

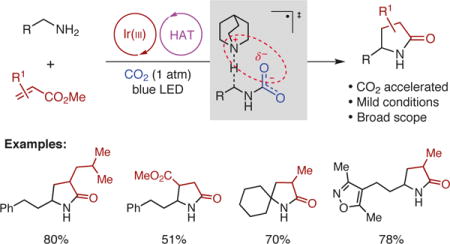
Abstract
Primary aliphatic amines are important building blocks in organic synthesis due to the presence of a synthetically versatile NH2 group. While N-functionalization of primary amines is well-established, selective C-functionalization of unprotected primary amines remains challenging. Here we report the use of CO2 as an activator for the direct transformation of abundant primary aliphatic amines into valuable γ-lactams under photoredox and hydrogen atom transfer (HAT) catalysis. Experimental and computational studies suggest that CO2 not only inhibits undesired N-alkylation of primary amines but also promotes selective intermolecular HAT by an electrostatically accelerated interaction between the in situ generated negatively charged carbamate and the positively charged quinuclidinium radical. This electrostatic attraction overwhelms the inherent bond dissociation energies which suggest HAT should occur unselectively. We anticipate that our findings will open up new avenues for amine functionalizations as well as selectivity control in HAT reactions.
Graphical abstract
Selective functionalization of aliphatic C−H bonds remains a considerable challenge1. Positions adjacent to functional groups tend to be activated towards functionalization, whether by classical deprotonation strategies or by emerging radical abstraction approaches. Among the latter, carbon-hydrogen bonds adjacent to alcohols, ethers and amides have been demonstrated to be susceptible to HAT chemistry with electrophilic agents such as thiyl radical2,3 and quinuclidinium radical cation4–6. These processes are selective because of a polarity match2 – a nucleophilic or hydridic hydrogen atom source is required. Amine activation is also an area of intense interest, dominated by tertiary amines which may be oxidized to the radical cation with subsequent proton loss to deliver an α-amino alkyl radical (Fig. 1a)7–10. Efficient α-functionalization of simple primary aliphatic amines is exceedingly rare, regardless of mechanism11–13. Indeed, primary aliphatic amines pose a significant challenge not just because of their much higher oxidation potential14 and propensity for overoxidation to the imine or nitrile (Fig. 1a)15, but also because of their pronounced nucleophilicity and basicity16,17.
Figure 1. Strategies for α–functionalization of aliphatic amines.
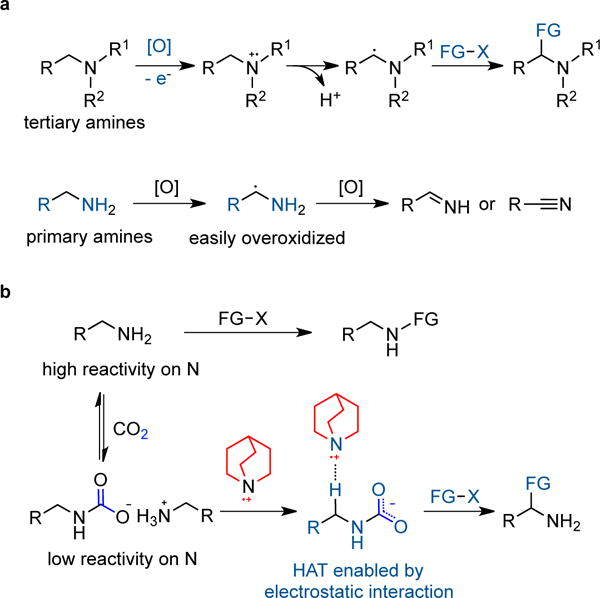
a, Generation and reactivity of α–amino radical from tertiary amines have been well establised but synthetic studies on α–amino radical of primary amines are rare as they can be easily overoxidized to imines or nitriles. b, Our hypothesis using CO2 as an activator for the α–functionalization of simple primary aliphatic amines relies on an electrostatic attraction between the in situ formed carbamate and quinuclidinium radical cation; CO2 also serves as a temporary protecting group to decrease the nucleophilicity of the NH2 group; counterion omitted for clarity. R, R1, R2, and X denote a general organic group. FG, functional group. HAT, hydrogen atom transfer.
It has long been recognized that primary alkyl amines react readily and reversibly with CO2 to form carbamates18. In the context of our recent effort in the area of selective amine functionalization19,20, we speculated that this equillibrium might be utilized to modulate the reactivity of primary alkyl amines as the formation of alkylammonium carbamate would dramatically diminish the nucleophilicity of the NH2 motif (Fig. 1b)21, thereby providing opportunities for functionalization of less reactive C(sp3)−H bonds. A central advantage of this strategy is that the free NH2 group would be restored via facile CO2 dissociation, making further synthetic manipulations possible without the need for protection and deprotection steps22–24. However, the installation of an electron-withdrawing group on nitrogen would also render the α-C−H bond less hydridic25,26 and would thus decelerate functionalization reactions using electrophilic reagents. We speculated, however, that if we used an electrophilic HAT catalyst that was cationic, such as quinuclidinium radical cation, we could rely on an electrostatic attraction27 with the carbamate oxygen to potentially facilitate the transformation (Fig. 1b).
Results and discussion
Our proposed mechanistic cycle for a CO2-promoted α–alkylation/lactamization of primary aliphatic amines is outlined in Fig. 2a. Irradiation of Ir(III) photocatalyst PC1 with visible light generates the long-lived excited state *Ir(III) 1, which is a strong oxidant (E1/2red [*IrIII/IrII] = +1.21 V vs saturated calomel electrode (SCE) in CH3CN)28 capable of oxidizing a HAT catalyst such as quinuclidine (E1/2ox = +1.1 V vs SCE in CH3CN)29 to form radical cation 4 and Ir(II) 2. Meanwhile, primary alkyl amine reacts readily with CO2 to form alkylammonium carbamate 6 (ref. 18). Facilitated by electrostatic attraction27, the electrophilic quinuclidinium radical 4 should selectively abstract α-C−H bond of the in situ generated alkylammonium carbamate 6 to produce carbon-centered radical 7, which adds to an electron-deficient acrylate to furnish alkyl radical 8. Single-electron reduction of the radical 8 by the Ir(II) species 2 followed by protonation4,19, CO2 dissociation, and intramolecular cyclization then affords the final γ-lactam product30.
Figure 2. Reaction development.
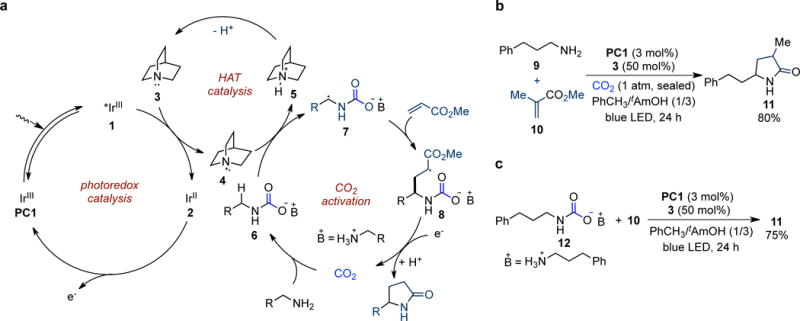
a, Proposed mechanism for the CO2-promoted α–alkylation/lactamization of primary aliphatic amines. One-electron oxidation of quinuclidine 3 by the excited photocatalyst 1 furnishes quinuclidinium radical cation 4, which undergoes selective HAT with the in situ formed alkylammonium carbamate 6 to afford radical 7. Addition into acrylate provides adduct 8, which is reduced by reduced photocatalyst 2 and protonated to give the final γ-lactam product after CO2 dissociation. b, Photocatalyst PC1 and quinuclidine are identified as the optimal catalyst combination in the model reaction using a mixture of toluene and tAmOH as the solvent. c, Reaction using preformed alkylamonium carbamate 12 in the absence of CO2 provides the desired product in 75% yield, which supports our proposal. Isolated yields are shown. tAmOH, tert-amyl alcohol.
We began our investigation by using 3-phenyl-1-propylamine 9 and methyl methacrylate 10 as the model substrates and found that α-alkylation/lactamization product 11 could be isolated in 80% yield using Ir[dF(CF3)ppy]2(dtbbpy)PF6 [dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-trifluorometh-ylpyridine; dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine] (PC1) as the photocatalyst and quinuclidine (3) as the HAT catalyst under an atmospheric pressure of CO2 with illumination by blue LEDs (Fig. 2b). Control experiments revealed that photocatalyst, visible light, quinuclidine, and CO2 were all necessary components for achieving high efficiency of this reaction (Supplementary Table 1). To verify if alkylammonium carbamate is involved in the reaction as proposed, carbamate 12 was synthesized31 and subjected to the optimal reaction conditions in the absence of CO2; in the event, product 11 was isolated in 75% yield (Fig. 2c), lending further support to our proposal.
With the optimal conditions in hand, we investigated the scope of the reaction and found that a diverse array of primary amines and acrylates are viable in this transformation, providing γ-lactams 13-57 in moderate to good yields and with diastereomeric ratios, where applicable, ranging from 1:1 to 2.3:1 (Fig. 3). Acrylates with or without an α or β substituent are tolerated, providing the desired products in reasonable to good yields (13-18, 41-81% yield). In addition to acrylate, methacrylonitrile may be coupled to give cyclic amidine with moderate efficiency (19, 54% yield). With respect to primary aliphatic amines, a broad range of simple amines with a linear or branched alkyl chain are succefully alkylated and cyclized to afford γ-lactams 20-28 in 50-75% yields. Amines with an α-substituent and cyclic primary amines are well tolerated, furnishing γ-lactams with a tetrasubstituted stereocenter in moderate to good yields (29-37, 43-70% yield). Sterically demanding primary amines are converted into the desired products 38 and 39 in 52% and 66% yields, respectively. Notably, primary alkyl amines with additional functionalities are also compatible with this protocol. For example, fluorinated amines (40, 41), TBS-protected alkanolamine (42, 43), β-Alanine ethyl ester (44), amines bearing a bisbenzylic hydrogen (45, 46) or an acetal functionality (47, 48) all underwent the reaction smoothly to afford the desired products in 45-75% yield. Moreover, heteroaryl groups commonly found in pharmaceuticals, such as pyridinyl (49), imidazolyl (50), isoxazolyl (51), and thiazolyl (52) could be incorporated into the γ-lactam products with equally high efficiency (62-80% yield). Comparable yields of 50 and 52 could be obtained with lower photocatalyst loading when the reactions were carried out on 1 or 3 mmmol scale. Alkene- or alkyne-containing amines (53, 54), mono-Boc protected diamines (55, 56), and L-lysine derivative (57) are also effective substrates. Of particular note is that when N-Boc-1,3-propanediamine was subjected to the standard reaction conditions, alkylation occurs selectively at the α position of the free NH2 group to give γ-lactam 55 in 64% yield, demonstrating that even subtle difference between N-Boc carbamate and the in situ generated alkylammonium carbamate can be differentiated under the reaction conditions. It is also noteworthy that the α-amino radical derived from the anionic carbamate is more nucleophilic than that derived from the N-Boc carbamate, leading to a faster alkylation.
Figure 3. Exploration of substrate scope.
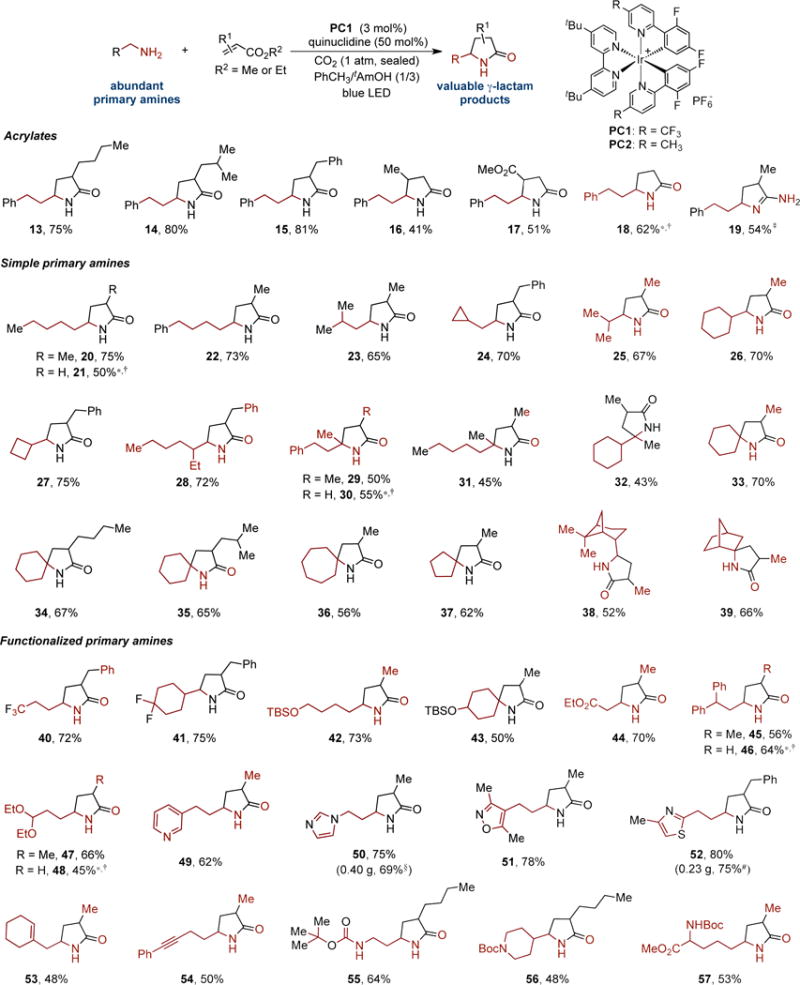
The scope of the reaction was found to be very broad, with a wide variety of primary amines and acrylates applicable in this reaction. Unless otherwise noted, all reactions were carried out on 0.2 mmol scale irradiated with a 34-W Kessil blue LED. Isolated yields are reported. All products are formed as racemates and the diastereomeric ratios (d.r.), where applicable, are between 1:1 and 2.3:1; see Supplementary Information for details. TBS, tert-butyldimethylsilyl; Boc, tert-butoxycarbonyl; *Reaction was performed using PC2 as the photocatalyst and toluene as the solvent;†The primary amine solution was stirred for 30 minutes under CO2 before adding the other reagents.‡methacrylonitrile was used in place of acrylate;§Reaction performed on 3 mmol scale with 1 mol% of PC1.#Reaction performed on 1 mmol scale with 2 mol% of PC1.
A series of mechanistic studies were performed to better understand the mechanism of the reaction. Stern-Volmer luminescence quenching experiments revealed that quinuclidine efficiently quenches the excited state of photocatalyst PC1 while primary amine 9 and the corresponding carbamate 12 do not (Supplementary Section 6). Careful monitoring of reaction between primary amine 9 and acrylate 58 in the presence and absence of CO2 revealed that acrylate consumption and product formation are much faster in the presence of CO2 (Fig. 4a). If the sole role of CO2 is to insulate the amine from undergoing N-alkylation via reversible formation of alkylammonium carbamate, the acrylate should be consumed faster in the absence of CO2. The fact that the reverse is true suggests that the in situ generated carbamate is activated towards productive HAT and subsequent alkylation by acrylate (Fig. 4b).
Figure 4. Mechanistic and computational studies.
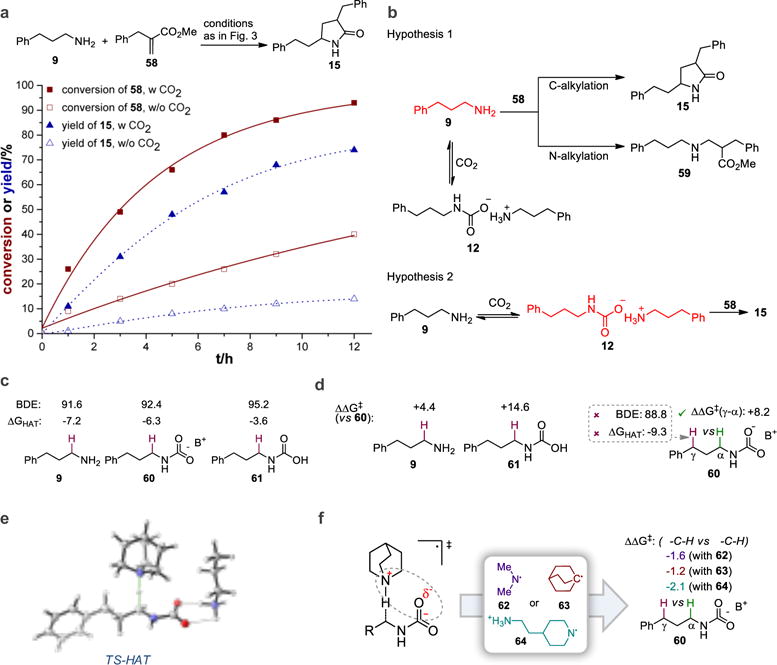
a, 1H nuclear magnetic resonance (NMR) studies show that both acrylate consumption and product formation are faster in the presence of CO2. b, Two hypotheses for the role of CO2 in the reaction. In hypothesis 1, where CO2 is merely a protecting group, acrylate consumption and product formation should be slower in the presence of CO2 due to the lower concentration of free amine 9. In hypothesis 2, where CO2 is an activator, acrylate consumption and product formation should be faster in the presence of CO2 due to the conversion of free amine into more reactive alkylammonium carbamate 12. c, Computed thermodynamic descriptors are unable to explain rate-acceleration by CO2 as well as high α-C−H selectivity. d, Selectivity and reactivity enhancement are achieved in the transition state (TS). e, Computed HAT transition state. f, Computational test for the role of electrostatics - removal or relocation of positive charge in the HAT reagent diminishes site-selectivity. Values are energies in kcal/mol. BDE, bond dissociation energy. B+ = n-C3H7NH3+.
We subsequently undertook computational studies to gain further insight on the mechanistic aspects of this transformation and the origins of selectivity (Fig. 4c). Using a high-level but affordable quantum chemical approach at the CPCM (Toluene) DLPNO-CCSD(T)/def2-TZVPD // CPCM (Toluene) M06L/6-31+G(d,p) level of theory (see Supplementary Section 9 for details), we initially assessed the effect of CO2 incorporation on bond dissociation energy (BDE) of various C−H sites in the substrate. This analysis revealed that there is no pronounced difference in BDEs of the reactive C−H bonds between the free amine 9 versus CO2-incorporated analogues, i.e. alkylammonium carbamate 60 or carbamic acid 61 (Fig. 4c). Moreover, for carbamate 60, BDEs predict the benzylic C−H site (γ) to be weakest, yet selective functionalization at the α-C−H site was observed experimentally. Thus, although commonly utilized as guidelines to rationalize and predict radical reactivities, thermodynamic descriptors, such as radical stabilities or bond strengths, do not allow rationalization of the observed site selectivity nor reactivity enhancement under CO2 conditions. By contrast, our calculations of the activation free energy barriers for hydrogen atom abstraction by a quinuclidinium radical cation were fully consistent with experimental observations. Firstly, abstraction of the hydrogen atom at the benzylic C−H site (γ) has a ΔΔG‡ = +8.2 kcal/mol higher activation free energy barrier than abstraction at the α-C−H site, in line with exclusive functionalization at the α site. We observed the same trend also with other substrates containing an activated γ-C−H site (e.g. the starting materials of 45, 49, 51 and 52, see Supplementary Fig. 8). Secondly, the carbamate 60 is predicted to be more reactive relative to the free amine 9 (ΔΔG‡ = +4.4 kcal/mol), consistent with the reactivity enhancement observed (Fig. 4a). Similar observations of counter-thermodynamic reactivity trends in HAT reactions have previously been made, with the underlying causes being ascribed to polarity-matching4,6, entropy-control32, and steric factors33. Our data suggest that the origin of the exquisite site selectivity is due primarily to stabilizing electrostatic interactions in the transition state between the reactive center α to the alkylammonium carbamate and the proximal positive charge of the quinuclidinium, displaying therefore the lowest activation barrier over alternative sites. In line with this, we predict decreased selectivities for HAT reagents for which the charge is absent (62, 63) or is located more distantly (64), regardless of the size of the reagent and the central atom (nitrogen vs carbon), rendering the benzylic C−H site (γ) more reactive in these cases (Supplementary Fig. 6).
Conclusions
In summary, we have developed a direct alkylation of primary amines using photoredox catalysis. The key finding is the use of CO2 to form the carbamate functionality in situ which accelerates the C−H bond activation by HAT. We present experimental and computational evidence in support of an electrostatic attraction again highlighting the unique role of the carbamate which is capable of undergoing accelerated HAT with the quinuclidinium radical cation in spite of its decreased hydridicity.
Methods
General procedure for the α–alkylation/lactamization of primary aliphatic amines
To a 25-mL oven-dried Schlenk sealing tube containing a magnetic stir bar were added [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (6.6 mg, 0.006 mmol), quinuclidine (11.1 mg, 0.1 mmol), primary alkyl amine (0.3 mmol), acrylate (0.2 mmol), and 0.5 mL of toluene and tAmOH mixture (1/3, v/v). The reaction tube was sealed, frozen by liquid nitrogen for 10 minutes, and evacuated under vacuum and backfilled with CO2 (balloon) three times through a three-way stopcock. Liquid nitrogen and the CO2 balloon were then removed. The reaction tube was sealed and allowed to stand at room temperature for 10 minutes, at which time the plug of the tube was slowly opened to release the excess of CO2 gas. The tube was then resealed and placed approximately 3 inches away from a Kessil® LED illuminator. The reaction mixture was stirred and irradiated for the indicated period of time. The internal temperature was measured to be approximately 40 °C using an infrared thermometer. The crude mixture was then concentrated in vacuo and purified by flash chromatography on silica gel with a 4 gram column on a Teledyne ISCO CombiFlash® Rf+ Lumen™ instrument using the indicated solvent system.
Data availability
All data generated and analysed during this study are included in this Article and its Supplementary Information, and are also available from the corresponding authors upon reasonable request.
Supplementary Material
Acknowledgments
Financial support from NIGMS (T.R.: GM125206) is acknowledged. Partial support from Catalysis Collaboratory for Light-activated Earth Abundant Reagents (C-CLEAR), which is supported by the National Science Foundation and the Environmental Protection Agency through the Network for Sustainable Molecular Design and Synthesis program (NSFCHE-1339674), is also acknowledged (T.R.). Calculations were performed with computing resources granted by JARA-HPC from RWTH Aachen University under project ‘jara0091’. We thank Professor J. Owen (Columbia University) for the use of his fluorometer.
Footnotes
Author Contributions
T.R. and J.Y. conceived the concept. T.R. directed the investigation. J.Y. performed the experiments and analysed the data. I.K. and F.S. carried out computational studies. J.Y., T.R., I.K. and F.S. collated the data, discussed the implications and prepared the manuscript.
Additional information
Supplementary information and chemical compound information are available in the online version of the paper. Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.He J, Wasa M, Chan KSL, Shao Q, Yu JQ. Palladium-catalyzed transformations of alkyl C–H bonds. Chem Rev. 2017;117:8754–8786. doi: 10.1021/acs.chemrev.6b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts BP. Polarity-reversal catalysis of hydrogen-atom abstraction reactions: concepts and applications in organic chemistry. Chem Soc Rev. 1999;28:25–35. [Google Scholar]
- 3.Qvortrup K, Rankic DA, MacMillan DWC. A general strategy for organocatalytic activation of C−H bonds via photoredox catalysis: direct arylation of benzylic ethers. J Am Chem Soc. 2014;136:626–629. doi: 10.1021/ja411596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeffrey JL, Terrett JA, MacMillan DWC. O–H hydrogen bonding promotes H-atom transfer from α C–H bonds for C-alkylation of alcohols. Science. 2015;349:1532–1536. doi: 10.1126/science.aac8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DWC. Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science. 2016;352:1304–1308. doi: 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le C, Liang Y, Evans RW, Li X, MacMillan DWC. Selective sp3 C–H alkylation via polarity-match based cross-coupling. Nature. 2017;547:79–83. doi: 10.1038/nature22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prier CK, Rankic DA, MacMillan DWC. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schultz DM, Yoon TP. Solar synthesis: prospects in visible light photocatalysis. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beatty JW, Stephenson CRJ. Amine functionalization via oxidative photoredox catalysis: methodology development and complex molecule synthesis. Acc Chem Res. 2015;48:1474–1484. doi: 10.1021/acs.accounts.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakajima K, Miyake Y, Nishibayashi Y. Synthetic utilization of α-aminoalkyl radicals and related species in visible light photoredox catalysis. Acc Chem Res. 2016;49:1946–1956. doi: 10.1021/acs.accounts.6b00251. [DOI] [PubMed] [Google Scholar]
- 11.Kubiak R, Prochnow I, Doye S. Titanium-catalyzed hydroaminoalkylation of alkenes by C–H bond activation at sp3 centers in the α-position to a nitrogen atom. Angew Chem Int Ed. 2009;48:1153–1156. doi: 10.1002/anie.200805169. [DOI] [PubMed] [Google Scholar]
- 12.Bexrud JA, Eisenberger P, Leitch DC, Payne PR, Schafer LL. Selective C–H activation α to primary amines. Bridging metallaaziridines for catalytic, intramolecular α-alkylation. J Am Chem Soc. 2009;131:2116–2118. doi: 10.1021/ja808862w. [DOI] [PubMed] [Google Scholar]
- 13.Das S, Kumar JSD, Thomas KG, Shivaramayya K, George MV. Photocatalyzed multiple additions of amines to α,β-unsaturated esters and nitriles. J Org Chem. 1994;59:628–634. [Google Scholar]
- 14.Adenier A, Chehimi MM, Gallardo I, Pinson J, Vilà N. Electrochemical oxidation of aliphatic amines and their attachment to carbon and metal surfaces. Langmuir. 2004;20:8243–8253. doi: 10.1021/la049194c. [DOI] [PubMed] [Google Scholar]
- 15.Largeron M. Protocols for the catalytic oxidation of primary amines to imines. Eur J Org Chem. 2013:5225–5235. [Google Scholar]
- 16.Calleja J, et al. A steric tethering approach enables palladium-catalysed C–H activation of primary amino alcohols. Nat Chem. 2015;7:1009–1016. doi: 10.1038/nchem.2367. [DOI] [PubMed] [Google Scholar]
- 17.Willcox D, et al. A general catalytic β-C–H carbonylation of aliphatic amines to β-lactams. Science. 2016;354:851–857. doi: 10.1126/science.aaf9621. [DOI] [PubMed] [Google Scholar]
- 18.Dell’Amico DB, Calderazzo F, Labella L, Marchetti F, Pampaloni G. Converting carbon dioxide into carbamato derivatives. Chem Rev. 2003;103:3857–3897. doi: 10.1021/cr940266m. [DOI] [PubMed] [Google Scholar]
- 19.Chu JC, Rovis T. Amide-directed photoredox-catalysed C–C bond formation at unactivated sp3 C–H bonds. Nature. 2016;539:272–275. doi: 10.1038/nature19810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi GJ, Zhu Q, Miller DC, Gu CJ, Knowles RR. Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer. Nature. 2016;539:268–271. doi: 10.1038/nature19811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peeters A, Ameloot R, De Vos DE. Carbon dioxide as a reversible amine-protecting agent in selective Michael additions and acylations. Green Chem. 2013;15:1550–1557. [Google Scholar]
- 22.Huang Z, Wang C, Dong G. Catalytic C(sp3)–H arylation of free primary amines with an exo directing group generated in situ. Angew Chem Int Ed. 2016;55:5299–5303. doi: 10.1002/anie.201604268. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Ge H. Site-selective C–H arylation of primary aliphatic amines enabled by a catalytic transient directing group. Nat Chem. 2017;9:26–32. [Google Scholar]
- 24.Wu Y, Chen YQ, Liu T, Eastgate MD, Yu JQ. Pd-catalyzed γ‐C(sp3)−H arylation of free amines using a transient directing group. J Am Chem Soc. 2016;138:14554–14557. doi: 10.1021/jacs.6b09653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee M, Sanford MS. Platinum-catalyzed, terminal-selective C(sp3)−H oxidation of aliphatic amines. J Am Chem Soc. 2015;137:12796–12799. doi: 10.1021/jacs.5b09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Howell JM, Feng K, Clark JR, Trzepkowski LJ, White MC. Remote oxidation of aliphatic C−H bonds in nitrogen-containing molecules. J Am Chem Soc. 2015;137:14590–14593. doi: 10.1021/jacs.5b10299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis HJ, Phipps RJ. Harnessing non-covalent interactions to exert control over regioselectivity and site-selectivity in catalytic reactions. Chem Sci. 2017;8:864–877. doi: 10.1039/c6sc04157d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowry MS, et al. Single-layer electroluminescent devices and photoinduced hydrogen production from an ionic iridium(III) complex. Chem Mater. 2005;17:5712–5719. [Google Scholar]
- 29.Liu WZ, Bordwell FG. Gas-phase and solution-phase homolytic bond dissociation energies of H−N+ bonds in the conjugate acids of nitrogen bases. J Org Chem. 1996;61:4778–4783. doi: 10.1021/jo950933r. [DOI] [PubMed] [Google Scholar]
- 30.Caruano J, Muccioli GG, Robiette R. Biologically active γ-lactams: synthesis and natural sources. Org Biomol Chem. 2016;14:10134–10156. doi: 10.1039/c6ob01349j. [DOI] [PubMed] [Google Scholar]
- 31.Masuda K, Ito Y, Horiguchi M, Fujita H. Studies on the solvent dependence of the carbamic acid formation from ω-(1-naphthyl)alkylamines and carbon dioxide. Tetrahedron. 2005;61:213–229. [Google Scholar]
- 32.Finn M, Friedline R, Suleman NK, Wohl CJ, Tanko JM. Chemistry of the t-butoxyl radical: evidence that most hydrogen abstractions from carbon are entropy-controlled. J Am Chem Soc. 2004;126:7578–7584. doi: 10.1021/ja0493493. [DOI] [PubMed] [Google Scholar]
- 33.Barham JP, John MP, Murphy JA. Contra-thermodynamic hydrogen atom abstraction in the selective C–H functionalization of trialkylamine N-CH3 groups. J Am Chem Soc. 2016;138:15482–15487. doi: 10.1021/jacs.6b09690. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated and analysed during this study are included in this Article and its Supplementary Information, and are also available from the corresponding authors upon reasonable request.