Figure 11. t-CSM peptide does not affect visceral pain.

Writhing characterized by abdominal stretching combined with a exaggerated extension of the hind limbs was induced by intraperitoneal injection of acetic acid in female mice. t-CSM peptide was administered i.t. 60 min prior the acetic acid injection. The scatter plot and bar graph represents the mean ± SEM of number of writhes obtained in 7–8 mice. No significant differences were detected between the conditions (Wilcoxon–Mann–Whitney nonparametric test).
3.10. t-CSM peptide does not induce typical motor deficit or sedation in rats
Because the current clinically used medications such as gabapentin result in severe sedation in humans, we determined whether our novel t-CSM peptide would result in similar sedative or motor impairment (ruling out false positives in pain behavioral tests based on motor reflex is of major importance). Naïve male SD rats were used to determine sedation/motor impairment so that results are not influenced by “states of injury”. Rats were trained to walk on a rotating rod with a maximal cutoff time of 180 seconds prior to administration of peptide or vehicle. To ensure availability of the peptide in the spinal cord, we administered the drug intrathecally in these experiments. Spinal delivery of t-CSM (20 μg/5 μL i.th.) resulted in no motor impairment or sedation (Fig. 12A) since all animals remained on the rotating rod until cut off (180 s) over a 24 hours period. In contrast, gabapentin, a drug known to produce severe sedation in humans, resulted in a decrease in the ability of naïve rats to remain on the rotating rod (data not shown). Motor performance was not affected by t-CSM administration in rats with SNI (Fig. 12B). Thus, using t-CSM in vivo is a safe strategy to target voltage-gated sodium channel NaV1.7 and reverse chronic pain.
Figure 12. The intrathecal administration of t-CSM did not result in significant deficit in motor coordination as tested using the rotarod performance test.

t-CSM (20 μg/5 μL, i.th.) was evaluated for motor deficits using the rotarod performance test using naïve (A) or SNI (B) rats. Vehicle treated animals and t-CSM treated animals remained on the rotarod for an average of 180 ± 0 seconds over a 24 hours period (p>0.01, two-way ANOVA, n=6–8). t-CSM did not affect motor performance at any time point measured in the SNI rats (p>0.01, two-way ANOVA, n=6–8).
4. Discussion
We examined CRMP2-dependent signaling mechanisms regulating NaV1.7 and peripheral pain transmission. We identified a novel interaction between the SUMO conjugating E2 enzyme Ubc9 and CRMP2 and used rational design to fashion a CRMP2 SUMOylation Motif (CSM) “decoy” peptide. We provide in vitro and in vivo evidence that interference of the Ubc9-CRMP2 interaction with this peptide reduces CRMP2 SUMOylation, leading to reduced NaV1.7 currents, and mitigation of nociception in neuropathic, but not visceral, pain.
During the last decade, NaV1.7 emerged as a high-value analgesic target. Mutations in SCN9A, the gene encoding NaV1.7, are associated with both lowered as well as heightened sensitivities to pain [2; 18; 38; 82]. Consequently, selectively inhibiting NaV1.7 is likely to be critical in modulating pain via pathways involving this channel. NaV1.7-selective compounds [13; 49; 85], peptide toxins [90] and neutralizing antibodies [45] cause only temporary partial analgesia, and some NaV1.7 gain-of-function mutation patients respond to pan-sodium channel blockers [12; 25]. The Waxman group has demonstrated a precision medicine approach, using genomics and molecular modeling, to treat pain in a handful of humans with gain-of-function mutations in NaV1.7 [7; 29]. Compounds developed by Pfizer (PF-0485624 [56]) and Genentech (GX-674, which demonstrated equipotent inhibition of NaV1.7 and NaV1.2, and only 4-fold less potency against NaV1.6 [1]) have not progressed to clinic. Thus, despite significant investment into NaV1.7 drug development programs, there has been only marginal success. We demonstrated that elimination of the CRMP2 SUMO modification site (i.e., K374) was sufficient to selectively decrease trafficking and therefore current density of NaV1.7, but not NaV1.1, 1.3, 1.5, 1.6, 1.8 or 1.9 [10; 19; 21]. We reported a 1.78Å resolution X-ray crystal structure of mouse CRMP2, which corroborated these findings [20]. The Wood group, using an epitope-tagged NaV1.7 mouse identified CRMP2 as part of the NaV1.7 complex in native conditions [41], independently confirming our findings. Congruent with these studies, here we identify a novel peptide, t-CSM, which recapitulates the reduction of sodium influx via NaV1.7. The heptamer peptide t-CSM bound to Ubc9 with nanomolar affinity is sufficient for in vitro SUMO-1 modification by Ubc9. This finding is in line with previous publications demonstrating that heptamer peptides, containing the SUMO-1 modification consensus sequence ψKXE, are sufficient for in vitro SUMO-1 modification catalyzed by Ubc9 [48; 73]. Thus, these data confirm the importance of CRMP2 SUMOylation as a regulator of NaV1.7 activity and advance targeting of the Ubc9-CRMP2 interaction as a novel means to achieve regulation of NaV1.7, setting the stage for future drug discovery campaigns to find selective CRMP2 SUMOylation inhibitors.
From a mechanistic viewpoint, targeting NaV1.7-mediated pain behavior takes advantage of robust channel expression on nociceptive afferents [17]. This allows reduction of NaV1.7 trafficking to selectively reduce the activity of ascending pain pathway neurons that promote perception of pain. In order to aid successful implementation of this strategy into clinical practice, future studies will explore long-term effects of antagonizing NaV1.7 trafficking on both ascending and descending pain pathways. Endogenous opioids function as effectors of descending pain pathways [80] and a recent study has linked knockout of DRG NaV1.7 expression and reduction of intracellular DRG sodium concentration to increased endogenous opioid mRNA expression [59]. The study utilized opioid receptor antagonists to conclude that endogenous opioid production contributes to NaV1.7 knockout-mediated congenital insensitivity to pain. Nav-endogenous opioid signaling pathways have not been completely elucidated but may permit NaV1.7 trafficking antagonists to engage a secondary response via endogenous opioid production. This arrangement could enhance the analgesic function of CRMP2 SUMOylation-targeting therapies.
Despite the importance of SUMOylation in regulating diverse biological systems and diseases, only a few small-molecule inhibitors of SUMOylation have been reported. These include mostly natural products such as ginkgolic acid [27], kerriamycin B [28], spectomycin B1, chaetochromin A, and viomellein [35]; however, none of these are commercially available. With respect to diseases, Ubc9 blockers have been utilized in cancer therapy because Ubc9 is overexpressed in several cancers [47; 60; 61] and many tumorigenesis associated proteins are reported to be SUMOylated [62]. As an example, human genome studies provide striking evidence for a link between the impaired SUMOylation of a single transcription factor and a predisposition to cancer [3; 92]. All hitherto reported strategies are therapeutically impractical because targeting the SUMOylation machinery – either E1 or E2 – would generate non-selective antagonists of essential biological processes. Our study is the first to design a SUMOylation inhibitor (i.e., the t-CSM peptide) against a singular protein-protein interaction (i.e., Ubc9-CRMP2) that is expected to allosterically modulate NaV1.7 activity. Because the modification of CRMP2 by SUMOylation to selectively regulate NaV1.7 membrane expression is conserved in human sensory neurons [19], finding a selective CRMP2 SUMOylation inhibitor would have a high likelihood of clinical translation.
Importantly, our approach does not target Ubc9, which is the necessary E2 conjugating enzyme for SUMOylation of several protein targets. Ubc9 knockout mice are embryonic lethal [68], and global decreases in SUMOylation are implicated in cognitive dysfunction [46]. Thus, in an effort to find a selective “druggable” pocket in CRMP2 close to the SUMOylation site on residue K374, we generated an in-silico model of CRMP2 with Ubc9-SUMO (Fig. 13A). The crystal structure of the mammalian guanosine triphosphate (GTP)ase-activating protein (RanGAP1), one of the first proteins reported to be covalently linked to SUMO [50], with Ubc9–SUMO2 (Protein Data Bank (PDB) ID: 5d2m [8]) was used as the starting model in which RanGAP1 was replaced with CRMP2 (PDB ID: 2gse [76]). Residue K374 was then used as an anchor to refine the complex using molecular minimization with the Schrodinger suite and is in proximity to Ubc9 active site C93 [48] (Fig. 13B, C). The identified potential ‘hot spot’ CRMP2 residues include: (i) K23; (ii) W366; (iii) V371 of CRMP2, which forms a carbonyl-backbone amide H-bond with A131 of Ubc9; (iv) M375; (v) E377; (vi) Q379; and (vii) R440 of CRMP2, which forms a salt bridge with E132 of Ubc9 (Fig. 13C). It has been shown that single mutations in A131 or E132 significantly affect Ubc9’s interaction with target proteins – the tumor suppressor protein p53, the ubiquitin-activating enzyme E1B, and promyelocytic leukemia protein – and reduce their SUMOylation [48]. Thus, targeting this CRMP2 pocket using in silico docking may prove useful in discovering small molecules disrupting CRMP2-Ubc9 interaction in the context of pain relief.
Figure 13. Mapping the interface between CRMP2 and Ubc9.

(A) Ribbon diagram of in silico model of CRMP2 (PDB ID: 2gse [76]) in gray with Ubc9 in white and SUMO-2 in black (PDB ID: 5d2m [8]). (B) View of CRMP2 K374 near the active site cysteine (C93) of Ubc9. (C) Hotspot residues in CRMP2 (carbon atoms in magenta) and Ubc9 (carbon atoms in teal). Dashed line indicates potential salt-bridge interaction between R440 and E132.
That CRMP2 is implicated in neuropathic pain can be surmised from recent work which demonstrates that there is a substantial increase in the amount of SUMOylated CRMP2 in sensory neurons and spinal cord dorsal horn in rodent models of chronic pain [10; 57; 64; 78]. This finding is congruent with transcriptomic data demonstrating an enrichment of CRMP2 in nociceptors [79], further underscoring the importance of targeting CRMP2 under these conditions. Validating the importance of SUMOylation of CRMP2 as a pathologic event in chronic pain, we previously observed that transient expression of a SUMO-null CRMP2, via spinal administration of plasmid encoding CRMP2 K374A, significantly attenuated pain behavior in SNI animals [64]. Here, we extend these findings by using a structurally guided, rationally designed CRMP2 SUMO “decoy” peptide that recapitulates observations made with the genetic manipulation with the reversal of thermal and tactile hypersensitivity when administered to animals with SNI. Notably, the peptide failed to reverse visceral pain, findings consonant with the reported lack of NaV1.7-dependency in this acute inflammatory model [36; 52]. These data consolidate the hypothesis that targeting CRMP2 SUMOylation to allosterically regulate NaV1.7, and by extension, NaV1.7-dependent acute, inflammatory, and neuropathic pain is a viable alternative strategy. A potential limitation of our study is that not all pain is NaV1.7 dependent; bone cancer pain and chemotherapy (i.e., oxaliplatin-evoked) mechanical and cold allodynia occur normally in NaV1.7 null mutant mice [58].
Not with standing these limitations, our work suggests that targeting CRMP2 modifications can be used to control ion channels implicated in neuropathic pain. Focusing on another CRMP2 modification, phosphorylation by cyclin dependent kinase 5 (Cdk5), we demonstrated that inhibiting phosphorylation by Cdk5 on residue S522 with (S)-lacosamide, an inactive stereoisomer of the clinically used antiepileptic drug (R)-lacosamide (Vimpat®), translated into a decrease of calcium influx via presynaptic N-type voltage-gated calcium channels [66]. This inhibition resulted in mitigation of thermal hypersensitivity and mechanical allodynia in model of postoperative, neuropathic, pain, and cephalic pain [63; 65]. (S)-lacosamide was also reported to affect NaV1.7-mediated sodium currents in a CRMP2 dependent manner, since CRMP2 SUMOylation is dependent on its prior phosphorylation by Cdk5 [67]. CRMP2 phosphorylation by Cdk5 at residue S522 plays other roles such as that in dendritic development [69]. Consequently, the strategy proposed here of disrupting CRMP2 SUMOylation, an event downstream of phosphorylation, may represent a more incisive approach for “fine-tuning” CRMP2 modifications to control ion channels implicated in pain.
The demonstration of a CRMP2 SUMOylation motif “decoy” peptide in reducing NaV1.7-mediated sodium influx and reversing SNI-related pain behaviors sets, combined with the structural modeling based identification of a pocket harboring CRMP2’s SUMOylation motif sets the stage for experimental (ALPHA, MST) and computational (in silico docking) screening of ligands/molecules that will biochemically and functionally target CRMP2’s SUMOylation to reduce NaV1.7 currents and reverse neuropathic pain.
Acknowledgments
This work was supported by National Institutes of Health awards (R01NS098772 from the National Institute of Neurological Disorders and Stroke to N.B. and R.K. and R01DA042852 from the National Institute on Drug Abuse to R.K.); and a Neurofibromatosis New Investigator Award from the Department of Defense Congressionally Directed Military Medical Research and Development Program (NF1000099); and a Children’s Tumor Foundation NF1 Synodos award (2015-04-009A) to R.K. A.M. was supported by a Young Investigator’s Award from the Children’s Tumor Foundation (2015-01-011). We thank Cynthia Madura for assistance with the writhing behavior experiments.
Footnotes
Conflict of interest – There is no conflict of interest for any of the authors.
References
- 1.Ahuja S, Mukund S, Deng L, Khakh K, Chang E, Ho H, Shriver S, Young C, Lin S, Johnson JP, Jr, Wu P, Li J, Coons M, Tam C, Brillantes B, Sampang H, Mortara K, Bowman KK, Clark KR, Estevez A, Xie Z, Verschoof H, Grimwood M, Dehnhardt C, Andrez JC, Focken T, Sutherlin DP, Safina BS, Starovasnik MA, Ortwine DF, Franke Y, Cohen CJ, Hackos DH, Koth CM, Payandeh J. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science. 2015;350(6267):aac5464. doi: 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- 2.Bennett DL, Woods CG. Painful and painless channelopathies. Lancet neurology. 2014;13(6):587–599. doi: 10.1016/S1474-4422(14)70024-9. [DOI] [PubMed] [Google Scholar]
- 3.Bertolotto C, Lesueur F, Giuliano S, Strub T, de Lichy M, Bille K, Dessen P, d’Hayer B, Mohamdi H, Remenieras A, Maubec E, de la Fouchardiere A, Molinie V, Vabres P, Dalle S, Poulalhon N, Martin-Denavit T, Thomas L, Andry-Benzaquen P, Dupin N, Boitier F, Rossi A, Perrot JL, Labeille B, Robert C, Escudier B, Caron O, Brugieres L, Saule S, Gardie B, Gad S, Richard S, Couturier J, Teh BT, Ghiorzo P, Pastorino L, Puig S, Badenas C, Olsson H, Ingvar C, Rouleau E, Lidereau R, Bahadoran P, Vielh P, Corda E, Blanche H, Zelenika D, Galan P, Aubin F, Bachollet B, Becuwe C, Berthet P, Bignon YJ, Bonadona V, Bonafe JL, Bonnet-Dupeyron MN, Cambazard F, Chevrant-Breton J, Coupier I, Dalac S, Demange L, d’Incan M, Dugast C, Faivre L, Vincent-Fetita L, Gauthier-Villars M, Gilbert B, Grange F, Grob JJ, Humbert P, Janin N, Joly P, Kerob D, Lasset C, Leroux D, Levang J, Limacher JM, Livideanu C, Longy M, Lortholary A, Stoppa-Lyonnet D, Mansard S, Mansuy L, Marrou K, Mateus C, Maugard C, Meyer N, Nogues C, Souteyrand P, Venat-Bouvet L, Zattara H, Chaudru V, Lenoir GM, Lathrop M, Davidson I, Avril MF, Demenais F, Ballotti R, Bressac-de Paillerets B French Familial Melanoma Study G. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011;480(7375):94–98. doi: 10.1038/nature10539. [DOI] [PubMed] [Google Scholar]
- 4.Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, Liu N, Xiong W, Ripsch MS, Wang Y, Fehrenbacher JC, Fitz SD, Khanna M, Park CK, Schmutzler BS, Cheon BM, Due MR, Brustovetsky T, Ashpole NM, Hudmon A, Meroueh SO, Hingtgen CM, Brustovetsky N, Ji RR, Hurley JH, Jin X, Shekhar A, Xu XM, Oxford GS, Vasko MR, White FA, Khanna R. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca(2)(+) channel complex. Nature medicine. 2011;17(7):822–829. doi: 10.1038/nm.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. The Journal of biological chemistry. 2009;284(45):31375–31390. doi: 10.1074/jbc.M109.009951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brittain MK, Brustovetsky T, Sheets PL, Brittain JM, Khanna R, Cummins TR, Brustovetsky N. Delayed calcium dysregulation in neurons requires both the NMDA receptor and the reverse Na+/Ca2+ exchanger. Neurobiology of disease. 2012;46(1):109–117. doi: 10.1016/j.nbd.2011.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao L, McDonnell A, Nitzsche A, Alexandrou A, Saintot PP, Loucif AJ, Brown AR, Young G, Mis M, Randall A, Waxman SG, Stanley P, Kirby S, Tarabar S, Gutteridge A, Butt R, McKernan RM, Whiting P, Ali Z, Bilsland J, Stevens EB. Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Science translational medicine. 2016;8(335):335ra356. doi: 10.1126/scitranslmed.aad7653. [DOI] [PubMed] [Google Scholar]
- 8.Cappadocia L, Pichler A, Lima CD. Structural basis for catalytic activation by the human ZNF451 SUMO E3 ligase. Nature structural & molecular biology. 2015;22(12):968–975. doi: 10.1038/nsmb.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. Journal of neuroscience methods. 1994;53(1):55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 10.Chew LA, Khanna R. CRMP2 and voltage-gated ion channels: potential roles in neuropathic pain. Neuronal Signaling. 2018;2:16. doi: 10.1042/NS20170220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chi XX, Schmutzler BS, Brittain JM, Wang Y, Hingtgen CM, Nicol GD, Khanna R. Regulation of N-type voltage-gated calcium channels (Cav2. 2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J Cell Sci. 2009;122(Pt 23):4351–4362. doi: 10.1242/jcs.053280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi JS, Zhang L, Dib-Hajj SD, Han C, Tyrrell L, Lin Z, Wang X, Yang Y, Waxman SG. Mexiletine-responsive erythromelalgia due to a new Na(v)1. 7 mutation showing use-dependent current fall-off. ExpNeurol. 2009;216(2):383–389. doi: 10.1016/j.expneurol.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 13.Chowdhury S, Chafeev M, Liu S, Sun J, Raina V, Chui R, Young W, Kwan R, Fu J, Cadieux JA. Discovery of XEN907, a spirooxindole blocker of NaV1. 7 for the treatment of pain. Bioorganic & medicinal chemistry letters. 2011;21(12):3676–3681. doi: 10.1016/j.bmcl.2011.04.088. [DOI] [PubMed] [Google Scholar]
- 14.Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1. 7 sodium channels in a painful inherited neuropathy. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24(38):8232–8236. doi: 10.1523/JNEUROSCI.2695-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87(2):149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 16.Diarra A, Sheldon C, Church J. In situ calibration and [H+] sensitivity of the fluorescent Na+ indicator SBFI. American journal of physiology Cell physiology. 2001;280(6):C1623–1633. doi: 10.1152/ajpcell.2001.280.6.C1623. [DOI] [PubMed] [Google Scholar]
- 17.Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The Na(V)1. 7 sodium channel: from molecule to man. Nature reviews Neuroscience. 2013;14(1):49–62. doi: 10.1038/nrn3404. [DOI] [PubMed] [Google Scholar]
- 18.Dib-Hajj SD, Yang Y, Waxman SG. Genetics and molecular pathophysiology of Na(v)1. 7-related pain syndromes. AdvGenet. 2008;63:85–110. doi: 10.1016/S0065-2660(08)01004-3. [DOI] [PubMed] [Google Scholar]
- 19.Dustrude ET, Moutal A, Yang X, Wang Y, Khanna M, Khanna R. Hierarchical CRMP2 posttranslational modifications control NaV1. 7 function. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(52):E8443–E8452. doi: 10.1073/pnas.1610531113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dustrude ET, Perez-Miller S, Francois-Moutal L, Moutal A, Khanna M, Khanna R. A single structurally conserved SUMOylation site in CRMP2 controls NaV1. 7 function. Channels. 2017;11(4):316–328. doi: 10.1080/19336950.2017.1299838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dustrude ET, Wilson SM, Ju W, Xiao Y, Khanna R. CRMP2 protein SUMOylation modulates NaV1. 7 channel trafficking. The Journal of biological chemistry. 2013;288(34):24316–24331. doi: 10.1074/jbc.M113.474924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eglen RM, Reisine T, Roby P, Rouleau N, Illy C, Bosse R, Bielefeld M. The use of AlphaScreen technology in HTS: current status. Current chemical genomics. 2008;1:2–10. doi: 10.2174/1875397300801010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emery EC, Luiz AP, Wood JN. Nav1. 7 and other voltage-gated sodium channels as drug targets for pain relief. Expert opinion on therapeutic targets. 2016;20(8):975–983. doi: 10.1517/14728222.2016.1162295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52(5):767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Fischer TZ, Gilmore ES, Estacion M, Eastman E, Taylor S, Melanson M, Dib-Hajj SD, Waxman SG. A novel Nav1. 7 mutation producing carbamazepine-responsive erythromelalgia. AnnNeurol. 2009;65(6):733–741. doi: 10.1002/ana.21678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Francois-Moutal L, Wang Y, Moutal A, Cottier KE, Melemedjian OK, Yang X, Wang Y, Ju W, Largent-Milnes TM, Khanna M, Vanderah TW, Khanna R. A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2. 2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain. 2015;156(7):1247–1264. doi: 10.1097/j.pain.0000000000000147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fukuda I, Ito A, Hirai G, Nishimura S, Kawasaki H, Saitoh H, Kimura K, Sodeoka M, Yoshida M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chemistry & biology. 2009;16(2):133–140. doi: 10.1016/j.chembiol.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Fukuda I, Ito A, Uramoto M, Saitoh H, Kawasaki H, Osada H, Yoshida M. Kerriamycin B inhibits protein SUMOylation. The Journal of antibiotics. 2009;62(4):221–224. doi: 10.1038/ja.2009.10. [DOI] [PubMed] [Google Scholar]
- 29.Geha P, Yang Y, Estacion M, Schulman BR, Tokuno H, Apkarian AV, Dib-Hajj SD, Waxman SG. Pharmacotherapy for Pain in a Family With Inherited Erythromelalgia Guided by Genomic Analysis and Functional Profiling. JAMA Neurol. 2016;73(6):659–667. doi: 10.1001/jamaneurol.2016.0389. [DOI] [PubMed] [Google Scholar]
- 30.Golebiowski F, Szulc A, Sakowicz M, Szutowicz A, Pawelczyk T. Expression level of Ubc9 protein in rat tissues. Acta biochimica Polonica. 2003;50(4):1065–1073. [PubMed] [Google Scholar]
- 31.Gonzalez-Cano R, Tejada MA, Artacho-Cordon A, Nieto FR, Entrena JM, Wood JN, Cendan CM. Effects of Tetrodotoxin in Mouse Models of Visceral Pain. Mar Drugs. 2017;15(6) doi: 10.3390/md15060188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. The Journal of biological chemistry. 1985;260(6):3440–3450. [PubMed] [Google Scholar]
- 33.Hay RT. SUMO: a history of modification. Molecular cell. 2005;18(1):1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 34.Henley JM, Craig TJ, Wilkinson KA. Neuronal SUMOylation: mechanisms, physiology, and roles in neuronal dysfunction. Physiological reviews. 2014;94(4):1249–1285. doi: 10.1152/physrev.00008.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirohama M, Kumar A, Fukuda I, Matsuoka S, Igarashi Y, Saitoh H, Takagi M, Shin-Ya K, Honda K, Kondoh Y, Saito T, Nakao Y, Osada H, Zhang KY, Yoshida M, Ito A. Spectomycin B1 as a Novel SUMOylation Inhibitor That Directly Binds to SUMO E2. ACS chemical biology. 2013;8(12):2635–2642. doi: 10.1021/cb400630z. [DOI] [PubMed] [Google Scholar]
- 36.Hockley JR, Gonzalez-Cano R, McMurray S, Tejada-Giraldez MA, McGuire C, Torres A, Wilbrey AL, Cibert-Goton V, Nieto FR, Pitcher T, Knowles CH, Baeyens JM, Wood JN, Winchester WJ, Bulmer DC, Cendan CM, McMurray G. Visceral and somatic pain modalities reveal NaV 1. 7-independent visceral nociceptive pathways. The Journal of physiology. 2017;595(8):2661–2679. doi: 10.1113/JP272837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inagaki N, Chihara K, Arimura N, Menager C, Kawano Y, Matsuo N, Nishimura T, Amano M, Kaibuchi K. CRMP-2 induces axons in cultured hippocampal neurons. Nature neuroscience. 2001;4(8):781–782. doi: 10.1038/90476. [DOI] [PubMed] [Google Scholar]
- 38.Jarecki BW, Sheets PL, Jackson JO, Cummins TR. Paroxysmal extreme pain disorder mutations within the D3/S4–S5 linker of Nav1. 7 cause moderate destabilization of fast inactivation. JPhysiol. 2008;586(Pt 17):4137–4153. doi: 10.1113/jphysiol.2008.154906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S. Molecular interaction studies using microscale thermophoresis. Assay and drug development technologies. 2011;9(4):342–353. doi: 10.1089/adt.2011.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ju W, Li Q, Allette YM, Ripsch MS, White FA, Khanna R. Suppression of pain-related behavior in two distinct rodent models of peripheral neuropathy by a homopolyarginine-conjugated CRMP2 peptide. Journal of neurochemistry. 2012;124(6):869–879. doi: 10.1111/jnc.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanellopoulos AH, Koenig J, Huang H, Pyrski M, Millet Q, Lolignier S, Morohashi T, Gossage SJ, Jay M, Linley JE, Baskozos G, Kessler B, Cox JJ, Zufall F, Wood JN, Zhao J. Mapping protein interactions of sodium channel NaV1.7 using epitope-tagged gene targeted mice. 2017 doi: 10.15252/embj.201796692. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khanna R, Wilson SM, Brittain JM, Weimer J, Sultana R, Butterfield A, Hensley K. Opening Pandora’s jar: a primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future Neurol. 2012;7(6):749–771. doi: 10.2217/FNL.12.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laedermann CJ, Cachemaille M, Kirschmann G, Pertin M, Gosselin RD, Chang I, Albesa M, Towne C, Schneider BL, Kellenberger S, Abriel H, Decosterd I. Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. The Journal of clinical investigation. 2013;123(7):3002–3013. doi: 10.1172/JCI68996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee CH, Ruben PC. Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels. 2008;2(6):407–412. doi: 10.4161/chan.2.6.7429. [DOI] [PubMed] [Google Scholar]
- 45.Lee JH, Park CK, Chen G, Han Q, Xie RG, Liu T, Ji RR, Lee SY. A monoclonal antibody that targets a NaV1. 7 channel voltage sensor for pain and itch relief. Cell. 2014;157(6):1393–1404. doi: 10.1016/j.cell.2014.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Lee L, Dale E, Staniszewski A, Zhang H, Saeed F, Sakurai M, Fa M, Orozco I, Michelassi F, Akpan N, Lehrer H, Arancio O. Regulation of synaptic plasticity and cognition by SUMO in normal physiology and Alzheimer’s disease. Scientific reports. 2014;4:7190. doi: 10.1038/srep07190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H, Niu H, Peng Y, Wang J, He P. Ubc9 promotes invasion and metastasis of lung cancer cells. Oncology reports. 2013;29(4):1588–1594. doi: 10.3892/or.2013.2268. [DOI] [PubMed] [Google Scholar]
- 48.Lin D, Tatham MH, Yu B, Kim S, Hay RT, Chen Y. Identification of a substrate recognition site on Ubc9. The Journal of biological chemistry. 2002;277(24):21740–21748. doi: 10.1074/jbc.M108418200. [DOI] [PubMed] [Google Scholar]
- 49.London C, Hoyt SB, Parsons WH, Williams BS, Warren VA, Tschirret-Guth R, Smith MM, Priest BT, McGowan E, Martin WJ, Lyons KA, Li X, Karanam BV, Jochnowitz N, Garcia ML, Felix JP, Dean B, Abbadie C, Kaczorowski GJ, Duffy JL. Imidazopyridines: a novel class of hNav1. 7 channel blockers. Bioorganic & medicinal chemistry letters. 2008;18(5):1696–1701. doi: 10.1016/j.bmcl.2008.01.047. [DOI] [PubMed] [Google Scholar]
- 50.Mahajan R, Gerace L, Melchior F. Molecular characterization of the SUMO-1 modification of RanGAP1 and its role in nuclear envelope association. The Journal of cell biology. 1998;140(2):259–270. doi: 10.1083/jcb.140.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Majava V, Loytynoja N, Chen WQ, Lubec G, Kursula P. Crystal and solution structure, stability and post-translational modifications of collapsin response mediator protein 2. FEBS J. 2008;275(18):4583–4596. doi: 10.1111/j.1742-4658.2008.06601.x. [DOI] [PubMed] [Google Scholar]
- 52.Marcil J, Walczak JS, Guindon J, Ngoc AH, Lu S, Beaulieu P. Antinociceptive effects of tetrodotoxin (TTX) in rodents. British journal of anaesthesia. 2006;96(6):761–768. doi: 10.1093/bja/ael096. [DOI] [PubMed] [Google Scholar]
- 53.Martin S, Nishimune A, Mellor JR, Henley JM. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature. 2007;447(7142):321–325. doi: 10.1038/nature05736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin S, Wilkinson KA, Nishimune A, Henley JM. Emerging extranuclear roles of protein SUMOylation in neuronal function and dysfunction. Nature reviews Neuroscience. 2007;8(12):948–959. doi: 10.1038/nrn2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martinez V, Thakur S, Mogil JS, Tache Y, Mayer EA. Differential effects of chemical and mechanical colonic irritation on behavioral pain response to intraperitoneal acetic acid in mice. Pain. 1999;81(1–2):179–186. doi: 10.1016/s0304-3959(99)00008-1. [DOI] [PubMed] [Google Scholar]
- 56.McCormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, Krambis MJ, Antonio BM, Zellmer SG, Printzenhoff D, Padilla KM, Lin Z, Wagoner PK, Swain NA, Stupple PA, de Groot M, Butt RP, Castle NA. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(29):E2724–2732. doi: 10.1073/pnas.1220844110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melemedjian OK, Yassine HN, Shy A, Price TJ. Proteomic and functional annotation analysis of injured peripheral nerves reveals ApoE as a protein upregulated by injury that is modulated by metformin treatment. Molecular pain. 2013;9:14. doi: 10.1186/1744-8069-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Minett MS, Falk S, Santana-Varela S, Bogdanov YD, Nassar MA, Heegaard AM, Wood JN. Pain without nociceptors? Nav1. 7-independent pain mechanisms. Cell Rep. 2014;6(2):301–312. doi: 10.1016/j.celrep.2013.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minett MS, Pereira V, Sikandar S, Matsuyama A, Lolignier S, Kanellopoulos AH, Mancini F, Iannetti GD, Bogdanov YD, Santana-Varela S, Millet Q, Baskozos G, MacAllister R, Cox JJ, Zhao J, Wood JN. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1. 7. Nature communications. 2015;6:8967. doi: 10.1038/ncomms9967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mo YY, Yu Y, Theodosiou E, Ee PL, Beck WT. A role for Ubc9 in tumorigenesis. Oncogene. 2005;24(16):2677–2683. doi: 10.1038/sj.onc.1208210. [DOI] [PubMed] [Google Scholar]
- 61.Moschos SJ, Jukic DM, Athanassiou C, Bhargava R, Dacic S, Wang X, Kuan SF, Fayewicz SL, Galambos C, Acquafondata M, Dhir R, Becker D. Expression analysis of Ubc9, the single small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, in normal and malignant tissues. Human pathology. 2010;41(9):1286–1298. doi: 10.1016/j.humpath.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 62.Moschos SJ, Mo YY. Role of SUMO/Ubc9 in DNA damage repair and tumorigenesis. Journal of molecular histology. 2006;37(5–7):309–319. doi: 10.1007/s10735-006-9030-0. [DOI] [PubMed] [Google Scholar]
- 63.Moutal A, Chew LA, Yang X, Wang Y, Yeon SK, Telemi E, Meroueh S, Park KD, Shrinivasan R, Gilbraith KB, Qu C, Xie JY, Patwardhan A, Vanderah TW, Khanna M, Porreca F, Khanna R. (S)-lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. Pain. 2016;157(7):1448–1463. doi: 10.1097/j.pain.0000000000000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moutal A, Dustrude ET, Largent-Milnes TM, Vanderah TW, Khanna M, Khanna R. Blocking CRMP2 SUMOylation reverses neuropathic pain. Molecular psychiatry. 2017 doi: 10.1038/mp.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moutal A, Eyde N, Telemi E, Park KD, Xie JY, Dodick DW, Porreca F, Khanna R. Efficacy of (S)-Lacosamide in preclinical models of cephalic pain. Pain reports. 2016;1(1) doi: 10.1097/PR9.0000000000000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moutal A, Francois-Moutal L, Perez-Miller S, Cottier K, Chew LA, Yeon SK, Dai J, Park KD, Khanna M, Khanna R. (S)-Lacosamide Binding to Collapsin Response Mediator Protein 2 (CRMP2) Regulates CaV2. 2 Activity by Subverting Its Phosphorylation by Cdk5. Molecular neurobiology. 2016;53(3):1959–1976. doi: 10.1007/s12035-015-9141-2. [DOI] [PubMed] [Google Scholar]
- 67.Moutal A, Yang X, Li W, Gilbraith KB, Luo S, Cai S, Francois-Moutal L, Chew LA, Yeon SK, Bellampalli SS, Qu C, Xie JY, Ibrahim MM, Khanna M, Park KD, Porreca F, Khanna R. CRISPR/Cas9 editing of Nf1 gene identifies CRMP2 as a therapeutic target in neurofibromatosis type 1-related pain that is reversed by (S)-Lacosamide. Pain. 2017;158(12):2301–2319. doi: 10.1097/j.pain.0000000000001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nacerddine K, Lehembre F, Bhaumik M, Artus J, Cohen-Tannoudji M, Babinet C, Pandolfi PP, Dejean A. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Developmental cell. 2005;9(6):769–779. doi: 10.1016/j.devcel.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 69.Niisato E, Nagai J, Yamashita N, Nakamura F, Goshima Y, Ohshima T. Phosphorylation of CRMP2 is involved in proper bifurcation of the apical dendrite of hippocampal CA1 pyramidal neurons. Developmental neurobiology. 2013;73(2):142–151. doi: 10.1002/dneu.22048. [DOI] [PubMed] [Google Scholar]
- 70.Piekarz AD, Due MR, Khanna M, Wang B, Ripsch MS, Wang R, Meroueh SO, Vasko MR, White FA, Khanna R. CRMP-2 peptide mediated decrease of high and low voltage-activated calcium channels, attenuation of nociceptor excitability, and anti-nociception in a model of AIDS therapy-induced painful peripheral neuropathy. Molecular pain. 2012;8(1):54. doi: 10.1186/1744-8069-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Podolsky AT, Sandweiss A, Hu J, Bilsky EJ, Cain JP, Kumirov VK, Lee YS, Hruby VJ, Vardanyan RS, Vanderah TW. Novel fentanyl-based dual mu/delta-opioid agonists for the treatment of acute and chronic pain. Life sciences. 2013;93(25–26):1010–1016. doi: 10.1016/j.lfs.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SA. Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell. 2005;121(1):37–47. doi: 10.1016/j.cell.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 73.Rodriguez MS, Dargemont C, Hay RT. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. The Journal of biological chemistry. 2001;276(16):12654–12659. doi: 10.1074/jbc.M009476200. [DOI] [PubMed] [Google Scholar]
- 74.Schmalhofer WA, Calhoun J, Burrows R, Bailey T, Kohler MG, Weinglass AB, Kaczorowski GJ, Garcia ML, Koltzenburg M, Priest BT. ProTx-II, a selective inhibitor of NaV1. 7 sodium channels, blocks action potential propagation in nociceptors. Molecular pharmacology. 2008;74(5):1476–1484. doi: 10.1124/mol.108.047670. [DOI] [PubMed] [Google Scholar]
- 75.Seidel SA, Dijkman PM, Lea WA, van den Bogaart G, Jerabek-Willemsen M, Lazic A, Joseph JS, Srinivasan P, Baaske P, Simeonov A, Katritch I, Melo FA, Ladbury JE, Schreiber G, Watts A, Braun D, Duhr S. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods. 2013;59(3):301–315. doi: 10.1016/j.ymeth.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stenmark P, Ogg D, Flodin S, Flores A, Kotenyova T, Nyman T, Nordlund P, Kursula P. The structure of human collapsin response mediator protein 2, a regulator of axonal growth. JNeurochem. 2007;101(4):906–917. doi: 10.1111/j.1471-4159.2006.04401.x. [DOI] [PubMed] [Google Scholar]
- 77.Strickland IT, Martindale JC, Woodhams PL, Reeve AJ, Chessell IP, McQueen DS. Changes in the expression of NaV1.7, NaV1.8 and NaV1. 9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur J Pain. 2008;12(5):564–572. doi: 10.1016/j.ejpain.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 78.Sui P, Watanabe H, Ossipov MH, Bakalkin G, Artemenko KA, Bergquist J. Proteomics of Neuropathic Pain: Proteins and Signaling Pathways Affected in a Rat Model. Journal of proteome research. 2014 doi: 10.1021/pr500241q. [DOI] [PubMed] [Google Scholar]
- 79.Thakur M, Crow M, Richards N, Davey GI, Levine E, Kelleher JH, Agley CC, Denk F, Harridge SD, McMahon SB. Defining the nociceptor transcriptome. Frontiers in molecular neuroscience. 2014;7:87. doi: 10.3389/fnmol.2014.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vanegas H. To the descending pain-control system in rats, inflammation-induced primary and secondary hyperalgesia are two different things. Neuroscience letters. 2004;361(1–3):225–228. doi: 10.1016/j.neulet.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 81.Wang Y, Park KD, Salome C, Wilson SM, Stables JP, Liu R, Khanna R, Kohn H. Development and characterization of novel derivatives of the antiepileptic drug lacosamide that exhibit far greater enhancement in slow inactivation of voltage-gated sodium channels. ACS chemical neuroscience. 2011;2(2):90–106. doi: 10.1021/cn100089b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Waxman SG. Nav1. 7, its mutations, and the syndromes that they cause. Neurology. 2007;69(6):505–507. doi: 10.1212/01.wnl.0000268068.02343.37. [DOI] [PubMed] [Google Scholar]
- 83.Waxman SG. Painful Na-channelopathies: an expanding universe. Trends in molecular medicine. 2013;19(7):406–409. doi: 10.1016/j.molmed.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 84.Wilkinson KA, Henley JM. Mechanisms, regulation and consequences of protein SUMOylation. The Biochemical journal. 2010;428(2):133–145. doi: 10.1042/BJ20100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Williams BS, Felix JP, Priest BT, Brochu RM, Dai K, Hoyt SB, London C, Tang YS, Duffy JL, Parsons WH, Kaczorowski GJ, Garcia ML. Characterization of a new class of potent inhibitors of the voltage-gated sodium channel Nav1. 7. Biochemistry. 2007;46(50):14693–14703. doi: 10.1021/bi7018207. [DOI] [PubMed] [Google Scholar]
- 86.Wilson SM, Moutal A, Melemedjian OK, Wang Y, Ju W, Francois-Moutal L, Khanna M, Khanna R. The functionalized amino acid (S)-Lacosamide subverts CRMP2-mediated tubulin polymerization to prevent constitutive and activity-dependent increase in neurite outgrowth. Frontiers in Cellular Neuroscience. 2014:8. doi: 10.3389/fncel.2014.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wilson SM, Schmutzler BS, Brittain JM, Dustrude ET, Ripsch MS, Pellman JJ, Yeum TS, Hurley JH, Hingtgen CM, White FA, Khanna R. Inhibition of transmitter release and attenuation of anti-retroviral-associated and tibial nerve injury-related painful peripheral neuropathy by novel synthetic Ca2+ channel peptides. The Journal of biological chemistry. 2012;287(42):35065–35077. doi: 10.1074/jbc.M112.378695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xiao Y, Pollack D, Nieves E, Winchell A, Callaway M, Vigodner M. Can your protein be sumoylated? A quick summary and important tips to study SUMO-modified proteins. Analytical biochemistry. 2015;477:95–97. doi: 10.1016/j.ab.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiology & behavior. 1976;17(6):1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- 90.Yang S, Xiao Y, Kang D, Liu J, Li Y, Undheim EA, Klint JK, Rong M, Lai R, King GF. Discovery of a selective NaV1. 7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(43):17534–17539. doi: 10.1073/pnas.1306285110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, Wu G, Jin J, Ding B, Zhu X, Shen Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. Journal of medical genetics. 2004;41(3):171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yokoyama S, Woods SL, Boyle GM, Aoude LG, MacGregor S, Zismann V, Gartside M, Cust AE, Haq R, Harland M, Taylor JC, Duffy DL, Holohan K, Dutton-Regester K, Palmer JM, Bonazzi V, Stark MS, Symmons J, Law MH, Schmidt C, Lanagan C, O’Connor L, Holland EA, Schmid H, Maskiell JA, Jetann J, Ferguson M, Jenkins MA, Kefford RF, Giles GG, Armstrong BK, Aitken JF, Hopper JL, Whiteman DC, Pharoah PD, Easton DF, Dunning AM, Newton-Bishop JA, Montgomery GW, Martin NG, Mann GJ, Bishop DT, Tsao H, Trent JM, Fisher DE, Hayward NK, Brown KM. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature. 2011;480(7375):99–103. doi: 10.1038/nature10630. [DOI] [PMC free article] [PubMed] [Google Scholar]