

Abstract
Background
Cell type-specific ribosome-pulldown has become an increasingly popular method for analysis of gene expression. It allows for expression analysis from intact tissues and monitoring of protein synthesis in vivo. However, while its utility has been assessed, technical aspects related to sequencing of these samples, often starting with a smaller amount of RNA, have not been reported. In this study, we evaluated the performance of five library prep protocols for ribosome-associated mRNAs when only 250 pg-4 ng of total RNA are used.
Results
We obtained total and RiboTag-IP RNA, in three biological replicates. We compared 5 methods of library preparation for Illumina Next Generation sequencing: NuGEN Ovation RNA-Seq system V2 Kit, TaKaRa SMARTer Stranded Total RNA-Seq Kit, TaKaRa SMART-Seq v4 Ultra Low Input RNA Kit, Illumina TruSeq RNA Library Prep Kit v2 and NEBNext® Ultra™ Directional RNA Library Prep Kit using slightly modified protocols each with 4 ng of total RNA. An additional set of samples was processed using the TruSeq kit with 70 ng, as a ‘gold standard’ control and the SMART-Seq v4 with 250 pg of total RNA. TruSeq-processed samples had the best metrics overall, with similar results for the 4 ng and 70 ng samples. The results of the SMART-Seq v4 processed samples were similar to TruSeq (Spearman correlation > 0.8) despite using lower amount of input RNA. All RiboTag-IP samples had an increase in the intronic reads compared with the corresponding whole tissue, suggesting that the IP captures some immature mRNAs. The SMARTer-processed samples had a higher representation of ribosomal and non-coding RNAs leading to lower representation of protein coding mRNA. The enrichment or depletion of IP samples compared to corresponding input RNA was similar across all kits except for SMARTer kit.
Conclusion
RiboTag-seq can be performed successfully with as little as 250 pg of total RNA when using the SMART-Seq v4 kit and 4 ng when using the modified protocols of other library preparation kits. The SMART-Seq v4 and TruSeq kits resulted in the highest quality libraries. RiboTag IP RNA contains some immature transcripts.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-5066-2) contains supplementary material, which is available to authorized users.
Keywords: RiboTag, Library preparation kits, Low-input RNA-seq, RNA-seq, Coverage bias
Background
Considerable scientific effort has been dedicated to understanding cell type-specific expression profiles from complex tissues, such as brain, liver, pancreas, testes, eye or ear [1–7]. To overcome the issue of cellular heterogeneity within complex tissues, two methods have been traditionally used in mice: Laser-Capture Microdissection (LCM) [8, 9] and Fluorescence Activated Cell Sorting (FACS) [10, 11]. However, LCM is a laborious and time-consuming procedure with low yield of mRNA; and FACS requires tissue dissociation – which may lead to changes in gene expression – and requires dedicated equipment [12]. More recently, single cell RNA-seq has been introduced. However, this technique too requires tissue dissociation and is currently limited by the number of genes detected per sequenced cell [13, 14]. To overcome these limitations, Translating Ribosome Affinity Purification (TRAP) [15] and RiboTag [16] have been recently developed to study cell type-specific transcriptome profiles. Both methods rely on immunoprecipitation of ribosome-attached RNA (also named ‘translatome’) by cell type-specific molecular targeting of the ribosomal proteins, often in a Cre-lox based system [15–17]. These methods have the advantage of not requiring tissue dissociation, thus allowing for cell type-specific translatome analysis from intact tissues.
While ribosome-attached RNA sequencing for expression analysis has been validated from a biological standpoint [18, 19], the technical aspects of its library construction and sequencing have not been studied. In instances where small complex tissues are studied, the amount of starting material after immunoprecipitation may be limited (e.g., less than 5 ng). When starting from low amounts of RNA, additional cycles of amplification using PCR are performed after adapter ligation to amplify the cDNA to generate enough material for sequencing. Multiple commercial kits are available in the market to build cDNA libraries from samples with low amounts of RNA, including kits from NuGEN, New England Biolabs (NEB), Illumina and TaKaRa. Standard protocols for library construction are commonly designed to start with more than 100 ng of total RNA [20, 21] and only a few studies have been conducted to compare the performance of library preparation kits using less than 5 ng of total RNA as their starting amount [22, 23]. In this study, we selected four of the commonly used library preparation kits that are also suitable for lower-input samples for comparison. We modified the standard protocols for NEB and Illumina library preparation kits to enable them to work with smaller amounts of RNA than the recommended amounts down to as little as 4 ng of total RNA. We included one kit, SMART-Seq v4, that was designed for single cell RNA-seq and tested it with 4 ng and 250 pg of total RNA. We evaluated the performance of the different kits based on duplication rate, percentage of intronic and exonic regions being detected, the evenness of coverage of transcripts and ribosomal RNA read-count in comparison to total reads. We also compared the reproducibility of the enrichment or depletion effect based on ribotag-translatome profile for the first time.
Methods
Animals
The Gfi1-Cre knock-in mice generated by Dr. Lin Gan (University of Rochester) were kindly provided by Dr. Jian Zuo of the Developmental Neurobiology Department at St. Jude Children’s Research Hospital. RiboTag mice generated by Dr. Paul S Amieux (University of Washington) were kindly provided by Dr. Mary-Kay Lobo of the Department of Neurobiology and Anatomy at University of Maryland Baltimore. B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J mice (also referred to as Ai14) were purchased from the Jackson Laboratory (stock #007914, Bar Harbor, ME). Experimental animals for RNA-seq, Gfi1Cre/+;RiboTagHA/HA, were obtained by crossing RiboTag mice with Gfi1-Cre mice. Animals for immunostaining, Gfi1Cre/+;Ai14, were obtained by crossing Gfi1-Cre mice with Ai14 mice [24]. All procedures involving animals were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and have been approved by the Institutional Animal Care and Use Committee at the University of Maryland, Baltimore (protocol numbers 1015003 and 0915006).
Ribosome immunoprecipitation and RNA extraction
Three 30-day old Gfi1Cre/+;RiboTagHA/HA mice were euthanized by CO2 asphyxiation followed by cervical dislocation. Livers were harvested and immediately frozen on dry ice. Equal amounts of liver were used for input RNA extractions (RNeasy Plus Micro kit, QIAGEN USA, Germantown, MD, USA) or futher processed for ribosome immunoprecipitation (5 μg of purified anti-HA.11, BioLegend, San Diego, CA, USA) followed by RNA extraction as previously decribed in Sanz et al., 2009 [16]. The RNeasy Plus Micro kit is optimized for the removal of genomic DNA through a combination of high salt buffer and the gDNA Eliminator spin column. Quality of the RNA was assessed on an Agilent Technologies Bioanalyzer 2100 RNA pico chip as per the manufacturer’s instructions (Agilent Technologies, Palo Alto, CA, USA). All samples had a RIN score of 10 and no evidence of DNA contamination in the form of a high molecular weight DNA band. All RNA was equally aliquoted to test for the performance of five commercial kits and seven protocols.
Real-time RT-PCR
Efficiency of the ribosome immunoprecipitation was assessed by reverse transcription followed by real time PCR. One nanogram of total RNA from the input and the IP samples was used for reverse transcription using the Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Thermo Fisher Scientific, Waltham, MA, USA). The real time PCR was performed on an Applied Biosystems® StepOnePlus™ Real-Time PCR System with the Maxima SYBR Green/ROX qPCR Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) and the following primers: Gapdh-Fw 5’-GGAGAAACCTGCCAAGTATGA-3′; Gapdh-Rv 5′- TCCTCAGTGTAGCCCAAGA-3′; Gfi1-Fw 5′- AATGCAGCAAGGTGTTCTC-3′; Gfi1-Rv 5′- CTTACAGTCAAAGCTGCGT-3′.
Immunostaining
Progeny from a cross between Gfi1Cre/+ mice and TdTomato reporter mice Ai14 were euthanized at P1 and their livers were harvested. Following fixation in 4% paraformaldehyde overnight at 4 °C, livers were cryoprotected through incubation in PBS with increasing amount of sucrose before being embedded in O.C.T. compound (Scigen, Gardena, CA, USA). Ten μm cryosections were permeabilized with PBS supplemented with 0.2% Tween-20 for 1 h at room temperature and incubated with Alexa Fluor™ 488 Phalloidin (1/800, Thermo Fisher Scientific, Waltham, MA, USA) and DAPI (1/20,000, Thermo Fisher Scientific, Waltham, MA, USA). Samples were mounted with ProLong Gold antifade reagent (Thermo Fisher Scientific, Waltham, MA, USA). Images were acquired using a Nikon Eclipse E600 microscope (Nikon, Tokyo, Japan) equipped with a Lumenera Infinity 3 camera (Lumenera, Ottawa, ON).
RNA-Seq library construction
Below are the experimental methods for RNA-Seq library construction. We followed the manufacturer’s instructions with minor modifications, as noted below. The shearing approach was not altered and remains different between kits.
Ovation® RNA-Seq system V2 combined with TruSeq RNA library prep kit v2
We performed a hybrid library preparation by using Ovation® RNA-Seq System V2 (NuGEN, San Carlos, CA, USA) to synthesize cDNA and the TruSeq RNA Library Preparation Kit v2 to construct the sequencing library (Illumina, San Diego, CA, USA), consistent with the NuGEN manufacturer protocol (See Additional file 1: Table S1). Briefly, 4 ng of total RNA or RiboTag IP RNA were used to synthesize cDNA following the NuGEN’s instructions. Subsequently, 200 ng of cDNA were sheared to an average size of 300 bp with a Covaris E220 Focused-Ultrasonicator (Covaris Inc., Woburn, MA, USA). Following the manufacturer protocol, the library was prepared from the sheared cDNA using the Illumina TruSeq RNA Library Prep Kit with 8 cycles of PCR.
SMARTer® stranded total RNA-Seq kit-Pico input mammalian
Four nanograms of RNA were used as input material and libraries were prepared by following the SMARTer Stranded Total RNA-Seq kit-Pico Input Mammalian user manual (Takara Bio USA, Mountain View, CA, USA). In brief, samples were fragmented at 94 °C for 4 min prior to first-strand synthesis. Illumina adaptors and indexes were added to single-stranded cDNA via 5 cycles of PCR. Libraries were hybridized to R-probes for fragments originating from ribosomal RNA to be cleaved by ZapR. The resulting ribo-depleted library fragments were amplified with 15 cycles of PCR.
SMART-Seq® v4 ultra® low input RNA kit for sequencing
Two types of libraries were prepared by using 4 ng or 250 pg RNA from each sample. Libraries were prepared by following the SMART-Seq v4 Ultra Low Input RNA Kit (Takara Bio USA, Mountain View, CA, USA) user manual. The cDNA was amplified with 11 cycles of PCR. Nextera XT kit (Nextera XT DNA Library Preparation Kit (Illumina, San Diego, CA) was used to make cDNA libraries suitable for Illumina sequencing.
TruSeq RNA library prep kit v2
Two types of libraries were prepared by using 70 ng or 4 ng RNA from each sample. The 70 ng libraries were built using TruSeq RNA Library Prep Kit v2 (Illumina, San Diego, CA, USA) according to the manufacturer protocol. Size selection was performed using SPRIselect beads (Beckman Coulter, Indianapolis, IN, USA) and in-house calibration values (first round selection to select the upper or right limit of the distribution), salt unit equals to 0.427 and second round selection to select the lower or left limit of the distribution, salt unit = 0.455). The cDNA was amplified with 19 cycles of PCR. Libraries were prepared using 4 ng of RNA with modifications to the standard protocol by reducing the end-repair reaction to 1/2 the recommended amounts of enzyme mix and sample volume. In addition A-tail ligation followed the standard protocol without the use of internal control mixes. Due to the low input amount, no size selection was applied to the 4 ng libraries. The cDNA was amplified with 22 cycles of PCR. Libraries prepared using 70 ng of RNA were prepared following the standard protocol and cDNA was amplified with 14 cycles as suggested by manufacture protocol.
NEBNext® ultra™ directional RNA library prep kit for Illumina
Four nanograms of total RNA were used for NEBNext® Ultra™ Directional RNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA). Poly-A selection and cDNA synthesis were performed according to NEB protocol. The adaptors were diluted with a 1:30 ratio instead of the recommended 1:10 ratio. Size selection was performed using SPRIselect beads. (Beckman Coulter, Indianapolis, IN, USA) with in-house calibration values. The cDNA was amplified with 22 cycles of PCR.
Sequencing
Samples prepared by TruSeq, NEB, NuGEN and SMARTer were sequenced at the Institute for Genome Sciences (IGS) Genomics Resource Center (Baltimore, MD) on a HiSeq 4000 using 75 base read lengths in paired-end mode. Samples prepared by SMART-Seq v4 were sequenced by the Genomics and Computational Biology Core (GCBC) at the National Institute on Deafness and Other Communication Disorders (NIDCD/NIH) on a HiSeq 1500 using a read length of 126 bases in paired-end mode.
RNA-Seq analyses
The Illumina adapters used during the library construction were removed from the reads using Trimmomatic [1]. In order to reduce the impact of lower quality reads on the alignment, all reads were trimmed to 60 bp using the FASTX Toolkit v-0.0.13 [25] resulting in a Phred-Quality-Score greater than 30. The reads generated for each RNA sample were analyzed and compared using an Ergatis-based RNA-Seq analysis pipeline [26] where sequencing reads were aligned and annotated to the UCSC mouse reference genome (mm10, GRCm38.84) from Ensembl (http://www.ensembl.org) using TopHat v-2.0.8 [27] (maximum number of mismatches = 2; segment length = 30; maximum multi-hits per read = 25; maximum intron length = 50,000) and the number of reads that aligned to the predicted coding regions were determined using HTSeq [28]. Bedtools (v-2.7.1) [29] was used to count the reads mapping to exons according to Ensembl gene annotations (March 2016, Mus_musculus.GRCm38.84, with 47,729 genes annotated). Read counts per million mapped reads values (CPM) [28] or reads per kilobase of transcripts per million mapped reads (RPKM) [30] were calculated and used for downstream analyses. 5′-3′ exonic coverage was calculated using CollectRnaSeqMetrics component of picard-tools (v-1.60, https://broadinstitute.github.io/picard/), and duplication rate was calculated using EstimateLibraryComplexity of Picard-tools.
Statistical analysis
All plots were generated using R (v-3.2.4), including the following R packages ggplot2, ComplexHeatmap for producing bar plots or heat maps, and limma to generate Venn diagrams. The difference among groups in boxplots was evaluated based on the overlapping of the notch region [31]. The notch is defined as median m ± 1.58IQR/√n [31]. The significance test is evaluated using a non-parametric Wilcoxon test with p < 0.05.
Accession number
All of the processed gene expression data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) under accession number GSE104213.
Results
Sample preparation for sequencing
In order to evaluate the efficiency of different library preparation kits with low amounts of RNA obtained after ribosome immunoprecipitation, we crossed RiboTag mice with Gfi1-Cre mice to obtain progeny that expressed HA-tagged ribosomes in cells with Gfi1 expression. We obtained RNA from liver because it is a tissue that, at least during embryogenesis, expresses Gfi1, thus allowing for early induced recombination in a subset of the liver cells for permanent expression of a reporter gene [32] (Additional file 2: Figure S1a). Additionally, the size of the liver would provide enough material to test five different kits with varying amounts of starting RNA from individually processed samples. Livers were processed for HA-tagged ribosome immunoprecipitation (IP) followed by RNA extraction as previously described [16]. Prior to sequencing, the efficacy of the IP was confirmed by comparing the level of Gfi1 transcripts in the input and IP samples using real time RT-PCR (Additional file 2: Figure S1b). The profiles generated by the five different commercial library preparition kits, from four different manufacturers, were compared in this study (See Additional file 1: Table S1). NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina (NEB) with 4 ng of RNA,NuGEN Ovation® RNA-Seq System V2 with 4 ng of RNA (NuGEN) with 4 ng of RNA, TaKaRa SMARTer® Stranded Total RNA-Seq Kit-Pico Input Mammalian with 4 ng of RNA (SMARTer), TaKaRa SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing with 4 ng and 0.25 ng of RNA (SMARTseq4 and SMARTseq0.25) and Illumina TruSeq RNA Library Prep Kit v2 with 4 ng and 70 ng of RNA (TruSeq4 and TruSeq70). Of these kits only the SMARTer kit produced strand specific libraries and we therefore did not analyze the data for strandness.
Comparison of mapping efficiency and duplication rate
The number of reads varied widely among samples being prepared by different library preparation kits. Input RNA samples generated 14.7 to 122 million pair-end reads (2 × 60 bp) and IP RNA samples generated 12 to 108 million pair-end reads (2 × 60 bp). Overall, fewer raw/mapped reads were generated when using the NuGEN kit. Of the raw reads, 12.5 to 111 million reads mapped to mouse genome for input RNA samples while 9.2 to 94.6 million reads mapped for IP RNA samples (Fig. 1a).
Fig. 1.

Descriptive characteristics of raw and mapped reads. a Total number of raw reads and number of reads mapped to the mouse genome (mm10, GRCm38.84). b Percentage of reads mapped to exonic, intronic and intergenic regions. NEB: NEBNext® Ultra™, NuG: NuGEN Ovation®, SMTer: SMARTer® Stranded; Tru4: TruSeq using 4 ng of RNA; Tru70: TruSeq using 70 ng of RNA. SMTseq4: SMART-Seq® v4 using 4 ng of RNA; SMTseq0.25: SMART-Seq® v4 using 250 pg of RNA
In order to evaluate the expression profile composition and library complexity, we assessed the duplication rate of the read pairs (Table 1) as lower duplication rates usually indicate a higher complexity of the sample and better representation of RNA present in a sample [20]. In this study, duplication rate ranged from 26 to 99% (Table 1). However, the duplication we observed was not well correlated with the numbers of PCR cycles and was more dependent on the library prep kit. For instance, while NEB and TruSeq4 samples both had the highest number of PCR cycles (22), their duplication rates differed (Table 1). Indeed, NEB-input samples had the highest duplication rate of 99% with the overall largest number of reads duplicated more than 200 times while the TruSeq4 samples had a duplication rate of 72% with a substantially smaller number of reads with greater than 200 duplications (Table 1 and Additional file 3: Figure S2).
Table 1.
Duplication rate of libraries prepared by different kits
| Kit | Sample type | PCR cycles for lib prep | Percent_Duplication (%) | |||
|---|---|---|---|---|---|---|
| replicate | avg | |||||
| 1 | 2 | 3 | ||||
| NEBa | Input | 22 | 97 | 99 | 97 | 97 |
| IP | 99 | 99 | 99 | 99 | ||
| NuG/Trub | Input | 8 | 52 | 53 | 52 | 53 |
| IP | 36 | 31 | 26 | 31 | ||
| SMTerc | Input | 15 | 83 | 86 | 85 | 85 |
| IP | 86 | 83 | 68 | 79 | ||
| Tru4d | Input | 22 | 67 | 77 | 71 | 72 |
| IP | 85 | 82 | 99 | 89 | ||
| Tru70e | Input | 19 | 90 | 94 | 92 | 92 |
| IP | 98 | 97 | 96 | 97 | ||
| SMTseq4f | Input | 11 | 40 | 38 | 40 | 40 |
| IP | 60 | 49 | 48 | 52 | ||
| SMTseq0.25f | Input | 11 | 37 | 37 | 41 | 38 |
| IP | 59 | 50 | 47 | 52 | ||
aNEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina
bNugen Ovation® RNAseq System v2
cSMARTer® Stranded Total RNA-Seq Kit-Pico Input Mammalian User
dTruSeq® RNA sample preparation v2,4 ng
eTruSeq® RNA sample preparation v2,70 ng
fSMART-Seq v4 Ultra Low Input RNA Kit
Detection of exonic, intergenic and intronic regions
Among the mapped reads, SMARTer samples showed the lowest alignment to exonic regions. The percentage of reads aligned to exonic regions was greater than 85% in samples prepared with NEB, TruSeq and SMARTseq library kits and less than 70% in samples prepared with the SMARTer and NuGEN kits (Fig. 1b). As expected, the overall percentage of reads aligning to intronic regions detected in input samples was less than 10% for most samples, except for samples prepared by the SMARTer kit, where more than 20% of the reads align to intronic regions. IP samples had roughly twice as many reads aligning to intronic regions, or 10% more intronic reads compared with the corresponding input RNA samples, which may suggest that the IP captures some immature mRNAs. In particular, the percentage of intronic reads from the SMARTer samples increased from 22% for the input RNA to 41% for the IP RNA. The percentage of intronic reads for the NuGEN samples ranged from 8% for the input RNA and 22% for the IP RNA.
Number of genes being detected as expressed
Because of the differences in sequencing efficiency and library complexity, we examined the number of features detected in samples prepared with each library kit. After removing ambiguous reads or reads mapped to multiple features using HTSeq, we detected between 10,184 to 21,161 genes where the CPM values were greater than zero (Table 2). The corresponding average raw read counts ranged from 0.76 to 37 million reads. Fewer features were detected in SMARTer and NEB samples. All of the annotated genes (47,729) were binned into 6 groups (RPKM≤1, 1 < RPKM≤10, 10 < RPKM≤100, 100 < RPKM≤1000, 1000 < RPKM≤10,000 and RPKM> 10,000. Fig. 2). SMARTer, NuGEN, and NEB samples had more genes that were entirely missed or had low expression levels (RPKM≤1) in comparison to the other kits in the input samples (Fig. 2a). The SMARTer and NEB samples had more genes with a lower expression levels (RPKM≤1) also in the IP (Fig. 2c). The number of genes within RPKM range (100–10,000) was relatively low in SMARTer and NuGEN samples (Fig. 2b, d). Conversely, SMARTer samples contained more highly expressed genes (RPKM> 10,000) than others samples, but the majority of these were rRNA genes or genes encoding for hypothetical proteins (Fig. 2b, d and See Additional file 1: Table S2).
Table 2.
Number of features with CPM > 0
| Kit | Sample type | Number of features with coverage (CPM > 0) | Number of reads of all expressed features (CPM > 0) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Replicate | avg | Replicate | avg | ||||||
| 1 | 2 | 3 | 1 | 2 | 3 | ||||
| NEB | Input | 13,543 | 12,464 | 11,419 | 12,475 | 22,373,163 | 26,292,017 | 22,305,929 | 23,657,036 |
| IP | 11,059 | 12,165 | 14,331 | 12,518 | 16,668,355 | 27,323,849 | 15,122,257 | 19,704,820 | |
| NuG | Input | 16,946 | 16,458 | 15,624 | 16,343 | 5,529,417 | 7,615,866 | 7,482,819 | 6,876,034 |
| IP | 17,715 | 17,841 | 18,198 | 17,918 | 3,223,153 | 5,319,712 | 5,141,329 | 4,561,398 | |
| SMTer | Input | 12,281 | 10,184 | 10,929 | 11,131 | 759,221 | 762,562 | 750,159 | 757,314 |
| IP | 12,422 | 11,168 | 15,684 | 13,091 | 701,059 | 645,414 | 464,199 | 603,557 | |
| Tru4 | Input | 19,906 | 19,863 | 19,631 | 19,800 | 35,368,451 | 45,466,334 | 31,299,550 | 37,378,112 |
| IP | 17,467 | 18,899 | 21,161 | 19,176 | 36,785,680 | 28,571,125 | 18,031,264 | 27,796,023 | |
| Tru70 | Input | 15,957 | 15,577 | 16,082 | 15,872 | 32,103,296 | 33,797,108 | 25,797,283 | 30,565,896 |
| IP | 14,430 | 15,213 | 15,680 | 15,108 | 31,150,532 | 29,799,544 | 27,902,766 | 29,617,614 | |
| SMTseq4 | Input | 19,111 | 19,287 | 19,630 | 19,343 | 23,573,272 | 24,615,731 | 30,217,161 | 26,135,388 |
| IP | 19,835 | 20,551 | 20,488 | 20,291 | 14,407,184 | 19,577,709 | 17,262,557 | 17,082,483 | |
| SMTseq0.25 | Input | 16,834 | 16,742 | 16,117 | 16,564 | 23,815,261 | 24,347,672 | 22,951,893 | 23,704,942 |
| IP | 16,827 | 16,870 | 16,527 | 16,741 | 13,971,566 | 15,047,219 | 14,691,099 | 14,569,961 | |
Fig. 2.
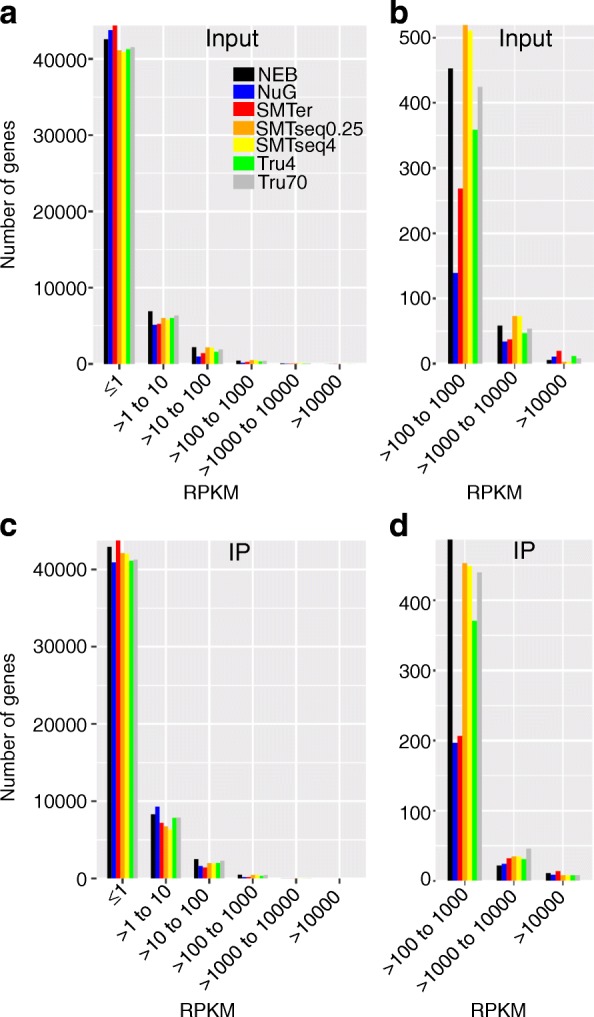
Detected transcripts binned by RPKM. There were 33,399 transcripts detected in at least one input sample, 35,395 transcripts detected in at least one IP sample and all of these genes were used for the binning. Y-axis: number of transcripts detected in at least one replicate. a and c Number of genes binned by RPKM as detected by the different kits in the input and IP, respectively. b and d These plots are subsets of (a) and (c) focusing only on highly expressed transcripts (RPKM> 100)
In order to compare the similarity of expression profiles of the different kits, we compared genes with at least 1 CPM in at least one replicate across all the samples. More than 60% of the genes were co-detected by all kits (Fig. 3). The median CPM for shared genes across all samples was 28 for input samples (Fig. 3a, c) and 36 for IP samples (Fig. 3b, d). Meanwhile, less than 10% of features were uniquely detected in NEB, NuGEN and TruSeq input samples, but over 20% of features were uniquely detected from the SMARTer samples. The median CPM of uniquely detected genes in SMARTer input samples was around 10, while the median for other kits was less than 3. A similar trend is observed in IP samples (Fig. 3b, d and See Additional file 1: Table S3).
Fig. 3.

Venn diagrams of identified features in the different libraries. The features with CPM ≥ 1 in at least one out of 3 replicates were used to generate these plots. a and c represent input samples and b and d represent IP samples. Most transcripts were detected by all kits tested. However, a higher rate of agreement is seen between the NEB, TruSeq and SMART-Seq prepared samples. NEB: NEBNext® Ultra™, NuG: NuGEN Ovation®, SMTer: SMARTer® Stranded; Tru4: TruSeq using 4 ng of RNA; Tru70: TruSeq using 70 ng of RNA. SMTseq4: SMART-Seq® v4 using 4 ng of RNA; SMTseq0.25: SMART-Seq® v4 using 250 pg of RNA
We also grouped all of the detected features into ribosomal RNA, non-coding (ribosomal RNA, lincRNA, microRNA) and protein-coding groups. The average CPM of ribosomal RNA and non-coding gene groups were 2-fold higher in NuGEN and SMARTer samples than in other samples (Table 3). Conversely, the average CPMs for the protein-coding group were similar across most samples, except for SMARTer prepared samples (Table 4). By comparing IP samples with input samples, it is interesting that the CPMs of IP samples are relatively lower than input samples, except for NuGEN prepared samples.
Table 3.
Descriptive statistic of non-coding detected features (CPM > =1)
| Kit | Sample type | Total features of ribosomal RNA | Average CPM of ribosomal RNA | Total features of ribosomal RNA, lincRNA, microRNA | Average CPM of ribosomal RNA, lincRNA, microRNA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Replicate | Replicate | Replicate | Replicate | ||||||||||
| 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | ||
| NEB | Input | 3 | 3 | 3 | 73.87 | 67.33 | 72.15 | 233 | 186 | 140 | 46.01 | 29.22 | 28.34 |
| IP | 3 | 3 | 4 | 14.24 | 6.15 | 7.80 | 128 | 157 | 268 | 78.05 | 28.33 | 34.25 | |
| NuG | Input | 8 | 11 | 10 | 5533.70 | 5571.81 | 5603.65 | 552 | 488 | 415 | 272.73 | 274.17 | 276.54 |
| IP | 13 | 11 | 17 | 1803.34 | 820.73 | 863.91 | 583 | 593 | 637 | 97.91 | 48.26 | 51.37 | |
| SMTer | Input | 8 | 5 | 6 | 4282.62 | 7858.83 | 4641.15 | 440 | 359 | 377 | 247.75 | 429.65 | 261.04 |
| IP | 12 | 6 | 17 | 4880.27 | 7906.14 | 3936.78 | 467 | 411 | 599 | 282.51 | 429.64 | 240.08 | |
| Tru4 | Input | 17 | 17 | 13 | 232.50 | 120.94 | 169.47 | 804 | 786 | 767 | 13.96 | 7.59 | 10.29 |
| IP | 8 | 7 | 19 | 415.93 | 129.19 | 168.59 | 546 | 650 | 927 | 26.49 | 8.94 | 11.45 | |
| Tru70 | Input | 6 | 4 | 4 | 102.39 | 28.55 | 58.50 | 410 | 363 | 406 | 6.34 | 2.08 | 3.81 |
| IP | 3 | 3 | 4 | 141.53 | 54.91 | 65.21 | 265 | 295 | 341 | 9.24 | 3.97 | 5.10 | |
| SMTseq4 | Input | 13 | 13 | 21 | 214.51 | 159.76 | 185.17 | 750 | 764 | 803 | 11.78 | 8.93 | 10.54 |
| IP | 7 | 11 | 10 | 851.62 | 63.75 | 92.92 | 844 | 881 | 889 | 46.39 | 5.91 | 8.73 | |
| SMTseq0.25 | Input | 6 | 7 | 3 | 224.50 | 191.46 | 235.20 | 531 | 515 | 461 | 12.32 | 10.52 | 13.04 |
| IP | 6 | 6 | 7 | 886.27 | 73.72 | 99.28 | 476 | 502 | 457 | 48.03 | 6.43 | 8.88 | |
Table 4.
Descriptive statistic of protein-coding detected features (CPM ≥ 1)
| Kit | Sample type | Total features of protein-coding RNA | Average CPM of protein-coding RNA | ||||
|---|---|---|---|---|---|---|---|
| Replicate | Replicate | ||||||
| 1 | 2 | 3 | 1 | 2 | 3 | ||
| NEB | Input | 11,316 | 10,551 | 9789 | 36.51 | 37.77 | 37.83 |
| IP | 8740 | 10,252 | 12,435 | 34.13 | 37.84 | 37.39 | |
| NuG | Input | 10,921 | 10,828 | 10,656 | 19.61 | 19.50 | 19.32 |
| IP | 12,970 | 13,142 | 13,318 | 32.65 | 36.35 | 36.12 | |
| SMTer | Input | 11,136 | 9368 | 10,019 | 21.47 | 7.90 | 20.48 |
| IP | 11,826 | 11,860 | 11,824 | 18.88 | 7.90 | 22.02 | |
| Tru4 | Input | 11,826 | 11,860 | 11,824 | 38.90 | 39.38 | 39.18 |
| IP | 12,360 | 12,842 | 12,941 | 37.97 | 39.28 | 39.09 | |
| Tru70 | Input | 11,690 | 11,691 | 11,611 | 39.47 | 39.79 | 39.66 |
| IP | 11,996 | 12,493 | 12,660 | 39.26 | 39.65 | 39.56 | |
| SMTseq4 | Input | 11,659 | 11,499 | 11,578 | 39.07 | 39.28 | 39.16 |
| IP | 12,443 | 12,579 | 12,587 | 36.50 | 39.51 | 39.01 | |
| SMTseq0.25 | Input | 11,500 | 11,475 | 11,384 | 39.03 | 39.16 | 38.97 |
| IP | 12,648 | 12,846 | 12,940 | 36.38 | 39.47 | 39.29 | |
Coverage uniformity relative to 5′ and 3′ ends
The evenness of transcript coverage was calculated by dividing the mean coverage of first (last) 100 bases (5′ or 3′) of transcripts divided by the mean coverage of all bases across the corresponding transcript (Fig. 4). The median was calculated and plotted for the 1000 most highly expressed transcripts. Most of the input RNA samples showed even coverage from 5′ to 3′ end, except for all NuGEN samples which had pronounced increase in coverage at the 3′ end. Additionally, consistently higher coverage at the 3′ end was observed among IP RNA samples, except for SMARTer samples with even coverage across 5′ and 3′ extremities.
Fig. 4.
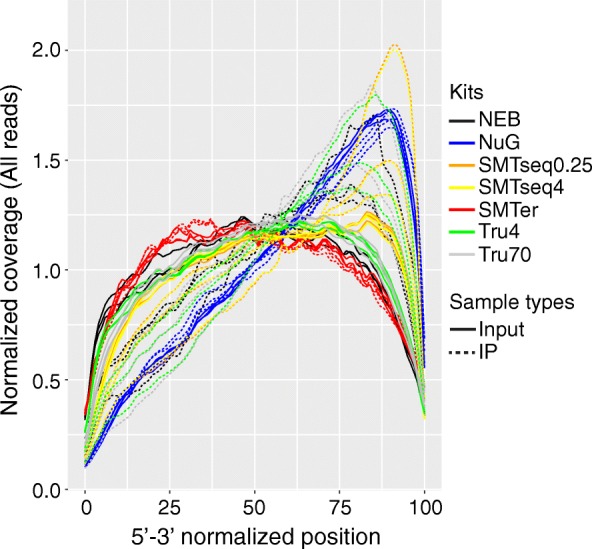
Distribution of normalized mean expression of the first (last) 100 bases of transcripts (in 5′- > 3′-orientation). X axis represents the 5′-3′ normalized position; Y axis represents normalized coverage. NEB: NEBNext® Ultra™, NuG: NuGEN Ovation®, SMTer: SMARTer® Stranded; Tru4: TruSeq using 4 ng of RNA; Tru70: TruSeq using 70 ng of RNA. SMTseq4: SMART-Seq® v4 using 4 ng of RNA; SMTseq0.25: SMART-Seq® v4 using 250 pg of RNA. Yellow and orange: SMTseq samples; Red: SMTer samples; Black: NEB samples; Blue: NuG samples; Green and grey: TruSeq samples. Solid: Input samples. Dotted: Ribo-IP samples
Similarity of expression profiles
In order to assess the similarity of expression profiles being generated by different library preparation kits, we applied Spearman correlation coefficients as a measure of similarity. The Spearman coefficient was calculated based on the rank of the CPM value as opposed to using the absolute values. This was done to accommodate the difference in CPM values due to differences in duplication rates observed among the kits (Fig. 5). The correlation coefficient for input samples ranged from 0.5 to 0.9, where SMARTseq profiles were better correlated with TruSeq70 than others (Spearman correlation coefficient ≥ 0.9). SMARTer samples had the lowest correlation (0.5) with the control library TruSeq70 (Fig. 5 and See Additional file 1: Table S4). Overall, as expected, input profiles are less correlated to corresponding IP profiles (See Additional file 1: Table S4). When we compared input samples with corresponding IP samples for each individual kit respectively, all input samples were clustered separately from IP samples except for the SMARTer samples (Additional file 4: Figure S3).
Fig. 5.

Hierarchical clustering of expression levels, based on the rank of the count of exon per million mapped reads (CPM). Dendrogram represents Spearman correlation coefficients between pairs of samples. NEB: NEBNext® Ultra™, NuG: NuGEN Ovation®, SMTer: SMARTer® Stranded; Tru4: TruSeq using 4 ng of RNA; Tru70: TruSeq using 70 ng of RNA. SMTseq4: SMART-Seq® v4 using 4 ng of RNA; SMTseq0.25: SMART-Seq® v4 using 250 pg of RNA. Yellow and orange: SMTseq samples; Red: SMTer samples; Black: NEB samples; Blue: NuGEN samples; Green and grey: TruSeq samples.. Color scale: Spearman correlation coefficients
Although two different amounts of RNA were used for the TruSeq library kit, TruSeq 4 ng samples were well correlated with TruSeq 70 ng samples (Spearman correlation coefficient was 0.96 ± 0.002 for input; 0.946 ± 0.01 for IP). Similarly, the SMARTseq samples with 0.25 ng and 4 ng were highly correlated (Spearman correlation coefficient was 0.95 ± 0.004 for input and 0.95 ± 0.005 for IP) (See Additional file 1: Table S4).
Transcript enrichment is better represented than transcript depletion in the IP samples
We evaluated the robustness of different kits for detecting enrichment (IP/input RNA > 1) or depletion (IP/input RNA < 1) of transcripts in the translatome (IP samples) compared to the transcriptome (input samples). Features with raw read counts ≥20 in input samples and with an enrichment or depletion factor ≥ 2 were included as enriched (IP/Input ≥2) or depleted transcripts (IP/Input ≤0.5). Of note, more transcripts were enriched than depleted (Fig. 6a and Additional file 5: Figure S4). NuGEN produced the greatest number of enriched transcripts (mean 4270) and smallest number of depleted transcripts (mean 74) as compared with other kits (Fig. 6b). Among the enriched transcripts from NuGEN, 60% were enriched less than 4-fold whereas only 25% of transcripts prepared by other kits were enriched less than 4-fold (Fig. 6b). NEB samples had the highest percentage of enriched/depleted transcripts (log2 (IP/INPUT) > 5 or log2(INPUT/IP) > 5) when compared to samples obtained from the other kits (Fig. 6b, Fig. 7a). Conversely, the enrichment profile of the SMARTer samples showed fewer enriched or depleted transcripts compared with the rest of the samples. Indeed, when plotting for the top 50 enriched transcripts (Fig. 7b), the median enrichment value for the SMARTer profile was significantly lower than other profiles (p < 0.05).
Fig. 6.

Descriptive characteristic of enrichment or depletion profiles as generated by the different library preparation kits. Genes which have at least 20 raw reads in the input samples and a ratio of IP/Input ≥2 or Input/IP ≥2 were used to generate the plots. a Total number of transcripts enriched or depleted. b Percentage of enriched or depleted transcripts grouped into different bins. X-axis: log2(IP/input), Y-axis: percentage of genes in each bin over whole population. NEB: NEBNext® Ultra™, NuG: NuGEN Ovation®, SMTer: SMARTer® Stranded; Tru4: TruSeq using 4 ng of RNA; Tru70: TruSeq using 70 ng of RNA. SMTseq4: SMART-Seq® v4 using 4 ng of RNA; SMTseq0.25: SMART-Seq® v4 using 250 pg of RNA. Yellow and orange: SMTseq samples; Red: SMTer samples; Black: NEB samples; Blue: NuGEN samples; Green and grey: TruSeq samples
Fig. 7.

Enrichment profiles and top 50 enriched transcripts. a Enrichment factor of transcripts are sorted in decreasing order based on log2 (IP/input). X-axis:transcripts, Y-axis:log2 value of enrichment (IP/Input). b Boxplot of top 50 enriched transcripts. NEB: NEBNext® Ultra™, NuG: NuGEN Ovation®, SMTer: SMARTer® Stranded; Tru4: TruSeq using 4 ng of RNA; Tru70: TruSeq using 70 ng of RNA. SMTseq4: SMART-Seq® v4 using 4 ng of RNA; SMTseq0.25: SMART-Seq® v4 using 250 pg of RNA. Yellow and orange: SMTseq samples; Red: SMTer samples; Black: NEB samples; Blue: NuGEN samples; Green and grey: TruSeq samples
We also compared the number of transcripts being enriched or depleted across samples (Additional file 6: Figure S5). NuGEN had the highest number of uniquely enriched transcripts that were detected (accounting for 25% of its total enriched transcripts, 95% of which are protein-coding genes). TruSeq4 and TruSeq70 had around 5% uniquely enriched transcripts (Additional file 6: Figure S5a,b).
We also clustered all the transcripts based on the rank of enrichment factor or depletion factor greater than 2 in at least one sample (Fig. 8). As expected, the profiles for TruSeq4 and TruSeq70 were most similar to each other (Spearman correlation coefficient > 0.7). The same is true for SMARTseq4 and SMARTseq0.25. On the other hand, the enrichment/depletion profile for SMARTer was the least similar to the other profiles (Spearman correlation coeffienct < 0.2).
Fig. 8.
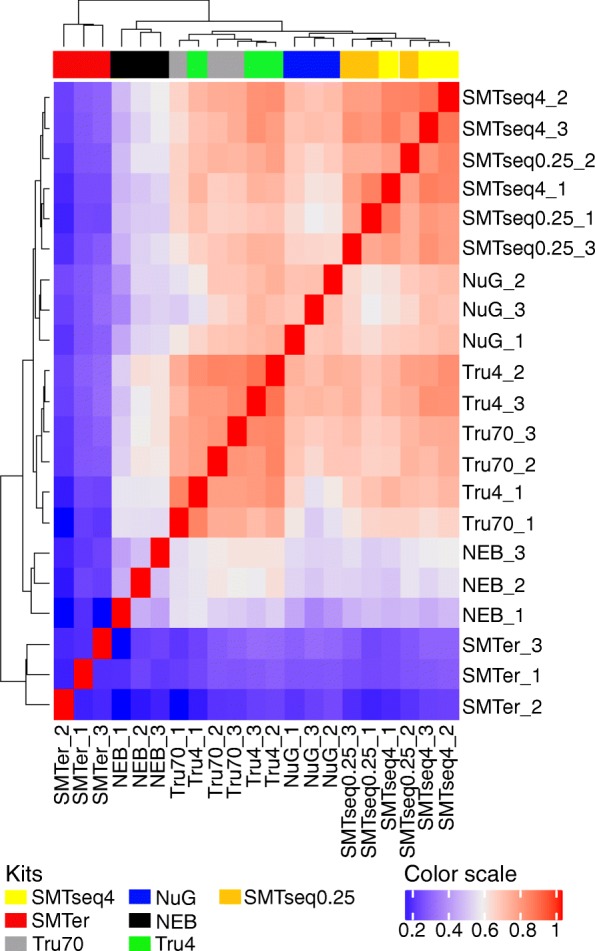
Hierarchical clustering based on the rank of IP/input value. Dendrogram represents Spearman correlation coefficients between pairs of samples. NEB: NEBNext® Ultra™, NuG: NuGEN Ovation®, SMTer: SMARTer® Stranded; Tru4: TruSeq using 4 ng of RNA; Tru70: TruSeq using 70 ng of RNA. SMTseq4: SMART-Seq® v4 using 4 ng of RNA; SMTseq0.25: SMART-Seq® v4 using 250 pg of RNA. Yellow and orange: SMTseq samples; Red: SMTer samples; Black: NEB samples; Blue: NuGEN samples; Green and grey: TruSeq samples
Transcript-length dependent enrichment/depletion
We examined whether the enrichment or depletion effects observed in the translatome were affected by the transcript length. Based on size distribution of the enriched/depleted transcripts (the majority being between 0.5 and 10 kb, Additional file 7: Figure S6), we grouped the transcripts into four bins (≤0.5 kb, 0.5–1 kb, 1 kb–10 kb, and > 10 kb) (Fig. 9). The median enrichment for transcripts was relatively higher in the longer transcript (> 10 kb) except in TruSeq70 samples (Fig. 9a). Within each transcript length bin, the median enrichment effect from NuGEN and SMARTer samples was much higher than TruSeq70 samples for transcripts less than 10 kb (Fig. 9b). For longer transcripts (> 10 kb), NEB, NuGEN, SMARTer and SMARTseq samples had a median enrichment that is much higher than those of TruSeq70 (Fig. 9b). Additionally, the enrichment effect for NEB samples distributed wider than all the other samples (Fig. 9).
Fig. 9.

Boxplot and violin plots of enriched features over different transcript lengths. a Boxplots of enrichment factor of different transcripts within each kit. b Boxplots of enrichment factor of different kits at each transcript length bin. The notch represents the median ± 1.58IQR/√n. The width of boxplot is proportion to sample size of each group
A similar trend was also observed in depleted transcripts. Across all transcript lengths, the range of depletion effect for NuGEN and SMARTer samples was less than for other samples (Fig. 10a). For NEB samples, the depletion effect distribution was wider than all the other samples (Fig. 10b). For longer transcripts (> 10 kb), NEB, NuGEN, SMARTer and SMARTseq samples showed fewer depletion effects than those from TruSeq70 (Fig. 10b).
Fig. 10.

Boxplot and violin plots of depletion features over different transcript lengths. a Boxplots of depletion factor of different transcripts within each kit. b Boxplots of depletion factor of different kits at each transcript length bin. The notch represents the median ± 1.58IQR/√n. The width of boxplot is proportion to sample size of each group
Discussion
In this study, we compared five library-preparation kits for RNA-seq, using low-quantity input RNA or RiboTag IP RNA, by applying a comprehensive set of quality measures. One of the major differences among library preparation kits was whether oligo (dT) is used to select mRNA. Among the kits tested, the NEBNext® Ultra™, the Illumina TruSeq® and the TaKaRa SMART-Seq® v4 Ultra® use oligo-dT primers to select for polyA mRNA. Conversely, the TaKaRa SMARTer® kit depends on locked nucleic acid (LNA) technology and random primers to capture both products with classical long polyA(+) and those with short poly(A) tails or polyA(−) transcripts and employs a ribosomal depletion step. Although the NuGEN Ovation® V2 kit uses a combination of semi-random hexamers and a poly-dT chimeric primer for 1st strand cDNA amplification in an effort to mitigate bias, 3′ end bias was still observed. Shanker, et al.(2015), also observed 3′ end bias using the NuGEN Ovation V2 kit with low input samples [22]. Interesingly, we observed greater 3′ end bias in IP samples (except for SMARTer prepared samples) than in the input samples, possibly suggesting some degradation of the RNA.
A higher percentage of reads mapped to the intronic and intergenic region in the samples derived from the TaKaRa SMARTer® kit in comparison to samples derived from the other five kits. Adiconis, et al., 2013 [23], also found a similar difference by comparing the SMARTer® kit to the TruSeq® kit. In our study, among the top-100 highly expressed transcripts in SMARTer samples, 30% were miRNA, lincRNA and rRNA. It is known that the source of miRNA or lincRNA is mainly from intergenic or intronic region, and that certain ribosomal RNAs generated by RNA polymerase I and III are without polyA tails. Therefore, we propose that the SMARTer® kit may be useful for studies which aim to focus on poly(A) negative transcripts or transcripts derived from non-exon-coding regions.
We observed lower duplication rates for the SMART-Seq® v4 Ultra (SMARTseq) and NuGEN prepared samples as compared to samples prepared with the other kits. A possible explanation to this observed lower duplication rate may relate to the protocols of these two kits. The mRNA is pre-amplifed to cDNA, before fragmentation, making the duplication rate resulting from the amplification harder to identify based on mapping position. Conversely, in other methods including the TruSeq kit, mRNA is fragmented first and the amplification only happens during the library construction step, making it easier to identify duplication based on the mapping position. For this study, the comparisons among different kits were achieved by using the rank-based method without removal of duplicate reads. Parekh et al., also showed that removal of duplicates improved neither accuracy nor precision and can actually worsen the power and the False Discovery Rate (FDR) for differential gene expression [33].
While Combs et al., (2015) [34] reported the use of the TruSeq kit with 100 ng of RNA, our modification of the TruSeq protocol provides the possibility to use the kit with RNA amounts as low as 4 ng. Indeed, our study, for the first time, shows that with protocol modifications, TruSeq with 4 ng of RNA performs similarly to the TruSeq with 70 ng of RNA with respect to the number of genes being captured and overall profile composition.
Comparing translatome (IP samples) against corresponding transcriptome (input samples), we find a relative higher intronic percentage in the translatome profiles, which might indicate that some non-mature RNA are precipitated during the IP process. Overall, we detected more enriched transcripts than depleted transcripts in the IP samples. Roh et al., report a similar result although the fold-change was greater in the depleted genes than the enriched genes [35]. This difference may result from the different tissues being used, and more specifically the percent of cells that express the tagged ribosomes. Among enriched transcripts, we observed an enrichment effect bias toward longer transcripts (> 10 kb) (Fig. 9a). This may relate to the nature of RiboTag IP since it is a method to detect polysome profiles during translation [16]. It is possible that the higher number of ribosomes on longer transcripts leads to a higher enrichment. In addition, the greater enrichment effect of longer transcript is slightly higer for samples prepared by SMARTer and SMARTseq kits. This may be related to the template-switch oligonucleotide with one locked nucleic acid (LNA) technique applied in these kits, which is aimed to improve the hybridization between the template-switch oligonucleotide and the cDNA product [36, 37], increasing full coverage for longer transcripts.
Conclusion
Amongst the kits and library prep protocols analyzed in this manuscript, SMART-Seq v4 and TruSeq offer the best sequencing results for libray preparation from smaller amounts of RNA as starting material. Indeed, the overall profile of 250 pg/4 ng samples from SMART-Seq v4 was similar to the TruSeq 70, here used as a gold-standard control. SMARTer Stranded Total RNA-Seq Kit might be a good choice to study both polyA(+) mRNA and non-polyA mRNA, especially non-coding RNAs. Since there is a coverage bias towards 3′ for IP samples and more enrichment for longer transcripts, correction should be included during comparison among samples, for example, using the bias correction function in Cufflink [38]. Finally, IP RNA from RiboTag samples is likely to include a higher rate of immature RNAs, given the observed increase in intronic sequences in the IP samples across all library prep approaches. Overall, we were able to observe both enriched and depleted transcripts of translatome profiles using all kits. Greater enrichment effects were detected than depletion, however this may be related to the percent of tagged ribosomes in the tissue and therefore tissue and Cre-driver specific. In summary, by considering the eveness of coverage, number of detected features, low CPM of non-coding genes, and similar enrichment profiles comparing to standard TruSeq70 prepared samples, the SMARTseq and NEB kits performed the best in comparison to the other kits tested. However, the SMARTseq kit had a lower duplication rate and allows reactions to start with as little as 250 pg, significantly decreasing the necessary amount of starting material. In addition, the modified TruSeq4 protocol provides good results based on the relative high number of detected features, low CPM of non-coding genes, and similarity of the enrichment profile to the standard TruSeq70.
Additional files
Table S1. Library preparation performed in this study. Table S2 Gene list with RPKM greater than 10,000. Table S3 Median of genes from unique and shared regions of Venn diagram. Table S4 Spearman correlation coefficients between different profiles. (XLSX 28 kb)
Figure S1. Cre recombination in liver cells expressing Gfi1. (a) Cryosection of liver from a cross between a Gfi1-Cre mouse and a TdTomato reporter mouse (Ai14) stained with DAPI and phalloidin. TdTomato is found in a subset of cells in the liver that is likely consistent with Kupffer or endothelial cells. (b) Enrichment of Gfi1 transcripts in the IP samples as compared to the input samples was assessed by reverse-transcription followed by real time PCR. (PDF 2947 kb)
Figure S2. Duplication rate of each library. X-axis: duplication rate, Y-axis: log10 of reads at different duplication rates. (PDF 622 kb)
Figure S3. Hierarchical clustering of expression levels, based on the rank of the count of exon per million mapped reads (CPM). Dendrograms represent Spearman correlation coefficients between pairs of samples that is 3 replicates for input and 3 replicates for IP. (PDF 233 kb)
{kind=link}
Figure S4. Bland–Altman plot (MA plot) of translatome (IP samples) and transcriptome (input samples) profiles for each kit. The red lines represent the boundary cutoff [− 1,1]. Dots above or below the red line represent the enriched or depleted transcripts. (JPG 587 kb)
Figure S5. Venn diagram of enriched/depleted transcripts (CPM ≥ 20 in at least one replicate, mean ratio of enrichement/depletion of the three replicates). The mean ratio IP/input is ≥2 or input/IP is ≥2. (PDF 1063 kb)
Figure S6. Histogram of length distribution for enriched or depleted transcripts. (PDF 353 kb)
Acknowledgements
We would like to thank Maggie Matern and Benjamin Shuster for critically reviewing this manuscript. This work was supported by R01DC03544 (NIDCD), R01DC013817 (NIDCD) and MR130240 (CDMRP/DoD).
Funding
This work was supported by R01DC03544 (NIDCD), R01DC013817 (NIDCD) and MR130240 (CDMRP/DoD). The funding sources provided the support for effort and materials, and did not have a role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the NCBI Gene Expression Omnibus (GEO) repository under accession number GSE104213 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104213.
The datasets supporting the conclusions of this article are included within the article and its additional files (Additional files 1, 2, 3, 4, 5, 6, and 7).
Abbreviations
- NEB
NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina with 4 ng of RNA
- NuGEN
NuGEN Ovation® RNA-Seq System V2 with 4 ng of RNA
- SMARTer
TaKaRa SMARTer® Stranded Total RNA-Seq Kit-Pico Input Mammalian with 4 ng of RNA
- SMARTseq4 and SMARTseq0.25
TaKaRa SMART-Seq® v4 Ultra® Low Input RNA Kit for Sequencing with 4 ng and 0.25 ng of RNA
- TruSeq4 and TruSeq70
Illumina TruSeq RNA Library Prep Kit v2 with 4 ng and 70 ng of RNA
Authors’ contributions
YS, BM, ETB, SO, XZ performed the experiments, YS, AS and AM performed the analysis, YS, BM and RH wrote the manuscript, AM, RJM, LT, LS and RH designed and oversaw the project. All the authors read and approved the final manuscript.
Ethics approval and consent to participate
All procedures involving animals were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and have been approved by the Institutional Animal Care and Use Committee at the University of Maryland, Baltimore (protocol numbers 1,015,003 and 0915006).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yang Song, Email: ysong@som.umaryland.edu.
Beatrice Milon, Email: BMilon@som.umaryland.edu.
Sandra Ott, Email: sott@som.umaryland.edu.
Xuechu Zhao, Email: XuZhao@som.umaryland.edu.
Lisa Sadzewicz, Email: LSadzewicz@som.umaryland.edu.
Amol Shetty, Email: AShetty@som.umaryland.edu.
Erich T. Boger, Email: erich.boger@nih.gov
Luke J. Tallon, Email: ljtallon@som.umaryland.edu
Robert J. Morell, Email: morellr@nidcd.nih.gov
Anup Mahurkar, Email: amahurkar@som.umaryland.edu.
Ronna Hertzano, Phone: 410-328-1892, Email: rhertzano@som.umaryland.edu.
References
- 1.JM S: Using fluorescence activated cell sorting to examine cell-type-specific gene expression in rat brain tissue. J Vis Exp 2015, 99:e52537. [DOI] [PMC free article] [PubMed]
- 2.Jung J, Jung H. Methods to analyze cell type-specific gene expression profiles from heterogeneous cell populations. Animal Cells and Systems. 2016;20:113–117. doi: 10.1080/19768354.2016.1191544. [DOI] [Google Scholar]
- 3.Kim T, Lim CS, Kaang BK. Cell type-specific gene expression profiling in brain tissue: comparison between TRAP, LCM and RNA-seq. BMB Rep. 2015;48(7):388–394. doi: 10.5483/BMBRep.2015.48.7.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Borozan I, Sun J, Guindi M, Fischer S, Feld J, Anand N, Heathcote J, Edwards AM, McGilvray ID. Cell-type specific gene expression signature in liver underlies response to interferon therapy in chronic hepatitis C infection. Gastroenterology. 2010;138(3):1123–1133. doi: 10.1053/j.gastro.2009.10.046. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Klughammer J, Farlik M, Penz T, Spittler A, Barbieux C, Berishvili E, Bock C, Kubicek S. Single-cell transcriptomes reveal characteristic features of human pancreatic islet cell types. EMBO Rep. 2016;17(2):178–187. doi: 10.15252/embr.201540946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li M, Jia C, Kazmierkiewicz KL, Bowman AS, Tian L, Liu Y, Gupta NA, Gudiseva HV, Yee SS, Kim M, et al. Comprehensive analysis of gene expression in human retina and supporting tissues. Hum Mol Genet. 2014;23(15):4001–4014. doi: 10.1093/hmg/ddu114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hertzano R, Elkon R, Kurima K, Morrisson A, Chan SL, Sallin M, Biedlingmaier A, Darling DS, Griffith AJ, Eisenman DJ, et al. Cell type-specific transcriptome analysis reveals a major role for Zeb1 and miR-200b in mouse inner ear morphogenesis. PLoS Genet. 2011;7(9):e1002309. doi: 10.1371/journal.pgen.1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng L, Zhang S, MacLennan GT, Williamson SR, Davidson DD, Wang M, Jones TD, Lopez-Beltran A, Montironi R. Laser-assisted microdissection in translational research: theory, technical considerations, and future applications. Appl Immunohistochem Mol Morphol. 2013;21(1):31–47. doi: 10.1097/PAI.0b013e31824d0519. [DOI] [PubMed] [Google Scholar]
- 9.Asano T, Masumura T, Kusano H, Kikuchi S, Kurita A, Shimada H, Kadowaki K. Construction of a specialized cDNA library from plant cells isolated by laser capture microdissection: toward comprehensive analysis of the genes expressed in the rice phloem. Plant J. 2002;32(3):401–408. doi: 10.1046/j.1365-313X.2002.01423.x. [DOI] [PubMed] [Google Scholar]
- 10.Smalley MJ, Titley J, O'Hare MJ. Clonal characterization of mouse mammary luminal epithelial and myoepithelial cells separated by fluorescence-activated cell sorting. In Vitro Cell Dev Biol Anim. 1998;34(9):711–721. doi: 10.1007/s11626-998-0067-0. [DOI] [PubMed] [Google Scholar]
- 11.Birnbaum K, Shasha DE, Wang JY, Jung JW, Lambert GM, Galbraith DW, Benfey PN. A gene expression map of the Arabidopsis root. Science. 2003;302(5652):1956–1960. doi: 10.1126/science.1090022. [DOI] [PubMed] [Google Scholar]
- 12.Graham M, Richardson JL, Macara IG. Does facs change gene expression. cytometry. 2015;87:166–175. doi: 10.1002/cyto.a.22608. [DOI] [PubMed] [Google Scholar]
- 13.Streets AM, Huang Y. How deep is enough in single-cell RNA-seq? Nat Biotechnol. 2014;32(10):1005–1006. doi: 10.1038/nbt.3039. [DOI] [PubMed] [Google Scholar]
- 14.Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF, et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods. 2014;11(1):41–46. doi: 10.1038/nmeth.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135(4):749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A. 2009;106(33):13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135(4):738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brar GA, Weissman JS. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol. 2015;16(11):651–664. doi: 10.1038/nrm4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin JQ, Amieux PS, Holt CE. Dynamic axonal translation in developing and mature visual circuits. Cell. 2016;166(1):181–192. doi: 10.1016/j.cell.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods. 2010;7(9):709–715. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faherty SL, Campbell CR, Larsen PA, Yoder AD. Evaluating whole transcriptome amplification for gene profiling experiments using RNA-Seq. BMC Biotechnol. 2015;15:65. doi: 10.1186/s12896-015-0155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shanker S, Paulson A, Edenberg HJ, Peak A, Perera A, Alekseyev YO, Beckloff N, Bivens NJ, Donnelly R, Gillaspy AF, et al. Evaluation of commercially available RNA amplification kits for RNA sequencing using very low input amounts of total RNA. J Biomol Tech. 2015;26(1):4–18. doi: 10.7171/jbt.15-2601-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adiconis X, Borges-Rivera D, Satija R, DeLuca DS, Busby MA, Berlin AM, Sivachenko A, Thompson DA, Wysoker A, Fennell T, et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat Methods. 2013;10(7):623–629. doi: 10.1038/nmeth.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matern M, Vijayakumar S, Margulies Z, Milon B, Song Y, Elkon R, Zhang X, Jones SM, Hertzano R. Gfi1Cre mice have early onset progressive hearing loss and induce recombination in numerous inner ear non-hair cells. Sci Rep. 2017;7:42079. doi: 10.1038/srep42079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pearson WR, Wood T, Zhang Z, Miller W. Comparison of DNA sequences with protein sequences. Genomics. 1997;46(1):24–36. doi: 10.1006/geno.1997.4995. [DOI] [PubMed] [Google Scholar]
- 26.Orvis J, Crabtree J, Galens K, Gussman A, Inman JM, Lee E, Nampally S, Riley D, Sundaram JP, Felix V, et al. Ergatis: a web interface and scalable software system for bioinformatics workflows. Bioinformatics. 2010;26(12):1488–1492. doi: 10.1093/bioinformatics/btq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 31.Krzywinski M, Altman N. Visualizing samples with box plots. Nat Methods. 2014;11(2):119–120. doi: 10.1038/nmeth.2813. [DOI] [PubMed] [Google Scholar]
- 32.Yang H, Gan J, Xie X, Deng M, Feng L, Chen X, Gao Z, Gan L. Gfi1-Cre knock-in mouse line: a tool for inner ear hair cell-specific gene deletion. Genesis. 2010;48(6):400–406. doi: 10.1002/dvg.20632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parekh S, Ziegenhain C, Vieth B, Enard W, Hellmann I. The impact of amplification on differential expression analyses by RNA-seq. Sci Rep. 2016;6:25533. doi: 10.1038/srep25533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Combs PA, Eisen MB. Low-cost, low-input RNA-seq protocols perform nearly as well as high-input protocols. PeerJ. 2015;3:e869. doi: 10.7717/peerj.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roh HC, Tsai LT, Lyubetskaya A, Tenen D, Kumari M, Rosen ED. Simultaneous transcriptional and Epigenomic profiling from specific cell types within heterogeneous tissues in vivo. Cell Rep. 2017;18(4):1048–1061. doi: 10.1016/j.celrep.2016.12.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10(11):1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 37.Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9(1):171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 38.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Library preparation performed in this study. Table S2 Gene list with RPKM greater than 10,000. Table S3 Median of genes from unique and shared regions of Venn diagram. Table S4 Spearman correlation coefficients between different profiles. (XLSX 28 kb)
Figure S1. Cre recombination in liver cells expressing Gfi1. (a) Cryosection of liver from a cross between a Gfi1-Cre mouse and a TdTomato reporter mouse (Ai14) stained with DAPI and phalloidin. TdTomato is found in a subset of cells in the liver that is likely consistent with Kupffer or endothelial cells. (b) Enrichment of Gfi1 transcripts in the IP samples as compared to the input samples was assessed by reverse-transcription followed by real time PCR. (PDF 2947 kb)
Figure S2. Duplication rate of each library. X-axis: duplication rate, Y-axis: log10 of reads at different duplication rates. (PDF 622 kb)
Figure S3. Hierarchical clustering of expression levels, based on the rank of the count of exon per million mapped reads (CPM). Dendrograms represent Spearman correlation coefficients between pairs of samples that is 3 replicates for input and 3 replicates for IP. (PDF 233 kb)
Figure S4. Bland–Altman plot (MA plot) of translatome (IP samples) and transcriptome (input samples) profiles for each kit. The red lines represent the boundary cutoff [− 1,1]. Dots above or below the red line represent the enriched or depleted transcripts. (JPG 587 kb)
Figure S5. Venn diagram of enriched/depleted transcripts (CPM ≥ 20 in at least one replicate, mean ratio of enrichement/depletion of the three replicates). The mean ratio IP/input is ≥2 or input/IP is ≥2. (PDF 1063 kb)
Figure S6. Histogram of length distribution for enriched or depleted transcripts. (PDF 353 kb)
Data Availability Statement
The datasets supporting the conclusions of this article are available in the NCBI Gene Expression Omnibus (GEO) repository under accession number GSE104213 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104213.
The datasets supporting the conclusions of this article are included within the article and its additional files (Additional files 1, 2, 3, 4, 5, 6, and 7).