

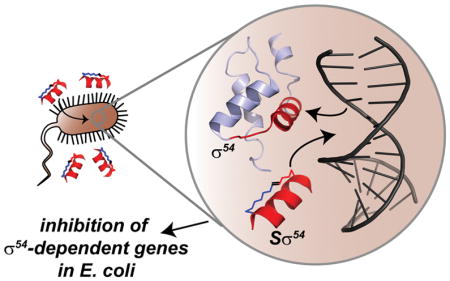
Summary
In response to environmental and other stresses, the σ54 subunit of bacterial RNA polymerase (RNAP) controls expression of several genes which play a significant role in the virulence of both plant and animal pathogens. Recruitment of σ54 to RNAP initiates promoter-specific transcription via the dsDNA denaturation mechanism of the cofactor. The RpoN box, a recognition helix found in the C-terminal region of σ54, has been identified as the component necessary for major groove insertion at the -24 position of the promoter. We employed the hydrocarbon stapled peptide methodology to design and synthesize stapled σ54 peptides capable of penetrating Gram-negative bacteria, binding the σ54 promoter, and blocking the interaction between endogenous σ54 and its target DNA sequence, thereby reducing transcription and activation of σ54 response genes.
Keywords: stapled peptides, antibiotic resistance, σ factor 54, nitrogen starvation, DNA-binding proteins, cell penetrating peptides, bacterial transcription
eTOC Blurb
The bacterial transcriptional regulator σ54 is involved in the virulence of pathogenic bacteria. Stapled peptides blocking the interaction between σ54 and its target DNA promoter inhibit activation of σ54-dependent genes in bacteria. These compounds provide an avenue to target bacterial pathogens and circumventing therapeutic drug resistance.
Introduction
Transcription factors lie at the crux of cellular homeostasis in all living systems. The transcription of genes into messenger RNA ultimately leads to the expression of proteins responsible for the execution of cellular functions with a high degree of temporal and spatial specificity; thus, disruptions in gene transcription can have deleterious consequences in the regulation of biological processes necessary for cell survival. Alterations such as the overexpression or deletion of a transcription factor are responsible for numerous disease states, underscoring the widespread interest in them as therapeutic targets in human disease, most notably in the cancer field. Targeting a transcription factor is fraught with numerous challenges, not the least of which is the difficulty of designing small molecules capable of disrupting weak interactions that span wide surfaces.
Much like in eukaryotic systems, gene transcription in prokaryotes follows a process involving several macromolecules working in concert. The initiation of bacterial transcription requires complexation of the RNA polymerase (RNAP) core enzyme with a sigma (σ)factor. σ-factors bind to the catalytic core of RNAP to initiate gene transcription after recognition of a promoter sequence and melting of dsDNA, and are functionally divided into housekeeping σ-factors, which control most prokaryotic genes, and alternative σ-factors, which bind to specific promoter sequences in response to environmental and physiological stresses (Cannon et al., 1993). The alternative factor σ54 directly regulates the transcription of mostly non-essential genes involved in nitrogen metabolism (Reitzer and Schneider, 2001). In several species of pathogenic bacteria such as Pseudomonas aeruginosa, Vibrio cholera, Borrelia burgdorferi, and Helicobacter pylori, the σ54 regulon controls the production of virulence factors involved in toxin release, quorum sensing, flagellar biosynthesis, and biofilm formation (Kazmierczak et al., 2005).
The structural features of σ54 have been characterized by NMR and provide a window into the intricacies of its cellular function (Doucleff et al., 2007). σ54 contains leucine zipper motifs that enable it to melt DNA in addition to hydrophobic regions which make contacts with RNAP. The C-terminal DNA binding domain of σ54, or RpoN box, binds to a specific promoter region leading to the formation of a complex activated by an ATP-dependent enhancer protein (Buck et al., 2000). σ54 engages its target promoter, located between positions –12 and –24 base pairs (bp) upstream of the transcription start site, via a conserved sequence of residues folded into an α-helix. Multiple hydrogen bonds and van der Waals interactions are essential for specific binding of the RpoN box to the major groove of DNA (Figure 1A) and subsequent activation of gene transcription. Given its role in the pathogenicity of many bacterial species, we hypothesized that blocking the interaction between endogenous σ54 and its promoter element would downregulate the expression of virulence genes.
Figure 1.

(A) NMR model of the C-terminal domain of σ54 interacting with DNA, with the RpoN box highlighted in red, and the −24 and −12 promoter sequences highlighted in blue. Residues essential for binding which also make key contacts with DNA are highlighted in red on the RpoN sequence. (B) Helical wheel of the RpoN box with staple positions indicated. (C) Linear peptide sequence of Sσ54. X = (S)-2-(4′-pentenyl)-alanine, Z = (R)-2-(7′-octenyl)-alanine, B = norleucine. All peptides contain β-alanine in the first position as a spacer (Supplementary Table 1). (D) Circular dichroism spectra of Sσ54 peptides.
Owing to a lack of any appreciable binding pocket, it has been extremely difficult to develop small molecules that target DNA specifically. There have been successes in this arena, most notably by Dervan and co-workers, who have developed programmable polyamides that bind specific DNA sequences and modulate eukaryotic gene expression (Dervan et al., 2005). Previously, our group and others (Walensky and Bird, 2014) have been involved in the development of hydrocarbon stapled peptides for their use as α-helix mimetics to target numerous protein-protein interactions involved in human disease. In many cases, these have led to compounds with potential therapeutic use; however, they have never been used to block a protein-DNA interaction successfully. Taking advantage of the helical shape of the RpoN box, we designed hydrocarbon stapled peptides that target the interaction between σ54 and DNA. Stapled σ54 peptides permeate Gram-negative bacteria and inhibit the transcription of genes activated by nitrogen depletion. Our data suggest that stapled peptides targeting bacterial transcription factors have potential for use as therapeutics to target the virulence properties regulated by σ54 in pathogenic bacteria.
Results
Design and Synthesis of Stapled σ54 Peptides
Gene expression in bacteria is accomplished by RNA polymerase (RNAP) in concert with a σ factor. σ factors guide RNAP to specific promoters and assist in melting dsDNA to initiate gene transcription. Bacteria possess multiple σ factors, each controlling specific responses to different environmental stimuli. Unlike other σ factors, σ54 requires the help of upstream ATP-dependent activator proteins to trigger transcription. A solution structure of σ54 bound to its promoter shows that a helix-loop-helix segment on the C-terminal portion of σ54 interacts with a region in the major groove of DNA located between −24 and −12 bp upstream of the gene transcription start site (Doucleff et al., 2007). Specifically, binding of σ54 to DNA takes place via the RpoN box α-helix, which is conserved across multiple bacterial species (Doucleff et al., 2005) (Figure 1A). Mutational studies have shown that several residues around the entire span of the RpoN α-helix are involved in a complex hydrogen bond network in which DNA wraps around the helix and facilitates binding and activation of transcription (Wang and Gralla, 2001). A helical wheel analysis (Figure 1B) shows the amphipathic character of the RpoN box with mostly hydrophobic residues on one face of the helix and polar amino acids arranged on the opposite side. With these design considerations in mind, we synthesized a family of stapled σ54 peptides (Sσ54) in which we varied the size and location of the hydrocarbon crosslink and kept intact the residues responsible for binding to DNA (Figure 1C). We examined their secondary structure by circular dichroism and found that, while the same length wild type σ54 peptide is unstructured, all Sσ54 peptides adopt an α-helical secondary structure in solution (Figure 1D).
Sσ54 Peptides Bind to the σ54 Promoter Element
With compounds in hand, we investigated whether Sσ54 peptides bind to a DNA oligo containing a target promoter sequence. We employed a fluorescence polarization binding assay to evaluate the ability of Sσ54 peptides to engage a fluorescein-tagged 30 bp oligo containing the sequence for the glnA σ54-dependent promoter sequence (glnAp2) (Smith and Bell, 2016) (Figure 2A). While the unstructured, wild type peptide did not bind appreciably to the target DNA sequence, all Sσ54 peptides bound to DNA with similar affinities (Kd = 0.86–5.98 μM). Testing a scrambled peptide in the same assay revealed only a weak binding affinity (Figure S1A). Additionally, none of the peptides bound to a FITC-tagged scrambled 30-mer to any appreciable extent (Figure S1B). We corroborated these results by performing gel shift assays using the FITC-glnAp2 probe. Exposure of stapled peptide Sσ54-2 to FITC-glnAp2 showed a significant reduction in the amount of free probe (Figure 2B, lanes 1 and 2). We likewise performed the experiment in the presence of the C-terminal fragment of σ54 (residues 338-398) which contains the RpoN box, and we observed that σ54(338-398) binds to the probe (lane 3), and Sσ54-2 disrupts this binding interaction effectively (lane 4) in a dose-dependent fashion (Figure S1C). Performing the experiment using a scrambled DNA probe (Figure S1D) shows that none of the stapled peptides can interact with the probe, corroborating the fluorescence polarization results (Figure S1B).
Figure 2.

(A) Fluorescence polarization binding analysis of Sσ54 peptides with FITC-glnAp2 30-mer. (B) Gel shift assay showing the association of Sσ54-2 with FITC-glnAp2 and the disruption of σ54(338-398) bound to FITC-glnAp2. (C) Polyacrylamide gel showing digestion of a 5′-6-FAM glnAp2 218-mer with DNase I. V = vehicle. (D) Densitometric quantification of remaining DNA relative to starting material (lane 3) from (B). Data are represented as mean ± SEM.
To further confirm the ability of Sσ54 peptides to bind to DNA, we performed a DNase digestion assay by combining a 6-FAM-labeled dsDNA oligo containing Escherichia coli glnAp2 with peptide and exposing the mixture to DNase I. The reaction mixtures were separated by polyacrylamide gel electrophoresis and analyzed by fluorescence scanning (Figure 2C). Treatment with the wild type σ54 peptide (lane 5) displayed a significant amount of DNA degradation as observed in vehicle-treated DNA (lane 4), confirming the fluorescence polarization results showing a lack of binding by this peptide. On the other hand, Sσ54 peptides were far more effective at protecting DNA from enzymatic degradation. There were differences in the ability of each Sσ54 peptide to shield DNA from cleavage, with Sσ54-1 and -2 (lanes 6 and 7) being the most effective (Figure 2D). Notably, the binding affinities correlate with the ability of the peptides to protect DNA from degradation by an endonuclease. The differential binding of the peptides suggests that each of these compounds may result in varying degrees of activity in a biological setting.
Sσ54 Peptides Enter Gram-Negative Bacteria by Diffusion
Armed with the knowledge that Sσ54 peptides can bind to a target promoter, we investigated whether the compounds penetrated bacterial cells. While studies have been done on the ability of stapled peptides to enter human cells (Bird et al., 2016), there are no reports showing whether they can penetrate bacterial cells. Entry of hydrophobic compounds into Gram-negative bacteria is met with several physical barriers. In particular, the lipopolysaccharide (LPS) envelope surrounding the outer membrane of the cell is responsible for the inability of many organic compounds to penetrate Gram-negative bacteria (Denyer and Maillard, 2002). To evaluate whether Sσ54 peptides enter bacterial cells, we treated BW25113 E. coli cells and PA01 P. aeruginosa cells with FITC-Sσ54 peptides. Uptake was measured by flow cytometry (Figure 3A). In both bacterial species, all FITC-Sσ54 peptides penetrated bacterial cells while the unstructured wild type FITC-σ54 peptide did not. Bacterial cells also possess the ability to control transport of macromolecules through ATP-dependent transporters (Mousa and Bruner, 2016), so to rule out that internalization of Sσ54 peptides is dependent on this system, we repeated the flow cytometry experiment with the addition of sodium azide as an ATPase inhibitor. Flow cytometry analysis showed no change in peptide permeability, suggesting that these compounds enter bacterial cells by diffusion. We confirmed the flow cytometry results by fluorescence confocal microscopy (Figure 3B). The images show that FITC-Sσ54 peptides accumulate in the cytoplasm of bacterial cells while the unstructured wild type compound is not permeable. Taken together, these data show that stapled peptides are capable of penetrating Gram-negative bacteria.
Figure 3.

(A) Flow cytometry of BW25113 E. coli and PA01 P. aeruginosa treated with 4 μM FITC-Sσ54 peptides. Data are represented as mean ± SD. (B) Fluorescence microscopy of BW25113 E. coli. FM 4-64: plasma membrane stain, FITC: Sσ54. The scale bars represent 10 μm.
Stapled σ54 Peptides Block the Upregulation of σ54-dependent Genes
In E. coli, σ54 is primarily responsible for triggering transcription in response to nitrogen starvation. We hypothesized that exposing E. coli cells to growth media deficient in nitrogen would initiate σ54-mediated transcription of nitrogen metabolism genes, and subsequent treatment with Sσ54 peptides would reverse or inhibit that transcriptional activation. To examine this, we cultured MG1655 E. coli cells in Gutnick minimal media (Brown, 2014) to induce nitrogen starvation (Figure S3). Cells were then treated with peptide, and RNA was later isolated and processed for RT-qPCR analysis (Figure 4A). We quantified RNA for the σ54-dependent nitrogen starvation response genes glnA, yeaG and nac, which are involved in nitrogen fixation, metabolism, and assimilation, respectively (Reitzer and Schneider, 2001). We also examined pspA, a σ54-dependent gene that responds to cell membrane disruptions and is not responsive to nitrogen levels, and we used the housekeeping gene cysG as a control. Exposure to nitrogen-deficient conditions activates transcription of glnA by a factor of 10, yeaG 100-fold, and nac close to 500-fold, while pspA was unaffected by nitrogen depletion. Treatment with the wild type σ54 peptide showed no difference in mRNA levels when compared to the untreated control; however, exposure to Sσ54 peptides, particularly Sσ54-2 and Sσ54-3, restricted the mRNA levels of the nitrogen response genes to the basal levels found in cells at the nitrogen-rich state (N+, Figure 4A). Some Sσ54 peptides also increased the levels of pspA transcripts, which is not surprising given the involvement of this gene in responding to changes in the integrity of the bacterial envelope and membranes. Because nitrogen depletion with concomitant blockade of the transcription of nitrogen metabolism genes is likely to be toxic to bacterial cells, we verified that E. coli cells are alive at the dose used in the RT-qPCR experiment via cell viability (Figure S2B) demonstrating that the low levels of nitrogen metabolism gene transcripts detected in Sσ54-treated cells are not due to cell death. We similarly evaluated whether the compounds were toxic to human cells, and the data show that, at these doses, Sσ54 peptides are not toxic to human cells (Figure S2C) despite the ability of the peptides to penetrate human cells (Figure S3).
Figure 4.

(A) Relative normalized expression of σ54-controlled genes after treatment with Sσ54. Data are represented as mean ± SEM. N+ denotes nitrogen-rich media while N- indicates nitrogen-deficient media. (B) Glutamine synthetase transferase assay with A540 values normalized to OD600. Data are represented as mean ± SEM. (C) RNA-Seq heat map for transcripts that are differentially expressed in MG1655 E. coli in response to nitrogen starvation and treatment with peptides. RNA of E. coli grown under nitrogen-rich conditions (N+) was used as a control.
We also sought to confirm that inhibition of σ54-dependent gene transcription led to a decrease in the nitrogen sensitivity response. We measured the intracellular activity of glutamine synthetase, the gene product of glnA, in a colorimetric assay (Rothstein et al., 1980), and found no appreciable enzymatic activity in cells treated with Sσ54-2 and Sσ54-3 (Figure 4B), which is consistent with our observations from the RT-qPCR experiment.
To understand more broadly the full-scale effects of stapled σ54 peptides on the transcription of activated σ54-dependent genes, we performed an RNA-Seq experiment to analyze the transcriptome of E. coli. MG1655 E. coli cells were grown in Gutnick media to induce nitrogen starvation, were treated with either DMSO, WT σ54, Sσ54-1, or Sσ54-2, and RNA was isolated and processed for RNA-Seq. Comparison of the data set for Sσ54-2 treatment versus DMSO control showed differential expression of 2,191 of the 4,273 genes detected, 1047 of which were positively regulated and 1140 were negatively regulated by a factor of 2 or greater. Gene set enrichment analysis (GSEA) (Subramanian et al., 2005) with all 155 E. coli gene sets with fewer than 500 genes per set from gene2go (Powell, 2014) showed that 6 gene sets were positively regulated with a false discovery rate of less 5%. These included carbohydrate and polysaccharide biosynthesis, amide biosynthesis and catabolism, and metal ion transport. 6 out of 155 gene sets were negatively regulated with a false discovery rate of less than 5%, including amino acid, organic acid, and organonitrogen catabolism. These results are consistent with the Sσ54 having minimal impact on gene sets that describe a common function.
When GSEA was performed using gene sets for the RpoN (σ54) and RpoH (σ32) regulons, the RpoN regulon (Bonocora et al., 2015) was shown to be negatively regulated with a false discovery rate of less 0.1% (Figure S4). This result is consistent with Sσ54-2 binding to the σ54 promoter preventing transcription of genes under σ54 control. In contrast, the RpoH regulon (Nonaka et al., 2006), which is regulated by the σ32 alternative sigma factor as part of the heat shock response, was neither positively nor negatively regulated by Sσ54-2 (Figure S4). Similarly, GSEA of Sσ54-1 versus DMSO showed the RpoN regulon was negatively regulated and the RpoH was not significantly impacted. Hierarchical clustering of the transcriptome data for genes under σ54 (RpoN) and σ32 (RpoH) control (Figure 4C) clearly showed that genes from the RpoN regulon cluster together and were negatively regulated in the positive control (i.e., growth in nitrogen rich media) and Sσ54-2 and Sσ54-1 treatment, but not with WT σ54 treatment. These data are consistent with and strongly support the selective, negative regulation of the RpoN regulon by Sσ54.
Discussion
The emergence of antibiotic resistance along with the decreasing numbers of new antimicrobials available in the clinic underscores the need for creative ways to treat bacterial infections. We hypothesized that a direct approach targeting the transcriptional machinery of pathogenic bacteria might mitigate their virulence. We have previously synthesized stapled peptides that target transcription factors in human cells and restore the function of a tumor suppressor through the inhibition of a protein-protein interaction (Bernal et al., 2010). The present work takes this concept further by directly inhibiting the interaction between a transcriptional regulatory protein and its promoter. Targeting gene transcription with precision has been a longstanding goal in the search for potential therapeutics that minimize collateral damage. The complexity of eukaryotic genomes has made executing this strategy challenging (Edwards et al., 2016); thus, a study in a simpler system provides a springboard for the development of agents that control gene transcription with specificity. This strategy, however, is not without risks. σ54 recognition sites are small and lend themselves to significant redundancy, increasing the likelihood of unexpected phenotypic responses. Here we present a case in which we target a σ54-mediated cellular response to a specific environmental stress. Sσ54 peptides successfully blocked the response without any appreciable side effects. Our work suggests that the inhibition of nitrogen sensitivity genes by treatment with Sσ54 peptides is directly related to specific targeting of genes with active σ54 promoters. It is likely that Sσ54 peptides bind to other regions of the bacterial genome, raising the concerns about compound specificity; and if the compounds are nonselective, we would expect to see varied transcriptional responses. Nevertheless, our transcriptional profiling data strongly indicate that, even if the Sσ54 peptides bound elsewhere, these other putative interactions are uneventful and do not affect the desired phenotypic outcome of the treatment. These results pave the way towards development of analogs to fine-tune the selective targeting of activated σ54 promoters.
One of the bigger questions arising from this work was to determine whether stapled peptides could penetrate the multiple physical barriers erected by Gram-negative bacteria, the most notorious being the LPS envelope surrounding the outer membrane (Wiese et al., 1999). Hydrophilic compounds may enter Gram-negative bacteria through porins embedded in the outer membrane, but these channels limit the size of molecules that can go through to 600 Da or less, smaller than any of the compounds tested. Hydrophobic compounds can penetrate the LPS matrix, but its dense composition significantly slows their diffusion rates. Lipid A, a phosphorylated glycosamine, is an integral component of the LPS envelope, and its phosphate groups are linked to each other and held in place by bridging divalent cations. Some effective agents against Gram-negative bacteria are the polymyxins, polycationic antibiotics which function partly by destabilizing the envelope and promoting their own entry (Hancock and Chapple, 1999). The Sσ54 peptides have charges of +3 and +4 at physiological pH, so it is likely that charge plays a role in their ability to penetrate E. coli and P. aeruginosa cells. Charge alone, however, is not sufficient to promote permeability, as the wild type σ54 peptide does not enter cells despite also having a charge of +4. Sσ54 peptides have a rigid α-helical shape with discrete hydrophobic and hydrophilic faces, and this likely allows them entry via disruption of the LPS matrix. This work demonstrates the potential of stapled peptides as antibiotics with the ability to access targets in the bacterial cytoplasm.
The search for agents to treat bacterial infections must keep up with the pace at which resistant pathogens are emerging. Antimicrobial peptides, in particular cationic host defense peptides, have shown promise as potential therapeutics (Overhage et al., 2008). Our work has shown that incorporating structural stability is essential in achieving bacterial cell permeability. Targeting the virulence properties of pathogenic bacteria opens a new therapeutic avenue that takes advantage of the hosts’ environment. By combining structural features that allow for permeability along with a defined genomic target controlling virulence, we are now able to target Gram-negative pathogens through a novel mechanism.
Significance
The emergence of bacterial drug resistance underscores the need for novel antibiotics. Gram-negative bacteria possess many physical barriers that make them impervious to most therapeutics. Additionally, virulence properties such as biofilm formation and toxin secretion make it significantly more difficult to target pathogens with synthetic agents. Disrupting virulence at the transcriptional level provides a new avenue to render bacteria vulnerable, but targeting protein-DNA interactions has been notoriously difficult. We show that using structurally defined hydrocarbon stapled peptides based on a DNA-interacting α-helix of σ54 can block specific gene transcription in response to an environmental stress. These stapled peptides readily enter Gram-negative bacteria by diffusion and target σ54 promoters in cells. This class of compounds will be developed for the treatment of infections by Gram-negative pathogens.
STAR Methods
Contact for Reagents and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Federico Bernal (federico.bernal@nih.gov).
Experimental Model and Subject Details
All overnight bacterial cultures were either grown in LB broth (Fisher) or Gutnick media containing 10 mM NH4Cl.
Method Details
Peptide Synthesis
Peptides were synthesized on a Tetras Peptide Synthesizer (Advanced ChemTech, Louisville, KY) using Fmoc-based solid phase peptide chemistry on Rink amide resin (30 μmol peptide per reaction). In brief, the synthesis protocol consists of removal of the Fmoc protective group with 25% piperidine in NMP, washing with NMP, and subsequent amino acid coupling using 10 equiv. amino acid (1 mL, 0.3 M), HCTU (0.99 mL, 0.3 M) and DIPEA (2.0 mL, 0.3 M) in NMP for 60 minutes before draining and washing. All peptide sequences include an N-terminal β-alanine spacing residue. Stapled peptides underwent a further ring-closing metathesis reaction on the resin-bound N-terminal Fmoc-protected peptide. After washing with NMP and 1,2-dichloroethane, the resin was exposed to three 6-hour cycles of bis(tricyclohexylphosphine)benzylidine ruthenium (IV) dichloride (Grubbs’ I catalyst, 3 mL, 1 mM) at room temperature in 1,2-dichloroethane. Peptides were primarily N-terminal acetylated for use in biochemical and cell-based assays. The acetylation reaction consisted of deprotection of the Fmoc group as outlined above, followed by reaction with neat acetic anhydride (1 mL) and DIPEA (2 mL, 0.3 M in NMP) for 1 hour. Other experiments made use of N-terminal fluoresceinated peptides. To this end, Fmoc-deprotected peptides were exposed to fluorescein isothiocyanate (2.8 mL, 25 mM) and DIPEA (0.2 mL, 0.3 M) in DMF for 12 hours. Final peptides were cleaved from the resin and fully deprotected by exposure to a solution containing 95% TFA, 2.5% water, and 2.5% triisopropyl silane for 2.5–3 hours. The cleaved peptides were precipitated into ice-cold 1:1 methyl-tert-butyl ether (MTBE), re-suspended in water and lyophilized as crude peptides overnight. The lyophilized peptides were purified by C-18 reverse phase HPLC on an Agilent 1200 HPLC system (Santa Clara, CA).
Circular Dichroism (CD) Spectroscopy
Acetylated compounds were dissolved in water to concentrations ranging from 60 to 75 μM. Final compound concentrations were determined by measuring sample absorbance at 205 nm using a NanoDrop2000 spectrophotometer (ThermoScientific, Wilmington, DE). Spectra were obtained on an Aviv Circular Dichroism Spectrometer, Model 420 (Aviv Biomedical, Inc, Lakewood, New Jersey) at 25°C. The spectra were collected using a 0.1 cm path length quartz cuvette (Hellma Analytics, Germany) with the following measurement parameters: wavelength, 240–190 nm; step resolution, 0.50 nm; averaging time, 5.0 sec per step. Spectra were processed using Aviv CDS Program software and converted to mean residue molar ellipticity using the cuvette path length (0.1 cm), the measured concentration, and the number of amino acids in the peptide (cross-linking amino acids and β-alanine cap were included as amino acids in this count).
Protein Purification
The C-terminal RpoN domain (CTD) of σ54 (residues 338–398) from Klebsiella pneumoniae was cloned into the pET28b expression vector by PCR (Q5 Site-directed Mutagenesis Kit; New England Biolabs) and confirmed through Sanger sequencing. The plasmid was then transformed into E. coli Rosetta 2 (DE3) cells. The cells were grown at 37°C in 500 mL of LB medium and induced with 1 mM IPTG at OD600: ~0.6. Four hours after induction cells were harvested via centrifugation and resuspended in 1X PBS supplemented with 1X Protease and Phosphatase Inhibitor Cocktail (ThermoScientific). Cells were lysed through sonication and the CTD was purified through Ni-affinity chromatography (Qiagen) and size exclusion chromatography (Superdex 75 10/300 GL; GE Healthcare).
Electrophoretic Mobility Shift Assays (EMSA)
FITC-glnAP2 DNA (5.625 nM) was initially incubated with our library of stapled peptides (20 μM) for 20 minutes at room temperature in the dark for 20 minutes, with DMSO as our negative control. The complexes were formed in 25 mM HEPES, 500 mM NaCl, 10% glycerol, pH 6.9. Next, the CTD protein (32 μM) was added to the mixture and further incubated at room temperature for 20 minutes. 12% polyacrylamide gels (29:1 acrylamide:bisacrylamide) were pre-run for 40 minutes at 4°C at 70 V. Samples were the n electrophoresed through the gels for approximately two hours at 10 V using 1X Tris-borate buffer. DNA was detected by fluorescence imaging on a Typhoon FLA 7000 (GE Healthcare).
Induction of Nitrogen Starvation
The genotypes and origins of all bacterial strains can be found in the key resources table. Nitrogen starvation was induced in one of two ways: MG1655 E. coli were grown in LB broth, then switched to M9 minimal media (1X M9 salts, MgSO4, CaCl2 and 20% (w/v) glycerol) containing either 40 mM (nitrogen rich) or 100 μM (nitrogen deficient) NH4Cl. Conversely, overnight cultures were also grown in Gutnick minimal media (33.8 mM KH2PO4, 77.5 mM K2HPO4, 5.74 mM K2SO4, 0.41 mM MgSO4) containing Ho-LE trace elements, 0.4% (w/v) glycerol and 10 mM NH4Cl. The following day, cultures were prepared using Gutnick media with 3 mM NH4Cl, a concentration that allows for complete depletion of nitrogen during 4 hours of growth. In Gutnick media, “nitrogen rich” is the point when OD600 = 0.3, and “nitrogen deficient” being approximately 30 minutes after cultures have reached stationary phase.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat polyclonal anti-Glutamine Synthetase | Abcam | Cat# ab6585 |
| Mouse monoclonal Anti-E coli RNA Sigma 54 (clone 6RN3) | BioLegend | Cat# 663303 |
| Mouse monoclonal anti-GAPDH (clone GA1R) | Thermo Fisher | Cat# MA5-15738 |
| Bacterial and Virus Strains | ||
| E. coli: MG1655 | Coli Genetic Stock Center – Yale University | Cat# CGSC 6300 |
| E. coli: BW25113 | Coli Genetic Stock Center – Yale University | Cat# CGSC 7636 |
| E. coli: Rosetta 2 (DE3) | Novagen | Cat# 71397 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| B-PER II | Thermo Fisher | Cat# 78260 |
| 2-methylimidazole | Alfa Aesar | Cat# A11949 |
| 2,4-dimethylimidazole | Sigma Aldrich | Cat# M50850 |
| Adenosine diphosphate sodium salt | Sigma Aldrich | Cat# A2754 |
| Hydroxylamine Hydrochloride | Sigma Aldrich | Cat# 159417 |
| Sodium Tartrate | Sigma Aldrich | Cat# SX0795 |
| Cobalt chloride hexahydrate | Sigma Aldrich | Cat# C2644 |
| Sodium molybdate dihydrate | Sigma Aldrich | Cat# 221848 |
| Copper chloride dehydrate | Sigma Aldrich | Cat# 459097 |
| SYBR Select Master Mix | Applied Biosystems | Cat# 4472942 |
| Critical Commercial Assays | ||
| CellTiter-Glo | Promega | Cat# G7571 |
| BacTiter-Glo | Promega | Cat# G8231 |
| Ammonia Quantification Assay | Sigma Aldrich | Cat# AA0100 |
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| Superscript III First strand cDNA synthesis system | Invitrogen | Cat# 18080051 |
| Deposited Data | ||
| Deep sequencing data | GEO | Acc# GSE111317 |
| Experimental Models: Cell Lines | ||
| Human: WS1 Fibroblast cells | ATCC | Cat# CRL-1502 |
| Oligonucleotides | ||
| See Table S1 for DNA oligo and primer sequences | This study | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7.01 | GraphPad Software | https://www.graphpad.com/ |
| SnapGene 3.0.3 | GSL Biotech | www.snapgene.com |
| ImageJ | National Institutes of Health | https://imagej.net/NIH_Image |
| ChemDraw 15.1 | PerkinElmer | http://www.cambridgesoft.com/software/overview.aspx |
Flow Cytometry
BW25113 E. coli or PA01 P. aeruginosa cells were grown in LB media to an OD600 of 0.4–0.6. 1.0 × 106 cells were washed twice with PBS and treated with the FITC-labeled stapled-α-helical peptide (4 μM) and incubated at 37°C for 90 min. Samples were washed once with PBS and incubated at 4°C as pellets for 30 min. Trypan Blue (0.04% w/v) was added to all samples and removed and washed with PBS. The measurements were performed using a Beckman-Coulter Gallios Flow Cytometer in which a 488 nm laser was used to excite FITC and fluorescence was detected using the FL1 channel.
Confocal Fluorescence Microscopy
Bacterial Cells
MG1655 E. coli cells were grown to an OD600 of 0.5–0.7 in LB media. The culture was split into separate 1 mL aliquots, centrifuged and resuspended in PBS. FITC-labeled compounds were added to a final concentration of 4 μM. Samples were incubated at 37°C in the dark for 90 minutes and then centrifuged to pellet the cells. The supernatant was discarded and washed twice with PBS. After centrifugation, the samples were incubated on ice as pellets in the dark for 30 minutes. FM 4-64 was added to a concentration of 2 μg mL−1 and incubated for 5 min at 37°C in the dark. After cent rifugation and resuspension in PBS, 4 μL of each sample was pipetted onto the center of a square, 1% (w/v) agarose pad. Using a scalpel, the pad was placed directly atop a glass-bottom culture dish (35 mM, poly-lysine coated, MatTek Corporation). The cells were then visualized and images acquired on a Zeiss LSM780 confocal scanning microscope (Zeiss).
Human Cells
WS1 normal human fibroblasts (7.5 × 105 cells) were seeded onto a white, clear 6-well plate in DMEM. Before seeding, a round glass coverslip was washed for 4 hours in 100% methanol, subsequently washed in ddH2O, and placed inside each well. The cells were then treated with FITC-labeled peptides at a final concentration of 5 μM for 90 minutes at 37°C and 5% CO2. The supernatant was discarded and the cells were fixed with formaldehyde solution (1.5% in 1X PBS) for 7 minutes. The formaldehyde solution was discarded and cells were subsequently washed for 30 min with 1X PBS. PBS was then aspirated, and the dsDNA was stained with 150 μL solution of DAPI (1:5000 in 1X PBS) (ThermoScientific) and incubated for 1 min. After aspirating the DAPI solution, a drop of antifade reagent in glycerol/PBS (ThermoScientific) was placed on a clean slide and the coverslip was gently placed cell down onto the slide. The edges were coated with nail polish and let dry for 30 min in the dark. The cells were then visualized and images acquired on a Leica TCS SP8 confocal laser scanning microscope (Leica Microsystems).
Fluorescence Polarization Binding Assays
FP assays were performed as described. Briefly, to determine the dissociation constants for peptide-DNA interactions, FITC-labeled oligonucleotides (5.625 nM) were incubated with different concentrations of stapled peptide (40 μM - 9.8 nM) at 25°C for 10 min in FPA buffer (50 mM Tris-HCl pH 7.5, 1 mM DTT, 0.1 mg mL−1 BSA) in a black, opaque 96-well plate, and fluorescence polarization measured at equilibrium on a SpectraMax M5 microplate reader (Molecular Devices).
DNase I Digestion
A 216 bp oligonucleotide containing the glnA σ54 promoter (glnAp2) was PCR amplified from pure, genomic DNA isolated from MG1655 E. coli. The resulting product was used as template in a subsequent PCR containing one 5′-6-FAM labeled primer and one unlabeled primer. The fluorescent PCR product was run on a 1% (w/v) agarose gel and gel purified. Reactions were performed as follows: 5 μL stapled peptide (Cfinal = 20 μM) and 35 μL 6-FAM DNA (250 ng) were incubated for 15 min at 37°C. 5 μL of DNase I (150 mU μL−1) was added and reactions were incubated for 5 min at 37°C. Reactions were termina ted with 5 μL EDTA (500 mM) and vortexed immediately. The digestion products were purified via PCR Purification (Qiagen) according to the manufacturer’s protocol. The total, purified digestion products were run on a 12% polyacrylamide-TBE gel and imaged on a Typhoon Imager (GE Life Sciences). FIJI was used to quantitate remaining starting material relative to DMSO-treated samples.
Cell Viability Assays
Bacterial Cells
MG1655 E. coli cells were grown to an OD600 of 1.0 and seeded onto a white, opaque 96-well plate in LB broth. The cells were incubated with compounds at the indicated doses for 1 h. Cell viability was assayed by addition of BacTiter-Glo™ bioluminescence reagent according to the manufacturer’s protocol (Promega) and luminescence measured using a Spectramax M5 microplate reader (Molecular Devices). Data are normalized to vehicle-treated controls.
Human Cells
WS1 normal fibroblasts (1.5 × 104 cells) were seeded onto a white, opaque 96-well plate. The cells were incubated with compounds at the indicated doses for 24 h. in Opti-MEM (Gibco). Cell viability was assayed by addition of CellTiter-Glo™ bioluminescence reagent according to the manufacturer’s protocol (Promega) and luminescence measured using a Spectramax M5 microplate reader (Molecular Devices). Data are normalized to vehicle-treated controls.
Quantitative RT-PCR
MG1655 E. coli were grown in Gutnick minimal media to induce nitrogen starvation. At OD600 0.5, cells were treated with 10 μM stapled peptide. RNA was stabilized after 80 minutes after treatment using the RNAprotect Bacteria Reagent (Qiagen), and purified using the RNeasy Mini Kit and RNase-free DNase (Qiagen). Purified RNA was stored at −80°C in nuclease-free water. 0.5 μg of total RNA was used for cDNA synthesis, following manufacturer protocols (SuperScript III First-strand cDNA Synthesis System, Invitrogen). qPCR reactions were performed as follows: 1 μL cDNA, 5 μL SYBR-Select Master Mix (Applied Biosystems) and 250 nM forward and reverse primer (see Table S2 for sequences) were combined to a final volume of 10 μL. Plates were briefly centrifuged (1 min, 1,000 RPM, RT) before target amplification was performed on a C1000 Touch Real-Time PCR System (Bio-Rad) using the following conditions: 50°C (2 min), followed by 95°C (2 min), followed by 40 cycles of 95°C (15 s) and 60°C (1 min). A melting curve was obtained at the end of each experiment with the following conditions: 65°C (5 sec), followed by 60 × 5 sec cycles with increases of 0.5°C per cycle. All experiments were performed in triplicate and cysG was used as a reference gene. Threshold-cycle (Ct) values were automatically calculated for each replicate and used to determine the relative expression of the gene of interest relative to cysG for both treated and untreated samples by the 2−ΔΔCt method (Livak and Schmittgen, 2001). p-values were calculated using one-way ANOVA analysis.
Quantification of Glutamine Synthetase Activity
MG1655 E. coli cells grown to an optical density (OD600) of approximately 0.15 were suspended in 40 mL of M9 minimal media containing either 40 mM NH4Cl (nitrogen-rich conditions) or 100 μM NH4Cl (nitrogen-deficient conditions). Compounds were added to the cultures to a concentration of 10 μM and the cells were incubated in a shaker at 37°C for 4 h. The samples were then removed from the incubator and cetyltrimethylammonium bromide (CTAB) was added to a concentration of 1.5 mg mL−1. Samples were incubated at 37°C for 2 min and subs equently centrifuged (3,500 RPM, 4°C) for 15 min. The supern atant was discarded, and the samples were washed with KCl (1% w/v) and centrifuged a second time. After discarding the supernatant, pellets were resuspended in 150 μL of KCl (1% w/v). An assay mixture (40 mM L-glutamine, 40 mM NH2OH·HCl, 800 μM Na-ADP, 40 mM potassium arsenate, 100 mM mixed imidazole buffer (0.33 M each of imidazole, 2-methylimidazole, and 2,4-dimethylimidazole, pH 7.15), 600 μM MnCl2, and 0.2 mg mL−1 CTAB, pH 7.15) was added, and samples were incubated at 37°C for 30 minutes. After incubation with termination buffer (3.33% FeCl3, 2% trichloroacetic acid, and 250 mM HCl) for 15 min, particulate matter was pelleted by centrifugation (3.500 RPM, RT) for 15 minutes. The absorbance at a wavelength of 540 nm was quantified using a Spectramax M5 plate-reader (Molecular Devices) to determine the amount of gamma glutamyl hydroxamate, indicative of active enzyme.
RNA-Seq
RNA for the RNA-Seq experiment was obtained exactly as described previously for the quantitative RT-PCR experiment. RNA libraries were prepared for sequencing using standard Illumina protocols. Sequenced reads were trimmed for adaptor sequence, masked for low-complexity or low-quality sequence, and then mapped to the E. coli K-12 genome using bowtie2 with default parameters. Reads were counted by feature count and reads-per-megabase of library size (CPM) were generated and normalized by the limmaVoom pipeline. All data are publicly available through GEO (accession # GSE111317).
Gene Set Enrichment Analysis
Gene set enrichment analysis was performed using GSEAPreranked version 6 module from GenePattern (Reich et al., 2006). The ranked list of genes based on fold change data from RNA-seq was used as the ranked list input for GSEAPreranked. A database containing 155 E. coli gene sets with fewer than 500 gene members each was obtained from gene2go (Powell, 2014). The gene set for rpoN regulon was generated by selecting all the genes with an intergenic sense rpoN consensus sequenced that were identified by ChIP-seq. The gene set for the rpoH regulon has been previously disclosed (Nonaka et al., 2006) and was used for this experiment. The rpoN and rpoH gene sets were combined to generate an alternative sigma factor gene set database. 1000 permutations of the gene set were used to assess the significance of the enrichment score for each gene set.
Hierarchical clustering analysis
Hierarchical clustering was performed using Heatmapper (Babicki et al., 2016) The distance measurement method used was Euclidean and clustering methods used was average linkage.
Quantification and Statistical Analysis
Unless otherwise specified, data are represented as the means of three, independent experiments (n=3) with error bars reflecting the standard error of the mean (SEM). p-values were determined with a 1-way ANOVA test compared to vehicle treatment (DMSO): (* = ≤ 0.05; ** = ≤ 0.005; *** = ≤ 0.0005; **** = ≤ 0.00005). All statistical analyses were performed with GraphPad Prism 7.01.
Data and Software Availability
Deep sequencing data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE111317.
Supplementary Material
Highlights.
Hydrocarbon crosslinks in σ54 RpoN box-derived peptides confer increased helicity.
Stapled σ54 (Sσ54) peptides bind to σ54 promoters in vitro.
Sσ54 peptides enter Gram-negative cells by diffusion.
Sσ54 peptides block transcriptional activation of σ54-dependent genes.
Acknowledgments
We are grateful to Dr. Don Court (NCI) for helpful discussions and the staff of the Biophysics Resource Facility in the Structural Biophysics Laboratory, the Optical Microscopy and Analysis Laboratory, and the CCR Sequencing Facility (NCI-Frederick) for assistance with instrumentation. This research was supported by a grant from the National Interagency Confederation for Biological Research (to S.R.P.) and the Intramural Research Program of the NIH and the National Cancer Institute.
Footnotes
Author Contributions
S.R.P. and D.I.P. contributed equally to this work. F.B. and C.N.B. conceived the idea, conceptualized the study, and supervised the work. A.L.W. synthesized all compounds. S.R.P., D.I.P., Y.K., B.M.P., and C.M. executed all experimental work. F.B., C.N.B., S.R.P., A.L.W., and D.I.P. analyzed data and wrote the manuscript.
Declaration of Interest
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, Wishart DS. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Research. 2016;44:W147–W153. doi: 10.1093/nar/gkw419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, Wahl GM, Walensky LD. A Stapled p53 Helix Overcomes HDMX-Mediated Suppression of p53. Cancer Cell. 2010;18:411–422. doi: 10.1016/j.ccr.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GH, Mazzola E, Opoku-Nsiah K, Lammert MA, Godes M, Neuberg DS, Walensky LD. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat Chem Biol. 2016;12:845–852. doi: 10.1038/nchembio.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonocora RP, Smith C, Lapierre P, Wade JT. Genome-Scale Mapping of Escherichia coli sigma54 Reveals Widespread, Conserved Intragenic Binding. PLoS Genet. 2015;11:e1005552. doi: 10.1371/journal.pgen.1005552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M, Gallegos MT, Studholme DJ, Guo Y, Gralla JD. The bacterial enhancer-dependent sigma(54) (sigma(N)) transcription factor. J Bacteriol. 2000;182:4129–4136. doi: 10.1128/jb.182.15.4129-4136.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon W, Claverie-Martin F, Austin S, Buck M. Core RNA polymerase assists binding of the transcription factor sigma 54 to promoter DNA. Mol Microbiol. 1993;8:287–298. doi: 10.1111/j.1365-2958.1993.tb01573.x. [DOI] [PubMed] [Google Scholar]
- Denyer SP, Maillard JY. Cellular impermeability and uptake of biocides and antibiotics in Gram-negative bacteria. Journal of Applied Microbiology. 2002;92:35S–45S. [PubMed] [Google Scholar]
- Dervan PB, Doss RM, Marques MA. Programmable DNA Binding Oligomers for Control of Transcription. Current Medicinal Chemistry - Anti-Cancer Agents. 2005;5:373–387. doi: 10.2174/1568011054222346. [DOI] [PubMed] [Google Scholar]
- Doucleff M, Malak LT, Pelton JG, Wemmer DE. The C-terminal RpoN Domain of σ54 Forms an Unpredicted Helix-Turn-Helix Motif Similar to Domains of σ70. Journal of Biological Chemistry. 2005;280:41530–41536. doi: 10.1074/jbc.M509010200. [DOI] [PubMed] [Google Scholar]
- Doucleff M, Pelton JG, Lee PS, Nixon BT, Wemmer DE. Structural basis of DNA recognition by the alternative sigma-factor, sigma54. J Mol Biol. 2007;369:1070–1078. doi: 10.1016/j.jmb.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AL, Meijer DH, Guerra RM, Molenaar RJ, Alberta JA, Bernal F, Bird GH, Stiles CD, Walensky LD. Challenges in Targeting a Basic Helix–Loop–Helix Transcription Factor with Hydrocarbon-Stapled Peptides. ACS Chemical Biology. 2016;11:3146–3153. doi: 10.1021/acschembio.6b00465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock REW, Chapple DS. Peptide Antibiotics. Antimicrobial Agents and Chemotherapy. 1999;43:1317–1323. doi: 10.1128/aac.43.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak MJ, Wiedmann M, Boor KJ. Alternative sigma factors and their roles in bacterial virulence. Microbiol Mol Biol Rev. 2005;69:527–543. doi: 10.1128/MMBR.69.4.527-543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mousa JJ, Bruner SD. Structural and mechanistic diversity of multidrug transporters. Natural Product Reports. 2016;33:1255–1267. doi: 10.1039/c6np00006a. [DOI] [PubMed] [Google Scholar]
- Nonaka G, Blankschien M, Herman C, Gross CA, Rhodius VA. Regulon and promoter analysis of the E. coli heat-shock factor, σ32, reveals a multifaceted cellular response to heat stress. Genes & Development. 2006;20:1776–1789. doi: 10.1101/gad.1428206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overhage J, Campisano A, Bains M, Torfs ECW, Rehm BHA, Hancock REW. Human Host Defense Peptide LL-37 Prevents Bacterial Biofilm Formation. Infection and Immunity. 2008;76:4176–4182. doi: 10.1128/IAI.00318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JAC. GO2MSIG, an automated GO based multi-species gene set generator for gene set enrichment analysis. BMC Bioinformatics. 2014;15:146. doi: 10.1186/1471-2105-15-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nature Genetics. 2006;38:500. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- Reitzer L, Schneider BL. Metabolic context and possible physiological themes of sigma(54)-dependent genes in Escherichia coli. Microbiol Mol Biol Rev. 2001;65:422–444. doi: 10.1128/MMBR.65.3.422-444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein DM, Pahel G, Tyler B, Magasanik B. Regulation of expression from the glnA promoter of Escherichia coli in the absence of glutamine synthetase. Proceedings of the National Academy of Sciences. 1980;77:7372–7376. doi: 10.1073/pnas.77.12.7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Bell CE. Domain Structure of the Redβ Single-Strand Annealing Protein: the C-terminal Domain is Required for Fine-Tuning DNA-binding Properties, Interaction with the Exonuclease Partner, and Recombination in vivo. Journal of Molecular Biology. 2016;428:561–578. doi: 10.1016/j.jmb.2016.01.008. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Bird GH. Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. Journal of Medicinal Chemistry. 2014;57:6275–6288. doi: 10.1021/jm4011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gralla JD. Roles for the C-terminal Region of Sigma 54 in Transcriptional Silencing and DNA Binding. Journal of Biological Chemistry. 2001;276:8979–8986. doi: 10.1074/jbc.M009587200. [DOI] [PubMed] [Google Scholar]
- Wiese A, Brandenburg K, Ulmer AJ, Seydel U, Müller-Loennies S. The Dual Role of Lipopolysaccharide as Effector and Target Molecule. Biological Chemistry. 1999:767. doi: 10.1515/BC.1999.097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.