

Abstract
Secretion of proteins lacking leader sequence was deemed rare and unconventional, only accountable for the export of a limited number of clients by mechanisms that are poorly defined. However, recent studies have shown that many leaderless proteins misfolded in the cytoplasm can be selectively exported to extracellular milieu via an unconventional secretory path termed Misfolding-Associated Protein Secretion (MAPS). This process uses the surface of the endoplasmic reticulum (ER) as a platform to enrich abnormally folded polypeptides, and then transport them into the lumen of ER-associated late endosomes for subsequent secretion. Elimination of misfolded proteins via MAPS appears to serve a role in protein homeostasis maintenance, particularly for stressed cells bearing an excess of protein quality control (PQC) burden.
Keywords: Misfolding-associated protein secretion (MAPS), Unconventional protein secretion, USP19, HSC70, DNAJC5/CSP, α-Synuclein, cell-to-cell transmission, protein quality control, proteasome, neurodegenerative diseases
Despite being catalyzed by a plethora of chaperons in the cell, protein folding remains as an error-prone task; misfolding frequently occurs, imposing a threat to the protein homeostasis network, particularly in terminally differentiated cells such as neurons as these cells cannot use proliferation to dilute aberrant polypeptides below aggregation thresholds (Balch et al, 2008; Wolff et al, 2014). Upon aggregation, misfolded proteins can recruit and inactivate functional proteins to cause great damages. As a result, links between protein aggregation and neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) etc. have frequently been noted. In fact, in each of these disorders, aggregates formed by specific misfolded proteins were reported, which include Aβ peptide and microtubule-associated Tau in AD, α-Synuclein (α-Syn) in PD, Tau and TDP-43 in FTD, and TDP-43 in ALS etc. [1].
To cope with protein misfolding crisis, eukaryotic cells have crafted a sophisticated protein quality control (PQC) network, which is comprised of chaperones, protein disaggregases, and degradation pathways. Among them, the ubiquitin proteasome system is the most effective ‘garbage disposing’ mechanism [2], accountable for clearing a large fraction of misfolded proteins from diverse subcellular compartments ranging from mitochondria to nuclei. This system uses the small polypeptide ubiquitin to ‘flag’ aberrant proteins for degradation by the 26S proteasome. Given its importance in protein homeostasis regulation, defects in the ubiquitin proteasome system are commonly associated with proteotoxic stress. Additionally, aging-associated decline in the efficiency of the ubiquitin proteasome system has been suggested as a potential contributor for age-dependent neurodegenerative diseases [3, 4].
Given the complexity and dynamics of the proteome landscape in higher eukaryotes, there may exist other PQC mechanisms. Indeed, recent studies have uncovered a unique mechanism capable of exporting misfolded cytosolic proteins to the extracellular milieu via an unconventional protein secretion (UPS) pathway [5, 6]. Intriguingly, many neurodegenerative disease-associated neurotoxic proteins such as Tau and α-Syn are clients of this pathway, suggesting a link between protein secretion-based PQC and neuronal dysfunctions.
An overview of unconventional protein secretion
It has been well established that secretory proteins usually exploit an amino-terminal signal sequence (also termed leader sequence) to gain access to the endoplasmic reticulum (ER), from which they are sorted into COPII vesicles for secretion to cell exterior. However, it has also been noted that some proteins, despite lacking signal sequence, can still be exported from cells through an conventional secretion mechanism [7]. To date, only a few UPS cargos have been characterized [8–10]. These studies collectively delineated four routes through which secretion of a leaderless protein can occur [11]. Among them, FGF-2 [12, 13] and HIV TAT [14, 15] each form a homo-oligomer upon binding to the plasma membrane, which creates a path for its own secretion. This process is referred to as type I UPS. By contrast, many cargos seem to reach cell exterior using a membrane compartment characteristic of the so-called type III UPS. In addition, a secretory pathway resembling type III UPS is the protein release via extracellular vesicles, the best characterized being the exosomes. Exosomes are vesicles formed when membranes of late endosomes bud inwards, sequestering cytoplasmic materials in vesicles in the lumen of late endosomes. These vesicles are then released to cell exterior upon fusion of late endosomes with the plasma membrane.
At first glance, the membrane organelles involved in type III UPS seem different (e.g. autophagosome, late endosomes, lysosomes in mammalian cells [11] or in yeast a compartment termed CUPS (Compartment for Unconventional Protein Secretion) [16, 17]), resulting in what appears to be distinct forms of UPS [11, 18]. However, since most endo-vesicles converge on late endosomes or lysosomes [19], and because lysosomes and a pre-lysosomal compartment are known to form a secretory compartment in specialized secretory cells such as mast cells and lymphocytes [20, 21], it is possible that a subset of late endosomes or pre-lysosomes normally have a secretory function even in non-secretory cells [22]; distinct type III UPS mechanisms may simply reflect the different recruiting strategies that target distinct cargos to this endo-secretory compartment (see below).
Misfolding-associated protein secretion (MAPS)
The MAPS pathway is a recently discovered UPS mechanism, which exports primarily misfolded proteins from mammalian cells using a type III UPS pathway [5]. This process is initiated when misfolded proteins are recruited to the surface of the ER by an ER-associated deubiquitinase (DUB) named USP19. Subsequently, cargo proteins enter the lumen of late endosomes, which are usually located in proximity to the ER. Secretion takes place when these vesicles fuse with the plasma membrane (Figure 1). As a result, the secreted cargos are not associated with any extracellular vesicles.
Figure 1. Misfolding-associated protein secretion (MAPS).
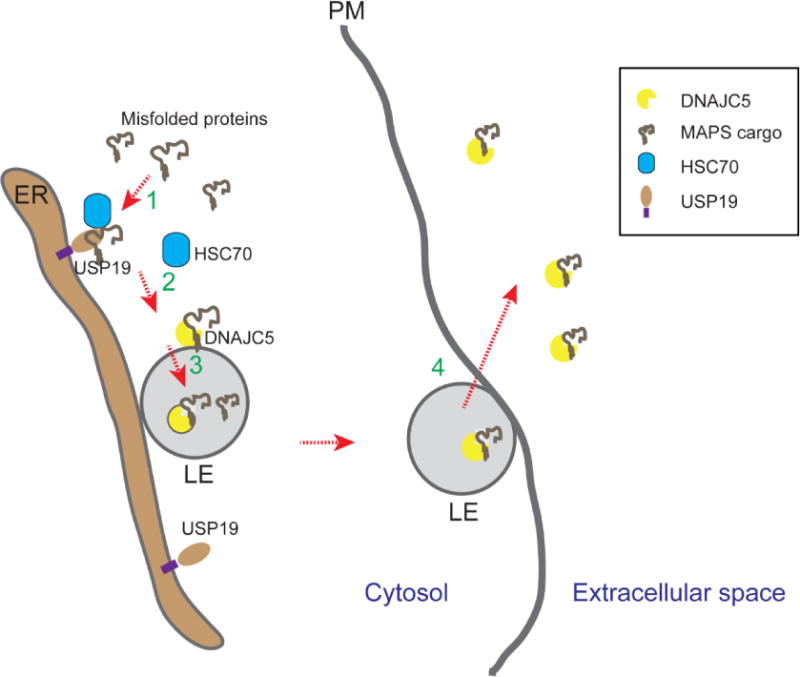
Misfolded cytosolic proteins can be secreted by MAPS following these steps. In step 1, misfolded proteins are enriched on the surface of endoplasmic reticulum (ER) by binding to a receptor (e.g. USP19) that has a chaperone activity. For USP19-mediated secretion, USP19-associated chaperone HSC70 may assist USP19 in substrate recruiting. In step 2, misfolded proteins are transferred to DNAJC5, a HSC70 co-chaperone that is associated with the membrane of late endosomes (LE) (Note that Les are generally in close contact with the ER). Next, the complex of DNAJC5 and misfolded proteins are moved into the lumen of late endosome via a mechanism that is unclear. In step 4, late endosomes contact and fuse with the plasma membranes (PM) to release misfolded proteins.
An important feature of the MAPS pathway is its selectivity towards misfolded proteins. In fact, even for wild-type proteins such as green fluorescence protein (GFP), upon overexpression a fraction of it appears to be misfolded, which can be secreted from mammalian cells [5, 23]. A truncated GFP variant that is more prone to misfolding is secreted much more efficiently than wild-type GFP. Likewise, an ectopically expressed unassembled (therefore misfolded) cytosolic protein is subject to USP19-mediated secretion, whereas the properly assembled endogenous counterpart is exempted [5]. Importantly, many neurodegenerative disease-associated proteins can all be secreted via MAPS, albeit with different efficiencies [5, 6]. The secretion efficiency of individual client seems to be at least in part regulated by the aggregation propensity of the substrate [24]. Accordingly, the MAPS pathway prefers soluble misfolded proteins or small aggregates, but exclude large protein aggregates.
The substrate specificity of MAPS can be in part attributed to an intrinsic chaperoning activity of USP19, which allows it to function as a membrane receptor to recruit misfolded polypeptides [5]. In addition, USP19 is also known to interact with two major cytosolic chaperones, Hsp90 and HSC70 [25]. While Hsp90 is dispensable for the secretion of the MAPS substrates tested to date [24], HSC70 serves a regulatory function in MAPS, but the precise role of HSC70 in this process remains to be determined. In one possible scenario, HSC70 may form a chaperone cascade with USP19, delivering substrates to a downstream effector for further processing (see below). Alternatively, HSC70 may be required to maintain the MAPS machinery in a function state.
Secretion of neurotoxic proteins has also been characterized by Fontaine and colleagues [6]. In a recent report, they demonstrated that misfolded Tau and α-Syn are both released into media from primary neurons and cultured cell lines via a mechanism depending on HSC70 and its co-chaperone DNAJC5. In addition, the secretion of these client proteins also requires a SNARE regulator, which normally governs intracellular membrane fusion [6]. These observations raise the possibility that USP19-regulated MAPS and DNAJC5-dependent secretion may occur via a common membrane compartment.
Indeed, it was recently demonstrated that USP19-stimulated protein secretion requires DNAJC5 acting downstream of USP19 [24]. Moreover, in DNAJC5-stimulated secretion, cargos could be detected entering a late endosome or pre-lysosomal compartment reminiscent of that in USP19-dependent secretion. Coincidentally, endogenous DNAJC5 is also localized to the surface of this compartment [24]. Although DNAJC5 is not a membrane protein, it contains a string of cysteine residues that can undergo palmitoylation [26]. This lipid modification facilitates the anchor of DNAJC5 into the membrane of late endosomes and lysosomes. Intriguingly, only the late endosome-associated DNAJC5 displays extensive co-localization with a MAPS substrate [24]. Collectively, these studies reveal a pathway in which misfolded proteins are initially enriched at the ER by interacting with USP19, and subsequently translocated to DNAJC5 on late endosomes for secretion (Figure 1).
Can MAPS helps to reduce the load of misfolded proteins and therefore promote protein homeostasis? Quantitative immunoblotting analyses suggested that although MAPS can export a wide spectrum of misfolded proteins, its capacity is limited; for any given substrate tested, only a small fraction (5-10%) ends up being secreted [25]. Intriguingly, many MAPS substrates were also reported to be degraded by the 26S proteasome [27, 28], suggesting that MAPS and proteasome represent two alternative paths for disposal of misfolded or unwanted proteins. In agreement with this idea, inhibition of proteasome enhances unconventional secretion of misfolded proteins through MAPS [25, 29]. However, it is unclear whether MAPS can be regulated by other cellular stress signals.
Although the ubiquitin proteasome system is more effective in eliminating misfolded proteins, the MAPS pathway clearly contributes to protein homeostasis because USP19 deficient cells are highly sensitive to proteasome inhibitor treatment [25]. This observation suggests that under normal conditions, MAPS occurs constitutively as a supplementary PQC mechanism as cells rely more on proteasome to eliminate misfolded proteins. The significance of the MAPS pathway only becomes recognizable when cells encounter an overwhelming load of unwanted proteins, exceeding the degradation capacity of the proteasome. Such scenario may occur during aging when the function of the proteasome declines or during cell differentiation when the cellular proteome landscape undergoes drastic reshaping. For instance, the differentiation of hematopoietic stem cell to red blood cells requires removing most cellular proteins but hemoglobin in a short period time. This is thought to be achieved by proteasomal degradation [30]. However, it remains possible that other mechanisms such as protein secretion may account for the elimination of some unwanted cytosolic proteins under this circumstance.
When misfolded proteins are secreted from individual cells, from the perspective of a multicellular organism, they remain associated with the individual, but only in a different compartment. Thus, misfolded proteins probably need to be chaperoned into cell exterior or to be rapidly removed after secretion, so aggregation in extracellular space can be avoided. Indeed, our recent work showed that the MAPS regulator DNAJC5 can be co-secreted together with some misfolded proteins [24]. Moreover, misfolded proteins released into media can be rapidly internalized by neighbor cells through receptor-mediated endocytosis [24]. The surface receptor(s) for misfolded proteins is unclear, but since recent studies have identified several receptors for protein aggregates formed by neurotoxic proteins such as Tau or α-Syn [31, 32], it is possible that cells may also have receptors for soluble misfolded proteins secreted by MAPS.
The secretory compartments in UPS pathways
Although both the MAPS pathway and exosome-mediated secretion use late endosome as a secretory intermediate, the two pathways have several key differences. Most importantly, in MAPS, substrates are translocated from the cytosol into the lumen of late endosomes, whereas in exosome-mediated secretion, cargos are engulfed into small intraluminal vesicles when the membrane of late endosomes invaginate. Accordingly, exosome formation requires a set of factors including ESCRT I, II, and III complexes, whereas for MAPS, at least ESCRT I is dispensable [5]. The secretory organelle in MAPS also differs from secretory autophagosomes since most late endosomes do not bear LC3, a major autophagosome marker [5]. However, as autophagosomes should eventually fuse with late endosomes or lysosomes [33–35], it is tempting to speculate that late endosomes or a pre-lysosomal compartment is the predominant secretory compartment in both type III UPS and exosome-mediated secretion, particularly as they are known to carry ‘fusogens’, which enable fusion with the plasma membrane. This model is consistent with the fact that even in non-secretory cells late endosome and lysosomes can fuse with the plasma membrane in a process critical for repairing damaged cell surface [22, 36]. In addition, in S. cerevisiae, the UPS cargo Acyl-CoA binding protein Acb1 was reported to translocate from the ER exit site to a compartment termed CUPS prior to secretion during nutrient starvation. Although it is unclear whether CUPS is equivalent to late endosomes in mammalian cells, the fact that the secretion of Acb1 is dependent on proteins involved in vesicle fusion is a strong indication that type III UPS may generally require the fusion of a common cargo-bearing compartment with the plasma membrane.
On the other hand, the mechanisms by which distinct cargos are targeted to the secretory compartment in different forms of UPS may vary significantly (Figure 2). Exosome-mediated secretion involves engulfing a portion of cytosol non-selectively into the so called intraluminal vesicles, whereas in autophagy-mediated secretion, cargoes are selectively imported into autophagosomes with the assistance of Hsp90 [35]. By contrast, MAPS cargoes use the USP19-DNAJC5 chaperone cascade and perhaps other chaperones to gain access to late endosomes. Interestingly, recent studies reported yet another UPS pathway (Type IV) that exports mutated ER integral membrane proteins to the plasma membrane without passing through the Golgi [37–39]. Intriguingly, the transport of misfolded pendrin to the plasma membrane in this type UPS is dependent on the HSC70 co-chaperone DNAJC14 [40], reminiscent of DNAJC5’s involvement in MAPS. However, whether vesicles carrying these UPS cargoes first fuse with late endosomes/lysosomes prior to cargo delivery to the cell surface remains to be examined.
Figure 2. Multiple pathways to generate secretory late endosome or lysosomes.
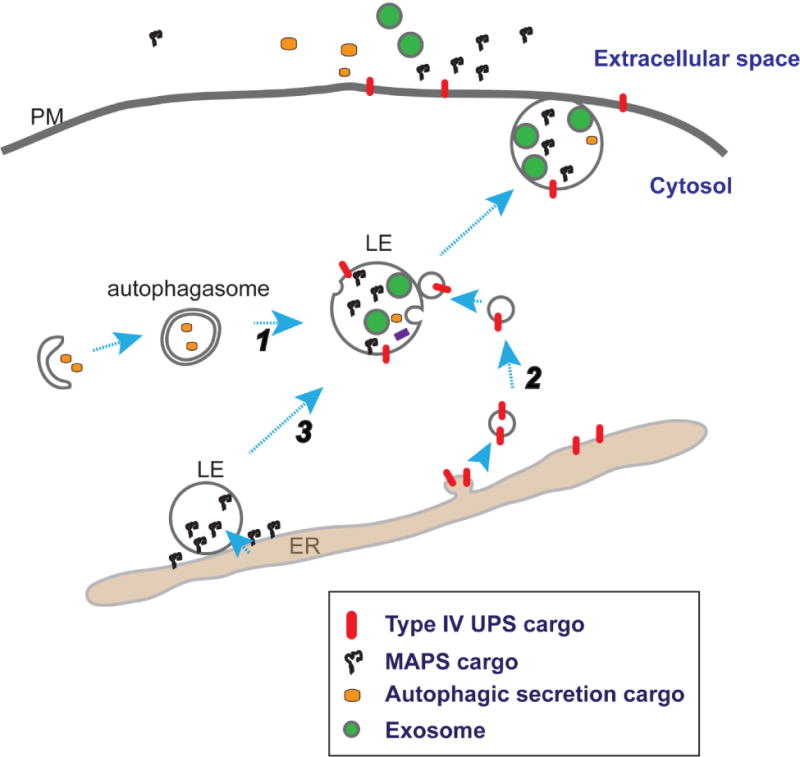
Different unconventional protein secretion pathways may only differ in the initial substrate recruiting step. Cytosolic cargos can enter a secretory late endosomes (LE) via autophagosome (1) or through a Golgi independent vesicular trafficking route between the endoplasmic reticulum (ER) and LE (2), or in the case of MAPS (3), through direct translocation into the LE lumen. Invagination of LE membrane also generates intraluminal vesicles (green circles). The fusion of late endosomes with the plasma membrane (PM) results in the secretion of both exosome-bearing and exosome independent cargos.
MAPS and chaperone-mediate autophagy
It was noteworthy that some cytosolic proteins can be selectively imported into lysosomes for degradation via a pathway termed chaperone-mediated autophagy (CMA) [41]. This process requires direct recognition of substrates bearing a penta-peptide by the cytosolic chaperone HSC70. Subsequently, HSC70 targets CMA substrates to lysosomes through an interaction with a lysosome-associated membrane protein named LAMP2A [42]. Despite having only one transmembrane segment, LAMP2A is thought to form a protein-conducting channel, allowing entry of CMA substrates into the lumen of lysosomes [41]. In mammalian cells, late endosomes mature to form lysosomes in the endocytic system. Consequently, a large population of late endosomes and lysosomes carry the same set of surface markers [19]. This, together with the fact that the recently characterized MAPS substrate α-Syn was also known as a CMA client protein [43], raises the question of whether these two pathways may use a common mechanism to deliver proteins into the lumen of a pre-lysosomal compartment. The answer is not entirely clear, but several distinctions between the two processes are noticeable. First, the MAPS pathway involves initial targeting of substrates to the ER surface, whereas in the CMA pathway, substrates are delivered directly to lysosomes by HSC70; secondly, misfolded MAPS substrates are recognized by USP19 owing to exposed hydrophobic segments, whereas CMA substrates use a specific penta-peptide for lysosomal targeting; thirdly, for CMA substrates, unfolding is a prerequisite for entry into lysosomes, whereas in MAPS, EGFP- or mCherry-tagged substrates were frequently detected in late endosomes by fluorescence microscopy, suggesting that at least a fraction of them enter late endosomes in a folded form. Lastly, even though DNAJC5 is localized to both late endosomes and lysosomes, its co-localization with a MAPS substrate is only observed in late endosomes but not in lysosomes [24]. Thus, it may be reasonable to assume that MAPS and CMA utilize two distinct paths to transport proteins into late endosomes or lysosomes.
MAPS and neurodegenerative diseases
Unconventional protein secretion has long been thought to play a role in neurodegenerative diseases. Several neurodegenerative disease-associated soluble proteins are known to accumulate in extracellular space in pathologic tissues [44]. The cause of their release from cells is largely unclear, neither is the underlying mechanism, but these proteins apparently need to be exported via an UPS mechanism since they do not possess any leader sequences. Intriguingly, some neurotoxic proteins can even undergo cell-to-cell transmission [45, 46]. These proteins are usually aggregation-prone. Thus, their transfer from cell to cell has been thought to facilitate the spreading of protein aggregates in diseases such as Alzheimer’s disease and Parkinson’s disease [47]. One sensible model in analogous to the prion hypothesis suggests that misfolded proteins upon entering recipient neurons may serve as seeds to cause more protein misfolding and aggregation [1, 48]. Accordingly, intercellular transmission of misfolded proteins is comprised of at least two steps: release of misfolded proteins from a donor neuron and their uptake by a recipient neuron. It is now well appreciated that neurons can use a variety of surface receptors to take up misfolded proteins from extracellular milieu [44]. By contrast, little is known about how misfolded proteins lacking leader sequence are exported from cells.
Several models have been proposed to explain the transfer of misfolded leaderless proteins between neurons (Figure 3). First, specialized membrane spikes could be formed at the plasma membranes, linking two neurons together. It was suggested that these so-called ‘nanotubes’ may transport misfolded proteins into recipient cells either directly or via lysosomes [49], but the mechanism of nanotube formation and its physiological relevance are not fully understood. Besides nanotubes, several studies have investigated the potential involvement of exosomes in secretion of misfolded Tau, SOD1, and α-Syn [29, 50–57]. The studies, while confirming that a small fraction of misfolded Tau and α-Syn are released from cultured cells by exosomes, also showed that most misfolded proteins are secreted via an exosome-independent mechanism [6, 29, 53]. This conclusion is not surprising because exosomes are formed when membranes of late endosome invaginate into the lumen, forming intraluminal vesicles (ILV) that carry a small portion of the cytosol. Consequently, this mechanism lacks the specificity required to concentrate selected cargos into the secretory late endosome. By contrast, MAPS uses chaperone such as USP19 and DNAJC5 to concentrate misfolded proteins in late endosomes. Therefore, it is conceivable that it should contribute significantly to the intercellular transmission of misfolded proteins between neurons.
Figure 3. Proposed models for intercellular transmission of misfolded proteins.
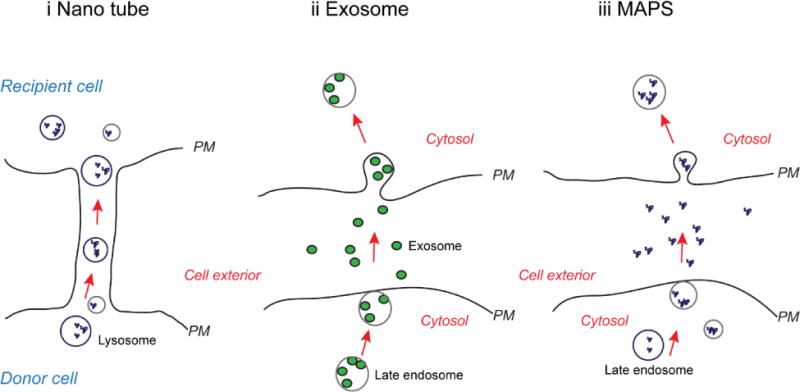
i, Misfolded proteins or protein aggregates are packed in to late endosome or lysosomes. These vesicles are moved from cell to cell through intercellular connections formed by the so called nano tube. PM, Plasma membrane. ii, In misfolding-associated secretion (MAPS), misfolded proteins are moved into late endosomes. These vesicles fuse with the plasma membrane, resulting in the secretion of misfolded proteins. Secreted proteins can enter a recipient cell by endocytosis. The mechanism(s) by which misfolded proteins enter lysosomes or late endosomes in model i and ii is unclear. iii, In exosome-mediated secretion, a fraction of the cytosol is engulfed into multi-vesicular body (MVB) during endosome maturation. When late endosomes containing MVB fuse with the plasma membrane, the cytosolic contents are released into the extracellular space in small vesicles termed exosome. Exosomes can then enter a recipient cell either by endocytosis or by fusion with the plasma membrane.
The fact that aberrant proteins secreted by MAPS can be taken up and degraded by other cells indicates a trans-cellular PQC program, which releases unwanted proteins from stressed cells and subsequent eliminates them by other cells in a multicellular organism. Uptake of misfolded proteins may usually be accomplished by specialized cells such as macrophage, which are well known for their roles in clearing extracellular garbage. Because misfolded proteins are escorted into the cell exterior by chaperones, which is followed by rapid removal through endocytosis, aggregation in extracellular space can be avoided despite constitutive secretion through MAPS. However, this balance may be disturbed during aging or under disease conditions, resulting in accumulation of protein aggregates in extracellular space. In fact, Alzheimer’s disease is a neurodegenerative disease associated with accumulation of extracellular misfolded protein amyloids that include the microtubule-binding protein Tau and the Aβ peptide. Intriguingly, recent studies reported that both ectopically expressed and endogenous Tau can be secreted from cell lines and primary neurons via MAPS [6], raising the possibility that MAPS, if uncoupled from endocytosis-mediated clearance, may contribute to the formation of Tau-containing fibrils in aging brain. The mechanism of Aβ secretion is not entirely clear, but a recent study suggested that it may also be exported via an UPS channel [58].
In Parkinson’s disease, the major component of the pathological Lewy body α-Syn can not only be released from neurons, but also undergo cell-to-cell transmission to spread α-Syn-containing aggregates [59, 60]. Mechanistically, it has been demonstrated that in vitro-formed α-Syn fibril upon injection into mice brains, can be transmitted from the injected region to a distal area to trigger phosphorylation of native α-Syn at Ser129, a hallmark of PD pathology [61]. Transmitted α-Syn fibrils can also provide a seeding template to cause misfolding of native α-Syn in recipient neurons, and thus accelerate the deterioration of neuronal functions [48]. Because therapeutic strategies aimed at reducing the entry of α-Syn into neurons alleviated disease symptoms in mouse models [31, 62], it is conceivable that agents inhibiting its release may also achieve a favorable effect, and therefore offer a new therapeutic strategy. In tissue culture cells, α-Syn secretion is largely mediated by MAPS. Thus, it is critical to determine whether this process contribute to the release of α-Syn from neurons in vivo.
Although the precise link between MAPS and PD pathology remains to be elucidated, this UPS process is clearly critical for neuronal functions because mutations in DNAJC5 have been linked to ceroid lipofuscinosis [26, 63], a neurodegenerative condition characterized by accumulation of auto-fluorescent ‘lysosome-like materials’. Interestingly, most lipfuscinosis-associated mutations affect the cysteine-rich segment that is required for localization of DNAJC5 to late endosomes and lysosomes. Thus, these lysosomal materials might be remnants of MAPS vesicles accumulated due to defects in secretion.
Perspectives
The discovery of MAPS has not only expanded the list of the currently known UPS substrates, but also reveals a back-up PQC strategy used by mammalian cells to promote protein homeostasis. The research on MAPS is clearly at its infancy. Future studies may significantly expand our knowledge on membrane trafficking events associated with MAPS in eukaryotic cells. For instance, how are certain misfolded proteins selected for secretion as opposed to degradation? How is this triaging decision made? How can a cytosolic protein enter the lumen of late endosomes? Is there a specific population of late endosomes that is dedicated for protein secretion? What is the machinery that catalyzes the fusion of late endosomes with the plasma membrane? Since this unique PQC strategy, while benefiting individual cells in short term, may impose a significant burden on cells that are responsible for clearing aberrant proteins from extracellular space, it can have a negative long-term impact on a multicellular organism. Thus, the pathway is likely tightly regulated. Indeed, several factors when overexpressed can significantly enhance the flux of the MAPS pathway, but physiological regulator of this process remains to be identified. With so many open questions, unconventional secretion of misfolded proteins will for sure offer an exciting niche in cell biology research for many years to come.
Acknowledgments
The research in my lab is supported by the intramural Research Program of the National Institute of Diabetes, Digestive, & Kidney Diseases in the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This review is part of a Special Issue of SCDB on “unconventional protein secretion” edited by Walter Nickel and Catherine Rabouille.
References
- 1.Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. Spreading of pathology in neurodegenerative diseases: a focus on human studies, Nature reviews. Neuroscience. 2015;16(2):109–20. doi: 10.1038/nrn3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2011;40(2):238–52. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Vernace VA, Arnaud L, Schmidt-Glenewinkel T, Figueiredo-Pereira ME. Aging perturbs 26S proteasome assembly in Drosophila melanogaster. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21(11):2672–82. doi: 10.1096/fj.06-6751com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292(5521):1552–5. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 5.Lee JG, Takahama S, Zhang G, Tomarev SI, Ye Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nature cell biology. 2016;18(7):765–76. doi: 10.1038/ncb3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fontaine SN, Zheng D, Sabbagh JJ, Martin MD, Chaput D, Darling A, Trotter JH, Stothert AR, Nordhues BA, Lussier A, Baker J, Shelton L, Kahn M, Blair LJ, Stevens SM, Jr, Dickey CA. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. The EMBO journal. 2016;35(14):1537–49. doi: 10.15252/embj.201593489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubartelli A, Cozzolino F, Talio M, Sitia R. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. The EMBO journal. 1990;9(5):1503–10. doi: 10.1002/j.1460-2075.1990.tb08268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malhotra V. Unconventional protein secretion: an evolving mechanism. The EMBO journal. 2013;32(12):1660–4. doi: 10.1038/emboj.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion, Nature reviews. Molecular cell biology. 2009;10(2):148–55. doi: 10.1038/nrm2617. [DOI] [PubMed] [Google Scholar]
- 10.Zhang M, Schekman R. Cell biology. Unconventional secretion, unconventional solutions. Science. 2013;340(6132):559–61. doi: 10.1126/science.1234740. [DOI] [PubMed] [Google Scholar]
- 11.Rabouille C. Pathways of Unconventional Protein Secretion. Trends in cell biology. 2017;27(3):230–240. doi: 10.1016/j.tcb.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 12.Steringer JP, Muller HM, Nickel W. Unconventional secretion of fibroblast growth factor 2–a novel type of protein translocation across membranes? Journal of molecular biology. 2015;427(6 Pt A):1202–10. doi: 10.1016/j.jmb.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 13.Steringer JP, Lange S, Cujova S, Sachl R, Poojari C, Lolicato F, Beutel O, Muller HM, Unger S, Coskun U, Honigmann A, Vattulainen I, Hof M, Freund C, Nickel W. Key steps in unconventional secretion of fibroblast growth factor 2 reconstituted with purified components. eLife. 2017;6 doi: 10.7554/eLife.28985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Debaisieux S, Rayne F, Yezid H, Beaumelle B. The ins and outs of HIV-1 Tat. Traffic. 2012;13(3):355–63. doi: 10.1111/j.1600-0854.2011.01286.x. [DOI] [PubMed] [Google Scholar]
- 15.Zeitler M, Steringer JP, Muller HM, Mayer MP, Nickel W. HIV-Tat Protein Forms Phosphoinositide-dependent Membrane Pores Implicated in Unconventional Protein Secretion. The Journal of biological chemistry. 2015;290(36):21976–84. doi: 10.1074/jbc.M115.667097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cruz-Garcia D, Curwin AJ, Popoff JF, Bruns C, Duran JM, Malhotra V. Remodeling of secretory compartments creates CUPS during nutrient starvation, The. Journal of cell biology. 2014;207(6):695–703. doi: 10.1083/jcb.201407119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curwin AJ, Brouwers N, Alonso YAM, Teis D, Turacchio G, Parashuraman S, Ronchi P, Malhotra V. ESCRT-III drives the final stages of CUPS maturation for unconventional protein secretion. eLife. 2016;5 doi: 10.7554/eLife.16299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rabouille C, Malhotra V, Nickel W. Diversity in unconventional protein secretion. Journal of cell science. 2012;125(Pt 22):5251–5. doi: 10.1242/jcs.103630. [DOI] [PubMed] [Google Scholar]
- 19.Huotari J, Helenius A. Endosome maturation. The EMBO journal. 2011;30(17):3481–500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blott EJ, Griffiths GM. Secretory lysosomes, Nature reviews. Molecular cell biology. 2002;3(2):122–31. doi: 10.1038/nrm732. [DOI] [PubMed] [Google Scholar]
- 21.van der Sluijs P, Zibouche M, van Kerkhof P. Late steps in secretory lysosome exocytosis in cytotoxic lymphocytes. Frontiers in immunology. 2013;4:359. doi: 10.3389/fimmu.2013.00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaiswal JK, Andrews NW, Simon SM. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. The Journal of cell biology. 2002;159(4):625–35. doi: 10.1083/jcb.200208154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanudji M, Hevi S, Chuck SL. Improperly folded green fluorescent protein is secreted via a non-classical pathway. Journal of cell science. 2002;115(Pt 19):3849–57. doi: 10.1242/jcs.00047. [DOI] [PubMed] [Google Scholar]
- 24.Xu Y, L C, Debello A, Wang L, Lee J, Saidi L, Lee JG, Ye Y. DNAJC5 facilitates USP19-dependent unconventional secretion of misfolded cytosolic proteins. Cell discovery. 2018 doi: 10.1038/s41421-018-0012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JG, Kim W, Gygi S, Ye Y. Characterization of the deubiquitinating activity of USP19 and its role in endoplasmic reticulum-associated degradation. The Journal of biological chemistry. 2014;289(6):3510–7. doi: 10.1074/jbc.M113.538934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgoyne RD, Morgan A. Cysteine string protein (CSP) and its role in preventing neurodegeneration. Seminars in cell & developmental biology. 2015;40:153–9. doi: 10.1016/j.semcdb.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Y, Anderson DE, Ye Y. The HECT domain ubiquitin ligase HUWE1 targets unassembled soluble proteins for degradation. Cell discovery. 2016;2:16040. doi: 10.1038/celldisc.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM. Degradation of alpha-synuclein by proteasome. The Journal of biological chemistry. 1999;274(48):33855–8. doi: 10.1074/jbc.274.48.33855. [DOI] [PubMed] [Google Scholar]
- 29.Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. Journal of neurochemistry. 2010;113(5):1263–74. doi: 10.1111/j.1471-4159.2010.06695.x. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen AT, Prado MA, Schmidt PJ, Sendamarai AK, Wilson-Grady JT, Min M, Campagna DR, Tian G, Shi Y, Dederer V, Kawan M, Kuehnle N, Paulo JA, Yao Y, Weiss MJ, Justice MJ, Gygi SP, Fleming MD, Finley D. UBE2O remodels the proteome during terminal erythroid differentiation. Science. 2017;357(6350) doi: 10.1126/science.aan0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, Umanah GE, Brahmachari S, Shin JH, Kang HC, Zhang J, Xu J, Chen R, Park H, Andrabi SA, Kang SU, Goncalves RA, Liang Y, Zhang S, Qi C, Lam S, Keiler JA, Tyson J, Kim D, Panicker N, Yun SP, Workman CJ, Vignali DA, Dawson VL, Ko HS, Dawson TM. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016;353(6307) doi: 10.1126/science.aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(33):E3138–47. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinseth MA, Anjard C, Fuller D, Guizzunti G, Loomis WF, Malhotra V. The Golgi-associated protein GRASP is required for unconventional protein secretion during development. Cell. 2007;130(3):524–34. doi: 10.1016/j.cell.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 34.Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. The Journal of cell biology. 2010;188(4):527–36. doi: 10.1083/jcb.200911154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang M, Kenny SJ, Ge L, Xu K, Schekman R. Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. eLife. 2015;4 doi: 10.7554/eLife.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca(2+)-regulated exocytosis of lysosomes. Cell. 2001;106(2):157–69. doi: 10.1016/s0092-8674(01)00421-4. [DOI] [PubMed] [Google Scholar]
- 37.Grieve AG, Rabouille C. Golgi bypass: skirting around the heart of classical secretion. Cold Spring Harb Perspect Biol. 2011;3(4) doi: 10.1101/cshperspect.a005298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabouille C, Linstedt AD. GRASP: A Multitasking Tether. Frontiers in cell and developmental biology. 2016;4:1. doi: 10.3389/fcell.2016.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gee HY, Lee MG. Seminars in cell & developmental biology. 2018 (this issue) [Google Scholar]
- 40.Jung J, Kim J, Roh SH, Jun I, Sampson RD, Gee HY, Choi JY, Lee MG. The HSP70 co-chaperone DNAJC14 targets misfolded pendrin for unconventional protein secretion. Nat Commun. 2016;7:11386. doi: 10.1038/ncomms11386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends in cell biology. 2012;22(8):407–17. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273(5274):501–3. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 43.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 44.Borland H, Vilhardt F. Prelysosomal Compartments in the Unconventional Secretion of Amyloidogenic Seeds. International journal of molecular sciences. 2017;18(1) doi: 10.3390/ijms18010227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nature reviews Neuroscience. 2010;11(3):155–9. doi: 10.1038/nrn2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nature reviews Molecular cell biology. 2010;11(4):301–7. doi: 10.1038/nrm2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo JL, Lee VM. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nature medicine. 2014;20(2):130–8. doi: 10.1038/nm.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee VM, Trojanowski JQ. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52(1):33–8. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 49.Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, Olivo-Marin JC, Melki R, Zurzolo C. Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. The EMBO journal. 2016;35(19):2120–2138. doi: 10.15252/embj.201593411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC, Alvarez VE, Lee NC, Hall GF. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. The Journal of biological chemistry. 2012;287(6):3842–9. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiology of disease. 2011;42(3):360–7. doi: 10.1016/j.nbd.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30(20):6838–51. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hasegawa T, Konno M, Baba T, Sugeno N, Kikuchi A, Kobayashi M, Miura E, Tanaka N, Tamai K, Furukawa K, Arai H, Mori F, Wakabayashi K, Aoki M, Itoyama Y, Takeda A. The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of alpha-synuclein. PloS one. 2011;6(12):e29460. doi: 10.1371/journal.pone.0029460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomes C, Keller S, Altevogt P, Costa J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neuroscience letters. 2007;428(1):43–6. doi: 10.1016/j.neulet.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 55.Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O’Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, Kumita JR, Luheshi LM, Yousefi M, Coleman BM, Hill AF, Plotkin SS, Mackenzie IR, Cashman NR. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(9):3620–5. doi: 10.1073/pnas.1312245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng J, Koutras C, Donnelier J, Alshehri M, Fotouhi M, Girard M, Casha S, McPherson PS, Robbins SM, Braun JEA. Neurons Export Extracellular Vesicles Enriched in Cysteine String Protein and Misfolded Protein Cargo. Scientific reports. 2017;7(1):956. doi: 10.1038/s41598-017-01115-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Record M, Carayon K, Poirot M, Silvente-Poirot S. Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochimica et biophysica acta. 2014;1841(1):108–20. doi: 10.1016/j.bbalip.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 58.Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC. Abeta secretion and plaque formation depend on autophagy. Cell reports. 2013;5(1):61–9. doi: 10.1016/j.celrep.2013.08.042. [DOI] [PubMed] [Google Scholar]
- 59.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338(6109):949–53. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. The Journal of experimental medicine. 2012;209(5):975–86. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nature cell biology. 2002;4(2):160–4. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 62.Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, Lee VM. Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell reports. 2014;7(6):2054–65. doi: 10.1016/j.celrep.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Noskova L, Stranecky V, Hartmannova H, Pristoupilova A, Baresova V, Ivanek R, Hulkova H, Jahnova H, van der Zee J, Staropoli JF, Sims KB, Tyynela J, Van Broeckhoven C, Nijssen PC, Mole SE, Elleder M, Kmoch S. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. American journal of human genetics. 2011;89(2):241–52. doi: 10.1016/j.ajhg.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]