

Abstract
Metastasis-associated lung adenocarcinoma transcript 1(MALAT1) is a highly conserved long non-coding RNA (lncRNA). Overexpression of MALAT1 has been demonstrated to related to poor prognosis of multiple myeloma(MM) patients. Here, we demonstrated that MALAT1 plays important roles in MM DNA repair and cell death. We found bone marrow plasma cells from patients with monoclonal gammopathy of undetermined significance (MGUS) and MM express elevated MALAT1 and involve in alternative-non-homozygous end joining (A-NHEJ) pathway by binding to PARP1 and LIG3, two key components of the A-NHEJ protein complex. Degradation of the MALAT1 RNA by RNase H using antisense gapmer DNA oligos in MM cells stimulated poly-ADP-ribosylation of nuclear proteins, defected the DNA repair pathway, and further provoked apoptotic pathways. Anti-MALAT1 therapy combined with PARP1 inhibitor or proteasome inhibitor in MM cells showed a synergistic effect in vitro. Furthermore, using novel single wall carbon nanotube (SWCNT) conjugated with anti-MALAT1 oligos, we successfully knocked down MALAT1 RNA in cultured MM cell lines and xenograft murine models. Most importantly, anti-MALAT1 therapy induced DNA damage and cell apoptosis in vivo, indicating that MALAT1 could serve as a potential novel therapeutic target for MM treatment.
Keywords: multiple myeloma, MALAT1, long non-coding RNA, A-NHEJ, antisense oligonucleotide drug
Introduction
Multiple myeloma (MM), a cancer of terminally differentiated plasma cells, is the second most frequently diagnosed hematologic cancer in the United States1. MM is nearly always preceded from an age-related progressive pre-malignant condition termed monoclonal gammopathy of undetermined significance (MGUS). The finding of long non-coding RNA (lncRNA) transcripts from genomic regions is one of the most unexpected findings of the genomics era. lncRNAs are a group of RNA transcripts longer than 200nt that do not encode proteins but are involved in various forms of gene expression regulation2. Rapidly accumulating evidences indicate that lncRNAs are involved in the initiation and progression of almost all kinds of cancer, including MM3–5. These findings collectively support the possibility that systematic investigation of lncRNA function in tumorigenesis will yield novel insights into diagnosis and treatment of cancers.
Metastasis-associated lung adenocarcinoma transcript 1(MALAT1), also known as nuclear-enriched noncoding transcript 2(NEAT2), is a highly conserved nuclear lncRNA (~8.7kb in human)6. MALAT1 was initially found to play an important role in nuclear speckles and to interact with pre-mRNA splicing factors in Hela cells7–10 through regulating a variety of biological process11. It was originally identified in metastatic non-small cell lung cancer (NSCLC)12 and over-expressed in different types of tumor such as hepatocellular carcinoma13, breast cancer14 and prostate cancer10. In MM, MALAT1 is reported to be the most highly expressed lncRNA and correlated with poor prognosis4, 15 and significantly unregulated in fatal course extramedullary MM compared with newly diagnosed MM patients16.
In our current study, we sought to determine the oncogenic role of MALAT1 and explore it as a possible therapeutic target for MM. We found that MALAT1 is highly expressed in MGUS, smoldering MM (SMM) and MM compared to normal plasma cells. We further identified the function of MALAT1 involving in alternative non-homologous end joining (A-NHEJ) pathway through binding with PARP1/ LIG3 complex, and regulated apoptosis via co-acting with PARP1. Finally, we developed a novel single-wall carbon nanotube (SWCNT)-conjugated anti-MALAT1 oligo, and used it in two MM xenograft murine models, and observed remarkable therapeutic outcomes.
Methods
Cell lines, plasmids and human MM tissues
Human MM cell lines including MM.1S, H929, RPMI8226 and HEK293T were obtained from ATCC. Bortezomib-resistant (RPMI8226/V10R) cell line is a kind gift of Dr. Robert Orlowski (The University of Texas M.D. Anderson Cancer Center, Houston, TX, USA). Melphalan-resistant (RPMI8226/LR5) and doxorubicin-resistant (RPMI8226/DOX40) cell lines are gift from Dr. William Dalton (Moffitt Cancer Center, Tampa, FL). Bone marrow (BM) plasma cells were isolated from 4 healthy donors (HDs) and 7 MM patients using CD138 magnetic beads (Miltenyi Biotec). Total RNA was isolated using Trizol reagent (Thermo Fisher Scientific). To establish A-NHEJ, NHEJ and homologous recombination (HR) DNA repair pathway reporter stable cell lines, pEJ2GFP-puro (#44025, Addgene)17, pimEJ5GFP (#44026, Addgene) 17 and pDRGFP (#26475, Addgene)18 vectors were transfected into HEK293T cells separately and selected with 2μg/mL puromycin. To construct the MALAT1 overexpression vector, full length of human MALAT1 cDNA was cloned into pCDH-MSCV-MCS-EF1-copGFP-T2A-Puro plasmid (System Biosciences), between NotI and SwaI sites. The packaging system was used according to the manufacturer’s protocol. MM.1S cells were infected by MALAT1 overexpression (V-MALAT1) or empty control virus (V-ctrl), and subjected to flow sorting by flow cytometry using green fluorescent protein copGFP as a marker.
Formalin-fixed paraffin-embedded (FFPE) BM blocks of 11 HDs and 9 MM patients were obtained from the myeloma tissue bank of the Cleveland Clinic Taussig Cancer Institute and the Norman Bethune International Peace Hospital. All participants signed informed consent forms. This study was approved by institutional review boards (IRB) of both Cleveland Clinic and Norman Bethune International Peace Hospital. MM tissue microarray (TMA) was purchased from US Biomax, lnc (T291b), which contained BM from 2 HDs and 4 MM patients.
RNA Antisense Purification (RAP)
Nuclear extracts were incubated with a 59bp biotin-labeled MALAT1 probe GTGCCTTTAGTGAGGGGTACCTGAAAAATCTTAAAAAAAGGCTTAGCGCCCACCTCACC/3Bio/ or sequence-scrambled probe TCAACCTTTACACCGATCTAGAATCGAATGCGTAGATTAGCCAGGTGCAAACCAAAAAT/3Bio/ and hybridized at 4°C for 2 hours. Hybridized material was captured with magnetic streptavidin beads (Thermo Fisher Scientific). Bound material was washed and eluted with RNaseH (New England Biolabs) as previously described11. The proteins were separated by SDS-PAGE and stained using Coomassie blue. Specific bands were isolated for whole proteomic mass spectrometry (MS) analysis.
Ribonucleoprotein immunoprecipitation (RIP)
2×107 H929 or MM.1S cells were rinsed with PBS and then irradiated with 150 mJ/cm2 at 254 nm using a UV cross-linker. Cell pellets were resuspended in 100 μL cytoplasmic extract (CE) buffer (10 mM HEPES, 60 mM KCl, 1 mM EDTA, 0.075% NP40, 1 mM DTT, pH 7.6). The cells were incubated on ice for 3 min and then centrifuged at 250g for 5 min. The cell nuclei were washed wit 500 μL CE buffer without NP40 and then resuspended in lysis buffer (150mM NaCl, 50mM Tris-HCl pH 7.5, 0.5% Triton X-100 supplemented with protease inhibitor cocktail and RNase inhibitor). The lysate was sonicated for 5 min with 30sec on/off intervals on ice, and then centrifuge at 14,000g for 10 minutes. The cell lysate was further diluted (1:5) with NT2 buffer (50 mM Tris-HCl pH7.4, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40 supplemented with fresh 200U RNaseOut, 400μM VRC, 1mM DTT, 20mM EDTA, and protease inhibitor cocktail).
Protein A/G magnetic beads (Pierce protein A/G magnetic beads; Thermo scientific) were washed with NT2 buffer 6 times and then pre-coated using 5% BSA NT2 buffer (1:5 v/v) at room temperature for 1 h. anti-PARP1 or anti-LIG3 antibody, 2μg, was added to 500 μL of the bead mixture and incubated at 4°C for 12 hours. The beads were washed in ice-cold NT2 buffer for 5 times and resuspended in 850 μL NT2 buffer.
The cell lysate was mixed with the antibody-coated beads, and an aliquot of the mixture was removed for total RNA and protein determination. The remaining lysate was incubated with beads at 4°C for 4 hours. After co-IP, the beads were washed as follows: twice with lysis buffer; thrice with the lysis buffer containing 900 mM NaCl and 1% NP-40; and twice more with lysis buffer. The beads were then transferred to a fresh tube and subjected to a final wash with the lysis buffer containing 0.05% NP-40. Following the washes, an aliquot of beads was removed from each sample and mixed with 2× LDS sample buffer for western blot analysis. Another aliquot of beads was used for RNA extraction.
Detailed description is provided in the Supplementary Materials and Methods.
Results
MALAT1 is the most highly expressed lncRNA in MGUS and MM
We first analyzed gene expression microarray datasets uploaded by 3 different groups, including Zhan dataset (GSE5900)19, Gutiérrez dataset (GSE16558)20 and López-Corral dataset (GSE47552)21. Analysis of all 3 datasets showed that MALAT1 expression was higher in MGUS, SMM and MM compare with healthy donors (HDs, Fig. S1A).
We next used in situ hybridization (ISH, Fig. S1B) and qRT-PCR (Fig. S1C) to detect MALAT1 in clinical MM samples and cell lines, and verified that MALAT1 was highly expressed in BM CD138+ cells from MM patients compared with HDs, which was consistent with microarray data. Furthermore, two groups have reported that MALAT1 overexpression was significantly correlated to poor prognosis in MM patients, including shorter progression-free survival (PFS) and overall survival (OS)15, 22.
MALAT1 over-expression accelerated proliferation and repressed apoptosis in MM
To explore the functions of over-expressed MALAT1 in MM, we infected V-MALAT1 or V-ctrl into MM.1S cells, and added puromycin for selection, then injected subcutaneously to the shoulders of SCID mice (Fig. 1A). Diameters of tumor were measured once a week, the growth of MM.1S-V-MALAT1 xenografts was significantly faster than controls (Fig. 1A). MALAT1 levels in MM.1S-V-MALAT1 xenografts were over-expressed confirmed by qRT-PCR (Fig. 1B). MM.1S-V-MALAT1 xenografts compared with the MM.1S-V-ctrl xenografts have higher proliferation and less apoptosis according to immunohistochemistry staining of Ki-67 and c-caspase3 (Fig. 1C).
Fig. 1. MALAT1 overexpression promoted the tumorigenesis of MM.

(A) 2×106 MALAT1 overexpressed or control MM.1S cells were injected subcutaneously to the shoulder of SCID mice. The sizes of xenograft were measured once a week. Mice were sacrificed 30 days after injection, and xenografts were weighted. (B) MALAT1 level was determined by qRT-PCR. (C) The levels of Ki-67 and c-Caspase3 were detected by immunohistochemistry. (*p<0.05, **p<0.01, ***p<0.001)
MALAT1 binds with PARP1/LIG3 complex in MM
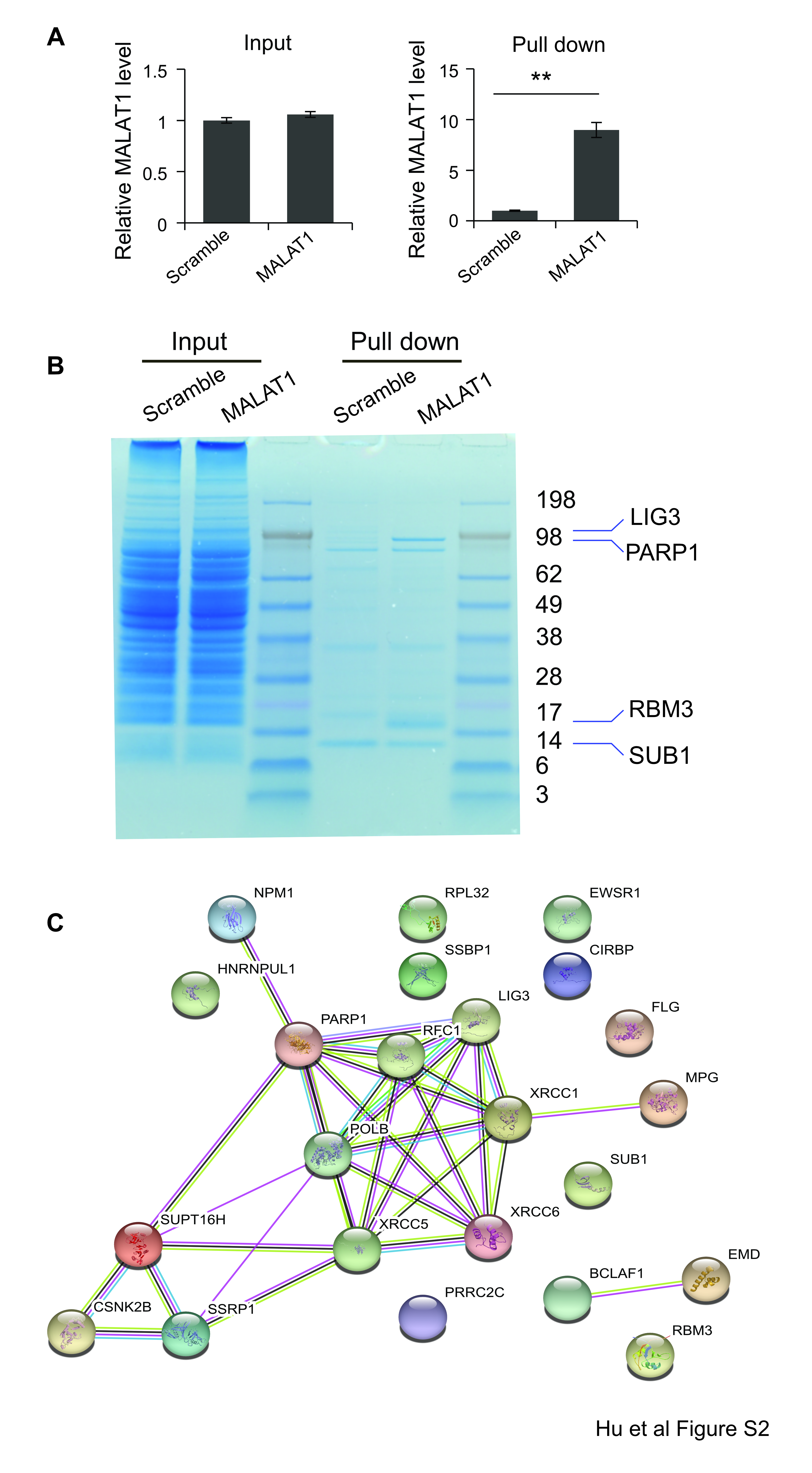
To investigate the co-factors binding to MALAT1 in MM cells, we used RNA antisense purification-mass spectrum (RAP-MS) to identify MALAT1 binding proteins (Fig. 2A). Biotin-labeled anti-MALAT1 DNA probe was used to pull-down MALAT1 in H929 cells, then MALAT1 pull-down sample was used to run a PAGE gel and subjected to MS analysis. (Fig. S2A–B). Using RAP-MS whole proteomic analysis, we identified 23 MALAT1 binding proteins (Table. S1). STRING database functional enrichment analysis revealed 10 of these proteins were related to DNA repair pathways (GO:0006281, false discovery rate 9.89e-08), including PARP1, LIG3, XRCC1, XRCC5, XRCC6, SUPT16H, NPM1, RFC1, SSRP1 and MPG (Fig. S2C). The notable proteins with strong signals, including PARP1, LIG3, and XRCC5 were further verified by western blot using MALAT1 pull-down protein lysate from H929, MM.1S and RPMI8226 cells, respectively (Fig. 2B). The co-localization between MALAT1 and PARP1 was further confirmed by immunofluorescence staining. As shown in Fig. 2C, more than 70% of the PARP1 signal was co-localized with MALAT1 signal in H929, MM.1S, and RPMI8226 cells.
Fig. 2. PARP1/LIG3 complex was identified as MALAT1 binding target by RAP-MS.

(A) Schematic diagram of the RAP-MS strategy used to identify MALAT1 binding proteins. (B) PARP1, LIG3 and XRCC5 were verified as MALAT1 binding proteins. (C) MALAT1 co-localized with PARP1 in H929, MM.1S and RPMI8226 cells (scale bar=1μM).
PARP1 was intensively investigated multiplefunctional protein which has been implicated in recognition of DNA single and double strand break (SSB and DSB)s during DNA repair and catalyzes PAR formation to induce cell apoptosis23. According to our RAP-MS results, MALAT1 pulled down PARP1, as well as other DNA repair proteins, thus we hypothesized that MALAT1 acts as a scaffold, to form functional complexes through bundling PARP1 and other proteins, then exerted its function in DNA repair pathway(s). To validate this hypothesis, we firstly used ribonucleoprotein immunoprecipitation (RIP) strategy to further prove the binding between MALAT1 and PARP1 in myeloma cells. As shown in Fig. 3A, the RNA-protein complexes in myeloma cells were first cross-linked by UV, then the cell lysate was incubated with PARP1 antibody-coated magnetic beads. After washing, total RNA was extracted from the precipitate, then the MALAT1 level was determined by qRT-PCR. We found PARP1 antibody-coated beads specifically enriched PARP1 signal (Fig. 3B), and MALAT1 was also enriched by PARP1 antibody-conjugated beads exclusively (Fig. 3C), which demonstrated direct interaction between MALAT1 and PARP1. Although there no RNA binding domain on LIG3, poly (ADP-ribose) polymerase and DNA-Ligase Zn-finger (zf-PARP) regions are present that can bind PARP124. PARP1 and LIG3 are critical molecules involved in the A-NHEJ DNA repair pathway25. Thus, we postulated that MALAT1 might play its role in A-NHEJ DNA repair by direct binding with PARP1 and indirect binding with LIG3.
Fig. 3. Verification of the binding between MALAT1 and PARP1 by RIP-PCR.

(A) Schematic diagram of the RIP-PCR assay. (B) The level of PARP1 before or after RIP was determined by immunoblotting. (C) MALAT1 pulled-down by PARP1 antibody-conjugated beads were determined by qRT-PCR (right panel), left panel is input (*p<0.05, **p<0.01, ***p<0.001).
MALAT1 inhibition induced DNA damage and apoptosis in MM
To demonstrate our postulation, we used two gapmer DNA antisense oligos targeting MALAT1 (anti-MALAT1-1/2) to knock-down MALAT1 expression and perform loss-of-function study of MALAT1 in MM cells. The gapmer DNA was flanked by blocks of 2′-OMe-modified RNAs, which would bind to MALAT1 RNA and induce cleavage of MALAT1 by RNase H26. qRT-PCR analysis showed MALAT1 was efficiently knocked-down in H929(Fig. 4A), MM.1S (Fig. 4B) and RPMI8226 cells (Fig. 4C). The frequency of DNA break increased substantially as revealed by immunofluorescence staining and western blot for γH2A.X. Interestingly, we found that PAR signal increased after MALAT1 knock-down indicating that MALAT1 antagonist did not inhibit, but enhanced PARP1 catalytic activity by releasing PARP1 from MALAT1/PARP1 complex, which would induce cell apoptosis directly27. Furthermore, defective DNA repair induced more cleavage of PARP1 and caspase3 (c-PARP1 and c-caspase3), which also contributed to cell apoptosis in MM cells. We also observed significantly increased apoptosis by flow cytometry analysis after anti-MALAT1 treatment (Fig. 4A–C).
Fig. 4. MALAT1 inhibition induced DNA damage and cell death in MM.

2′-OMe-modified anti-MALAT1 oligos or control oligos were transfected into H929(A), MM.1S(B) or RPMI8226(C) cells, respectively. At 48h after transfection, cells were collected and subjected to qRT-PCR, immunofluorescence staining for γH2A.X, immunoblotting of γH2A.X, PARP1, c-PARP1, caspase-3, and c-caspase3 and apoptosis assay. (*p<0.05, **p<0.01).
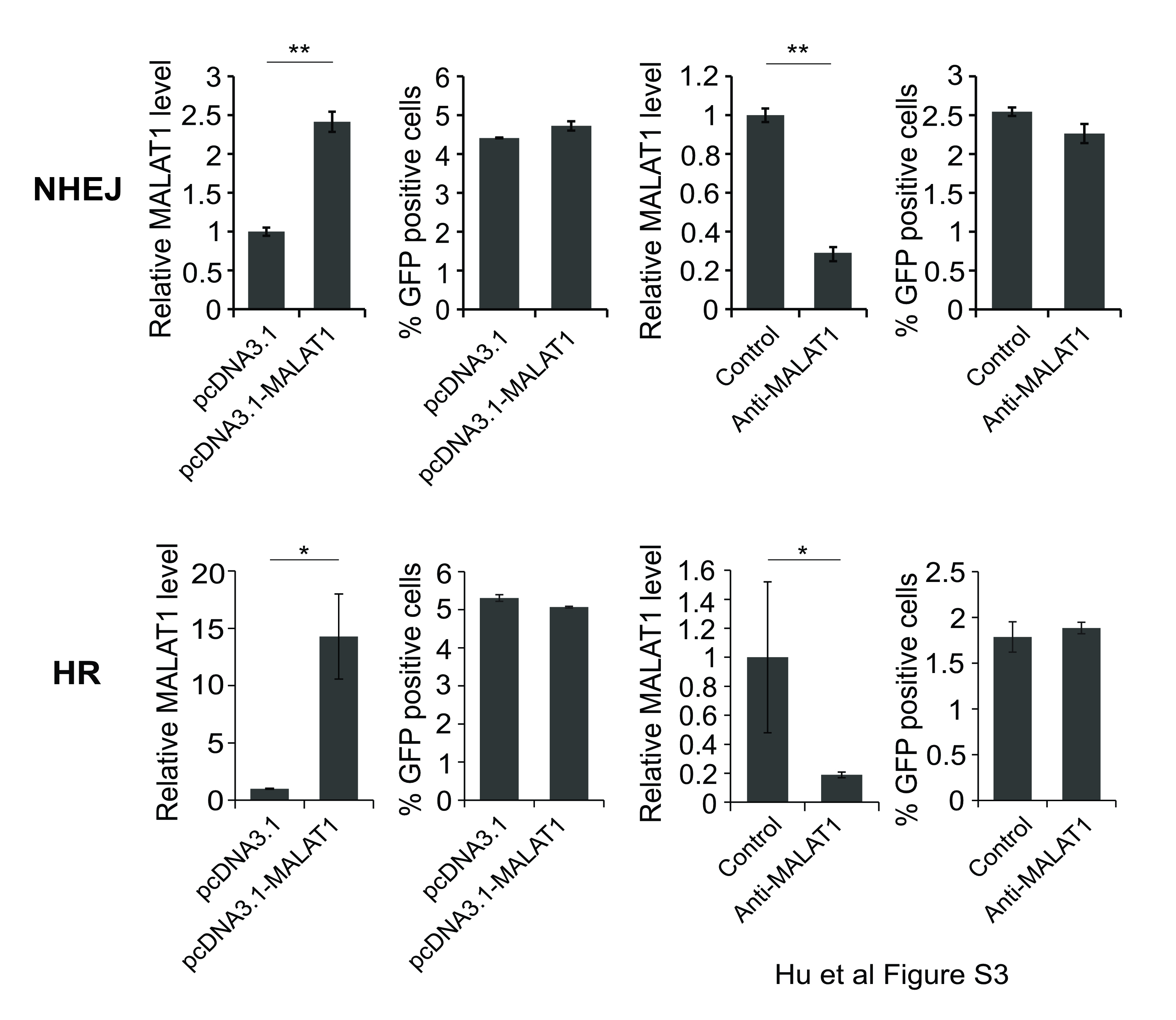
To determine whether MALAT1 specificly involved in A-NHEJ DNA repair pathway, we generated pEJ2GFP-puro (A-NHEJ reporter), pimEJ5GFP (NHEJ reporter) and pDRGFP (HR reporter) stable cell lines in HEK293T cells separately17, 18. I-SceI was used to generate DNA damages at I-SceI sites on these plasmids. MALAT1 overexpression vector or anti-MALAT1 gapmer DNA were used to up-/down-regulate the expression of MALAT1. All vectors would produce GFP once the plasmid DNAs were repaired by correspondent functions, thus we could evaluate which DNA repair pathway involved by examining GFP positive ratio by flow cytometry. As shown in Fig. 5A, HEK293T-EJ2GFP with over-expressed MALAT1 had significant increase of GFP positive cells, whereas HEK293T-EJ2GFP with knocked-down MALAT1 had significant decrease of GFP positive cells. However, no significant difference was found in the HEK293T-imEJ5GFP and HEK293T-DRGFP cells with up-/down-regulation of MALAT1(Fig. S3), indicating that MALAT1 specificly involved in A-NHEJ pathway. The result were further verified by immuofluoresence staining of LIG3/PARP1 and LIG3/γH2A.X in H929 cells after we knocked down MALAT1. We found MALAT1 knock-down had no influence on LIG3/PARP1 co-localization (Fig. 5B, Fig. S4), but interrupted the co-localization between LIG3 and γH2A.X (Fig. 5C, Fig. S4). These results demonstrated that MALAT1 is crucial in A-NHEJ pathway through recruiting LIG3 to γH2A.X loci on DSB.
Fig. 5. MALAT1 coordinated with PAPR1 inhibitor through inhibiting A-NHEJ.

(A) In A-NHEJ reporter plasmid (pEJ2-GFP-puro) stable expression 293T cells, pCBA-SceI was transient transfected with MALAT1 overexpression/empty vector or anti-MALAT1/control gapmer. Then GFP positive cells were determined by flow cytometry. (B) The co-localization between LIG3 and PARP1 or γH2A.X were determined by immunofluorescence staining (scale bar=5μM). (C) H929, MM.1S and RPMI8226 cells transfected with anti-MALAT1 or control oligos were treated with ABT-888. Cells were collected for WB. MALAT1 were determined by qRT-PCR.(*p<0.05, **p<0.01)
To further understand how does MALAT1 knock down affect other components of A-NHEJ DNA repair pathway, NHEJ pathway, and HR pathway, we detected the expression levels of proteins involved in these pathways including CtIP, MRE11, RAD50, NBS1, p-ATM, p-ATR, XRCC5 and XRCC6. We found that RAD50, pATM, or pATR upregulated in all three MM cell lines, MRE11 upregulated in RPMI8226 cell line, after MALAT1 was knockdown, but not CtIP, NBS1, XRCC5 and XRCC6 protein levels (Fig. S5A). To determin if MALAT1 inhibition will affect MRE11-RAD50-NBS1 (MRN) complex or XRCC5 and XRCC6 complex formation, we did immunoflurecence staining of MRE11 and NBS1, XRCC5 /XRCC6 complex in MM cell lines. We found that MALAT1 knock down didn’t influence the MRN complex and XRCC5/XRCC6 complex formation (Fig. S5B–C), indicating MALAT1 is dispensable for the initial DSB recognition of either A-NHEJ or NHEJ or HR pathways.
PARP1 inhibitor cooperated with MALAT1 antagonist to induce DNA damage and apoptosis in MM
To determine whether inhibiting the dissociated PARP1 catalytic activity induces additional DNA damage to further increase cell death after anti-MALAT1 treatment in MM cells, we used the PARP1 inhibitor ABT888 to specially inhibit PARP1 activity in MALAT1 knocked-down MM cells. We observed increased PAR signal in H929 and MM.1S when treated with anti-MALAT1 only, but saw dose-dependent decreased PAR signal in the same cell lines treated synchronously with anti-MALAT1 and ABT888. Combination treatment significantly increased the level of γH2A.X, cleaved PARP1, and cleaved caspase-3(Fig. 5C).
MALAT1 inhibition potentiates the cytotoxic effects of bortezomib in MM
Bortezomib treatment could induce “BRCAness” in MM and impair HR pathway28. Our results have demonstrated that repressing MALAT1 inhibited A-NHEJ activity, thus we postulated that MALAT1 antagonists might have synergistic effect with bortezomib by disabling both HR and A-NHEJ pathways, then provoked cell death by inducing severe DNA damages in MM. To verify our assumption, anti-MALAT1-1 or scrambled DNA oligos was transfected into H929, MM.1S and RPMI8226 cells and treated with various doses of bortezomib, then cells were collected for apoptosis assay. We found expression of BRCA1 and BRCA2 were dramatically down-regulated in all 3 cell lines received high-dose bortezomib treatment (Fig. 6A–C). Whereas γH2A.X signals and apoptosis ratio were increased by both high-/low-dose bortezomib treatment, and these effects were amplified by combining with anti-MALAT1 treatment. Meanwhile, anti-MALAT1 treated MM cells were more sensitive to bortezomib compared to untreated cells according to our cell viability assay, which showed the IC50 reduced from 4.9 nM to 3.6 nM in H929 cells, from 8.9 nM to 6.6 nM in MM.1S cells, and from 10.1 nM to 8.2 nM in sensitive to bortezomib compared with control cells (Fig. 6A–C). These results implied that bortezomib and anti-MALAT1 acted synergistically to induce MM cell death via promoting DNA damages.
Fig. 6. Synergistic effect of anti-MALAT1 and bortezomib in MM.

H929(A), MM.1S(B) and RPMI8226(C) cells were transfected with 1nM anti-MALAT1 or control oligos and treated with bortezomib. Cells were collected for immunoblotting, apoptosis assay and qRT-PCR. Cell viability was measured and IC50 was calculated before and after MALAT1 knockdown.( *p<0.05, **p<0.01, ***p<0.001)
To understand the role of MALAT1 in drug resistance in MM, we used bortezomib-, melphalan- and doxorubicin- resistance MM cell lines, RPMI8226/V10R, RPMI8226/LR5 and RPMI8226/DOX40 and their parental cell line RPMI8226 used as control. We found MALAT1 expression was significantly higher in these resistant MM cell lines compared with RPMI8226 cells (Fig. S6A). Furthermore, after MALAT1 level was knocked-down (Fig. S6B), an increased apoptotic cells numbers were observed in all three resistant cell lines (Fig. S6C). IC50 of RPMI8226/LR5 cells to melphalan decreased from 53.4 μM to 34.0 μM, RPMI8226/DOX40 cells to doxorubicin decreased from 2.46 μM to 1.48 μM and RPMI8226/V10R to bortezomib decreased from 193.8 nM to 143.9 nM, respectively (Fig. S7A–C). Those results indicated that anti-MALAT1 treatment resensitized resistant MM cells to their corresponding drugs again.
SWCNT-anti-MALAT1 oligo repressed MM proliferation and induced cell apoptosis in vivo
Our data indicated that MALAT1 antagonist was a robust tool to provoke DNA damage and apoptosis in MM. However, the efficient delivery of anti-MALAT1 oligos in vivo was the main obstacle that limits clinical application of this type of therapy. As a novel nanomaterial for drug delivery, SWCNT may deliver nucleic-acid drugs stablely and efficiently with good tolerability and minimal toxicity in vitro29 and in vivo30. To track the delivery visible, we conjugated SWCNT with Cy3-labeled-anti-MALAT1 oligos (SWCNT-anti-MALAT1-Cy3)(Fig. 7A)13, and then added it into culture medium of H929-GFP and MM.1S-GFP cells to validate delivery efficiency. As shown in Fig. 7B, SWCNT-anti-MALAT1-Cy3 was delivered into the nucleus of MM cells efficently and suppressed the endogenous MALAT1 level in both H929 and MM.1S cells significantly (Fig. 7C).
Fig. 7. SWCNT-anti-MALAT1 showed high delivery efficiency and minimal toxicity.

(A) Schematic diagram of SWCNT-anti-MALAT1-Cy3 gapmer oligos (Scale bars=100μM). (B) H929-GFP and MM.1S-GFP cells were co-cultured with SWCNT-anti-MALAT1-cy3 for 48h. (C) MALAT1 level was knocked-down successfully.(**p<0.01, ***p<0.001).
To further estimate the treatment potential of SWCNT-anti-MALAT1 in vivo, we subcutaneously injected MM.1S-Luc-GFP cells on the back of SCID mice to establish human MM xenograft murine model (Fig. 8A). At day 14 after tumor cell injection, SWCNT-anti-MALAT1 or SWCNT-ctrl oligos were injected directly into tumors and repeated at days 21, 24 and 28, respectively. We observed tumor burden with IVIS after luciferin injection, and found the luciferin signal was significant lower in SWCNT-anti-MALAT1 treated group compared with SWCNT-ctrl treated group. Then we measured MALAT1 level with RNA samples extracted from tumor xenografts and found that MALAT1 expression was significantly downregulated by SWCNT-anti-MALAT1 treatment, which indicated SWCNT delivered MALAT1 antisense oligo efficiently into MM cells in this human MM xenograft murine model. Western-blot results showed c-PARP1 increased in anti-MALAT1 treatment group. Immunohistochemistry results revealed decreased Ki-67 and increased c-caspase3 signals on SWCNT-anti-MALAT1 treated tumor sections.
Fig. 8. SWCNT-anti-MALAT1 treatment repressed myeloma growth in both xenograft and disseminated murine models.

(A) MM.1S-Luc-GFP cells were injected subcutaneously to SCID mice (5 mice each group). SWCNT-anti-MALAT1 or SWCNT-ctrl was injected into the tumors at the indicated days. Tumor growth was monitored by IVIS. Mice were sacrificed on day 30, tumor samples were subjected to qRT-PCR, WB and immunohistochemistry (Scale bars=100μM). (B) SCID mice (7 mice each group) were intravenously injected with MM.1S-Luc-GFP cells, then injected with SWCNT-anti-MALAT1 or SWCNT-ctrl once a week through tail veins. Kaplan-Meier analysis indicated SWCNT-anti-MALAT1 prolonged mouse lifespan significantly (P=0.04). (C) Proposed model of MALAT1 antagonist induces MM cell apoptosis.
MM is a systematic disease and malignant cells usually involve multiple organs in patient. To mimic this situation, we generated a disseminated MM murine model, and used SWCNT-anti-MALAT1 to test treatment effect on it (Fig. 8B). We firstly intravenously injected MM.1S-Luc-GFP cells into SCID mice through tail veins. At day 7 after tumor cell injection, SWCNT-anti-MALAT1 or SWCNT-ctrl oligos were injected through tail veins and repeated at days 14, 21, 28 and 35, respectively. We detected luciferin signal at day 35 and recorded their survival status, then we found SWCNT-anti-MALAT1 treatment not only reduced the tumor burden, but extended lifespan significantly (P=0.04) compare with SWCNT-ctrl treated group.
To further confirm the therapeutic effect of anti-MALAT1 in MM in vivo, we generated another murine xenograft model and disseminated model with H929-Luc-mCherry cells (Fig. S8). We found SWCNT-anti-MALAT1 inhibited H929 growth dramatically in both models. Meanwhile, SWCNT-anti-MALAT1 efficiently knocked-down MALAT1 expression, upregulated c-PARP1 and c-caspase3, and inhibited Ki-67 signal. In H929 disseminated model, SWCNT-anti-MALAT1 treatment extended mice lifespan significantly (P=0.009).
Discussion
This study is the first to elucidate the function of the lncRNA MALAT1 in MM. PARP1 and LIG3 are two key molecules required for the highly error-prone A-NHEJ31 DNA repair pathway. We demonstrated that MALAT1 is critical for PARP1/LIG3 complex to recognize DSBs γH2A.X loci on DNA, then activated A-NHEJ DNA repair in MM. LIG3 is upregulated in multiple myeloma32, chronic myeloid leukemia33, and breast cancer34. Strikingly, A-NHEJ is associated with frequent chromosome abnormalities such as deletions, translocations, inversions, and other complex rearrangements35. Thus over-expression of MALAT1 in MM may enhance A-NHEJ DNA repair pathway to induce secondary chromosome changes36, which may promote disease progression, but also induce drug resistance. Dissecting the mechanism of how MALAT1 directly or indirectly recruits LIG3 to γH2A.X loci, which represents DSBs, to favor A-NHEJ repair pathway will be our next focus by investigating the function of different domains on LIG3 and PARP1. Further gain-of-function studies of MALAT1 in normal or precursor cells and transgenic mice will be needed in the future to confirm our findings.
To evaluate MALAT1 as a possible therapeutic target, we pursued antisense inhibition and observed increased DNA damage and apoptosis in MM cells due to dissociation of the MALAT1/PARP1/LIG3 complex and deregulation of A-NHEJ pathway. We verified that knock-down MALAT1 in MM cells had synergistic effect with PARP1 inhibitor or bortezomib through inducing more cell apoptosis. Most of U.S. Food and Drug Administration (FDA)-approved PARP1 inhibitors are used to inhibit the catalytic activity of PARP1 and increase DNA damage in ovarian cancer with BRCA1/2 mutations, where the HR pathway is defected37–39. Unrepaired DNA damage will induce cell apoptosis. MALAT1 antagonist acts its role through PARP1 but the underlying mechanism is different from PARP1 inhibitors. In contrast, Anti-MALAT1 treatment disrupts MALAT1/PARP1/LIG3 DNA repair complex, then dissociated free PARP1 will induce polyADP-ribosylation in the nucleus, which will promote cell apoptosis directly. Anti-MALAT1 treatment will also impaired the A-NHEJ DNA repair pathway, which will further induce cell apoptosis due to unrepaired DSB DNA. When combined bortezomib and anti-MALAT1 therapy in MM, bortezomib repressed HR through reducing BRCA1/2 expression, meanwhile anti-MALAT1 inhibited A-NHEJ activity, thus apoptosis accumulated dramatically through synchronous dysfunction of two DSB repair pathways. These results provide new therapeutic strategy for MM patients.
To today, FDA has approved several antisense oligonucleotide drugs, including nusinersen for spinal muscular atrophy40, mipomersen for homozygous familial hypercholesterolemia41, fomivirsen for cytomegalovirus retinitis42, and eteplirsen for Duchenne muscular dystrophy43. We used gapmer DNA flanked by 2′-OMe-modified MALAT1 antisense oligonucleotides to achieve remarkable inhibition effects, and applied SWCNT as delivery system to improve affinity, stabilize the oligos and against nuclease degradation during delivery. To the best of our knowledge, this is the first report to use functionalized SWCNTs to deliver anti-sense oligos targeting lncRNAs in tumor. Due to their surface chemistry properties for delivery and large cargo capability, SWCNT represents a novel and useful nanomaterial for drug delivery, which not only stabilize nucleic acid molecule from digestion of nucleases, but increase penetration of DNA/RNA dramatically without toxicity29. In our study, SWCNT was functionalized covalently and then conjugated with anti-MALAT1, which allow anti-MALAT1 to be released with high concentration in MM cells and induced DNA damage and apoptosis effectively in both in vitro and in vivo experiments without bringing any toxicity in normal cells. Thus SWCNT-anti-MALAT1 is an ideal therapeutic method for the MM patients.
In conclusion, we have shown that MALAT1 exerted DNA protective and anti-apoptotic functions via binding to PARP1/LIG3 protein complexes, targeting MALAT1 induced DNA damage and apoptosis, therefore inhibited MM growth (Fig. 8C). Furthermore, we demonstrated that MALAT1 could be targeted via neutralization by antisense in vitro and in vivo, this treatment extended lifespan of MM-bearing mice significantly. Synergism of MALAT antisense with proteasome or PARP1 inhibitors further illustrated the potential therapeutic value of MALAT1 for MM patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank the Lerner Research Institute proteomic, genomic and imaging cores for their assistance and support. We thank Dr. Cassandra Talerico, a salaried employee of the Cleveland Clinic, for editorial assistance and helpful comments. Funding: This work was financially supported by NIH/NCI grant R00 CA172292 (to J.J.Z) and start-up funds(to J.J.Z) and the Clinical and Translational Science Collaborative(CTSC) of Case Western Reserve University Core Utilization Pilot Grant(to J.J.Z). The Orbitrap Elite instrument was purchased via an NIH shared instrument grant, 1S10RR031537-01. This work utilized the Leica SP8 confocal microscope that was purchased with funding from National Institutes of Health SIG grant 1S10OD019972-01.
Footnotes
Authorship:
Contribution: Y.H. and J.L. designed and performed experiments, analyzed data, and wrote the manuscript; H.F., J.F., C.L., W.C., and G.Z. performed experiments and analyzed data; O.S., M.J., S.L., R.F., and Q.Y. provided clinical samples. J.J.Z designed the project, performed experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors report no potential conflicts of interest.
References:Uncategorized References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013 Jan;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Ntziachristos P, Abdel-Wahab O, Aifantis I. Emerging concepts of epigenetic dysregulation in hematological malignancies. Nature immunology. 2016 Sep;17(9):1016–1024. doi: 10.1038/ni.3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans JR, Feng FY, Chinnaiyan AM. The bright side of dark matter: lncRNAs in cancer. The Journal of clinical investigation. 2016 Aug 01;126(8):2775–2782. doi: 10.1172/JCI84421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ronchetti D, Agnelli L, Taiana E, Galletti S, Manzoni M, Todoerti K, et al. Distinct lncRNA transcriptional fingerprints characterize progressive stages of multiple myeloma. Oncotarget. 2016 Mar 22;7(12):14814–14830. doi: 10.18632/oncotarget.7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong KY, Li Z, Zhang X, Leung GK, Chan GC, Chim CS. Epigenetic silencing of a long non-coding RNA KIAA0495 in multiple myeloma. Molecular cancer. 2015 Sep 26;14:175. doi: 10.1186/s12943-015-0444-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt LH, Spieker T, Koschmieder S, Schaffers S, Humberg J, Jungen D, et al. The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J Thorac Oncol. 2011 Dec;6(12):1984–1992. doi: 10.1097/JTO.0b013e3182307eac. [DOI] [PubMed] [Google Scholar]
- 7.Kato L, Begum NA, Burroughs AM, Doi T, Kawai J, Daub CO, et al. Nonimmunoglobulin target loci of activation-induced cytidine deaminase (AID) share unique features with immunoglobulin genes. Proceedings of the National Academy of Sciences of the United States of America. 2012 Feb 14;109(7):2479–2484. doi: 10.1073/pnas.1120791109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaudhry MA. Expression pattern of small nucleolar RNA host genes and long non-coding RNA in X-rays-treated lymphoblastoid cells. International journal of molecular sciences. 2013 Apr 25;14(5):9099–9110. doi: 10.3390/ijms14059099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011 Mar 24;471(7339):467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren S, Liu Y, Xu W, Sun Y, Lu J, Wang F, et al. Long noncoding RNA MALAT-1 is a new potential therapeutic target for castration resistant prostate cancer. The Journal of urology. 2013 Dec;190(6):2278–2287. doi: 10.1016/j.juro.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 11.West JA, Davis CP, Sunwoo H, Simon MD, Sadreyev RI, Wang PI, et al. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Molecular cell. 2014 Sep 04;55(5):791–802. doi: 10.1016/j.molcel.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003 Sep 11;22(39):8031–8041. doi: 10.1038/sj.onc.1206928. [DOI] [PubMed] [Google Scholar]
- 13.Luo JH, Ren B, Keryanov S, Tseng GC, Rao UN, Monga SP, et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology. 2006 Oct;44(4):1012–1024. doi: 10.1002/hep.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guffanti A, Iacono M, Pelucchi P, Kim N, Solda G, Croft LJ, et al. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC genomics. 2009 Apr 20;10:163. doi: 10.1186/1471-2164-10-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho SF, Chang YC, Chang CS, Lin SF, Liu YC, Hsiao HH, et al. MALAT1 long non-coding RNA is overexpressed in multiple myeloma and may serve as a marker to predict disease progression. BMC cancer. 2014 Nov 04;14:809. doi: 10.1186/1471-2407-14-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Handa H, Kuroda Y, Kimura K, Masuda Y, Hattori H, Alkebsi L, et al. Long non-coding RNA MALAT1 is an inducible stress response gene associated with extramedullary spread and poor prognosis of multiple myeloma. Br J Haematol. 2017 Nov;179(3):449–460. doi: 10.1111/bjh.14882. [DOI] [PubMed] [Google Scholar]
- 17.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008 Jun 27;4(6):e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999 Oct 15;13(20):2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhan F, Barlogie B, Arzoumanian V, Huang Y, Williams DR, Hollmig K, et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood. 2007 Feb 15;109(4):1692–1700. doi: 10.1182/blood-2006-07-037077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gutierrez NC, Sarasquete ME, Misiewicz-Krzeminska I, Delgado M, De Las Rivas J, Ticona FV, et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia. 2010 Mar;24(3):629–637. doi: 10.1038/leu.2009.274. [DOI] [PubMed] [Google Scholar]
- 21.Lopez-Corral L, Corchete LA, Sarasquete ME, Mateos MV, Garcia-Sanz R, Ferminan E, et al. Transcriptome analysis reveals molecular profiles associated with evolving steps of monoclonal gammopathies. Haematologica. 2014 Aug;99(8):1365–1372. doi: 10.3324/haematol.2013.087809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Handa H, Kuroda Y, Kimura K, Masuda Y, Hattori H, Alkebsi L, et al. Long non-coding RNA MALAT1 is an inducible stress response gene associated with extramedullary spread and poor prognosis of multiple myeloma. Br J Haematol. 2017 Aug 02; doi: 10.1111/bjh.14882. [DOI] [PubMed] [Google Scholar]
- 23.Huambachano O, Herrera F, Rancourt A, Satoh MS. Double-stranded DNA binding domain of poly (ADP-ribose) polymerase-1 and molecular insight into the regulation of its activity. The Journal of biological chemistry. 2011 Mar 04;286(9):7149–7160. doi: 10.1074/jbc.M110.175190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leppard JB, Dong Z, Mackey ZB, Tomkinson AE. Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair. Mol Cell Biol. 2003 Aug;23(16):5919–5927. doi: 10.1128/MCB.23.16.5919-5927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiruvella KK, Liang Z, Wilson TE. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol. 2013 May 01;5(5):a012757. doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lennox KA, Behlke MA. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic acids research. 2016 Jan 29;44(2):863–877. doi: 10.1093/nar/gkv1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simbulan-Rosenthal CM, Rosenthal DS, Iyer S, Boulares AH, Smulson ME. Transient poly(ADP-ribosyl)ation of nuclear proteins and role of poly(ADP-ribose) polymerase in the early stages of apoptosis. J Biol Chem. 1998 May 29;273(22):13703–13712. doi: 10.1074/jbc.273.22.13703. [DOI] [PubMed] [Google Scholar]
- 28.Neri P, Ren L, Gratton K, Stebner E, Johnson J, Klimowicz A, et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood. 2011 Dec 08;118(24):6368–6379. doi: 10.1182/blood-2011-06-363911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang X, Wang G, Liu R, Wang Y, Wang Y, Qiu X, et al. RNase non-sensitive and endocytosis independent siRNA delivery system: delivery of siRNA into tumor cells and high efficiency induction of apoptosis. Nanoscale. 2013 Aug 21;5(16):7256–7264. doi: 10.1039/c3nr01183f. [DOI] [PubMed] [Google Scholar]
- 30.Murakami T, Sawada H, Tamura G, Yudasaka M, Iijima S, Tsuchida K. Water-dispersed single-wall carbon nanohorns as drug carriers for local cancer chemotherapy. Nanomedicine (Lond) 2008 Aug;3(4):453–463. doi: 10.2217/17435889.3.4.453. [DOI] [PubMed] [Google Scholar]
- 31.Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell death & disease. 2015 Mar 19;6:e1697. doi: 10.1038/cddis.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herrero AB, San Miguel J, Gutierrez NC. Deregulation of DNA double-strand break repair in multiple myeloma: implications for genome stability. PLoS One. 2015;10(3):e0121581. doi: 10.1371/journal.pone.0121581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sallmyr A, Tomkinson AE, Rassool FV. Up-regulation of WRN and DNA ligase IIIalpha in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood. 2008 Aug 15;112(4):1413–1423. doi: 10.1182/blood-2007-07-104257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tobin LA, Robert C, Nagaria P, Chumsri S, Twaddell W, Ioffe OB, et al. Targeting abnormal DNA repair in therapy-resistant breast cancers. Molecular cancer research : MCR. 2012 Jan;10(1):96–107. doi: 10.1158/1541-7786.MCR-11-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muvarak N, Kelley S, Robert C, Baer MR, Perrotti D, Gambacorti-Passerini C, et al. c-MYC Generates Repair Errors via Increased Transcription of Alternative-NHEJ Factors, LIG3 and PARP1, in Tyrosine Kinase-Activated Leukemias. Molecular cancer research : MCR. 2015 Apr;13(4):699–712. doi: 10.1158/1541-7786.MCR-14-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soni A, Siemann M, Grabos M, Murmann T, Pantelias GE, Iliakis G. Requirement for Parp-1 and DNA ligases 1 or 3 but not of Xrcc1 in chromosomal translocation formation by backup end joining. Nucleic Acids Res. 2014 Jun;42(10):6380–6392. doi: 10.1093/nar/gku298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res. 2015 Oct 01;21(19):4257–4261. doi: 10.1158/1078-0432.CCR-15-0887. [DOI] [PubMed] [Google Scholar]
- 38.Balasubramaniam S, Beaver JA, Horton S, Fernandes LL, Tang S, Horne HN, et al. FDA Approval Summary: Rucaparib for the treatment of patients with deleterious BRCA mutation-associated advanced ovarian cancer. Clin Cancer Res. 2017 Jul 27; doi: 10.1158/1078-0432.CCR-17-1337. [DOI] [PubMed] [Google Scholar]
- 39.Scott LJ. Niraparib: First Global Approval. Drugs. 2017 Jun;77(9):1029–1034. doi: 10.1007/s40265-017-0752-y. [DOI] [PubMed] [Google Scholar]
- 40.Aartsma-Rus A. FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid Ther. 2017 Apr;27(2):67–69. doi: 10.1089/nat.2017.0665. [DOI] [PubMed] [Google Scholar]
- 41.Smith RJ, Hiatt WR. Two new drugs for homozygous familial hypercholesterolemia: managing benefits and risks in a rare disorder. JAMA Intern Med. 2013 Sep 09;173(16):1491–1492. doi: 10.1001/jamainternmed.2013.6624. [DOI] [PubMed] [Google Scholar]
- 42.Highleyman L. FDA approves fomivirsen, famciclovir, and Thalidomide. Food and Drug Administration. BETA. 1998 Oct 5; [PubMed] [Google Scholar]
- 43.Nelson SF, Miceli MC. FDA Approval of Eteplirsen for Muscular Dystrophy. JAMA. 2017 Apr 11;317(14):1480. doi: 10.1001/jama.2017.2601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.