

Abstract
Under the hypothesis that increased extracellular sodium induces sustained neuronal excitability with the onset and progression of migraine, this study evaluates dynamic in vivo 23Na fluxes in the brain of a preclinical rodent analogue of migraine. Ultra-high field 23Na MRI at 21.1 T has demonstrated potential to quantify sodium concentrations with good spatial and temporal resolution following the onset of central sensitization. Sprague-Dawley male rats with implanted intraperitoneal (IP) lines were studied by MRI before and after an in situ injection of 10 mg/kg of nitroglycerin (NTG) versus vehicle and saline controls. Slice-selective 23Na images were acquired using a multi-slice FID-based Chemical Shift Imaging sequence with resolution of 1.1 × 1.1 × 3 mm for a 9-min acquisition. A total of twenty-seven repeated scans were acquired over 1 h of baseline scanning and longitudinally up to 3 h post injection. Increases of 23Na MRI signal in the brainstem, extra-cerebral cerebrospinal fluid (CSF) and cisterna magna were evident almost immediately after NTG injection, gaining significance from controls in 36 min. The cerebellum and third ventricle also showed sustained trends of increased 23Na, with the former gaining significance at over 2 h post-NTG injection. The data provide evidence of an early change in sodium concentration, markedly in posterior fossa CSF and brainstem regions. Further study of fluctuations of sodium concentration and their modulation with treatments could help understand the dynamic features of migraine, location of a putative migraine generator, and guide development of therapeutic measures to correct the disturbance of sodium homeostasis.
Keywords: Migraine, 23Na MRI, Headache, Trigeminovascular System, Sodium Chemical Shift Imaging
1. Introduction
Migraine manifests due to a complex and heterogeneous interplay of genetic, environmental and hormonal factors that influence susceptibility. Although central sensitization and pain susceptibility is a major manifestation of migraine, activation and recruitment of neural structures is not well understood. As overt interictal symptoms and brain lesions are absent, migraine is predominantly considered a functional brain disorder. Because prevention or rescue are not universally successful [32,50], it remains important to elucidate neural components involved in migraine and their sequence of recruitment.
Ionic instability and biochemical imbalances are implicit in migraineurs. In clinical cohorts, altered neuronal excitability may stem from ionic imbalance during cortical spreading depression [20,37], which is considered the basis for visual auras (not reported by all migraineurs). Though not found commonly, genetic variations in the familial hemiplegic migraine mutation (FHM I-IV) alter ionic homeostasis in the brain because they involve mutation of genes that code for ion transporters and channels [17,40,46,55]. However, cerebrospinal fluid (CSF) during episodic migraine also revealed elevated sodium levels, whereas other ions (calcium, magnesium and potassium) were not impacted [23,25]. Plasma ionic levels of the same migraineurs were not altered [23]. Given that CSF is in direct contact with the interstitial brain, a choroid plexus or brain-based disturbance or source may be responsible for these altered sodium levels.
In 2011, Harrington and coworkers [24] reported elevated brain sodium in a two-dimensional 7-mm imaging slice of the in vivo rodent brain for a triggered migraine analogue. With a 20-min acquisition and volume averaging, this study was unable to identify anatomically localized changes that have potentially rapid onset. Accordingly, at higher spatial and temporal resolution, the current study aims to understand the evolution of localized ionic imbalances that potentially lead to a neuronal dysfunction following central sensitization. To probe brain sodium noninvasively, sodium magnetic resonance imaging (23Na MRI) is employed to evaluate a nitroglycerin (NTG) administered migraine analogue in rodents. NTG-triggered migraine is a well characterized clinical [2,7,16] and preclinical [9,14,24,28] model. Even with the existence of other preclinical models, including noxious stimulation and calcitonin gene related peptide (CGRP) infusion [47,49], the clinical phenotype of NTG-triggered migraine notably is regarded as indistinguishable from spontaneous migraine [11,16,27]. As well as a wealth of literature, NTG triggering also offers controlled implementation as a rational first step to elucidate the involvement of sodium in migraine generation and propagation.
For the present study, 23Na MRI at the ultra-high field of 21.1 T was employed to detect localized sodium changes at the millimolar (mM) level in the in vivo brain. To achieve the desired spatial and temporal resolution while maintaining high SNR and sensitivity, a slice-selective chemical shift imaging (CSI) approach was employed to acquire the free induction decay (FID) with an effectively zero echo time and short repetition times [8,26]. The high sensitivity accorded by imaging at 21.1 T allowed for a novel time course to identify and localize sodium at both baseline and onset of the migraine analogue, providing temporal mapping of dynamic changes over a three-hour period.
2. Materials and Methods
2.1 Animal Models
Animal procedures were approved by the Institutional Animal Care and Uses Committees at the Huntington Medical Research Institutes (HMRI) in Pasadena, CA and the Florida State University in Tallahassee, FL. While in the MRI scanner, a total of 15 Sprague-Dawley male rats (Harlan, Indianapolis, IN), weights between 170 and 250 g, were administered in situ with an intraperitoneal (IP) injection of either 10 mg/kg of NTG (n=6) to induce a migraine analogue, or saline (n=6) and vehicle (n=3) as controls. Male rats were evaluated in this study to correlate sodium changes with extensive published behavioral and cFos data with the institution of the same NTG dose, and partly to avoid the variability of hormonal variability implicit in the female cycle. Interestingly, the female threshold to NTG is much lower than males (unpublished studies). Rats were randomly assigned to all treatment groups. The vehicle formulation contains 30% ethanol, 30% propylene glycol made up in normal saline (0.9% sodium chloride injection, USP, Baxter Healthcare Corporation, Deerfield, IL) with the pH maintained at 7.4.
Animals were treated under anesthesia with the same procedure that was demonstrated to have behavioral responses to NTG (aversive threshold and eye squinting) in un-anesthetized rats and ex vivo cFos activation in the second order trigeminal neurons of the brainstem [24]. For this study, rats were induced at 4% isoflurane in O2 and maintained at 3% during surgical implantation of the IP line. For scanning, animals were maintained at approximately 2.5-1.5% isoflurane, adjusted to retain a level plane of anesthesia while in the magnet. Respiratory rate was monitored (SA Instruments, Stony brook, NY) and maintained between 30-50 breaths/min. The animals were kept at a constant temperature (30°C) in the MRI scanner by means of a regulated water supply. Animals were euthanized after experimentation.
2.2 MRI Protocol
All scans were performed at the 21.1-T, 900-MHz vertical MRI scanner designed and constructed at the National High Magnetic Field Laboratory in Tallahassee, FL [19]. The magnet is equipped with a Bruker Avance III console (Bruker-Biospin, Billerica, MA) and is operated using Paravision 5.1. A microimaging gradient system (Resonance Research, Inc, Billerica, MA) provides a peak gradient strength of 60 G/cm over a 64-mm diameter. For both excitation and detection, a home-built double tuned 23Na/1H radio frequency (RF) coil based on a linear birdcage design [45] was used to acquire in vivo 23Na and 1H data at resonance frequencies of 237 and 900 MHz, respectively. For imaging in a vertical 21.1-T magnet, rats were loaded into the RF coil and animal cradle in a supine position, and were positioned in a heads-up orientation during all scans. Respiratory rate was monitored pneumatically (SA Instruments, Stony Brook, NY) and maintained between 30-50 breaths/min.
For anatomical localization, the placement of the MRI slices was identified by making use of proton Rapid Acquisition with Relaxation Enhancement (RARE) sequences. For 23Na CSI (Fig. 1&2 A-D) and 1H references (Fig. 1&2 E-H), 3-mm thick coronal slices were located 8.5 mm anterior, 3.5 mm posterior, 6 mm posterior and 9.3 mm posterior with respect to bregma. These slices corresponded to an image focus on the eyes and olfactory bulb (combined slice #1), the third ventricle, neocortex and jaw muscle (combined slice #2), extra-cerebral anterior to the fourth ventricle CSF superior to the superior colliculus and the brainstem, cerebellum and the cisterna magna (combined slice #3), respectively. Slice-selective sodium images were acquired using a multi-slice FID-based Chemical Shift Imaging (CSI) sequence. From the four different slice locations, hamming filtered FID data were collected from a 3 × 3 cm field of view that was phase-encoded with a matrix of 26 × 26 to yield an in-plane acquisition resolution of 1.1 × 1.1 mm. A weighted Gaussian sampling profile was employed to phase-encode these 26 × 26 CSI voxels and provide filtering during the acquisition; 500 phase encodes were used. With 420 complex points recorded from a spectral bandwidth of 10 kHz, each 3-mm slice was acquired with a 500-μs Gaussian pulse with a 90° flip angle. This excitation yielded maximum signal for a repetition time (TR) of 180 ms and effective dwell time (time between the end of excitation and start of acquisition) less than 660 μs. Six averages yielded a total of 3000 repetitions for the complete data acquisition of a single slice, which permitted the complete dataset for four slices to be acquired in nine minutes and corresponds to the time course of a previous study that assessed metabolic changes in NTG-triggered rats [1] while still yielding a sodium SNR in excess of 30:1.
Figure 1. Representative sodium and proton MR images displaying relative acquisition locations in the rodent brain.
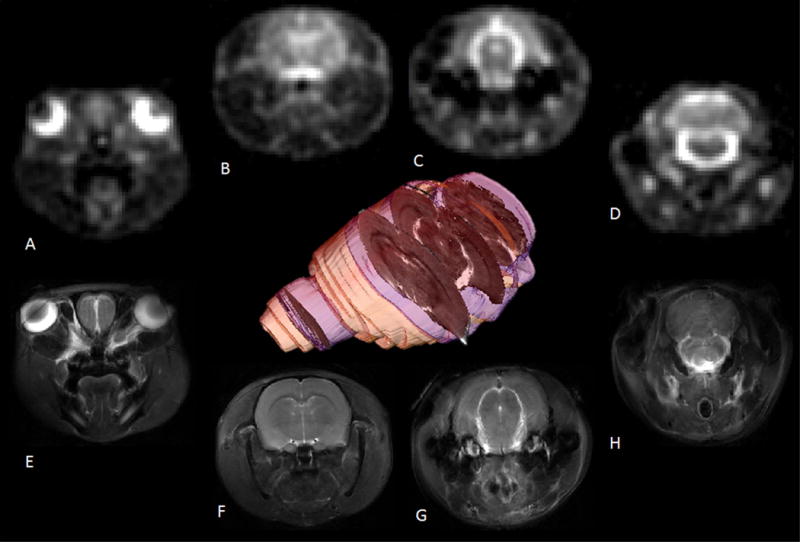
Coronal images (A–D) from a 2D multi-slice 23Na CSI acquired at 1-mm in-plane resolution with a 3-mm slice and (E-H) 117×117-mm 1H multi-slice fast spin-echo images acquired with the same FOV, slice thickness and slice locations. The 3D volume was rendered from a 1H multi-slice data to indicate the 23Na CSI slice locations and thicknesses (blue) relative to the brain (yellow) and a center-slice 1H anatomical reference (grayscale).
Figure 2. Representative coronal images for image segmentation 23Na CSI (top row) with 1H fast spin-echo anatomical references (bottom row).
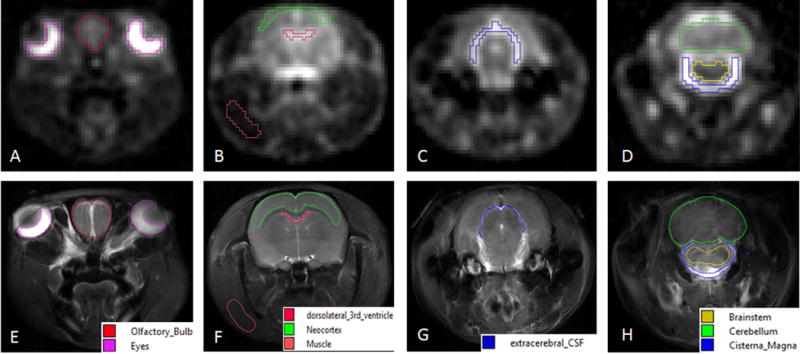
Regions of interest identified were: (A&E) eyes & olfactory bulb; (B & F) dorsolateral third ventricle, neocortex and muscle; (C & G) extracerebral CSF; (D & H) cisterna magna, brainstem and cerebellum.
A total of 27 repeated scans were acquired starting from pre-injection (at 11:00 am on each experiment day), with 1 hour of baseline scanning, followed by 3-h post injection scanning (initiated at 12:00 pm on each experiment day). This longitudinal time course allowed for investigation of bulk sodium changes during the onset and propagation of central sensitization, while controlling for any cyclical or circadian variations in biological sodium fluxes.
2.3 Phantom Measurements
A phantom was utilized to generate a standard curve to calibrate the mean signal for total sodium signal to absolute values for bulk sodium concentration changes (Supplementary Fig. 5). The phantom consisted of four pipettes filled starting with 7.4-pH phosphate buffered saline and titrated to yield sodium concentrations of 137, 68, 34 and 8 mM. An identical protocol to the in vivo CSI was used to acquire this data to calibrate 23Na signal to sodium concentration. Corresponding well to bulk 23Na relaxation in the CSF, T1 and T2* relaxation of this phantom were determined to be 56 and 51 ms, respectively. Although relaxation shortens in brain tissue (apparent T1 = 41 ms and apparent fast T2* = 33 ms), this phantom calibration was applied to both CSF and brain sodium concentrations due to the long TR and zero echo time of the FID-based CSI acquisition, which emphasizes bulk sodium and mitigates impacts from slightly different relaxation times.
2.4 Data Analysis
Prior to image reconstruction, the 2D CSI datasets were zero-filled to 512 points in the spectral dimension and a 64 × 64 image matrix in the spatial dimensions to achieve an image resolution of 0.47×0.47×3 mm. During fast Fourier transform, 20-Hz exponential line broadening was applied in the spectral dimension. CSI data sets were imported into an image rendering, quantification and analysis software package (AMIRA 5.4.3, FEI Visualization Sciences Group, Boudreaux, FR). Reconstructed 23Na images were analyzed using a 100-point range centered around the transmit frequency. No additional filtering or windowing was applied to achieve visualization of the 23Na CSI data.
The high SNR and image resolution did not require registration of 23Na images to the associated reference 1H scans for segmentation of specific anatomical regions. Images were manually segmented via anatomical landmarks identified with aid of the scalable rat brain atlas [41,44]. The regions of interest (ROI) selected were the eyes, olfactory bulb, third ventricle, neocortex, fourth ventricle, cerebellum, brainstem and cisterna magna along with a muscular region in the rat jaw to serve as an extracranial control for 23Na signal (Fig. 2). The mean signal intensities from each ROI were calibrated to the phantom measurements to determine total sodium concentration. All values from pre-injection baseline scans were averaged together to determine a pre-injection value. To determine trends and significances, individual time points in the cohorts were further converted to, and analyzed as, percent change with respect to the pre-injection baseline average.
2.5 Statistical Analysis
All datasets were analyzed using IBM SPSS 20.0 for Windows (IBM, Armonk, NY, USA). For ROI analysis, the relative values from each scan time series were compared, between groups and time points, using a mixed model ANOVA with repeated measures (P<0.05) and are presented as mean ± standard deviation. Tukey’s post hoc test for multiple comparisons was used to assess between group effects, with P<0.05 deemed to be statistically significant. For repeated measures to assess within group effects, a least significance difference (LSD) post-hoc test was used for pairwise comparison; temporal assessments within groups were deemed significant for P<0.05.
3. Results
As is evident, 23Na CSI provides improved image quality with spatial resolution compared to conventional acquisition methods (Fig. 1), albeit at the limited expense of acquisition time. The high SNR and image resolution, accordingly, did not necessitate registration to associated 1H reference images (Fig. 1 & Fig. 2) for anatomical segmentation.
In preclinical rodent models of migraine, NTG-induced central sensitization is a well-characterized model that has demonstrated increased brain sodium levels [24], hypersensitivity to pain based on behavioral response [18,24,28,31,53], altered cerebral blood flow [22,27], increased cFos expression in the trigeminal nucleus caudalis (TNC) [24,54], increased expression of nitric oxide synthase [21,42] and abnormalities in brainstem auditory evoked potentials (BAEPs) of rats [4]. In clinical cohorts, NTG administration triggers a mild immediate headache, which is stronger in migraine sufferers than in healthy subjects and derives from the acute vasodilatory effects of nitric oxide (NO) impacting the cerebral vasculature. Not evident in non-migraineurs, the NTG-triggered headache develops into a delayed migraine headache in migraineurs. It is not clear how NTG triggers the onset of this migraine, but the central sensitization of this effect is paralleled in the rodent analogue [5,11]. The behavioral onset of central sensitization in the NTG-triggered rodent model has been reported 1-h post administration [24].
The effect of the onset and progression of central sensitization on total 23Na concentrations in comparison to the saline and vehicle controls is evident in Figures 3 (A-H). Administration of NTG appears to initiate a progressive and sustained sodium increase in specific anatomical locations compared to control cohorts. Significances are reported as early as 45-min post injection in the brainstem (p = 0.004) and extra-cerebral CSF (p = 0.003) (Fig. 3 A-D), followed by the cisterna magna (p = 0.030) (Fig. 3 E-F) and cerebellum (p = 0.042) (Fig. 3 G-H). Compared to saline baseline, at 36-min post injection, the first significant percent change of total sodium is evident in the NTG-triggered brainstem, with a 4.65 ± 2 mM increase; the brainstem observed a maximal change of 22 ± 5 mM over 2-h post injection. The cisterna magna peaked at 13 ± 7 mM, and the extracerebral CSF displayed a peak difference from the saline control at 9 ± 3 mM at 1.5-h post injection.
Figure 3. Temporal plots for percent change in 23Na concentration.
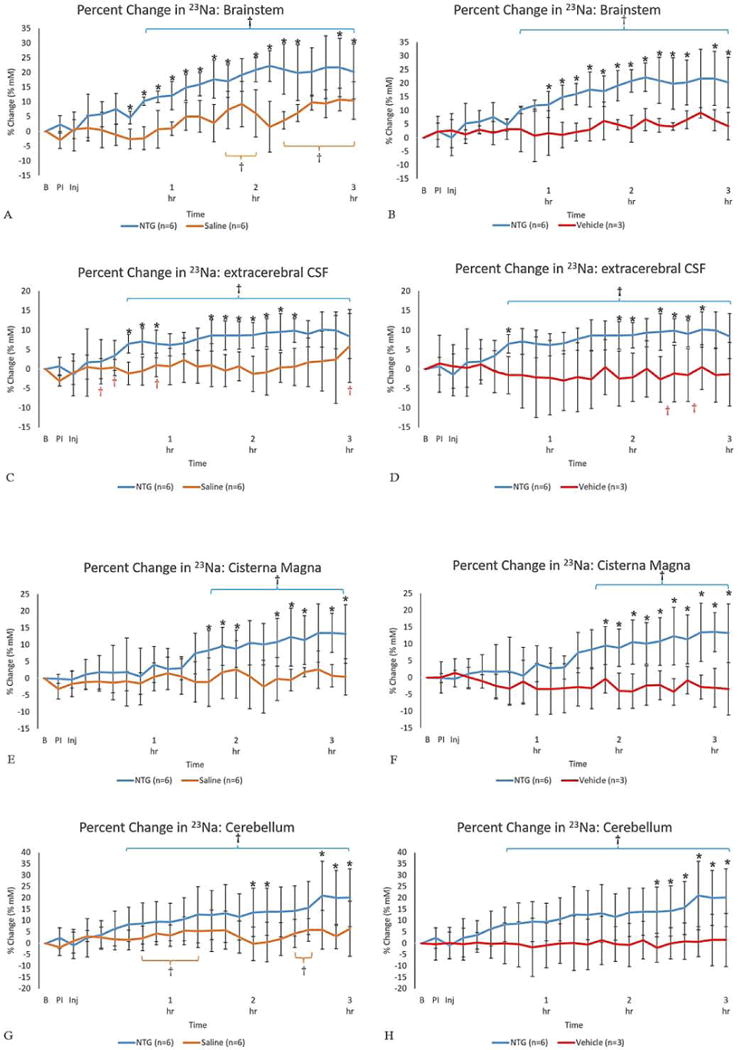
Percent change in 23Na concentration (% change ± SD) for (A-B) brainstem, (C-D) extracerebral CSF, (E-F) cisterna magna and (G-H) cerebellum as a function of time after injection. Signals were normalized to a pre-injection average baseline, with t=0 representative of the pre-injection scan acquired just before the IP injection. Utilizing a mixed model ANOVA, statistical significances are *p<0.05 (Tukey’s post-hoc) for comparisons between (left columns) NTG and saline as control & (right columns) NTG and vehicle as an additional control. For temporal comparisons within the individual cohorts’ statistical significances are identified by †p<0.05 (LSD). (Blue error brackets indicate NTG, whereas orange brackets indicate saline.)
Abbreviations: B, Baseline; PI, Pre-Injection; Inj, Injection point; IP, intraperitoneal; SD, Standard Deviation.
The third ventricle (Supplementary Fig. 1) and olfactory bulb (Supplementary Fig. 2) showed slight increases, but these paralleled, for the most part, the control cohorts. Due to volume averaging, the olfactory bulb segmentation encompasses the entire bulb along with associated CSF. Similarly, the dorso-lateral third ventricle segmentation may include potential averaging with some of the lateral ventricles. The eyes (Supplementary Fig. 3), neocortex (Supplementary Fig. 4) and muscle (Supplementary Fig. 4)—an extracranial control for this study—remained unaltered for the duration of the time course with no significant differences between the NTG, vehicle and saline cohorts. Based on the negligible temporal changes presented by saline and vehicle, sodium changes can be attributed to the impacts of NTG triggering and not due to other common experimental conditions, such as isoflurane anesthesia, animal positioning or dehydration. Rodent cohorts injected with vehicle were comparable to saline cohorts throughout the course of the study. Though ethanol was not assumed to be inert, the findings for NTG triggering appear to be indicative of central sensitization mediated through NTG actions and not via ethanol’s effect on central nervous system (CNS) functions. Thus, for this acute model, comparisons to saline serve as an appropriate control, though this might not be reflected in more chronic NTG-triggered migraine models that require repeated injections.
4. Discussion
The present study is the first to report sodium alterations localized with this degree of spatial and temporal resolution in an acutely triggered central sensitization model. Using naïve rodents, the current study probed acute ionic imbalances instituted during the latency period before perceived pain, identified as <45 min post-NTG administration in rodents, as well as longitudinally for up to 3 h during the onset and consolidation of central sensitization. The impacts of NTG-triggered central sensitization were combined in Figure 4 for all ROI to outline an anatomical basis and temporal course of impact. The temporal 23Na MRI map reveals elevated sodium in localized anatomical regions as early as 36-min post-NTG administration, with the brainstem and extra-cerebral CSF demonstrating rapid and sustained increases reaching significance with a 5-mM increase over baseline. The prodromal recruitment of the brainstem prior to the onset of behavioral sensitization provides evidence of its early involvement, likely impacting second order neurons of the trigeminovascular system (TGVS) in the trigeminal nucleus caudalis (TNC). CSF demonstrates a gradient of progressive effects both in time and concentration. Interestingly, extra-cerebral CSF proximal and anterior to the brainstem demonstrate early increases in sodium that mirror brainstem changes, reaching significance at 87 min in the cisterna magna.
Figure 4. Temporally significant sodium increases suggest a hypothesized cascade of neurophysiological events leading to central sensitization.
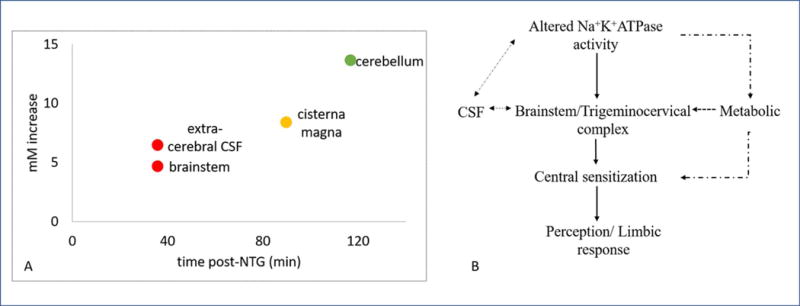
A) Onset of temporal significance for specific brain regions, post administration of NTG to trigger a migraine analogue in rodents. B) Potential neurophysiological cascade of events, elaborated in text, that lead to the onset and progression of NTG triggered acute migraine. The dashed arrows are representative of the hypothesized chain of events that lead to or attenuate the cascade whereas the solid lines are representative of the functional changes taking place.
A key TGVS player, the brainstem modulates nociceptive input beyond its primary role in receiving environmental stimuli, integrating such information and distributing it to cortical regions [10,43]. Cerebral blood vessels and meninges are surrounded by peripheral branches of axons from the trigeminal ganglion [3,20]. Second order neurons in the TNC receive nociceptive input from the trigeminal ganglion, which combine with cervical afferents in the trigeminocervical complex (TCC). Under modulations by the brainstem, the TCC conveys stimuli to the thalamus, hypothalamus, and somatosensory cortex via ascending and descending pathways [20,51]. The combined cervical and trigeminal afferents and their higher order projections potentially explain the distribution of migraine pain over the occipital and frontal regions [3] as well as increased sensitization to stimuli such as light and sound. Aptly, the brainstem is an integration center for the ascending and descending pathways from and to the cortex, thalamus and hypothalamus, along with neural projections associated with the TCC [2].
The brainstem previously has been implicated in PET and fMRI studies in chronic migraineurs using NTG-triggered [2,7,13] and spontaneous migraine [57]. Weiller and colleagues reported persistence of brainstem activation even following sumatriptan, concluding that activation is not just a nociceptive response [57]. The functional and behavioral manifestations strongly indicate a major role for the brainstem in migraine propagation. These studies relied on glucose metabolism (PET) and oxygenated blood flow changes (fMRI), thereby utilizing secondary markers of migraine onset. Furthermore, these clinical studies constituted evaluations of chronic migraineurs, for which long-term glucose metabolism and blood flow may be potentially altered, thereby obscuring acute changes that may be evident during the initial onset of migraine. Afridi et al. [2] and Weiller et al. [57] analyzed the laterality of brainstem activation, but also hypothesized potential brainstem dysfunction in chronic migraineurs, suggesting a distinct metabolic or structural alteration compared to non-migraineurs. Instituted in otherwise naïve rats, the current NTG-triggered study implicates early brainstem involvement in central sensitization. Replicating a similar pattern of activation using sodium as the probing agent, the current study calls into question whether a pre-existing metabolic or structural dysfunction is at the root of chronic migraine.
The cerebellum is the last to gain significance at greater than 117 min after NTG injection. Given the downstream association of the cerebellum with inputs from the TGVS and somatosensory cortex, the delayed but higher (13 mM) sodium impacts observed may reflect nociceptive and potential vestibular impacts of migraine in this acute model. A number of studies hint at cerebellar dysfunction in migraine: gray and white matter abnormalities [15,36], cerebral function tests to assess spatiotemporal and motion processing [30,33,34,39], ASL perfusion effects [29,56] and a recent fMRI study [35] tracing connectivity between the cerebellum and rostral pons, thalamus and aqueduct have demonstrated differences in the cerebella of chronic migraineurs in the interictal state and with the onset of nociceptive pain. Moulton and coworkers [38] demonstrated BOLD activation in multiple cerebellar areas as a response to aversive sensory input and nociceptive stimulus processing. Corresponding well with these earlier findings, the present sodium data implies that the cerebellum plays more than just a passive role in motor response, potentially mediating noxious stimuli processing.
The question of a “migraine generator” has evoked tremendous interest and both a brainstem and hypothalamic origin have been suggested [12,48]. Sodium increases that precede behavioral changes support the theory of sodium flux as potentially causative. Spatially unique changes of sodium that arise first in the brainstem and extra-cerebral CSF are more in line with a posterior fossa located migraine generator, but a diffuse territory for pleiotropic effects may arise from different CSF locations. The presented data is supportive of this interpretation but not definitive, as different sodium concentrations may have differential effects anatomically. Further studies are actively investigating the mechanism for changes in sodium. Not surprisingly, future studies also will need to evaluate other migraine triggers (CGRP among these) to determine the ubiquity of sodium involvement.
The present study has certain limitations. The 2D multi-slice 23Na acquisition is subject to volume averaging in the third dimension, which limits higher order segmentations in functionally connected neuroanatomical regions of the TGVS. In-depth segmentation of the ventricular system also is warranted given the choroid plexus hypothesis. Future studies will address these targets. The current study also interrogates total sodium concentration, but does not seek to differentiate between intracellular versus extracellular sodium (beyond regional segmentation) or the neural versus vascular origins of the 23Na signal. Sodium signal increases could originate from sodium transport between these compartments rather than a total increase. However, based on relaxation, acquisition parameters and relatively fast exchange mechanisms, compartmental movement of sodium alone would not account for signal or concentration increases. Also, previous data from chronic migraineurs reported increases of 23Na in only the CSF and not plasma [23], supporting a brain basis rather than a systemic source.
The current data only indicate an increase in the net sodium, which in the CSF regions could suggest an increased production of CSF due to pump impairments at the choroid plexuses, increased sodium distribution from the CSF or reduced drainage of the CSF. The Na+/K+/ATPase transporter (NKAT), which is the primary regulator of brain sodium, consumes 50% of brain energy to maintain homeostasis [6,58], and is the likely mechanism available to induce the substantial extracellular sodium increases (>5 mM) observed in this study. NTG acts as a NO donor, regulating cGMP and CGRP, with NKAT modulation a downstream impact of these pathways. CGRP pathways mediate CAM-kinase II, PKA and PKC, potentially playing a role in central sensitization and the alteration of ionic channels. Tassorelli et al. made use of brain sections after systemic NTG injection to demonstrate increased cGMP immunoreactivity in the TNC and somatosensory cortex for a time course study (1-5 h post injection) [52].
It stands to reason that elevated extracellular sodium would increase neural excitability. Additional studies performed at 21.1 T in NTG-triggered rodents over a similar post-injection timeframe demonstrate that lactate production is enhanced early (35 min after NTG) in the sensitized brain [1]. The increasing [Na+] of the brainstem and extracerebral CSF reported in this study likely arises from or induces increased NKAT activity. As a probable consequence of increased NKAT and neural activity, elevated brain lactate is evident on the same timescale that recently was reported in the same model system [1]. Furthermore, osmoregulatory molecules—taurine being the most dominant—remain elevated in NTG-treated animals. These energetic and neuroprotective events implicate the potential involvement and eventual dysregulation of NKAT as a primary link in the eventual chain of migraine propagation. Based on the current study as well as metabolic MRS findings, Fig. 4B proposes an organization of neurophysiological events that leads to the onset and propagation of central sensitization. It is believed that neuronal excitability stems from sodium imbalances due to altered NKAT activity. These ionic imbalances trigger/activate the TCC highlighted by significant changes observed in the brainstem, which propagate via either (i) activation of associated structures and pain pathways or (ii) decreased modulation of control pathways, both leading to central sensitization.
Given the metabolic and CSF alterations observed, the time course of events suggests the involvement of the CSF as either a primary source or the main conduit by which sodium imbalances are propagated, with subsequent metabolic alterations promulgated downstream. Under this hypothesis, ionic imbalances would deplete energy and lead to metabolic strain that would modulate TGVS and pain pathways. As such, CSF sodium and its influence on osmoregulation and energetics have early and widespread impacts that initiate a cascade of dysregulation that results in migraine. The current study implicates unique, location-specific sodium imbalances in the cascade of central sensitization, and raises a fundamental question regarding these ionic imbalances: is the activation of the brainstem fundamental to migraine or is it a consequence of activation of trigeminovascular pathways?
Supplementary Material
Acknowledgments
This work was supported by the NIH (R01-NS072497 and R01-NS102395), HMRI and UCGP from the National High Magnetic Field Laboratory, which is funded by the NSF (DMR-1157490) and the State of Florida.
Footnotes
Author contributions: The authors of this manuscript have contributed in the following fashion: N.A.: Conceptualization, Methodology, Data acquisition & processing, Writing–original draft, review and editing; J.T.R.: Data processing, Writing–review & editing; D.C.H.: Data processing; M.G.H. & S.C.G.: Contributed equally in this work; Conceptualization, Funding acquisition, Methodology, Writing–review & editing
Conflict of interest statement
The authors have no conflict of interest to declare.
References
- 1.Abad N, Rosenberg JT, Roussel T, Grice DC, Harrington MG, Grant SC. Metabolic Assessment of a Migraine Model Using Relaxation-Enhanced 1H Spectroscopy at Ultrahigh Field. Magnetic Resonance in Medicine. 2018;79:1266–1275. doi: 10.1002/mrm.26811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afridi S, Matharu M, Lee L, Kaube H, Friston K, Frackowiak R, Goadsby P. A PET study exploring the laterality of brainstem activation in migraine using glyceryl trinitrate. Brain. 2005;128:932–939. doi: 10.1093/brain/awh416. [DOI] [PubMed] [Google Scholar]
- 3.Akerman S, Holland PR, Goadsby PJ. Diencephalic and brainstem mechanisms in migraine. Nat Rev Neurosci. 2011;12:570–584. doi: 10.1038/nrn3057. [DOI] [PubMed] [Google Scholar]
- 4.Arakaki X, Galbraith G, Pikov V, Fonteh AN, Harrington MG. Altered brainstem auditory evoked potentials in a rat central sensitization model are similar to those in migraine. Brain Res. 2014;1563:110–121. doi: 10.1016/j.brainres.2014.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashina M, Hansen JM, Olesen J. Pearls and pitfalls in human pharmacological models of migraine: 30 years’ experience. Cephalalgia. 2013;33:540–553. doi: 10.1177/0333102412475234. [DOI] [PubMed] [Google Scholar]
- 6.Astrup J, Sorensen P, Sorensen H. Oxygen and Glucose Consumption Related to Na+-K+ Transport in Canine Brain. Stroke. 1981;12:726–730. doi: 10.1161/01.str.12.6.726. [DOI] [PubMed] [Google Scholar]
- 7.Bahra A, Matharu M, Buchel C, Frackowiak R, Goadsby P. Brainstem activation specific to migraine headache. Lancet. 2001;357:1016–1017. doi: 10.1016/s0140-6736(00)04250-1. [DOI] [PubMed] [Google Scholar]
- 8.Baier S, Kraemer P, Grudzenski S, Fatar M, Kirsch S, Schad LR. Chlorine and sodium chemical shift imaging during acute stroke in a rat model at 9.4 Tesla. Magn Reson Mat Phys Biol Med. 2014;27:71–79. doi: 10.1007/s10334-013-0398-z. [DOI] [PubMed] [Google Scholar]
- 9.Bergerot A, Holland PR, Akerman S, Bartsch T, Ahn AH, MaassenVanDenBrink A, Reuter U, Tassorelli C, Schoenen J, Mitsikostas DD, van den Maagdenberg AMJM, Goadsby PJ. Animal models of migraine: looking at the component parts of a complex disorder. Eur J Neurosci. 2006;24:1517–1534. doi: 10.1111/j.1460-9568.2006.05036.x. [DOI] [PubMed] [Google Scholar]
- 10.Berridge C, Waterhouse B. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Rev. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- 11.Bes A, Kunkel R, Lance JW, Nappi G, Pfaffenrath V, Rose FC, Schoenberg BS, Soyka D, Tfelt-Hansen P, Welch KMA, Wilkinson M, Olesen J, Bousser M, Diener H, Dodick D, First M, Goadsby PJ, Goebel H, Lainez MJA, Lance JW, Lipton RB, Nappi G, Sakai F, Schoenen J, Silberstein SD, Steiner TJ, Olesen J, Bendtsen L, Dodick D, Ducros A, Evers S, First M, Goadsby PJ, Hershey A, Katsarava Z, Levin M, Pascual J, Russell MB, Schwedt T, Steiner TJ, Tassorelli C, Terwindt GM, Vincent M, Wang S, Olesen J, Evers S, Charles A, Hershey A, Lipton R, First M, Bolay H, Lanteri-Minet M, MacGregor EA, Takeshima T, Schytz HW, Ashina S, Goicochea MT, Hirata K, Holroyd K, Lampl C, Lipton RB, Mitsikostas DD, Schoenen J, Goadsby P, Boes C, Bordini C, Cittadini E, Cohen A, Leone M, May A, Newman L, Pareja J, Park J, Rozen T, Waldenlind E, Wang S, Ducros A, Evers S, Fuh J, Ozge A, Pareja JA, Pascual J, Peres M, Young W, Yu S, Schwedt T, Abu-Arafeh I, Gladstone J, Huang S, Jensen R, Lainez JMA, Obelieniene D, Sandor P, Scher AI, Ducros A, IHS The International Classification of Headache Disorders, 3rd edition (beta version) Cephalalgia. 2013;33:629–808. doi: 10.1177/0333102413485658. [DOI] [PubMed] [Google Scholar]
- 12.Burstein R, Noseda R, Borsook D. Migraine: Multiple Processes, Complex Pathophysiology. J Neurosci. 2015;35:6619–6629. doi: 10.1523/JNEUROSCI.0373-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao Y, Welch K, Aurora S, Vikingstad E. Functional MRI-BOLD of visually triggered headache in patients with migraine. Arch Neurol. 1999;56:548–554. doi: 10.1001/archneur.56.5.548. [DOI] [PubMed] [Google Scholar]
- 14.de Tommaso M, Libro G, Guido M, Difruscolo O, Losito L, Sardaro M, Cerbo R. Nitroglycerin induces migraine headache and central sensitization phenomena in patients with migraine without aura: a study of laser evoked potentials. Neurosci Lett. 2004;363:272–275. doi: 10.1016/j.neulet.2004.04.029. [DOI] [PubMed] [Google Scholar]
- 15.Demir BT, Bayram NA, Ayturk Z, Erdamar H, Seven P, Calp A, Sazak M, Ceylan HG. Structural Changes in the Cerebrum, Cerebellum and Corpus Callosum in Migraine Patients. Clin Invest Med. 2016;39:S21–S26. [PubMed] [Google Scholar]
- 16.Di Clemente L, Coppola G, Magis D, Gerardy P, Fumal A, De Pasqua V, Di Piero V, Schoenen J. Nitroglycerin sensitises in healthy subjects CNS structures involved in migraine pathophysiology: Evidence from a study of nociceptive blink reflexes and visual evoked potentials. Pain. 2009;144:156–161. doi: 10.1016/j.pain.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 17.Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari M, Herzog J, van den Maagdenberg A, Pusch M, Strom T. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371–377. doi: 10.1016/S0140-6736(05)66786-4. [DOI] [PubMed] [Google Scholar]
- 18.Ferrari LF, Levine JD, Green PG. Mechanisms mediating nitroglycerin-induced delayed-onset hyperalgesia in the rat. Neuroscience. 2016;317:121–129. doi: 10.1016/j.neuroscience.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu R, Brey W, Shetty K, Gor’kov P, Saha S, Long J, Grant S, Chekmenev E, Hu J, Gan Z, Sharma M, Zhang F, Logan T, Bruschweller R, Edison A, Blue A, Dixon I, Markiewicz W, Cross T. Ultra-wide bore 900 MHz high-resolution NMR at the National High Magnetic Field Laboratory. J Magn Reson. 2005;177:1–8. doi: 10.1016/j.jmr.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 20.Goadsby PJ, Charbit AR, Andreou AP, Akerman S, Holland PR. Neurobiology of Migraine. Neuroscience. 2009;161:327–341. doi: 10.1016/j.neuroscience.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 21.Greco R, Ferrigno A, Demartini C, Zanaboni A, Mangione AS, Blandini F, Nappi G, Vairetti M, Tassorelli C. Evaluation of ADMA-DDAH-NOS axis in specific brain areas following nitroglycerin administration: study in an animal model of migraine. J Headache Pain. 2015;16:74. doi: 10.1186/s10194-015-0560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greco R, Meazza C, Mangione AS, Allena M, Bolla M, Amantea D, Mizoguchi H, Sandrini G, Nappi G, Tassorelli C. Temporal profile of vascular changes induced by systemic nitroglycerin in the meningeal and cortical districts. Cephalalgia. 2011;31:190–198. doi: 10.1177/0333102410379887. [DOI] [PubMed] [Google Scholar]
- 23.Harrington M, Fonteh A, Cowan R, Perrine K, Pogoda J, Biringer R, Huhmer A. Cerebrospinal fluid sodium increases in migraine. Headache. 2006;46:1128–1135. doi: 10.1111/j.1526-4610.2006.00445.x. [DOI] [PubMed] [Google Scholar]
- 24.Harrington MG, Chekmenev EY, Schepkin V, Fonteh AN, Arakaki X. Sodium MRI in a rat migraine model and a NEURON simulation study support a role for sodium in migraine. Cephalalgia. 2011;31:1254–1265. doi: 10.1177/0333102411408360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrington MG, Fonteh AN, Arakaki X, Cowan RP, Ecke LE, Foster H, Huehmer AF, Biringer RG. Capillary Endothelial Na plus, K plus, ATPase Transporter Homeostasis and a New Theory for Migraine Pathophysiology. Headache. 2010;50:459–478. doi: 10.1111/j.1526-4610.2009.01551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heiler PM, Langhauser FL, Wetterling F, Ansar S, Grudzenski S, Konstandin S, Fatar M, Meairs S, Schad LR. Chemical Shift Sodium Imaging in a Mouse Model of Thromboembolic Stroke at 9.4 T. J Magn Reson Imaging. 2011;34:935–940. doi: 10.1002/jmri.22700. [DOI] [PubMed] [Google Scholar]
- 27.Iversen H, Olesen J, Tfelthansen P. Intravenous Nitroglycerin as an Experimental-Model of Vascular Headache - Basic Characteristics. Pain. 1989;38:17–24. doi: 10.1016/0304-3959(89)90067-5. [DOI] [PubMed] [Google Scholar]
- 28.Jansen-Olesen I, Tfelt-Hansen P, Olesen J. Animal Migraine Models for Drug Development: Status and Future Perspectives. CNS Drugs. 2013;27:1049–1068. doi: 10.1007/s40263-013-0121-7. [DOI] [PubMed] [Google Scholar]
- 29.Jayamahan J, Street M. Fatal Cerebellar Infarction in a Migraine Sufferer while Receiving Sumatriptan. Intensive Care Med. 1995;21:82–83. doi: 10.1007/BF02425161. [DOI] [PubMed] [Google Scholar]
- 30.Koppen H, Boele H, Palm-Meinders IH, Koutstaal BJ, Horlings CGC, Koekkoek BK, van der Geest J, Smit AE, van Buchem MA, Launer LJ, Terwindt GM, Bloem BR, Kruit MC, Ferrari MD, De Zeeuw CI. Cerebellar function and ischemic brain lesions in migraine patients from the general population. Cephalalgia. 2017;37:177–190. doi: 10.1177/0333102416643527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma Z, Wang S, Li C, Ma X, Gu T. Increased metabolite concentration in migraine rat model by proton MR spectroscopy in vivo and ex vivo. Neurol Sci. 2008;29:337–342. doi: 10.1007/s10072-008-0991-5. [DOI] [PubMed] [Google Scholar]
- 32.Marmura MJ, Silberstein SD, Schwedt TJ. The Acute Treatment of Migraine in Adults: The American Headache Society Evidence Assessment of Migraine Pharmacotherapies. Headache. 2015;55:3–20. doi: 10.1111/head.12499. [DOI] [PubMed] [Google Scholar]
- 33.McKendrick AM, Sampson GP. Low spatial frequency contrast sensitivity deficits in migraine are not visual pathway selective. Cephalalgia. 2009;29:539–549. doi: 10.1111/j.1468-2982.2008.01817.x. [DOI] [PubMed] [Google Scholar]
- 34.McKendrick A, Vingrys A, Badcock D, Heywood J. Visual dysfunction between migraine events. Invest Ophthalmol Vis Sci. 2001;42:626–633. [PubMed] [Google Scholar]
- 35.Mehnert J, Schulte L, Timmann D, May A. Activity and connectivity of the cerebellum in trigeminal nociception. Neuroimage. 2017;150:112–118. doi: 10.1016/j.neuroimage.2017.02.023. [DOI] [PubMed] [Google Scholar]
- 36.Messina R, Rocca MA, Colombo B, Teggi R, Falini A, Comi G, Filippi M. Structural brain abnormalities in patients with vestibular migraine. J Neurol. 2017;264:295–303. doi: 10.1007/s00415-016-8349-z. [DOI] [PubMed] [Google Scholar]
- 37.Moskowitz M. The Neurobiology of Vascular Head Pain. Ann Neurol. 1984;16:157–168. doi: 10.1002/ana.410160202. [DOI] [PubMed] [Google Scholar]
- 38.Moulton EA, Elman I, Pendse G, Schmahmann J, Becerra L, Borsook D. Aversion-Related Circuitry in the Cerebellum: Responses to Noxious Heat and Unpleasant Images. J Neurosci. 2011;31:3795–3804. doi: 10.1523/JNEUROSCI.6709-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen BN, McKendrick AM, Vingrys AJ. Abnormal inhibition-excitation imbalance in migraine. Cephalalgia. 2016;36:5–14. doi: 10.1177/0333102415576725. [DOI] [PubMed] [Google Scholar]
- 40.Ophoff R, Terwindt G, Vergouwe M, vanEijk R, Oefner P, Hoffman S, Lamerdin J, Mohrenweiser H, Bulman D, Ferrari M, Haan J, Lindhout D, vanOmmen G, Hofker M, Ferrari M, Frants R. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 41.Papp EA, Leergaard TB, Calabrese E, Johnson GA, Bjaalie JG. Waxholm Space atlas of the Sprague Dawley rat brain. Neuroimage. 2014;97:374–386. doi: 10.1016/j.neuroimage.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pardutz A, Krizbai I, Multon S, Vecsei L, Schoenen J. Systemic nitroglycerin increases nNOS levels in rat trigeminal nucleus caudalis. Neuroreport. 2000;11:3071–3075. doi: 10.1097/00001756-200009280-00008. [DOI] [PubMed] [Google Scholar]
- 43.Parvizi J, Damasio A. Neuroanatomical correlates of brainstem coma. Brain. 2003;126:1524–1536. doi: 10.1093/brain/awg166. [DOI] [PubMed] [Google Scholar]
- 44.Paxinos G, Watson C, Emson P. Ache-Stained Horizontal Sections of the Rat-Brain in Stereotaxic Coordinates. J Neurosci Methods. 1980;3:129–149. doi: 10.1016/0165-0270(80)90021-7. [DOI] [PubMed] [Google Scholar]
- 45.Qian C, Masad IS, Rosenberg JT, Elumalai M, Brey WW, Grant SC, Gor’kov PL. A volume birdcage coil with an adjustable sliding tuner ring for neuroimaging in high field vertical magnets: Ex and in vivo applications at 21.1 T. J Magn Reson. 2012;221:110–116. doi: 10.1016/j.jmr.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riant F, Roze E, Barbance C, Meneret A, Guyant-Marechal L, Lucas C, Sabouraud P, Trebuchon A, Depienne C, Tournier-Lasserve E. PRRT2 mutations cause hemiplegic migraine. Neurology. 2012;79:2122–2124. doi: 10.1212/WNL.0b013e3182752cb8. [DOI] [PubMed] [Google Scholar]
- 47.Schindler M. The role of CGRP and CGRP antagonists in migraine. Br J Pharmacol. 2001;133:U137–U137. [Google Scholar]
- 48.Schulte LH, May A. The migraine generator revisited: continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain. 2016;139:1987–1993. doi: 10.1093/brain/aww097. [DOI] [PubMed] [Google Scholar]
- 49.Schytz HW, Schoonman GG, Ashina M. What have we learnt from triggering migraine? Curr Opin Neurol. 2010;23:259–265. doi: 10.1097/WCO.0b013e328337b884. [DOI] [PubMed] [Google Scholar]
- 50.Smitherman TA, Burch R, Sheikh H, Loder E. The Prevalence, Impact, and Treatment of Migraine and Severe Headaches in the United States: A Review of Statistics From National Surveillance Studies. Headache. 2013;53:427–436. doi: 10.1111/head.12074. [DOI] [PubMed] [Google Scholar]
- 51.Tajti J, Szok D, Pardutz A, Tuka B, Csati A, Kuris A, Toldi J, Vecsei L. Where does a migraine attack originate? In the brainstem. J Neural Transm. 2012;119:557–568. doi: 10.1007/s00702-012-0788-9. [DOI] [PubMed] [Google Scholar]
- 52.Tassorelli C, Blandini F, Greco R, Nappi G. Nitroglycerin enhances cGMP expression in specific neuronal and cerebrovascular structures of the rat brain. J Chem Neuroanat. 2004;27:23–32. doi: 10.1016/j.jchemneu.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 53.Tassorelli C, Greco R, Wang D, Sandrini M, Sandrini G, Nappi G. Nitroglycerin induces hyperalgesia in rats - a time-course study. Eur J Pharmacol. 2003;464:159–162. doi: 10.1016/s0014-2999(03)01421-3. [DOI] [PubMed] [Google Scholar]
- 54.Tassorelli C, Joseph S. Systemic Nitroglycerin Induces Fos Immunoreactivity in Brain-Stem and Forebrain Structures of the Rat. Brain Res. 1995;682:167–181. doi: 10.1016/0006-8993(95)00348-t. [DOI] [PubMed] [Google Scholar]
- 55.Vanmolkot K, Kors E, Hottenga J, Terwindt G, Haan J, Hoefnagels W, Black D, Sandkuijl L, Frants R, Ferrari M, van den Maagdenberg A. Novel mutations in the Na+,K+-ATPase pump gene ATP1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol. 2003;54:360–366. doi: 10.1002/ana.10674. [DOI] [PubMed] [Google Scholar]
- 56.Vincent M, Hadjikhani N. The cerebellum and migraine. Headache. 2007;47:820–833. doi: 10.1111/j.1526-4610.2006.00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weiller C, May A, Limmroth V, Juptner M, Kaube H, Vonschayck R, Coenen H, Diener H. Brain-Stem Activation in Spontaneous Human Migraine Attacks. Nat Med. 1995;1:658–660. doi: 10.1038/nm0795-658. [DOI] [PubMed] [Google Scholar]
- 58.Whittam R. Dependence of Respiration of Brain Cortex on Active Cation Transport. Biochem J. 1962;82:205. doi: 10.1042/bj0820205. &. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.