

SUMMARY
Much of our understanding of chromosome segregation is based on cell culture systems. Here, we examine the importance of the tissue environment for chromosome segregation by comparing chromosome segregation fidelity across several primary cell types in native and nonnative contexts. We discover that epithelial cells have increased chromosome missegregation outside of their native tissues. Using organoid culture systems, we show that tissue architecture, specifically integrin function, is required for accurate chromosome segregation. We find that tissue architecture enhances the correction of merotelic microtubule-kinetochore attachments and this is especially important for maintaining chromosome stability in the polyploid liver. We propose that disruption of tissue architecture could underlie the widespread chromosome instability across epithelial cancers. Moreover, our findings highlight the extent to which extracellular context can influence intrinsic cellular processes and the limitations of cell culture systems for studying cells that naturally function within a tissue.
In brief
Tissue architecture and integrin function are critical factors that support chromosome segregation fidelity in epithelial tissues
Graphic Abstract
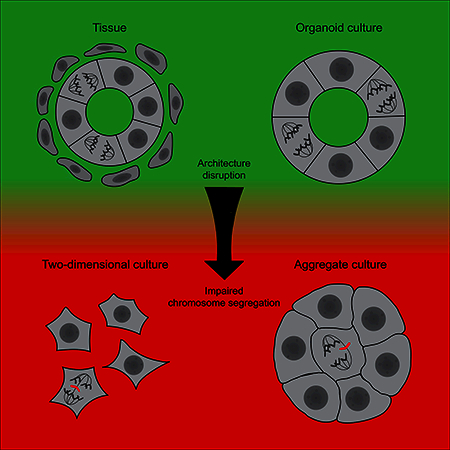
INTRODUCTION
Organism viability requires faithful chromosome segregation among germline and somatic cells. Chromosome segregation errors can lead to chromosome damage and aneuploidy. Aneuploidy is usually associated with impaired cellular fitness, and indeed when present throughout an organism typically results in embryonic lethality (Hassold and Jacobs, 1984; Williams et al., 2008). However, chromosome missegregation and aneuploidy are also prevalent in cancer, where aneuploidy is suspected to enhance cellular fitness (Knouse et al., 2017). Although these consequences of aneuploidy are paradoxical at the cellular level, they are uniformly adverse for the organism. As such, chromosome segregation is subject to meticulous execution and stringent regulation.
The central mediators of chromosome segregation are the microtubules of the mitotic spindle and the kinetochores of sister chromatids. Interactions between microtubules and kinetochores drive the partitioning of sister chromatids into separate daughter cells. In order for sister chromatids to segregate properly, their kinetochores must attach to microtubules emanating from opposite spindle poles. However, microtubules capture kinetochores randomly, making it possible for a given kinetochore to attach to microtubules emanating from the same spindle pole as the sister kinetochore, both spindle poles, or neither spindle pole. In contrast to proper attachments, which produce tension across sister kinetochores, most improper attachments fail to produce such tension. In order to eliminate aberrant attachments, the cell relies on the cooperative functions of error correction and the spindle assembly checkpoint. The error correction pathway severs microtubule-kinetochore attachments that are not under tension while the spindle assembly checkpoint prevents anaphase onset until all kinetochores are stably bound to microtubules (Cheeseman et al., 2002; DeLuca et al., 2006; Liu et al., 2009).
The most common chromosome segregation error occurs when a merotelic attachment, in which a kinetochore is bound by microtubules emanating from both spindle poles, persists into anaphase (Cimini et al., 2001). Although merotelic attachments can be a natural consequence of unbiased microtubule-kinetochore capture, they form more frequently when spindle formation is impaired, such as when centrosome separation is delayed or when a cell enters mitosis with supernumerary centrosomes (Ganem et al., 2009; Silkworth et al., 2012; 2009). Some merotelic attachments can be detected and corrected by the error correction pathway as mitosis progresses (Cimini et al., 2003; 2006). However, because a merotelically-attached kinetochore is bound by microtubules emanating from opposite spindle poles, it is possible for the kinetochore to be stretched sufficiently to escape error correction. As a result, these merotelic attachments can persist into anaphase. Depending on the ratio of microtubules from each spindle pole bound to a persistent merotelic attachment, the affected chromosome may lag behind the two main masses of segregating chromosomes during anaphase and potentially segregate into the wrong daughter cell (Cimini et al., 2004). If the lagging chromosome is separated sufficiently from the other segregating chromosomes, it will not be incorporated into the main nucleus but instead form a separate micronucleus during telophase. Regardless of whether or not the lagging chromosome segregates into the correct daughter cell, it can experience extensive DNA damage during cytokinesis or once entrapped in a micronucleus (Crasta et al., 2012; Hatch et al., 2013; Janssen et al., 2011; Zhang et al., 2015). Although lagging chromosomes and micronuclei are observed in less than 1% of mitoses in untransformed cell lines, their frequency can exceed 50% in many cancer cell lines (Cimini et al., 2001; Thompson and Compton, 2008; 2011).
Given that the key mediators of chromosome segregation all act within the cell, one might assume that this process is independent of the external environment. However, factors acting at the cell cortex and beyond are known to influence various aspects of chromosome segregation. For example, the pattern by which cells adhere to the extracellular matrix, transduced intracellularly via integrins, is known to influence the orientation of the mitotic spindle and the cell division axis in both cultured cells and tissues (Lechler and Fuchs, 2005; Théry et al., 2005). Additionally, many cultured cells round during mitosis (Stewart et al., 2010). Preventing this process through mechanical confinement or cytoskeletal perturbations can interfere with various aspects of chromosome segregation (Itabashi et al., 2012; Lancaster et al., 2013). There is also evidence to suggest that chromosome segregation fidelity is subject to higher regulation from the tissue environment. Hepatocytes are polyploid and harbor supernumerary centrosomes. Consistent with the established correlation between supernumerary centrosomes and chromosome missegregation in cultured cells, studies of hepatocytes in culture have shown high levels of chromosome missegregation and aneuploidy (Duncan et al., 2012; 2010). However, single cell sequencing of hepatocytes in the adult liver has revealed only low levels of aneuploidy (Knouse et al., 2014). In a similar vein, although many cancer cells have high levels of chromosome missegregation—a condition known as chromosome instability—there is no universal explanation for its presence. Indeed, many chromosomally unstable cancer cells have functional spindle assembly checkpoints (Haruki et al., 2001; Tighe et al., 2001). These observations warrant investigating chromosome segregation fidelity directly in tissues.
Here, we examine chromosome segregation fidelity across multiple mammalian cell types within their native tissues and as cultured cells. We find that epithelial cells have high chromosome segregation fidelity in tissues but that this is lost when tissue structure is disrupted. Using organoid culture systems, we show that tissue architecture, specifically integrin function, is required for chromosome segregation fidelity in epithelia. We find that tissue architecture enhances the correction of merotelic attachments, and that this is especially important for accurate chromosome segregation in the polyploid liver. Our results highlight the importance of external factors in governing chromosome segregation fidelity in epithelia, suggest that disruption of tissue architecture could underlie chromosome instability in cancer, and reveal the limitations of cell culture systems for faithful recapitulation of fundamental cellular processes.
RESULTS
Tissue architecture is required for chromosome segregation fidelity in epithelia
To evaluate chromosome segregation in primary cells, we isolated five proliferative tissues from mouse: mammary gland during pregnancy, embryonic skin, neonatal liver, embryonic brain, and lymph nodes during an immune response. We then analyzed the mitoses of mammary epithelial cells, keratinocytes, hepatocytes, neural progenitor cells, and T cells in their native tissues and after the cells had been dissociated and expanded in culture for up to 48 hours (Figure 1A). We quantified chromosomes missegregation in two different ways: (1) lagging chromosomes in anaphase and (2) micronuclei in telophase (Figure 1B). For mammary gland, skin, and neonatal liver, we never observed lagging chromosomes and micronuclei in the tissue. However, we observed significant increases in these defects when epithelial cells from these tissues were dissociated and expanded in culture (Figure 1B-C). For embryonic brain and lymph node, we observed a low frequency of chromosome segregation defects in the tissues and these defects increased slightly but not significantly when the cells were dissociated and expanded in culture (Figure 1B, note that we never observed micronuclei in lymphocytes which could be due to these cells having short anaphase spindles that favor the incorporation of lagging chromosomes into the main nuclei during telophase). Importantly, the increase in chromosome missegregation in dissociated cells was not secondary to polyploidization or supernumerary centrosomes, as there was little to no increase in the number of mitotic cells harboring supernumerary centrosomes within the first 48 hours of culture (Figure S1A). These results lead to the surprising conclusion that chromosome segregation fidelity depends on the tissue environment for several cell types.
Figure 1. Tissue architecture is required for chromosome segregation fidelity in epithelia.
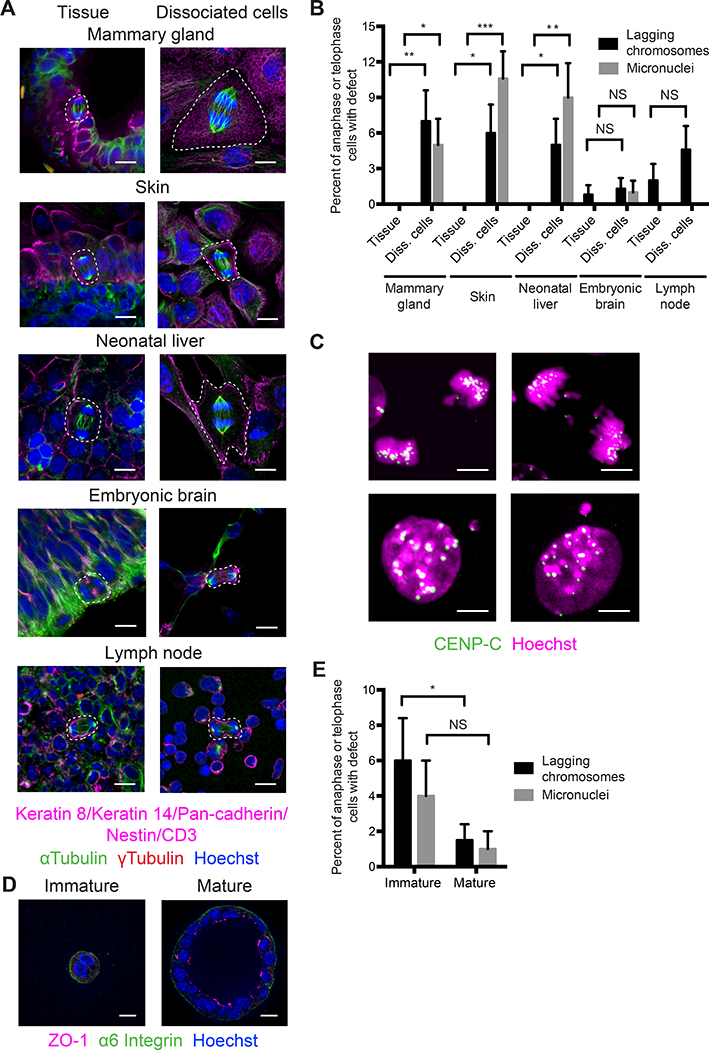
(A) Images of mitotic cells (dashed white outline) in tissue and dissociated cells from mammary gland, skin, neonatal liver, embryonic brain, and lymph node immunostained for epithelial or cell type-specific markers (magenta), αtubulin (green), and ϒtubulin (red). DNA is stained with Hoechst (blue). Scale bars, 10 μm.
(B) Prevalence of lagging chromosomes (black bars) and micronuclei (grey bars) in tissues and dissociated cells. Error bars represent standard deviation. P values are: p = 0.007 for lagging chromosomes in mammary gland tissue versus dissociated cells, p = 0.03 for micronuclei in mammary gland tissue versus dissociated cells, p = 0.01 for lagging chromosomes in skin tissue versus dissociated cells, p = 0.0004 for micronuclei in skin tissue versus dissociated cells, p = 0.03 for lagging chromosomes in neonatal liver tissue versus dissociated cells, and p = 0.002 for micronuclei in neonatal liver tissue versus dissociated cells by one-tailed Fisher’s exact test. NS, not significant. n ≥ 2 biological replicates totaling ≥ 100 anaphase cells and ≥ 100 telophase cells per condition.
(C) Images of lagging chromosomes (arrowheads) in a dissociated keratinocyte (top panel, left) and a neonatal hepatocyte (top panel, right) and micronuclei (arrowheads) in dissociated mammary epithelial cells (bottom panel) immunostained for the centromere (CENP-C, green). DNA is stained with Hoechst (magenta). Scale bars, 5 μm.
(D) Images of immature and mature spheroids after 48 and 96 hours of culture in Matrigel, respectively, immunostained for ZO-1 (magenta) and α6 integrin (green). DNA is stained with Hoechst (blue). Scale bars, 10 μm.
(E) Prevalence of lagging chromosomes (black bars) and micronuclei (grey bars) in immature and mature spheroids. Error bars represent standard deviation. p = 0.04 for lagging chromosomes in immature versus mature spheroids by one-tailed Fisher’s exact test. NS, not significant. n ≥ 2 biological replicates totaling ≥ 100 anaphase cells and ≥ 100 telophase cells per condition.
See also Figure S1.
The tissue and cell culture environments differ in many ways. When cells are dissociated from their tissues and expanded in culture they lose their canonical adhesions to other cells and the extracellular matrix, and consequently may lose their native shape and polarity. Beyond these structural changes, cells in culture are exposed to different extracellular signals and atmosphere compared to cells in tissues. Any or all of these alterations could in principle influence chromosome segregation. However, the observation that the tissue environment was important for chromosome segregation fidelity in some but not all cell types provided insight into the tissue dependence. The cell types that experienced increased chromosome missegregation upon dissociation— mammary epithelial cells, keratinocytes, and neonatal hepatocytes—naturally divide within an epithelium and maintain adhesions to neighboring cells and the extracellular matrix when doing so (Ragkousi and Gibson, 2014) (Figure 1A). In contrast, cell types for which the tissue environment contributed little to chromosomes segregation fidelity— neural progenitor cells and T cells—normally migrat e to less structured areas, the ventricular zone and lymph node, respectively, to divide (Kriegstein and Alvarez-Buylla, 2009; Neumann et al., 1992) (Figure 1A).
We hypothesized that disruption of epithelial architecture causes chromosome missegregation in cells that naturally divide within an epithelium. To test this, we cultured mammary epithelial cells in Matrigel as this produces spheroids that resemble the acini of the mammary epithelium (Jechlinger et al., 2009). After 48 hours in Matrigel, cells formed small clusters that had neither a central lumen nor defined apicobasal polarity (henceforth immature spheroids, Figure 1D). However, after 96 hours in Matrigel the majority of cells formed spheroids with a single cell layer surrounding a central lumen (henceforth mature spheroids, Figure 1D). These cells had apicobasal polarity as evidenced by the basal localization of the cell-matrix adhesion protein α6 integrin and the apical ribbon of the tight junction protein ZO-1 (Figure 1D). The immature and mature spheroids thus provided conditions that were equivalent in all respects except for the duration of culture in Matrigel and the absence or presence of epithelial architecture. Notably, cells in immature spheroids had similar levels of chromosome missegregation as dissociated cells (Figure 1E, 1B). In contrast, chromosome segregation fidelity was significantly improved in mature spheroids (Figure 1E). As was true for dissociated cells, the increased chromosome missegregation in immature spheroids was not secondary to polyploidization or supernumerary centrosomes (Figure S1B). We conclude that tissue architecture is required for accurate chromosome segregation in epithelia.
Integrin function is required for chromosome segregation fidelity
We next sought to determine which element of tissue architecture was important for chromosome segregation fidelity. Each epithelium is defined by distinct patterns of adhesions between adjacent cells and between cells and the extracellular matrix. These adhesions in turn impart shape and polarity to individual cells (Lee and Streuli, 2014). We tested whether changes in cell shape or polarity were responsible for chromosome missegregation upon tissue architecture disruption. To evaluate cell shape, we measured three orthogonal dimensions of interphase and mitotic cells in tissues, dissociated cells, and spheroids. We then calculated the centroid for a given group of cells and performed permutation tests to determine whether different groups of cells had significantly different dimensions. This analysis led to several conclusions. First, in tissues, we observed variable differences in the dimensions of interphase and mitotic cells. In the mammary gland and lymph node, interphase and mitotic cells did not have significantly different dimensions, whereas for skin, neonatal liver, and embryonic brain, the centroids of interphase and mitotic cells differed by approximately 3 μm (Figure 2A, S2A-D). Second, dramatic shape changes occurred when cells from all tissues except lymph node transitioned from the tissue to culture (Figure 2A, S2A-D). In these cases, the cells spread out and flattened, expanding in the x and y dimensions and decreasing in the z dimension. Once in culture, there was never a significant difference in the shape of interphase and mitotic cells across all cell types (Figure 2A, S2A-D). This is in contrast to commonly cultured cell lines which round up during mitosis (Lancaster et al., 2013; Stewart et al., 2010).
Figure 2. Integrin function is required for chromosome segregation fidelity.
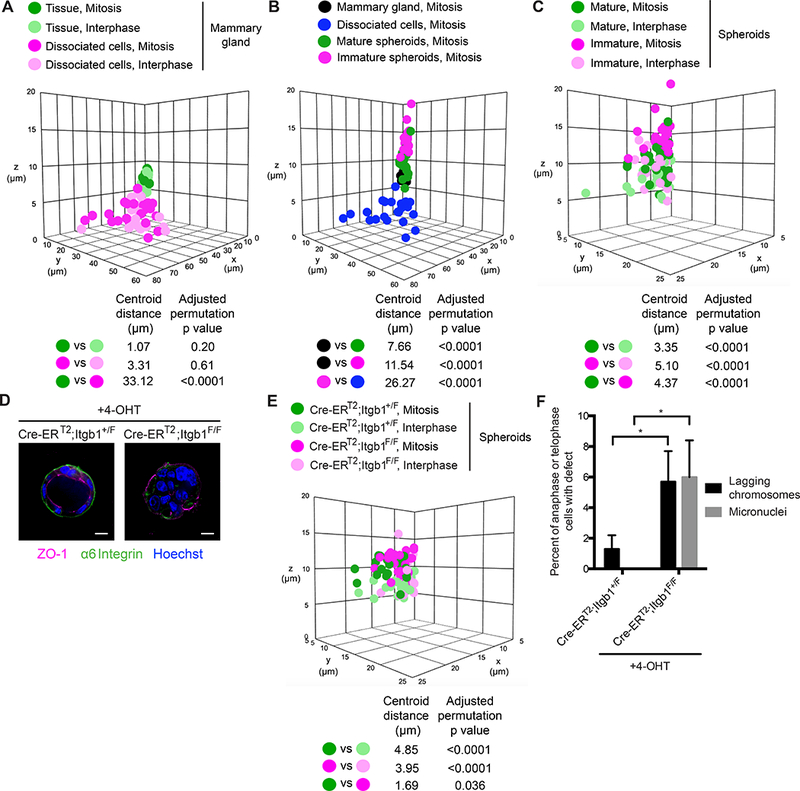
(A) Dimensions of mitotic (dark circles) and interphase (light circles) mammary epithelial cells in mammary gland (green circles) and as dissociated cells (pink circles). For dissociated cells, the z dimension always corresponds to the height of the cell relative to the coverslip. The distance between the centroids (in μm) of select pairs of conditions and the adjusted permutation p value using the Benjamini and Hochberg method for multiple comparisons are indicated below the graph. n = 2 biological replicates with ≥ 10 cells per replicate per condition.
(B) Dimensions of mitotic mammary epithelial cells in mammary gland (black circles), as dissociated cells (blue circles), in mature spheroids (green circles), and in immature spheroids (pink circles). n = 2 biological replicates with ≥ 10 cells per replicate per condition. The mitotic cells from mammary gland and dissociated cells are replotted from Figure 2A.
(C) Dimensions of mitotic (dark circles) and interphase (light circles) cells in mature (green circles) and immature (pink circles) spheroids. n = 2 biological replicates with ≥ 10 cells per replicate per condition. The mitotic cells from immature and mature spheroids are replotted from Figure 2B.
(D) Images of control (Cre-ERT2; Itgb1+/F) and β1 integrin knockout (Cre-ERT2; Itgb1F/F) spheroids treated with 4-hydroxytamoxifen (4-OHT) immunostained for ZO-1 (magenta) and α6 integrin (green). DNA is stained with Hoechst (blue). Scale bars, 10 μm.
(E) Dimensions of mitotic (dark circles) and interphase (light circles) cells in control and β1 integrin knockout spheroids treated with 4-hydroxytamoxifen (4-OHT). n = 2 biological replicates with ≥ 10 cells per replicate per condition.
(F) Prevalence of lagging chromosomes (black bars) and micronuclei (grey bars) in control and β1 integrin knockout spheroids treated with 4-hydroxytamoxifen (4-OHT). Error bars represent standard deviation. P values are: p = 0.047 for lagging chromosomes in control versus β1 integrin knockout spheroids and p = 0.01 for micronuclei in control versus β1 integrin knockout spheroids by one-tailed Fisher’s exact test. n = 2 biological replicates totaling ≥ 100 anaphase cells and ≥ 100 telophase cells per condition.
See also Figure S2.
Mammary epithelial cells in Matrigel were more similar in shape to cells in the mammary gland than to dissociated cells (Figure 2B). Among the spheroids, there were significant differences in the dimensions of mitotic cells in immature and mature spheroids, however the magnitude of the centroid difference (4.37 μm) was far less than that for mitotic cells in mammary gland compared to dissociated cells (33.12 μm; Figure 2A, C). That culturing cells as immature spheroids resulted in a dramatic rescue in cell shape compared to dissociated mammary epithelial cells, but did not improve chromosome segregation fidelity, argues against the cell shape changes that occur in the absence of tissue architecture as driving the observed chromosome segregation defects.
Across epithelia, integrin-mediated adhesion to the extracellular matrix plays a critical role in establishing intracellular polarity (Lee and Streuli, 2014). To test the importance of integrin-mediated adhesion and downstream polarity, we depleted β1 integrin in mature spheroids (Figure S2E). Depleting β1 integrin disrupted spheroid architecture, with cells occluding the lumen, and eliminated cell polarity, with loss of the apical ribbon of ZO-1 and basal localization of α6 integrin (Akhtar and Streuli, 2012) (Figure 2D). β1 integrin knockout also subtly altered cell shape, with centroids of mitotic cells in control and knockout spheroids differing by 1.69 μm (Figure 2E). Importantly, loss of β1 integrin caused chromosome segregation defects at a frequency similar to that of immature spheroids and dissociated mammary epithelial cells (Figure 2F, 1E, 1B). As with all other cases of disrupted tissue architecture, the increased chromosome missegregation was not due to supernumerary centrosomes (Figure S2F). These results indicate that integrin-mediated adhesion to the extracellular matrix is required for chromosome segregation fidelity in epithelia. Although loss of β1 integrin did subtly alter cell shape, making it possible that cell shape changes underlie the chromosome segregation defects, the small magnitude of this change relative to the overall dimensions of the cells makes this unlikely. These results, in conjunction with our observations in immature spheroids where β1 integrin is present but polarity has yet to be established, suggests that tissue architecture facilitates chromosome segregation fidelity through integrin-mediated establishment of cell polarity.
Tissue architecture enhances the correction of merotelic microtubule-kinetochore attachments
We next sought to determine how tissue architecture influences chromosome segregation. The frequency with which merotelic attachments persist as lagging chromosomes depends on their rate of formation and the extent to which they are eliminated by the coupled activity of error correction and the spindle assembly checkpoint (Cimini et al., 2003; 2006). We used immature and mature spheroids to gain mechanistic insight as they provide a genetically and environmentally equivalent system for testing the role of tissue architecture.
We first investigated whether tissue structure influences chromosome segregation via gene expression changes or post-transcriptional effects. To this end, we performed RNA sequencing on spheroids after 48 and 96 hours of culture in Matrigel, when the majority of spheroids are immature and mature, respectively. Gene expression analysis revealed that spheroids after 48 and 96 hours in Matrigel were transcriptionally more similar to each other than to dissociated mammary epithelial cells cultured for the same duration (Figure S3A). Gene set enrichment analysis revealed that genes involved in epithelial-mesenchymal transition were up-regulated in 48 hour spheroids, perhaps reflecting the absence of epithelial architecture (Figure S3B). Hallmark gene sets enriched in 96 hour spheroids included E2F targets, consistent with the observed increase in mitotic index at this time (Figure S3B). Indeed, the expression of genes that drive proliferation, such as Ccnb1, was 1.2-fold higher in spheroids after 96 hours compared to 48 hours (Figure 3A). Expression of chromosome segregation genes, those mediating microtubule-kinetochore attachment, error correction, and the spindle assembly checkpoint, was also elevated in 96 hour spheroids (Figure 3A). However, the fold increase in chromosome segregation genes was similar to that of proliferative genes, indicating that, per mitotic cell, the expression of genes regulating chromosome segregation is equivalent in the two types of spheroids (Figure 3A). This suggests that tissue architecture influences chromosome segregation in a post-transcriptional manner.
Figure 3. Tissue architecture enhances the correction of merotelic microtubule-kinetochore attachments.
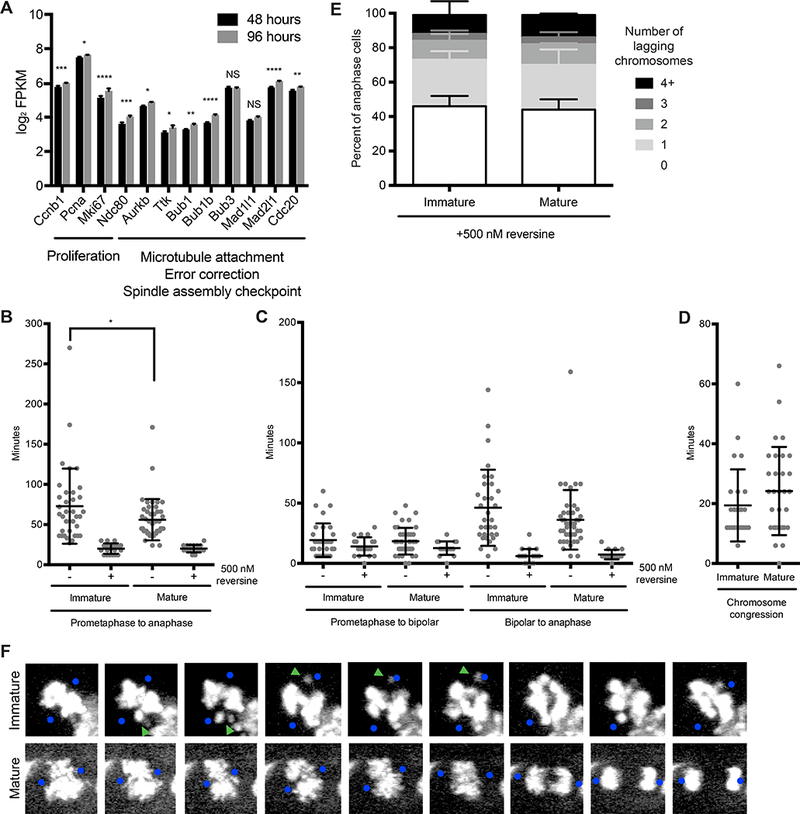
(A) Average expression (log2 FPKM) of genes involved in proliferation and chromosome segregation in spheroids cultured for 48 or 96 hours in Matrigel. Error bars represent standard deviation. P values are: p = 0.00046 (Ccnb1), 0.01 (Pcna), 2.1 × 10−8 (Mki67), 0.00044 (Ndc80), 0.01 (Aurkb), 0.039 (Ttk), 0.0047 (Bub1), 2.2 × 10−7 (Bub1b), 3.2 × 10−6 (Mad2l1), 0.0035 (Cdc20) by Wald test adjusted for multiple testing by the Benjamini and Hochberg method. NS, not significant. n = 3 biological replicates per condition.
(B) Time elapsed from prometaphase to anaphase in mitotic mammary epithelial cells in Centrin 2-GFP;H2B-mCherry immature and mature spheroids in the absence (−) and presence (+) of 500 nM reversine. Horizontal lines represent mean and standard deviation. p = 0.04 for immature and mature spheroids in the absence of reversine by two-tailed unpaired t-test. n ≥ 2 biological replicates totaling ≥ 16 mitoses per condition.
(C) Time elapsed from prometaphase to the establishment of a bipolar spindle and the establishment of a bipolar spindle to anaphase in mitotic mammary epithelial cells in Centrin 2-GFP;H2B-mCherry immature and mature spheroids in the absence (−) and presence (+) of 500 nM reversine. Horizontal lines represent mean and standard deviation. n ≥ 2 biological replicates totaling ≥ 16 mitoses per condition.
(D) Duration of chromosome congression in mitotic mammary epithelial cells in Centrin 2-GFP;H2B-mCherry immature and mature spheroids. Horizontal lines represent mean and standard deviation. n ≥ 2 biological replicates totaling > 25 mitoses per condition.
(E) Quantification of lagging chromosomes in anaphase of mammary epithelial cells in immature and mature spheroids in the presence of 500 nM reversine. Error bars represent standard deviation. n = 2 biological replicates with > 35 anaphase cells per replicate per condition.
(F) Time lapse images of mitotic mammary epithelial cells in Centrin 2-GFP;H2B-mCherry immature (top panel) and mature (bottom panel) spheroids. Blue dots represent the position of Centrin 2-GFP foci when they could be visualized. Green arrowheads mark chromosomes expelled from the metaphase plate in immature spheroids. Number indicates minutes since prometaphase onset. Only the H2B-mCherry signal (white) is shown.
See also Figure S3.
We then analyzed the mitoses in immature and mature spheroids to identify evidence of increased formation or reduced elimination of merotelic attachments in the absence of architecture. We first examined mitotic progression by live imaging of spheroids from mice expressing centrin 2-GFP and H2B-mCherry fusion proteins to reveal centrosome and chromosome movement, respectively. By analyzing chromosome movement we observed that the time from prometaphase to anaphase was significantly longer in immature compared to mature spheroids (Figure 3B, first and third columns, Movies S1–4). We then analyzed centrosome movement with respect to chromosome movement and found that much of the increased duration of mitosis in immature spheroids occurred after the centrosomes had moved to opposite sides of the condensed chromosomes, although this difference was not statistically significant (Figure 3C, fifth and seventh columns). This suggested that cells in immature spheroids were arresting in late prometaphase or metaphase, a signature of spindle assembly checkpoint activation. Indeed, the differences between immature and mature spheroids were eliminated by treatment with the spindle assembly checkpoint inhibitor reversine (Santaguida et al., 2010) (Figure 3B-C). These results suggest that the increase in lagging chromosomes in the absence of tissue architecture is not a consequence of an impaired checkpoint but rather due to an increased prevalence of merotelic attachments that triggers checkpoint activation and, because not all merotelic attachments can be corrected, increases the frequency of lagging chromosomes in anaphase.
Increased merotely and checkpoint activation could result from more merotelic attachments forming during early mitosis or reduced correction of these attachments as mitosis progresses. Impaired spindle formation, either from delayed centrosome separation or supernumerary centrosomes, can increase the frequency of merotely in early mitosis (Ganem et al., 2009; Silkworth et al., 2009; 2012). However, we did not observe any evidence of impaired spindle formation in the absence of tissue architecture. For one, centrosome number was not increased in dissociated cells, immature spheroids, or integrin knockout spheroids (Figure S1A-B, S2F). Moreover, live imaging of immature spheroids did not reveal an increase in the time for centrosomes to move to opposite sides of the condensed chromosomes or the time required for chromosomes to congress to the metaphase plate (Figure 3C, first and third columns, 3D). To more directly quantify merotelic attachments in early mitosis, we induced precocious anaphase using reversine and quantified lagging chromosomes in immature and mature spheroids. The prevalence of lagging chromosomes in immature and mature spheroids was equivalent in the presence of reversine (Figure 3E). Together, these results argue that disruption of tissue architecture does not increase the formation of merotelic attachments during early mitosis, leaving the possibility that loss of tissue architecture impairs their correction.
The error correction pathway severs erroneous microtubule-kinetochore attachments to produce an unbound kinetochore that activates the spindle assembly checkpoint and serves as a substrate for future microtubule binding. We observed that in 12% (3 of 26) of immature spheroid mitoses individual chromosomes were expelled from the metaphase plate and moved toward the spindle pole (Figure 3F, top panel, arrowheads). We never observed such events in mature spheroids (0 of 30, Figure 3F, bottom panel). The poleward movement of chromosomes could reflect a sudden loss of microtubule attachment from the opposite spindle pole (Lampson et al., 2004). This observation, coupled to the prolonged activation of the spindle assembly checkpoint, suggests that the error correction pathway is destabilizing erroneous attachments but that proper attachments are not readily established in immature spheroids. It remains to be determined how tissue architecture promotes error correction. We did not observe any significant differences in the shape of the mitotic spindle or the density of spindle microtubules that were conserved in both cases of disrupted spheroid architecture (immature spheroids and β1 integrin knockout spheroids) (Figure S3C-F). We suspect that tissue architecture influences another aspect of the spindle, such as tension, which in turn influences how easily proper attachments are established once chromosomes have congressed to the metaphase plate.
Tissue architecture is especially important for chromosome segregation fidelity in the polyploid liver
If tissue architecture indeed facilitates the correction of merotelic attachments, it should be especially important in situations of increased merotely. The liver provides an ideal tissue for testing this prediction as hepatocytes polyploidize during early postnatal development (Guidotti et al., 2003). To examine mitoses of polyploid hepatocytes, we harnessed the liver’s remarkable regenerative capacity and performed partial hepatectomies on mice to stimulate proliferation in the tissue. We then compared the mitoses of the regenerating (polyploid) adult liver to the growing (diploid) neonatal liver. We first confirmed that polyploid hepatocytes harboring supernumerary centrosomes proliferate during regeneration (Figure S4A). Consistent with reports of hepatocytes and other cell types with supernumerary centrosomes in culture, hepatocytes in regenerating livers entered mitosis with multipolar spindles but then clustered centrosomes to form a bipolar spindle by anaphase (Duncan et al., 2010; Ganem et al., 2009; Silkworth et al., 2009) (Figure 4A, S4B). However, in contrast to prior studies, we observed very few lagging chromosomes and micronuclei in regenerating livers (Figure 4B). Nonetheless, this high accuracy of chromosome segregation was dependent on tissue architecture as we observed high levels of chromosome missegregation when we dissociated adult hepatocytes and expanded them in culture (Figure 4B-D). Similarly, we observed a significant increase in lagging chromosomes in regenerating livers depleted of β1 integrin without an obvious alteration in cell shape, although the effect was not as dramatic as that for β1 integrin knockout in spheroids (Speicher et al., 2014) (Figure 4B, E). Although there is evidence that cell shape and extracellular matrix adhesion can influence centrosome clustering and thus chromosome segregation fidelity in polyploid cells, there was no significant increase in the prevalence of multipolar anaphases in either dissociated hepatocytes or β1 integrin knockout livers, arguing that the increased chromosome missegregation was not secondary to impaired centrosome clustering (Kwon et al., 2008) (Figure S4B). Thus, while polyploid hepatocytes retain the capacity to cluster centrosomes when tissue architecture is disrupted, they lose the ability to correct merotelic attachments. This supports our conclusion that tissue architecture facilitates the correction of merotelic attachments throughout epithelia, as we find that tissue architecture has its greatest impact on chromosome segregation fidelity in cells harboring supernumerary centrosomes.
Figure 4. Tissue architecture is especially important for chromosome segregation fidelity in the polyploid liver.
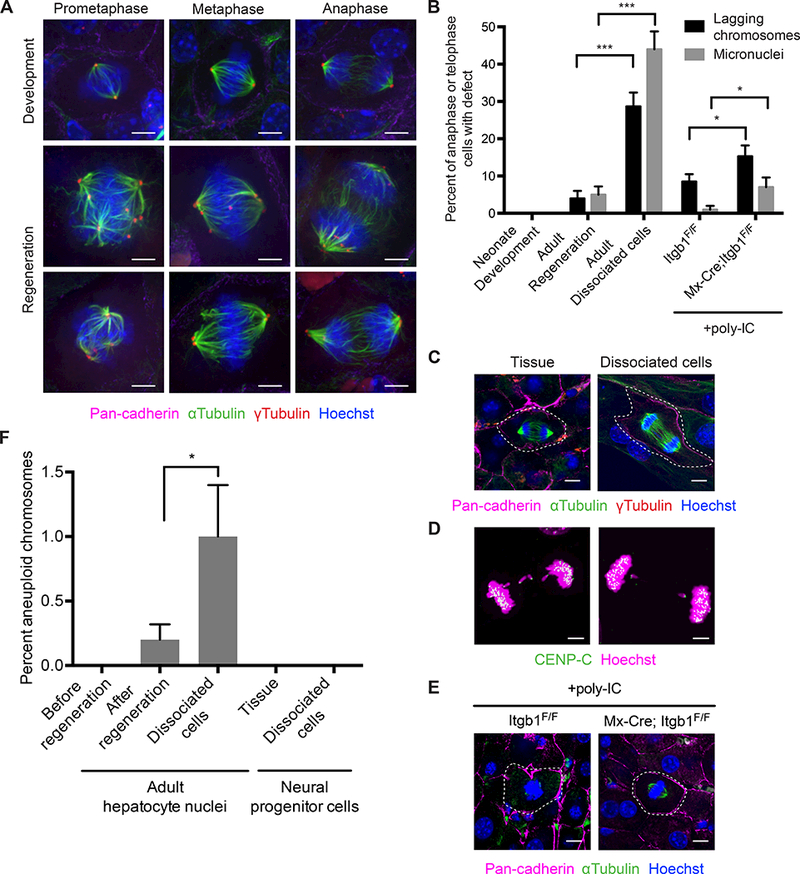
(A) Images of mitotic hepatocytes in prometaphase, metaphase, and anaphase in liver during neonatal development and adult regeneration immunostained for pan-cadherin (magenta), αtubulin (green), and ϒtubulin (red). DNA is stained with Hoechst (blue). Scale bars, 5 μm.
(B) Prevalence of lagging chromosomes (black bars) and micronuclei (grey bars) of hepatocytes in liver during neonatal development and adult regeneration, in dissociated cells from adult liver, and in control (Itgb1F/F) and β1 integrin knockout (Mx-Cre; Itgb1F/F) liver during adult regeneration after treatment with poly-IC. Error bars represent standard deviation. P values are: p = 0.0001 for lagging chromosomes in adult regeneration versus dissociated cells, p = 0.0001 for micronuclei in adult regeneration versus dissociated cells, p = 0.035 for lagging chromosomes in control versus β1 integrin knockout liver, and p = 0.03 for micronuclei in control versus β1 integrin knockout liver by one-tailed Fisher’s exact test. n ≥ 2 biological replicates totaling ≥ 100 anaphase cells and ≥ 100 telophase cells per condition.
(C) Images of mitotic hepatocytes (dashed white outline) from adult liver during regeneration and dissociated cells from adult liver immunostained for pan-cadherin (magenta), αtubulin (green), and ϒtubulin (red). DNA is stained with Hoechst (blue). Scale bars, 10 μm.
(D) Images of lagging chromosomes (arrowheads) in dissociated adult hepatocytes immunostained for the centromere (CENP-C, green). DNA is stained with Hoechst (magenta). Scale bars, 5 μm.
(E) Images of mitotic hepatocytes (dashed white outline) from control and β1 integrin knockout liver during adult regeneration immunostained for pan-cadherin (magenta), and αtubulin (green). DNA is stained with Hoechst (blue). Scale bars, 10 μm.
(F) Percent of aneuploid chromosomes in adult hepatocyte nuclei from liver before regeneration, after regeneration, and after expansion as dissociated cells, and neural progenitor cells from embryonic brain and after expansion as dissociated cells as determined by single nucleus or cell sequencing. Data for liver before regeneration and embryonic brain are from Knouse et al. 2014. p = 0.025 for adult hepatocyte nuclei after regeneration versus after expansion as dissociated cells by one-tailed Fisher’s exact test. n ≥ 1 biological replicate totaling > 25 nuclei or cells per condition.
See also Figure S4.
To test whether the chromosome segregation defects we observed led to genomic alterations, we sequenced single hepatocyte nuclei from hepatocytes after liver regeneration and from dissociated hepatocytes that were expanded in culture for an equivalent number of population doublings. Compared to liver before regeneration, there was a slight but insignificant increase in the frequency of aneuploid chromosomes after regeneration (Knouse et al., 2014) (Figure 4F). However, expanding hepatocytes in culture led to a five-fold increase in aneuploidy, nearly consistent with the six-fold increase in lagging chromosomes (Figure 4F, B). Neural progenitor cells expanded in culture for an equivalent number of population doublings did not accumulate any aneuploidy, consistent with them not manifesting high levels of chromosome missegregation as dissociated cells (Figure 4F, 1B). We conclude that tissue architecture, through enhanced correction of merotelic attachments, provides genomic stability even to polyploid epithelia. This resolves the conflicting reports of high levels of chromosome missegregation and aneuploidy in cultured hepatocytes but low levels of aneuploidy in the liver (Duncan et al., 2012; 2010; Knouse et al., 2014).
DISCUSSION
Control of chromosome segregation fidelity in epithelia
We investigated external influences on chromosome segregation fidelity by analyzing mitosis in several cell types in their native tissues and in culture. We find that for three epithelia—mammary gland, skin, and liver— the epithelial cells’ external environment influences chromosome segregation fidelity. A conserved feature of these tissues is that they require epithelial cells to adopt defined shape and polarity for the tissue to provide barrier or transport function. This shape and polarity are dictated by extracellular cues. The cells maintain these characteristics throughout cell division, presumably to maintain tissue function (Ragkousi and Gibson, 2014). We find that features imposed by the larger tissue also promote proper chromosome segregation. As a result, when tissue architecture is disrupted, and cells lose the extracellular cues that influence intrinsic features such as polarity and shape, these cells also lose chromosome segregation fidelity. This might explain in part why cell division and epithelial morphogenesis are temporally separated during development (Grosshans and Wieschaus, 2000).
We demonstrate that tissue architecture facilitates chromosome segregation fidelity by enhancing the correction of merotelic attachments. Our results suggest, but do not prove, that this occurs via integrin-mediated establishment of polarity and not through external regulation of cell shape. There are established links between integrins, polarity, and the mitotic machinery. Integrin-mediated polarity influences mitotic spindle orientation via the polarized proteins LGN and NuMA interacting with dynein to position astral microtubules (Bergstralh et al., 2013; Lechler and Fuchs, 2005). Thus, it is plausible that integrin-mediated establishment of polarity could influence the mitotic spindle in a way that promotes proper microtubule-kinetochore attachments. Although we did not observe defects in spindle formation or morphology in the absence of tissue architecture, it is possible that interactions between polarity factors at the cell cortex and astral microtubules increase tension exerted across the spindle, which would in turn facilitate the correction of merotelic attachments. Determining how factors at the cell cortex influence the formation of proper microtubule-kinetochore attachments is an important future direction. Of note, we tried to test the role of cell polarity directly by culturing primary epithelial cells on micropatterns but were unable to achieve sufficient adhesion and proliferation of primary epithelial cells on the patterns to investigate chromosome segregation.
Not all cells depend on their native environments for chromosome segregation fidelity. For tissues in which cells do not divide in the context of a defined architecture— embryonic brain and lymph node—we did not observe a significant increase in chromosome segregation defects when cells were dissociated and expanded in culture. This suggests that cells that divide outside of the context of epithelial architecture regulate chromosome segregation entirely via intrinsic mechanisms and this renders the fidelity of this process independent of the external environment. However, it is possible that these autonomous mechanisms are not as robust as those provided by the tissue environment. While we never observed lagging chromosomes in the mammary gland, skin, or neonatal liver, we did on occasion observe lagging chromosomes in the embryonic brain and lymph node. Whether these subtle differences in chromosome segregation defects translate to physiologically relevant differences in chromosome stability across tissues, and whether these trends hold for other cell types not investigated in this present study, remains to be determined. In this vein, we note that appreciable levels of chromosome missegregation have been observed in cleavage-stage embryos, where cells proliferate rapidly in the absence of tissue structure (Vanneste et al., 2009).
Disruption of tissue architecture as a source of chromosome instability in cancer
Elevated chromosome missegregation, a state known as chromosome instability, is a defining feature of epithelial tumors and believed to drive both tumor development and metastasis (Knouse et al., 2017). In spite of its ubiquity, there is no universal explanation for chromosome instability in cancer. Notably, disruption of tissue architecture occurs throughout epithelial tumorigenesis. Epithelial cells often lose polarity during their initial transformation and epithelial features are further lost when these cells undergo an epithelial-mesenchymal transition (EMT) and metastasize (Muthuswamy and Xue, 2012; Nieto et al., 2016). Inflammation could further disrupt tumor architecture throughout tumorigenesis. Our observation that disruption of epithelial architecture leads to chromosome missegregation raises the possibility that the disruption of architecture during carcinogenesis provides a universal path to chromosome instability regardless of the mutational background and in the presence of otherwise functional mitotic machinery. We highlight that the degree of chromosome instability caused by disruption of tissue architecture would be conducive to tumorigenesis. The frequency of lagging chromosomes resulting from disrupted architecture is moderate relative to that caused by mutations in the mitotic machinery. Indeed, prior studies have shown that moderate chromosome missegregation promotes tumorigenesis, whereas higher levels are incompatible with cell viability (Silk et al., 2013).
Directly testing whether architecture disruption underlies chromosome instability in cancer is challenging for several reasons. To test this hypothesis during primary tumor formation, one must exclude the possibility that any of the mutations that led to tumorigenesis have a direct effect on chromosome segregation. Moreover, although EMT can be induced in untransformed cells and thereby provide a genetically controlled means of testing this hypothesis, EMT inhibits the proliferation of untransformed cells. Nonetheless, there is circumstantial evidence supporting a causal relationship between EMT and chromosome instability. First, we note that immature spheroids exhibit a gene expression signature reminiscent of EMT, suggesting that they reflect similar tissue disruptions and that the chromosome missegregation occurring in immature spheroids could also take place during the EMT. Second, multiple studies have demonstrated a correlation between chromosome instability and EMT (Comaills et al., 2016; Roschke et al., 2008). While this correlation has led many to propose that chromosome instability promotes EMT, our observations suggest the opposite: that loss of epithelial architecture during EMT drives chromosome instability.
The importance of context
Cell culture systems have greatly advanced our understanding of many cellular processes. However, our finding that primary epithelial cells in culture do not undergo chromosome segregation with the same fidelity as in tissues has several implications. Given the intimate link between chromosome stability and cell viability, our observation stresses the importance of utilizing organotypic culture systems that recapitulate tissue architecture for any pursuits requiring the long-term or high-fold expansion of primary epithelial cells outside of their native tissue. More broadly, our findings show that a process as intracellular as chromosome segregation is still influenced by extracellular context. We expect that chromosome segregation is not the only process subject to such dependence. While cell culture systems can certainly provide a tractable means of investigating cell biology, consideration must also be given to the larger context.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Angelika Amon (angelika@mit.edu).
Experimental Model and Subject Details
Animals
C57BL/6J mice, Centrin 2-GFP mice, Cre-ERT2 mice, and Itgb1flox mice were purchased from the Jackson Laboratory. Mx-Cre and Itgb1flox mice were obtained from Reinhard Fässler (Max Planck Institute of Biochemistry, Martinsried, Germany). H2B-mCherry mice were obtained from the RIKEN Center for Life Science Technologies (CLST) (Kobe, Japan). The genders of the mice from which tissues were harvested are indicated below. All mice were group-housed with a 12-hour light-dark cycle (light from 7 AM to 7 PM, dark from 7 PM to 7 AM) in a specific-pathogen-free animal facility with unlimited access to food and water. All animal procedures were approved by the local animal welfare committees (Massachusetts Institute of Technology Committee on Animal Care and Kantonales Veterinäramt Zürich).
Tissues
Proliferative mammary glands were harvested from 8-week-old pregnant females at 4.5 days gestation. Proliferative skin was harvested from male and female embryos at 14.5 to 16.5 days gestation. Proliferative livers were harvested from male and female pups at 8 to 10 days of age (development) and from 8-week-old males 48 hours after partial hepatectomy (regeneration). Proliferative brains were harvested from male and female embryos at 12.5 to 14.5 days gestation. To stimulate T cell proliferation in the lymph node, 8-week-old males were injected with 25 μg of CD3ε antibody (clone 145–2C11, BD Biosciences) into the hock (Kamala, 2007). Ipsilateral inguinal lymph nodes containing proliferating T cells were harvested 24–48 hours later.
Method Details
Tissue dissociation
Mammary epithelial cells
Mammary glands from a single 6- to 8-week-old female mouse were harvested and digested in 10 mL DMEM-F12 with 2.5 mM L-glutamine, 25 mM HEPES, 100 U/mL and 100 μg/mL penicillin-streptomycin, 20 μg/mL Liberase TM (Roche), and 150 U/mL collagenase III at 37 °C with 5% CO2 for 16 hours. Digested tissue was resuspended with gentle trituration, diluted with 25 mL PBS, and pelleted at 1,000 RPM at room temperature for 5 minutes. The supernatant was aspirated to remove all but 5 mL of PBS and an equal volume of 0.25% trypsin-EDTA was added. Digested tissue was trypsinized at 37 °C with 5% CO2 for 45 minutes with occasional agitation. Trypsin was inactivated by adding 25 mL of DMEM-F12 with 2.5 mM L-glutamine, 25 mM HEPES, 100 U/mL and 100 μg/mL penicillin-streptomycin, and 10% FBS. Cells were treated with 100 μL of 1 mg/mL DNase and pelleted at 1,000 RPM at room temperature for 5 minutes. Cells were resuspended in phenol red-free Mammary Epithelial Basal Medium (MEBM) with growth factors (Lonza).
Keratinocytes
Neonatal (one-day-old) male and female mice were decapitated and cleaned with betadine followed by isopropyl alcohol. Skin was removed and floated on 0.25% trypsin in PBS at 4 °C for 16 hours. Epidermis was peeled from dermis, transferred to DMEM with 10% FBS, minced using a razor blade, and triturated using a serological pipette. Cells were centrifuged at 150 g at 4 °C for 5 minutes, resuspended in DMEM with 10% FBS, and passaged through at 70 μm mesh strainer. Cells were again pelleted, resuspended in Complete Defined Keratinocyte Serum-Free Medium (Thermo Fisher Scientific), and passaged through another 70 μm mesh strainer.
Neonatal hepatocytes
Livers were isolated from neonatal (8- to 10-day-old) male and female mice, cut into small pieces, and incubated in disruption solution (HBSS without calcium and magnesium, 25 mM HEPES, 0.5 mM EDTA, and 0.9 mM MgCl2, pH 7.4) at 37 °C with shaking for 10 minutes. This step was repeated twice with fresh disruption solution. Liver pieces were then incubated in digestion solution (HBSS with calcium and magnesium, 25 mM HEPES, and 0.05% collagenase IV, pH 7.4) at 37 °C with shaking for 5 minutes. Remaining liver pieces were transferred to fresh digestion solution for another 5 minutes and the two digestion solutions were combined. The suspension was triturated with a serological pipette, passed through a 70 μm filter, and diluted in hepatocyte medium (DMEM with 4.5 g/L glucose, 10% FBS, 1X MEM Non-Essential Amino Acids (Thermo Fisher Scientific). Cells were pelleted at 30 g at 4 °C for 3 minutes, resuspended in 5 mL hepatocyte medium, and combined with 5 mL Percoll-HBSS (4.5 mL Percoll, 0.5 mL 10X HBSS). Cells were pelleted at 150 g at 4 °C for 3 minutes, washed with hepatocyte media, and resuspended in SUM3 media (75% DMEM with 4.5 g/L glucose, 25% Waymouth’s MB 752/1, 0.5% FBS, 2 mM L-glutamine, 10 mM HEPES, 50 ng/mL epidermal growth factor, 1 μg/mL insulin, 30 nM sodium selenite, 10 μg/mL transferrin, 50 ng/mL somatotropin, 670 ng/mL triiodo-L-thyronine). Adult hepatocytes 8-week-old male mice were anesthetized with 2% isoflurane-oxygen delivered via nosecone. The liver was perfused with disruption solution for 10 minutes followed by digestion solution for 10 minutes by incising the portal vein with a 25G needle attached to a peristaltic pump. The inferior vena cava was incised immediately after perfusion began to allow fluid outflow. After perfusion was complete, the digested liver was transferred to 10 mL fresh digestion solution, shaken to liberate cells, triturated with a serological pipette, passaged through a 100 μm filter, and diluted with 10 mL hepatocyte medium. Cells were pelleted at 30 g at 4 °C for 3 minutes, resuspended in 12 mL hepatocyte medium, and combined with 12 mL Percoll-HBSS (10.8 mL Percoll, 1.2 mL 10X HBSS). Cells were pelleted at 150 g at 4 °C for 3 minutes, washed with hepatocyte medium, and resuspended in SUM3 media.
Neural progenitor cells
Brains were isolated from male and female embryos at 12.5 to 14.5 days gestation and minced into small pieces in PBS with 2% glucose. Brain pieces were transferred to 0.25% trypsin in EDTA and incubated at 37 °C for 15 minutes. An equal volume of NeuroCult proliferation medium with FBS (80% NeuroCult NSC Basal Medium, 10% NeuroCult NSC Proliferation Supplement, 10% FBS) (STEMCELL Technologies) was added to trypsin solution and tissue was triturated extremely gently using a P1000 pipette tip five times. Cells were centrifuged at 150 g at room temperature for 5 minutes. The pellet was triturated once in 1 mL NeuroCult proliferation medium with FBS, dissolved in an additional 2 mL NeuroCult proliferation medium with FBS, passaged through a 40 μm filter, and resuspended in NeuroCult proliferation medium with EGF (90% NeuroCult NSC Basal Medium, 10% NeuroCult NSC Proliferation Supplement, 20 ng/mL epidermal growth factor) (STEMCELL Technologies).
T cells
Spleens were harvested from 6- to 8-week-old male mice, homogenized through a 100 μm filter into 5 mL RPMI 1640 medium using a 1 mL syringe plunger, and centrifuged at 400 g at room temperature for 5 minutes. Cells were resuspended in 100 μL of supernatant, combined with 900 μL sterile water for 2 seconds to lyse erythrocytes, followed immediately by 100 μL of 10X PBS and 5 mL of RPMI 1640 medium. Cells were passed through a 40 μm filter, counted, and pelleted at 400 g at room temperature for 5 minutes. Cells were resuspended in 40 μL MACS buffer (2 mM EDTA and 0.5% BSA in PBS) per 107 cells. CD8 T cells were then isolated using the mouse CD8a+ T Cell Isolation Kit (Miltenyi Biotech) according to manufacturer instructions.
Cell culture
Mammary epithelial cells
To culture mammary epithelial cells as monolayers, dissociated cells were cultured in MEBM with supplements on dishes coated with 30 μg/mL collagen I at 37 °C with 5% CO2 for 24 hours. Cells were then washed with PBS three times and treated with 0.25% trypsin-EDTA at 37 °C for 45 seconds to remove contaminating fibroblasts. Remaining cells were then trypsinized with 0.25% trypsin-EDTA at 37 °C for 6 minutes and resuspended in DMEM-F12 with 2.5 mM L-glutamine, 25 mM HEPES, 100 U/mL and 100 μg/mL penicillin-streptomycin, and 10% FBS. Cells were pelleted at 1,000 RPM for 5 minutes, washed with PBS, resuspended in MEBM with supplements, 1 μM progesterone, 1 μg/mL prolactin, plated on coverslips coated with 30 μg/mL collagen I, and incubated at 37 °C with 5% CO2 for 24 hours. To culture mammary epithelial cells as spheroids, cells were trypsinized and washed as above and resuspended in MEBM with supplements at 7 × 105 cells per mL. Cells were then combined with four volumes of phenol red-free, growth factor-reduced Matrigel (Corning) and 100 μL gels were cast in individual wells of 24-well glass-bottom plates (Mat Tek). Gels were polymerized at 37 °C for 30 minutes before adding 500 μL of MEBM with supplements,1 μM progesterone, 1 μg/mL prolactin. Gels were cultured at 37 °C with 5% CO2 for 24 to 96 hours, with medium changed after 48 hours of culture. To deplete β1-integrin in spheroids, 100 nM 4-hydroxytamoxifen (4-OHT) was added to the medium of Cre-ERT2;Itgb1flox/flox or Cre-ERT2;Itgb1+/flox mammary epithelial cells for the 24 hours of culture in a monolayer and the first 48 hours of culture in Matrigel. To inhibit the spindle assembly checkpoint, 500 nM reversine (Cayman) was added to medium 2 hours prior to fixation or imaging.
Keratinocytes
Dissociated cells were cultured in Complete Defined Keratinocyte Serum-Free Medium (Thermo Fisher Scientific) on coverslips coated with 10 μg/mL fibronectin at 37 °C with 5% CO2 for 24 hours.
Hepatocytes
Dissociated cells were cultured in SUM3 medium on coverslips coated with 30 μg/mL collagen I at 37 °C with 5% CO2 for 48 hours.
Neural progenitor cells
Dissociated cells were cultured in NeuroCult proliferation medium with EGF on coverslips coated with 5 μg/mL CellAdhere Laminin-521 (STEMCELL Technologies) at 37 °C with 5% CO2 for 48 hours.
T cells
Individual wells of a 6-well plate were coated with 0.5 μg/mL CD3ε antibody (clone 145–2C11, BioLegend), 5 μg/mL CD28 antibody (clone 37.51, BioLegend) and 11.25 μg/mL Retronectin (Takara Bio) in 4 mL PBS at 4 °C overnight. Wells were then blocked with 2% BSA in PBS for 30 minutes and washed with PBS twice. Isolated T cells were cultured in T cell medium (90% RPMI 1640, 10% FBS, 2 mM glutamine, 50 U/mL and 50 μg/mL penicillin-streptomycin, 50 μM 2-mercaptoethanol, 1.7 μM insulin, 68.8 μM transferrin, and 38.7 nM sodium selenite) along with 0.5 ng/mL IL-7 (R&D Systems) and 20 ng/mL IL-2 (R&D Systems) at 2.5 × 106 cells per well at 37 °C with 5% CO2 for 36 hours.
Immunostaining
Tissues were fixed in 4% paraformaldehyde in PBS at room temperature for 16–24 hours. Fixed tissues were washed with PBS, cryoprotected with 30% sucrose, and frozen in O. C. T. Compound (Tissue-Tek). Slides with 6 to 30 μm-thick sections were prepared using a cryostat and stored at −80°C until use. Slides were dried at room temperature for 4–24 hours, rehydrated in PBS for 15 minutes and, if antigen retrieval was necessary, boiled in sodium citrate buffer (10 mM tri-sodium citrate dihydrate, 0.05% Tween 20, pH 6.0) for 20 minutes. Slides were washed with PBS, dried briefly, and sections outlined with a hydrophobic pen. Sections were incubated with extraction buffer (1% Triton X-100 in PBS) for 15 minutes followed by incubation in blocking solution (3% BSA, 0.3% Triton X-100 in PBS) for 1 hour at room temperature. Sections were incubated with primary antibodies diluted in blocking solution at room temperature for 16–24 hours. Sections were washed three times with blocking solution for 10 minutes each and then incubated with secondary antibodies diluted in blocking solution with 5 μg/mL Hoechst 33342 (Thermo Fisher Scientific) at room temperature for 1–2 hours. Sections were washed with blocking solution for 5 minutes twice and once with PBS for 5 minutes. Sections were mounted with ProLong Gold Antifade Reagent (Life Technologies). To immunostain dissociated cells, cells were washed with PBS and fixed with 4% PFA in PBS at room temperature for 10 minutes or 4% PFA in PBS at room temperature for 5 minutes followed by ice-cold methanol at – 20°C for 5 minutes. Fixed cells were washed with PBS for 5 minutes, permeabilized with 0.1% Triton X-100 in PBS (0.1% PBST) for 10 minutes, and blocked with 4% BSA in 0.1% PBST for 20 minutes. Primary antibodies were diluted in blocking solution and applied for 30 minutes. Cells were washed with 0.1% PBST for 5 minutes thrice. Secondary antibodies were diluted in blocking solution with 5 μg/mL Hoechst 33342 dye (Thermo Fisher Scientific) and applied for 30 minutes. Cells were washed with 0.1% PBST for 5 minutes twice followed by a PBS wash for 5 minutes. All incubations were performed at room temperature. Cells were mounted with ProLong Gold Antifade Mountant (Thermo Fisher Scientific).
To immunostain spheroids, gels were washed with PBS and fixed with 4% PFA in PBS at room temperature for 20 minutes or 4% PFA in PBS at room temperature for 10 minutes followed by ice-cold methanol at – 20 °C for 10 minutes. Fixed gels were washed with PBS for 5 minutes, permeabilized with 0.5% Triton X-100 in PBS (0.5% PBST) for 20 minutes, and blocked with 4% BSA in 0.5% PBST for 1 hour with rocking. Primary antibodies were diluted in blocking solution and applied for 16–24 hours with rocking. Gels were washed with 0.5% PBST for 10 minutes thrice. Secondary antibodies were diluted in blocking solution with 5 μg/mL Hoechst 33342 dye (Thermo Fisher Scientific) and applied for 1–2 hours. Gels were washed with 0.5% PBST for 10 minutes twice followed by PBS for 10 minutes. All incubations were performed at room temperature in the original glass-bottom wells. Gels were coated with ProLong Gold Antifade Mountant (Thermo Fisher Scientific).
The following primary antibodies were used: α-Tubulin-FITC (clone DM1A, 1:1,000, Sigma), Υtubulin (clone GTU88, 1:500, Sigma), keratin 14 (clone Poly19053, 1:1,000, BioLegend), cytokeratin 8 (clone EP1628Y, 1:1,000, Abcam), pan-cadherin (ab6529, 1:500, Abcam), nestin conjugated to AlexaFluor 647 (ab196693, 1:100, Abcam), CD3 (ab5690, 1:250, Abcam), CENP-C (gift from Iain Cheeseman, 1:10,000), ZO-1 (61–7300, 1:100, Thermo Fisher Scientific), and α6 integrin (clone MAB1378, 1:250, Millipore). All secondary antibodies were various AlexaFluor conjugates (1:1,000, Thermo Fisher Scientific). The Υtubulin antibody was directly conjugated using the AlexaFluor 568 Antibody Labeling Kit (Thermo Fisher Scientific). AlexaFluor 568 Phalloidin (1:500, Thermo Fisher Scientific) was added to the secondary antibody solution when desired to visualize F-actin. Images were acquired using a CSU-22 spinning disc confocal head (Yokogawa) with Borealis modification (Andor) mounted on an Axiovert 200M microscope (Zeiss) with a 63X oil immersion objective (Zeiss), an Orca-ER CCD camera (Hamamatsu), and MetaMorph acquisition software (Molecular Devices).
Image analysis
To quantify lagging chromosomes, anaphase cells were identified based on the presence of an anaphase spindle by α-tubulin immunostaining. The criterion for designating a chromosome as lagging was that the entire chromosome was fully separate from the main mass of segregating chromosomes and contained a centromere as judged by either differential Hoechst staining or by CENP-C immunofluorescence.
To quantify micronuclei, telophase cells were identified based on the presence of a midbody by α-tubulin immunostaining. Micronuclei were defined as Hoechst-positive foci that were distinct from the main nucleus and that also contained a centromere as judged by differential Hoechst staining or by CENP-C immunofluorescence. To quantify cell shape, samples were stained with phalloidin to label the cell cortex. In tissues, images of interphase and mitotic cells were acquired as z stacks with 0.5 μm spacing. The dimensions were then measured manually using Volocity (PerkinElmer). First, the z plane that was equidistant from either edge of the chromatin was selected.
From this position, the longest possible axis was measured and set as the x dimension. The next longest axis perpendicular to the x dimension was measured and set as the y dimension. Finally, the axis perpendicular to the x and y axis, which was necessarily the shortest axis, was measured and set as the z dimension. These dimensions were then graphed in three-dimensional space using Python and Plotly. For dissociated cells from mammary gland, skin, neonatal liver, and embryonic brain, the smallest dimension, and thus the z dimension, always corresponded to the height of the cell relative to the coverslip.
To analyze mitotic spindle morphology and microtubule density within the spindle, images of metaphase cells in which the mitotic spindle was parallel to the imaging plane were acquired as z stacks with 0.5 μm spacing. The z plane in the middle of the spindle was used for all measurements. To measure microtubule density, the mitotic spindle was outlined and the α-tubulin signal per μm was calculated. A region of cytoplasm was similarly outlined and the α-tubulin signal per μm was calculated. The background signal was subtracted from the spindle signal to arrive at the spindle intensity per μm. All spindle dimension and intensity measurements were acquired using Volocity (PerkinElmer). The spindle skew angle was measured using FIJI.
Immunoblotting
Spheroids were isolated from Matrigel by incubation with dispase (Corning) at 37°C for 1 hour. Digestion was terminated by addition of 5 mM EDTA and spheroids resuspended by trituration. Spheroids were pelleted at 200 g for 5 minutes and resuspended in RIPA buffer (50 mM Tris pH 8.0, 150 mM sodium chloride, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing Complete Mini Protease Inhibitor Cocktail (Roche). Lysates were homogenized using a rubber policeman, incubated on ice for 20 minutes, and centrifuged at 13,000 rpm at 4 °C for 20 minutes. Protein concentration of supernatants was measured using Bradford dye (Bio-Rad) on a spectrophotometer. Lysates were diluted in 5X sample buffer (250 mM Tris pH 6.8, 50% glycerol, 5% β-mercaptoethanol, 0.025% bromophenol blue, 5% SDS). Samples were separated on homemade polyacrylamide gels and transferred to Immobilon-P membranes (Millipore) via wet transfer. Membranes were blocked with 5% milk in TBST (50 mM Tris pH 8.0, 150 mM NaCl, 0.1% Tween-20) for 1 hour at room temperature. Membranes were incubated in primary antibodies diluted in 5% milk in TBST at 4°C with rocking for 16 hours and washed with TBST for 10 minutes thrice. Membranes were incubated in secondary antibodies diluted in TBST at room temperature with rocking for 1 hour and washed with TBST for 10 minutes thrice. Membranes were incubated in ECL Prime Western Blotting Detection Reagent (GE Healthcare) for 5 minutes and imaged on an ImageQuant LAS 4000 luminescent image analyzer (GE Healthcare). The following primary antibodies were used: β1 integrin (clone MAB1997, 1:1,000, Millipore) and β-actin (clone AC-74, 1:20,000, Sigma). For β1 integrin, β-mercaptoethanol was eliminated from the 5X sample buffer. All secondary antibodies were ECL horseradish peroxidase linked (1:5,000, GE Healthcare).
RNA sequencing
To isolate RNA from spheroids, RNA STAT-60 (Amsbio) was added to mammary cells cultured in 2D and mammary spheroids. Cells were then homogenized with a rubber policeman or 1 mL syringe plunger, and triturated with a P1000 pipette tip. RNA was purified by chloroform extraction according to RNA STAT-60 instructions. RNA libraries were prepared using TruSeq (Illumina) and sequenced on a HiSeq 2000 (Illumina). For quality control purposes, BEDTools (version 2.25.0) was used to count the reads falling into genes, coding regions, intronic regions, 5’ or 3’ UTRs, flanking 3 kb genic regions, and intergenic regions. Other basic statistics including mapping rates, ratio of sense versus anti-sense reads, and rRNA percentages were also collected for each sample. To quantify gene expression, RSEM (version 1.2.30) was used to estimate gene expression levels based on mm9 UCSC known gene annotations. The count table was imported into DESeq2 (version 1.10.1) for differential gene expression test. The gene expression (Log2FPKM) table was first filtered to remove lowly expressed and low variance genes (average <1 and variance <0.2), and the rest were used for clustering analysis using Ward’s method in SpotFire and GSEA (2.2.2) analysis against MSigDB hallmark gene sets.
Live imaging and analysis
Mammary epithelial cells from Centrin 2-GFP;H2B-mCherry mice were combined with Matrigel as described above and gels were cast in 35 mm glass-bottom dishes (Mat Tek). Dishes were imaged under an incubated stage set to 37 °C with 5% CO2 (Pathology Devices). Images were acquired using a CSU-22 spinning disc confocal head (Yokogawa) with Borealis modification (Andor) mounted on an Axiovert 200M microscope (Zeiss) with a 40X water immersion objective, an Orca-ER CCD camera (Hamamatsu), and MetaMorph acquisition software (Molecular Devices). Only cells that entered mitosis after imaging began were considered for analysis. If the cell did not enter anaphase before imaging ceased, it was analyzed only if it had been in mitosis for at least 60 minutes. In these cases, the duration of prometaphase to anaphase was the time interval from the start of mitosis to the end of imaging. Prometaphase was defined as the first frame when individual chromosomes could be observed beyond the spherical confines of the nucleus. Anaphase was defined as the first frame that chromosomes were pulled toward opposite poles. The establishment of a bipolar spindle was defined as the first frame when the Centrin 2-GFP foci formed a 180° angle with respect to one another based on a line drawn from one centrin focus through the center of the condensing chromatin. Chromosome congression was defined as the time elapsed between the appearance of individual chromosomes beyond the spherical confines of the nucleus and the formation of a metaphase plate.
Partial hepatectomies
Partial hepatectomies were performed on 8 week-old male mice as previously described with minor modifications (Mitchell and Willenbring, 2008). Instead of removing the entire median lobe and the gallbladder, only the right median lobe was removed while the left median lobe and gallbladder were spared. Mice were sacrificed 48 hours after surgery to process livers for immunofluorescence. For β1 integrin knockout experiments, Mx-Cre;Itgb1flox/flox or Itgb1flox/flox mice were injected intraperitoneally with poly-IC (250 mg, Amersham Biosciences) twice with a time interval of two days and partial hepatectomies were performed 14 days after the final injection.
Single nucleus or cell sequencing
To isolate nuclei from regenerated livers, livers were harvested from mice ten days after partial hepatectomy, pressed through a 40 μM filter into sucrose buffer (250 mM sucrose, 5 mM MgCl2, 10 mM Tris-Cl pH 7.4), centrifuged at 600 g at 4°C for 10 minutes, resuspended in sucrose buffer, and centrifuged and resuspended again. To isolate nuclei from hepatocytes expanded in culture, hepatocytes were cultured in SUM3 medium for four days, trypsinized, washed with PBS, incubated in 0.2X PBS on ice for 10 minutes, lysed with a dounce homogenizer for 8 strokes, centrifuged at 1,000 g at 4 °C for 10 minutes, resuspended in sucrose buffer, and centrifuged and resuspended again. To isolate neural progenitor cells, neural progenitor cells were cultured in NeuroCult proliferation medium with EGF for four days, washed with PBS, dissociated with StemPro Accutase (Thermo Fisher Scientifc), resuspended in NeuroCult proliferation medium, and gently triturated. To isolate single nuclei or neural progenitor cells, 0.25 × 104 nuclei or cells were added to 20 mL of sucrose buffer or medium in a 15 cm plate. Single cells and nuclei were isolated using a homemade microaspirator and transferred to 8 μL water in a 96-well plate. The microaspirator needle was cleaned with 10% (vol/vol) bleach followed by water after transferring each cell or nucleus. Cells and nuclei were lysed and genomic DNA amplified using the GenomePlex Single Cell Whole Genome Amplification Kit (Sigma). All reagents were added using aerosol-resistant pipette tips in a laminar flow hood. After amplification, 4 μL of each sample were analyzed by agarose gel electrophoresis. Samples producing a smear ranging from 100 to 1,000 bp were sequenced. Samples were cleaned with paramagnetic beads (Beckman Coulter), normalized to 0.2 ng/μL, and libraries prepared using NexteraXT (Illumina) preformed at 1/12 reaction volume on a Mosquito HV (TTP Labtech). Samples were sequenced using a HiSeq2000 (Illumina) using 40 nucleotide single-end reads. Sequence reads were trimmed to 40 nucleotides and aligned to the mouse genome (mm9) using BWA (0.6.1) (Li and Durbin, 2009) with default options. HMMcopy (0.1.1) (Ha et al., 2012) was used to detect copy number alterations by estimating copy number in 500-kb bins controlling for mappability [downloaded from UCSC Genome Bioinformatics (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/ or http://hgdownload.cse.ucsc.edu/goldenPath/mm9/encodeDCC/wgEncodeMapability/)] and GC content (calculated by HMMcopy gcCounter). Standard deviations (SDs) of the corrected read copies (log2 based) from HMMcopy were computed within sliding windows (30 adjacent 500-kb bins) for all chromosomes, and the average was calculated for each chromosome. The average SDs of the three autosomes with highest variability were averaged to generate a variability score (VS). Cells with a VS exceeding 0.34 were excluded from analysis. In order to detect aneuploidy in the context of a diploid or tetraploid cell or nucleus, a log2 ratio exceeding 0.25 was considered a chromosome gain and a log2 ratio below −0.3 was considered a chromosome loss.
Fluorescence in situ hybridization (FISH)
Liver sections were immunostained for pan-cadherin as described above. After final washes, sections were fixed with 2% paraformaldehyde in PBS for 10 minutes and washed three times with PBS for 5 minutes each. Hydrophobic ink was removed and slides were incubated in 2× SSC for 5 minutes, foll owed by incubation in 50% formamide in 2× SSC for 2 hours. Fluorescently labe led probes targeting different regions of mouse chromosome 16 (RP23–354F11 and RP23–18M23) (Empire Genomics) were diluted in hybridization buffer as per the manufacturer’s instructions and applied to sections. Sections were sealed with a coverslip and rubber cement and incubated in the dark at 45 °C for 2 hours to allow probes to infiltrate the section. This was followed by incubation at 85 °C for 5 minutes t o denature the DNA. Hybridization was performed in the dark at 37 °C for 48 hours. Sl ides were then washed with 0.4× SSC containing 0.3% NP-40 for 2 minutes at 73 °C, f ollowed by 2× SSC containing 0.1% NP-40 for 1 minutes at room temperature. Slides were incubated in 0.05 μg/mL DAPI in 2× SSC for 30 minutes and mounted with ProL ong Gold Antifade Reagent (Life Technologies). Images were acquired on a spinning disk confocal microscope (PerkinElmer) and analyzed using the Volocity software package (PerkinElmer).
Quantification and Statistical Analysis
General statistics
The number of biological replicates, the number of cells analyzed per biological replicate, and the statistical test performed are indicated in the figure legends. A biological replicate was defined as tissue or cells harvested from a single mouse. All statistical tests except those for cell shape were performed using Prism (GraphPad). For all statistical tests, a cutoff of p < 0.05 was used to indicate significance.
Statistics for chromosome segregation defects
To determine the prevalence of lagging chromosomes and micronuclei in cells, the final percentage and standard deviation were calculated across pooled replicates. This was necessary because of significant variability in the number of mitotic cells obtained across biological replicates. Notably, in all cases where the prevalence of lagging chromosomes or micronuclei exceeded 2%, lagging chromosomes and micronuclei were observed across multiple replicates.
Statistics for cell shape
To determine whether the dimensions of cells in any two conditions were significantly different, permutation tests of the group centroids were performed in Python. First, the centroid for each condition was determined and the distance between the centroids for the two different conditions was calculated. To calculate the centroid distances that could be observed under the null hypothesis of no difference between the two conditions, the measurements (each measurement being a set of x, y, and z values) from all cells in the two conditions were randomly assigned to two different groups containing the same number of cells as the two original conditions. The difference between the centroids of these two new groups was calculated. This randomization was repeated 10,000 times. The permutation p value indicates the proportion of times the randomized centroid distances were greater than or equal to that of the centroid distance between the two conditions. This p value was then adjusted for multiple hypothesis testing using the Benjamini and Hochberg method.
Data and Software Availability
The three-dimensional plots from Figure 2 and Supplemental Figure 2 and live imaging files from Figure 3 have been uploaded to Mendeley Data (https://data.mendeley.com/datasets/9wp8t8c38s/draft?a=fe6dd007–5685-4ce0-a9b4–82085ffdc906). The RNA sequencing data have been deposited in the NCBI Gene Expression Omnibus under accession number GSE111078. The single cell sequencing data have been deposited in the NCBI Sequence Read Archive under accession number SRP133432.
Supplementary Material
Movie S1. Live imaging of mitosis in an immature spheroid, Related to Figure 3 Live imaging of a mitotic mammary epithelial cell in a Centrin 2-GFP;H2B-mCherry immature spheroid. The Centrin 2-GFP and H2B-mCherry signals are shown in red and green, respectively. The duration of the movie covers 210 minutes of imaging.
Movie S2. Live imaging of mitosis in an immature spheroid, Related to Figure 3 Live imaging of a mitotic mammary epithelial cell in another Centrin 2-GFP;H2BmCherry immature spheroid. The Centrin 2-GFP and H2B-mCherry signals are shown in red and green, respectively. The duration of the movie covers 210 minutes of imaging.
Movie S3. Live imaging of mitosis in a mature spheroid, Related to Figure 3 Live imaging of a mitotic mammary epithelial cell in a Centrin 2-GFP;H2B-mCherry mature spheroid. The Centrin 2-GFP and H2B-mCherry signals are shown in red and green, respectively. The duration of the movie covers 210 minutes of imaging.
Movie S4. Live imaging of mitosis in a mature spheroid, Related to Figure 3 Live imaging of a mitotic mammary epithelial cell in another Centrin 2-GFP;H2BmCherry mature spheroid. The Centrin 2-GFP and H2B-mCherry signals are shown in red and green, respectively. The duration of the movie covers 210 minutes of imaging.
Supplemental Figure 1. Centrosome number in mitotic epithelial cells in the presence and absence of tissue architecture, Related to Figure 1
(A) Quantification of centrosome number (γtubulin foci) in prometaphase cells in tissue and dissociated cells from mammary gland, skin, neonatal liver, embryonic brain, and lymph node. n = 1 biological replicate with 50 prometaphase cells per condition.
(B) Quantification of centrosome number (γtubulin foci) in prometaphase mammary epithelial cells in immature and mature spheroids. n = 1 biological replicate with 50 prometaphase cells per condition.
Supplemental Figure 2. Measurement of cell shape across contexts and depletion of integrin in spheroids, Related to Figure 2
(A) Dimensions of mitotic (dark circles) and interphase (light circles) keratinocytes in skin (green circles) and as dissociated cells (pink circles). For dissociated cells, the z dimension always corresponds to the height of the cell relative to the coverslip. The distance between the centroids (in μm) of select pairs of conditions and the adjusted permutation p value using the Benjamini and Hochberg method for multiple comparisons are indicated below the graph. n = 1 biological replicate with ⩾ 10 cells per condition.
(B) Dimensions of mitotic (dark circles) and interphase (light circles) hepatocytes in neonatal liver (green circles) and as dissociated cells (pink circles). The distance between the centroids (in μm) of select pairs of conditions and the adjusted permutation p value using the Benjamini and Hochberg method for multiple comparisons are indicated below the graph. n = 1 biological replicate with ⩾ 10 cells per condition.
(C) Dimensions of mitotic (dark circles) and interphase (light circles) neural progenitor cells in embryonic brain (green circles) and as dissociated cells (pink circles). The distance between the centroids (in μm) of select pairs of conditions and the adjusted permutation p value using the Benjamini and Hochberg method for multiple comparisons are indicated below the graph. n = 1 biological replicate with ⩾ 10 cells per condition.
(D) Dimensions of mitotic (dark circles) and interphase (light circles) T cells in lymph node (green circles) and as dissociated cells (pink circles). The distance between the centroids (in μm) of select pairs of conditions and the adjusted permutation p value using the Benjamini and Hochberg method for multiple comparisons are indicated below the graph. n = 1 biological replicate with ⩾ 10 cells per condition.
(E) Western blot of whole cell lysates from control (Cre-ERT2; Itgb1+/F) or β1 integrin knockout (Cre-ERT2; Itgb1F/F) spheroids treated with 4-hydroxytamoxifen (4-OHT). β-actin was used as a loading control.
(F) Quantification of centrosome number (γtubulin foci) in prometaphase mammary epithelial cells in control (Cre-ERT2; Itgb1+/F) and β1 integrin knockout (Cre-ERT2; Itgb1F/F) spheroids treated with 4-hydroxytamoxifen (4-OHT). n = 1 biological replicate with 50 prometaphase cells per condition.
Supplemental Figure 3. Comparison of gene expression and spindle morphology across spheroid conditions, Related to Figure 3
(A) Ward’s clustering of mammary epithelial cells grown as dissociated cells and as spheroids for 48 and 96 hours based on expression (log2 FPKM) of protein-coding genes by RNA sequencing.
(B) Hallmark gene sets enriched in spheroids after 48 and 96 hours of culture in Matrigel using a false discovery rate (FDR) cutoff of 5%. The FDR for each gene set is indicated in parentheses.
(C) Images of metaphase mammary epithelial cells in immature (first panel) and mature (second panel) spheroids, and control (Cre-ERT2; Itgb1+/F, third panel) and β1 integrin knockout (Cre-ERT2; Itgb1F/F, fourth panel) spheroids treated with 4-hydroxytamoxifen (4-OHT), immunostained for atubulin (green) and γtubulin (red). DNA is stained with Hoechst (blue). Scale bars, 5 μm.
(D) Measurement of the cell length, spindle length (pole-pole distance), spindle width (at spindle midzone), cell-spindle ratio, and pole-cortex distance in metaphase mammary epithelial cells in immature and mature spheroids, and control (Cre-ERT2; Itgb1+/F) and β1 integrin knockout (Cre-ERT2; Itgb1F/F) spheroids treated with 4-hydroxytamoxifen (4-OHT). P values are: p = 0.02 for cell length in immature versus mature spheroids, p = 0.01 for spindle width in immature versus mature spheroids, and p = 0.02 for pole-cortex distance in immature versus mature spheroids by two-tailed unpaired t-test. n = 2 biological replicates with ⩾ 9 metaphase cells per replicate per condition.
(E) Measurement of spindle skew in metaphase mammary epithelial cells in immature and mature spheroids, and control (Cre-ERT2; Itgb1+/F) and β1 integrin knockout (Cre-ERT2; Itgb1F/F) spheroids treated with 4-hydroxytamoxifen (4-OHT). A diagram to the right of the graph depicts the angle being measured. n = 2 biological replicates with ⩾ 9 metaphase cells per replicate per condition.
(F) Measurement of α-tubulin intensity (background subtracted) per area of the mitotic spindle in metaphase mammary epithelial cells in immature and mature spheroids, and control (Cre-ERT2; Itgb1+/F) and β1 integrin knockout (Cre-ERT2; Itgb1F/F) spheroids treated with 4-hydroxytamoxifen (4-OHT). n = 2 biological replicates with ⩾ 9 metaphase cells per replicate per condition.
Supplemental Figure 4. Polyploid hepatocytes proliferate during adult liver regeneration and undergo bipolar mitoses, Related to Figure 4
(A) Ploidy of hepatocytes in interphase, prometaphase, and anaphase in liver during neonatal development and adult regeneration as inferred by chromosome 16 FISH (interphase) and centrosome number (γtubulin foci, prometaphase and anaphase). n = 1 biological replicate totaling 100 cells for interphase FISH, > 50 mitoses for γtubulin foci in prometaphase, and > 25 mitoses for γtubulin foci in anaphase.
(B) Quantification of bipolar and multipolar anaphases in hepatocytes in liver during neonatal development and adult regeneration, as dissociated cells from adult liver, and in control (Itgb1F/F) and β1 integrin knockout (Mx-Cre; Itgb1F/F) liver during adult regeneration after treatment with poly-IC. n ⩾ 1 biological replicate totaling > 50 anaphases per condition.
HIGHLIGHTS.
Primary epithelial cells missegregate chromosomes when cultured as monolayers
Organoid culture rescues mitotic defects in an integrin-dependent manner
Tissue architecture promotes correction of merotelic kinetochore attachments
Disruption of tissue architecture could underlie chromosome instability in cancer
ACKNOWLEDGMENTS
We thank Sarah Paracha for technical assistance, Konstantina Rowald and Rocio Sotillo for help with establishing spheroid culture, Wendy Salmon for imaging guidance, Summer Morrill and Eghosa Eke for computational assistance, Jie Wu and Duan Ma of the Barbara K. Ostrom (1978) Bioinformatics and Computing Facility for bioinformatics support, and Stefano Santaguida, Buzz Baum, and Iain Cheeseman for helpful discussions. This work was supported by NIH Grant CA206157–22 to A. A., the Kathy and Curt Marble Cancer Research Fund, and the Koch Institute Support Grant P30-CA14051 from the National Cancer Institute. A. A. is an investigator of the Howard Hughes Medical Institute, the Paul F. Glenn Center for Biology of Aging Research at MIT, and the Ludwig Institute for Cancer Research. K.A.K. was supported by the NIGMS Training Grant T32GM007753.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Akhtar N, and Streuli CH (2012). An integrin--ILK--microtubule network orients cell polarity and lumen formation in glandular epithelium. Nature Publishing Group 15, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstralh DT, Haack T, and St Johnston D (2013). Epithelial polarity and spindle orientation: intersecting pathways. Philos. Trans. R. Soc. Lond., B, Biol. Sci 368, 20130291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Anderson S, Jwa M, Green EM, Kang J-S, Yates JR III, Chan CSM, Drubin DG, and Barnes G (2002). Phospho-Regulation of Kinetochore-Microtubule Attachments by the Aurora Kinase Ipl1p. Cell 111, 163–172. [DOI] [PubMed] [Google Scholar]
- Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, and Salmon ED (2001). Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. Journal of Cell Biology 153, 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D, Cameron LA, and Salmon ED (2004). Anaphase spindle mechanics prevent mis-segregation of merotelically oriented chromosomes. Current Biology 14, 2149–2155. [DOI] [PubMed] [Google Scholar]
- Cimini D, Moree B, Canman JC, and Salmon ED (2003). Merotelic kinetochore orientation occurs frequently during early mitosis in mammalian tissue cells and error correction is achieved by two different mechanisms. Journal of Cell Science 116, 4213–4225. [DOI] [PubMed] [Google Scholar]
- Cimini D, Wan X, Hirel CB, and Salmon ED (2006). Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Current Biology 16, 1711–1718. [DOI] [PubMed] [Google Scholar]
- Comaills V, Kabeche L, Morris R, Buisson R, Yu M, Madden MW, LiCausi JA, Boukhali M, Tajima K, Pan S, et al. (2016). Genomic Instability Is Induced by Persistent Proliferation of Cells Undergoing Epithelial-to-Mesenchymal Transition. CellReports 17, 2632–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, and Pellman D (2012). DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca JG, Gall WE, Ciferri C, Cimini D, Musacchio A, and Salmon ED (2006). Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell 127, 969–982. [DOI] [PubMed] [Google Scholar]
- Duncan AW, Newell AEH, Smith L, Wilson EM, Olson SB, Thayer MJ, Strom SC, and Grompe M (2012). Frequent Aneuploidy Among Normal Human Hepatocytes. Gastroenterology 142, 25–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Taylor MH, Hickey RD, Newell AEH, Lenzi ML, Olson SB, Finegold MJ, and Grompe M (2010). The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 467, 707–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, and Pellman D (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans J, and Wieschaus E (2000). A genetic link between morphogenesis and cell division during formation of the ventral furrow in Drosophila. Cell 101, 523–531. [DOI] [PubMed] [Google Scholar]
- Guidotti J-E, Bregerie O, Robert A, Debey P, Brechot C, and Desdouets C (2003). Liver Cell Polyploidization: A Pivotal Role for Binuclear Hepatocytes. Journal of Biological Chemistry 278, 19095–19101. [DOI] [PubMed] [Google Scholar]
- Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, et al. (2012). Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Research 22, 1995–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki N, Saito H, Harano T, Nomoto S, Takahashi T, Osada H, and Fujii Y (2001). Molecular analysis of the mitotic checkpoint genes BUB1, BUBR1 and BUB3 in human lung cancers. Cancer Lett. 162, 201–205. [DOI] [PubMed] [Google Scholar]
- Hassold TJ, and Jacobs PA (1984). Trisomy in Man. Annu. Rev. Genet 18, 69–97. [DOI] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, and Hetzer MW (2013). Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell 154, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itabashi T, Terada Y, Kuwana K, Kan T, Shimoyama I, and Ichiwata S (2012). Mechanical impulses can control metaphase progression in a mammalian cell. Proceedings of the National Academy of Sciences 109, 7320–7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJPL, and Medema RH (2011). Chromosome Segregation Errors as a Cause of DNA Damage and Structural Chromosome Aberrations. Science 333, 1895–1898. [DOI] [PubMed] [Google Scholar]
- Jechlinger M, Podsypanina K, and Varmus H (2009). Regulation of transgenes in three-dimensional cultures of primary mouse mammary cells demonstrates oncogene dependence and identifies cells that survive deinduction. Genes & Development 23, 1677–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamala T (2007). Hock immunization: a humane alternative to mouse footpad injections. J. Immunol. Methods 328, 204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouse KA, Davoli T, Elledge SJ, and Amon A (2017). Aneuploidy in Cancer: Seq-ing Answers to Old Questions. Annu. Rev. Cancer Biol 1, 335–354. [Google Scholar]
- Knouse KA, Wu J, Whittaker CA, and Amon A (2014). Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. U.S.a 111, 13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, and Alvarez-Buylla A (2009). The Glial Nature of Embryonic and Adult Neural Stem Cells. Annu. Rev. Neurosci 32, 149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, and Pellman D (2008). Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes & Development 22, 2189–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson MA, Renduchitala K, Khodjakov A, and Kapoor TM (2004). Correcting improper chromosome-spindle attachments during cell division. Nature Cell Biology 6, 232–237. [DOI] [PubMed] [Google Scholar]
- Lancaster OM, Le Berre M, Dimitracopoulos A, Bonazzi D, Zlotek-Zlotkiewicz E, Picone R, Duke T, Piel M, and Baum B (2013). Mitotic Rounding Alters Cell Geometry to Ensure Efficient Bipolar Spindle Formation. Developmental Cell 25, 270–283. [DOI] [PubMed] [Google Scholar]
- Lechler T, and Fuchs E (2005). Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature 437, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, and Streuli CH (2014). Integrins and epithelial cell polarity. Journal of Cell Science 127, 3217–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Vader G, Vromans MJM, Lampson MA, and Lens SMA (2009). Sensing chromosome bi-orientation by spatial separation of aurora B kinase from kinetochore substrates. Science 323, 1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell C, and Willenbring H (2008). A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc 3, 1167–1170. [DOI] [PubMed] [Google Scholar]
- Muthuswamy SK, and Xue B (2012). Cell polarity as a regulator of cancer cell behavior plasticity. Annu. Rev. Cell Dev. Biol 28, 599–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann CM, Oughton JA, and Kerkvleit NI (1992). Anti-CD3-induced T-cell activation in vivo: Flow cytometric analysis of dose-responsive, time-dependent, and cyclosporin A-sensitive parameters of CD4+ and CD8+ cells from the draining lymph nodes of C57Bl/6 mice. Int. J. Immunopharmac 14, 1295–1304. [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RY-J, Jackson RA, and Thiery JP (2016). EMT: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Ragkousi K, and Gibson MC (2014). Cell division and the maintenance of epithelial order. The Journal of Cell Biology 207, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roschke AV, Glebov OK, Lababidi S, Gehlhaus KS, Weinstein JN, and Kirsch IR (2008). Chromosomal Instability Is Associated with Higher Expression of Genes Implicated in Epithelial-Mesenchymal Transition, Cancer Invasiveness, and Metastasis and withLower Expression of Genes Involved in Cell Cycle Checkpoints, DNA Repair, and Chromatin Maintenance. Neoplasia 10, 1222–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D’Alise AM, Taylor SS, and Musacchio A (2010). Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. The Journal of Cell Biology 190, 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Meth 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, and Weaver BA (2013). Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. U.S.a 110, E4134–E4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth WT, Nardi IK, Paul R, Mogilner A, and Cimini D (2012). Timing of centrosome separation is important for accurate chromosome segregation. Molecular Biology of the Cell 23, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth WT, Nardi IK, Scholl LM, and Cimini D (2009). Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS ONE 4, e6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speicher T, Siegenthaler B, Bogorad RL, Ruppert R, Petzold T, Padrissa-Altes S, Bachofner M, Anderson DG, Koteliansky V, Fässler R, et al. (2014). Knockdown and knockout of β1-integrin in hepatocytes impairs liver regeneration through inhibition of growth factor signalling. Nat Commun 5, 3862. [DOI] [PubMed] [Google Scholar]
- Stewart MP, Helenius J, Toyoda Y, Ramanathan SP, Muller DJ, and Hyman, (2010). Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature 469, 226–230. [DOI] [PubMed] [Google Scholar]
- Théry M, Racine V, Pépin A, Piel M, Chen Y, Sibarita J-B, and Bornens M (2005). The extracellular matrix guides the orientation of the cell division axis. Nature Cell Biology 7, 947–953. [DOI] [PubMed] [Google Scholar]
- Thompson SL, and Compton DA (2008). Examining the link between chromosomal instability and aneuploidy in human cells. The Journal of Cell Biology 180, 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, and Compton DA (2011). Chromosome missegregation in human cells arises through specific types of kinetochore–microtubule attachment errors. Proceedings of the National Academy of Sciences 108, 17974–17978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe A, Johnson VL, Albertella M, and Taylor SS (2001). Aneuploid colon cancer cells have a robust spindle checkpoint. EMBO Rep. 2, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanneste E, Voet T, Le Caignec C, Ampe M, Konings P, Melotte C, Debrock S, Amyere M, Vikkula M, Schuit F, et al. (2009). Chromosome instability is common in human cleavage-stsage embryos. Nat Med 15, 577–583. [DOI] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, and Amon A (2008). Aneuploidy Affects Proliferation and Spontaneous Immortalization in Mammalian Cells. Science 322, 703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, and Pellman D (2015). Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Live imaging of mitosis in an immature spheroid, Related to Figure 3 Live imaging of a mitotic mammary epithelial cell in a Centrin 2-GFP;H2B-mCherry immature spheroid. The Centrin 2-GFP and H2B-mCherry signals are shown in red and green, respectively. The duration of the movie covers 210 minutes of imaging.
Movie S2. Live imaging of mitosis in an immature spheroid, Related to Figure 3 Live imaging of a mitotic mammary epithelial cell in another Centrin 2-GFP;H2BmCherry immature spheroid. The Centrin 2-GFP and H2B-mCherry signals are shown in red and green, respectively. The duration of the movie covers 210 minutes of imaging.
Movie S3. Live imaging of mitosis in a mature spheroid, Related to Figure 3 Live imaging of a mitotic mammary epithelial cell in a Centrin 2-GFP;H2B-mCherry mature spheroid. The Centrin 2-GFP and H2B-mCherry signals are shown in red and green, respectively. The duration of the movie covers 210 minutes of imaging.
Movie S4. Live imaging of mitosis in a mature spheroid, Related to Figure 3 Live imaging of a mitotic mammary epithelial cell in another Centrin 2-GFP;H2BmCherry mature spheroid. The Centrin 2-GFP and H2B-mCherry signals are shown in red and green, respectively. The duration of the movie covers 210 minutes of imaging.
Supplemental Figure 1. Centrosome number in mitotic epithelial cells in the presence and absence of tissue architecture, Related to Figure 1
(A) Quantification of centrosome number (γtubulin foci) in prometaphase cells in tissue and dissociated cells from mammary gland, skin, neonatal liver, embryonic brain, and lymph node. n = 1 biological replicate with 50 prometaphase cells per condition.
(B) Quantification of centrosome number (γtubulin foci) in prometaphase mammary epithelial cells in immature and mature spheroids. n = 1 biological replicate with 50 prometaphase cells per condition.
Supplemental Figure 2. Measurement of cell shape across contexts and depletion of integrin in spheroids, Related to Figure 2
(A) Dimensions of mitotic (dark circles) and interphase (light circles) keratinocytes in skin (green circles) and as dissociated cells (pink circles). For dissociated cells, the z dimension always corresponds to the height of the cell relative to the coverslip. The distance between the centroids (in μm) of select pairs of conditions and the adjusted permutation p value using the Benjamini and Hochberg method for multiple comparisons are indicated below the graph. n = 1 biological replicate with ⩾ 10 cells per condition.
(B) Dimensions of mitotic (dark circles) and interphase (light circles) hepatocytes in neonatal liver (green circles) and as dissociated cells (pink circles). The distance between the centroids (in μm) of select pairs of conditions and the adjusted permutation p value using the Benjamini and Hochberg method for multiple comparisons are indicated below the graph. n = 1 biological replicate with ⩾ 10 cells per condition.
(C) Dimensions of mitotic (dark circles) and interphase (light circles) neural progenitor cells in embryonic brain (green circles) and as dissociated cells (pink circles). The distance between the centroids (in μm) of select pairs of conditions and the adjusted permutation p value using the Benjamini and Hochberg method for multiple comparisons are indicated below the graph. n = 1 biological replicate with ⩾ 10 cells per condition.
(D) Dimensions of mitotic (dark circles) and interphase (light circles) T cells in lymph node (green circles) and as dissociated cells (pink circles). The distance between the centroids (in μm) of select pairs of conditions and the adjusted permutation p value using the Benjamini and Hochberg method for multiple comparisons are indicated below the graph. n = 1 biological replicate with ⩾ 10 cells per condition.
(E) Western blot of whole cell lysates from control (Cre-ERT2; Itgb1+/F) or β1 integrin knockout (Cre-ERT2; Itgb1F/F) spheroids treated with 4-hydroxytamoxifen (4-OHT). β-actin was used as a loading control.
(F) Quantification of centrosome number (γtubulin foci) in prometaphase mammary epithelial cells in control (Cre-ERT2; Itgb1+/F) and β1 integrin knockout (Cre-ERT2; Itgb1F/F) spheroids treated with 4-hydroxytamoxifen (4-OHT). n = 1 biological replicate with 50 prometaphase cells per condition.
Supplemental Figure 3. Comparison of gene expression and spindle morphology across spheroid conditions, Related to Figure 3
(A) Ward’s clustering of mammary epithelial cells grown as dissociated cells and as spheroids for 48 and 96 hours based on expression (log2 FPKM) of protein-coding genes by RNA sequencing.
(B) Hallmark gene sets enriched in spheroids after 48 and 96 hours of culture in Matrigel using a false discovery rate (FDR) cutoff of 5%. The FDR for each gene set is indicated in parentheses.
(C) Images of metaphase mammary epithelial cells in immature (first panel) and mature (second panel) spheroids, and control (Cre-ERT2; Itgb1+/F, third panel) and β1 integrin knockout (Cre-ERT2; Itgb1F/F, fourth panel) spheroids treated with 4-hydroxytamoxifen (4-OHT), immunostained for atubulin (green) and γtubulin (red). DNA is stained with Hoechst (blue). Scale bars, 5 μm.
(D) Measurement of the cell length, spindle length (pole-pole distance), spindle width (at spindle midzone), cell-spindle ratio, and pole-cortex distance in metaphase mammary epithelial cells in immature and mature spheroids, and control (Cre-ERT2; Itgb1+/F) and β1 integrin knockout (Cre-ERT2; Itgb1F/F) spheroids treated with 4-hydroxytamoxifen (4-OHT). P values are: p = 0.02 for cell length in immature versus mature spheroids, p = 0.01 for spindle width in immature versus mature spheroids, and p = 0.02 for pole-cortex distance in immature versus mature spheroids by two-tailed unpaired t-test. n = 2 biological replicates with ⩾ 9 metaphase cells per replicate per condition.
(E) Measurement of spindle skew in metaphase mammary epithelial cells in immature and mature spheroids, and control (Cre-ERT2; Itgb1+/F) and β1 integrin knockout (Cre-ERT2; Itgb1F/F) spheroids treated with 4-hydroxytamoxifen (4-OHT). A diagram to the right of the graph depicts the angle being measured. n = 2 biological replicates with ⩾ 9 metaphase cells per replicate per condition.
(F) Measurement of α-tubulin intensity (background subtracted) per area of the mitotic spindle in metaphase mammary epithelial cells in immature and mature spheroids, and control (Cre-ERT2; Itgb1+/F) and β1 integrin knockout (Cre-ERT2; Itgb1F/F) spheroids treated with 4-hydroxytamoxifen (4-OHT). n = 2 biological replicates with ⩾ 9 metaphase cells per replicate per condition.
Supplemental Figure 4. Polyploid hepatocytes proliferate during adult liver regeneration and undergo bipolar mitoses, Related to Figure 4
(A) Ploidy of hepatocytes in interphase, prometaphase, and anaphase in liver during neonatal development and adult regeneration as inferred by chromosome 16 FISH (interphase) and centrosome number (γtubulin foci, prometaphase and anaphase). n = 1 biological replicate totaling 100 cells for interphase FISH, > 50 mitoses for γtubulin foci in prometaphase, and > 25 mitoses for γtubulin foci in anaphase.
(B) Quantification of bipolar and multipolar anaphases in hepatocytes in liver during neonatal development and adult regeneration, as dissociated cells from adult liver, and in control (Itgb1F/F) and β1 integrin knockout (Mx-Cre; Itgb1F/F) liver during adult regeneration after treatment with poly-IC. n ⩾ 1 biological replicate totaling > 50 anaphases per condition.