

Abstract
Hydrogen sulfide (H2S) and nitric oxide (NO) are now recognized as important regulators in the cardiovascular system, although they were historically considered as toxic gases. As gaseous transmitters, H2S and NO share a wide range of physical properties and physiological functions: they penetrate into the membrane freely; they are endogenously produced by special enzymes, they stimulate endothelial cell angiogenesis, they regulate vascular tone, they protect against heart injury, and they regulate target protein activity via posttranslational modification. Growing evidence has determined that these two gases are not independent regulators but have substantial overlapping pathophysiological functions and signaling transduction pathways. H2S and NO not only affect each other's biosynthesis but also produce novel species through chemical interaction. They play a regulatory role in the cardiovascular system involving similar signaling mechanisms or molecular targets. However, the natural precise mechanism of the interactions between H2S and NO remains unclear. In this review, we discuss the current understanding of individual and interactive regulatory functions of H2S and NO in biosynthesis, angiogenesis, vascular one, cardioprotection, and posttranslational modification, indicating the importance of their cross-talk in the cardiovascular system.
1. Introduction
Hydrogen sulfide (H2S) and nitric oxide (NO) are considered as toxic gases and environmental pollutants for many years. However, recent studies investigate that they play a key role in physiological activities in many organ systems. NO, as the first gaseous transmitter, can regulate vascular tone, heart function, endothelial cell angiogenesis, and so on [1, 2]. H2S is identified as the third gaseous transmitter due to its biological functions, alongside carbon monoxide (CO), the second transmitter [3].
There are many similar biological characteristics for H2S and NO. For example, they are produced by specific enzymes, they penetrate into the membrane freely, and they are sensitive to reactive oxygen species (ROS). Except these features, both molecules regulate many physiological functions through similar signal pathways in the cardiovascular system [4, 5]. Although the interactions between NO and H2S are previously considered independently, there is growing evidence of cross-talk between these two gaseous transmitters. In 2009, first experimental evidences reported that there was a cross-talk between NO and H2S [6]. Since then, many studies have shown that the biological regulations are dependent on not only NO but also H2S. These two molecules can change each other's activities and the interactions alter related proteins' functions [5–7]. The therapeutic potential of NO and H2S is very immense and explored through preclinical and clinical studies [8].
Due to the physiological importance of NO and H2S, this review discusses the protective effects of NO and H2S and the signaling mechanisms under their interactions in the cardiovascular system.
2. Physical Properties, Biosynthesis, and Reactivity of H2S
2.1. Physical Properties of H2S
H2S is a strong reduced colorless gas with an odor of rotten eggs. H2S is easily oxidized to yield some sulfur-containing substances. In aqueous solution, it is hydrolyzed to hydrogen sulfide ions (HS−) and sulfide ions (S2−), which are in dynamic equilibrium in the following sequential reactions:
| (1) |
More than one third of H2S is undissociated and the others existed as HS− and S2−. The application of H2S is most studied in bacteria [9]. Since the discovery of H2S generation from mammalian cells, many researchers focus on the biological functions of H2S in this emerging field. It is important to investigate the levels of H2S in blood and tissue for its physiological functions. There are several analytical methods to detect H2S concentration, such as colorimetry [10], fluorescent probes [11], liquid chromatography-mass spectrometry [12], spectrophotometric analysis [13], silver sulfide or polarographic sensor [14, 15], and headspace gas determination [16]. Different analysis technologies got the different H2S concentrations. It has been reported that the level of H2S in Wistar rats blood is ~10 μmol/L, which was detected by colorimetry method [17], while ~46 μmol/L H2S is in Sprague Dawley rat plasma, which was measured by colorimetry method [3]. The plasma level of H2S in human is 10–100 μmol/L, which was identified by ion chromatography method [18]. The enzymatic capacity method was used to determine that the physiological plasma level of H2S in the brain is 50–160 μmol/L [19]. However, other researchers got different results. Furne et al. used enzymatic capacity method and reported that the H2S concentrations in the brain and liver were ~15 nmol/L [20]. Ishigami et al. also showed that the H2S concentration was at a low level in the brain, which was detected by gas chromatography method [15]. A new liquid chromatography-mass spectrometry method was developed by Tan et al., and they found that there was ~0.4 μmol/mg protein H2S in rat cardiac ventricular myocytes and there was 1.5 μmol/mg protein H2S in mice heart [12]. The reason of this inconsistency is that not only H2S is easily oxidized but also the disadvantages of analysis technologies are often in question, such as complex preparation processes, low sensitivity and specificity, and time-consuming procedures. Striking different H2S concentrations may cause uncertainty for the exact mechanistic role of H2S in physiological and pathological processes. Therefore, it is very essential to develop a new method to detect H2S concentration in cells, blood, and tissue.
2.2. Biosynthesis and Reactivity of H2S
Endogenous H2S is generated in mammalian tissues through enzymatic and nonenzymatic pathways. Two pyridoxal-5′-phosphate- (PLP-) dependent enzymes, cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS), use L-cysteine or homocysteine as substrates to synthesize H2S [21, 22]. CSE and CBS are expressed in different specific tissues and both of them are needed to produce H2S. The expression of CBS is mainly expressed in neurons and astrocytes of the central nervous system (CNS) [19], while CSE is most located in the kidney and liver [23], the cardiovascular system (CVS), especially in cardiomyocytes [24], vascular smooth muscle cells [25], and endothelial cells [26, 27]. In addition, H2S is synthesized by 3-mercaptopyruvate sulfurtransferase (3-MST) with cysteine aminotransferase (CAT), which is a PLP-independent pathway for H2S formation [28]. The expression of 3-MST is found in the liver, heart, kidney, and brain [29]. CSE and CBS only express in cytosol, while 3-MST expresses not only in cytosol but also in mitochondria. The production of H2S from CBS is primary responsible in regulating the nervous system. The H2S generation from CSE protects against injuries in the cardiovascular system. CBS catalyzes homocysteine and L-cysteine to generate cystathionine and H2S [30]. L-cysteine is catalyzed by CSE into thiocysteine and pyruvate, and then thiocysteine is lysed to produce cysteine and H2S [31]. α-Ketoglutarate acid and L-cysteine can be catalyzed by CAT to synthesize 3-mercaptopyruvate. 3-Mercaptopyruvate is desulfurated by 3-MST to generate thiosulfate and then thiosulfate is reduced to produce H2S [32] (Figure 1). Some specific inhibitors are available to attenuate the activity of CSE and CBS to reduce the generation of H2S, such as D,L-propargylglycine (PAG), β-cyano-L-alanine (BCA), aminooxyacetate (AOAA), and hydroxylamine (HA) [33, 34]. PAG can only inhibit the activity of CSE, whereas BCA and AOAA can reduce both activities of CSE and CBS. When at a low concentration, HA is not only an inhibitor of CSE but also an attenuator of the activity of CBS [35–37].
Figure 1.
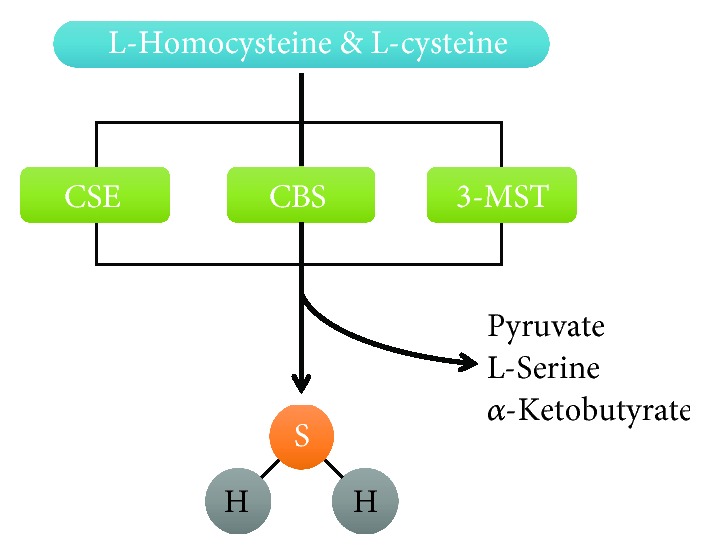
Biosynthesis of H2S. L-Homocysteine and L-cysteine are oxidized by three different enzymes, CSE, CBS, and 3-MST, to generate H2S.
2.2.1. Role of H2S in Angiogenesis
Ischemic heart disease (IHD) is the main cause of death in the world. There are some weak points in traditional therapeutic methods [38]. H2S can stimulate endothelial cell angiogenesis, which is a new potential therapeutic application for IHD. Cai et al. reported that H2S could dose-dependently increase the cell number, migration, and tube formation through the Akt pathway [39]. In line with this result, the microvessel formation was obviously inhibited in CSE knockout mice and H2S enhanced angiogenesis through the mitogen-activated protein kinase (MAPK) pathway [40]. As a molecular switch, H2S specifically broke cys1045-cys1024 disulfide bond in vascular endothelial growth factor receptor 2 (VEGFR2) and stimulated its conformation for angiogenesis [41]. Kan et al. also identified that H2S enhanced the activity of signal transducer and activator of transcription 3 (STAT3) through the VEGFR2 pathway [42]. H2S increased the mammalian target of rapamycin (mTOR) phosphorylation through VEGFR2 and then stimulated endothelial cell proliferation [43]. However, because of the proangiogenic effect of H2S, high concentrations of H2S in atherosclerotic plaques will be a potential risk for plaque vulnerability [44] (Figure 2).
Figure 2.
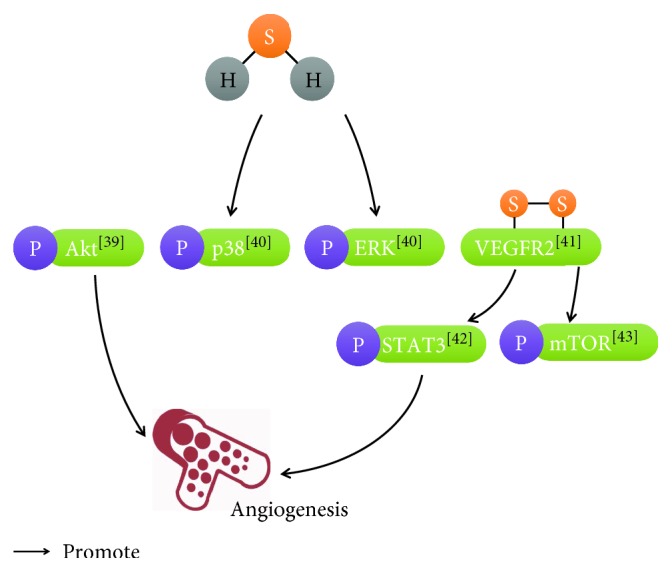
Regulatory role of H2S in endothelial cell angiogenesis. [39]: Cai et al.; [40]: Papapetropoulos et al.; [41]: Tao et al.; [42]: Kan et al.; [43]: Zhou et al.
2.2.2. Protein S-Sulfhydration by H2S
S-sulfhydration is one main posttranslational modification of proteins. Mustafa et al. found that glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was S-sulfhydrated at cys150 by H2S to increase catalytic activity [45]. H2S S-sulfhydrated Keap1 at cys151 and regulated Nrf2 activation to protect against cellular aging induced by oxidative stress [46]. H2S production from CSE S-sulfhydrated the p65 subunit of NF-κB at cys38 to mediate its antiapoptotic actions [47]. Cheung and Lau reported that S-sulfhydrated proteins were identified by proteomic approach in mice aorta, which would be a major step towards understanding the mechanistic role of H2S in atherosclerosis. H2S also induced S-sulfhydration of glutathione peroxidase 1 and further reduced lipid peroxidation and increased antioxidant defense in the aorta by prompting glutathione synthesis [48]. Endogenous H2S, produced by CSE, directly S-sulfhydrated Sirt1 that enhanced Sirt1 binding to zinc ion and then promoted its deacetylation activity and reduced atherosclerotic plaque formation [49]. ATP synthase, the mitochondrial inner membrane protein, regulated mitochondrial bioenergetics, and the α subunit (ATP5A1) of ATP synthase was S-sulfhydrated by H2S at cys244 and cys294 [50]. H2S regulates Krüppel-like factor 5 (KLF5) transcription activity via specificity S-sulfhydration at cys664 to prevent myocardial hypertrophy [51]. S-sulfhydration of specificity protein 1 (Sp1) by H2S at cys68 and cys755 plays an important role in maintaining vascular health and function [52]. H2S attenuated DNA damage in human endothelial cells and fibroblasts by S-sulfhydrating mitogen-activated protein kinase kinase 1 (MEK1) at cys341, which led to poly[ADP-ribose] polymerase 1 (PARP-1) activation [53]. H2S S-sulfhydrated p66Shc at cys59 residue and prevented H2O2-induced phosphorylation of p66Shc and then inhibited mitochondrial ROS production [54]. Protein tyrosine phosphatases (PTPs) regulate many signal transduction pathways. PTP1B, the one member of PTPs, was reversibly inactivated by H2S via S-sulfhydration at cys215 residue [55]. Moreover, H2S could S-sulfhydrate and inhibit protein phosphatase 2A (PP2A) to activate 5′ adenosine monophosphate-activated protein kinase (AMPK) in the heart, which resulted to the decrease of mitochondrial biogenesis [56] (Table 1).
Table 1.
Selected protein targets of H2S.
| Selected protein activity | Activity |
|---|---|
| GAPDH | Increased [45] |
| Keap1 | Decreased [46] |
| NF-κB | Increased [47] |
| Glutathione peroxidase 1 | Increased [48] |
| Sirt1 | Increased [49] |
| ATP5A1 | Increased [50] |
| KLF5 | Decreased [51] |
| Sp1 | Increased [52] |
| MEK1 | Increased [53] |
| p66Shc | Decreased [54] |
| PTP1B | Decreased [55] |
| PP2A | Decreased [56] |
2.2.3. Methods Used to Detect Protein S-Sulfhydration
Based on the critical role of S-sulfhydration, various methods are developed to detect the new posttranslational modification of proteins. Biotin-switch assay was firstly used to detect protein nitrosylation, and then this method was modified to determine levels of protein S-sulfhydration. In this assay, S-methyl methanethiosulfonate (MMTS) was used to block thiol group, and N-[6-(biotinamido)hexyl]-30-(20-pyridyldithio) propionamide reacted with the persulfide group. The biotinylated proteins were pulled down by streptavidin agarose beads and detected by Western blot. Using this method, Mustafa et al. reported that about 10–25% proteins were S-sulfhydrated under physiological condition in the liver [45]. However, Pan and Carroll found that MMTS could not block all free thiol groups and caused a false positive [57]. Protein S-sulfhydration was also identified by mass spectrometry (MS) assay. Briefly, target protein was immunoprecipitated and tryptic digested and then injected into the spectrometer [45]. Longen et al. improved MS assay and named it as qPerS-SID (quantitative Persulfide Site Identification). Firstly, trichloroacetic acid was used to dissolve cell completely and ensure cysteine modification stability. Secondly, both thiols and persulfides were labeled by the thiol-reactive reagent, Indoacetyl-PEG2-Biotin, and enriched by streptavidin agarose beads. And then samples were reduced by tris(2-carboxyethyl)phosphine (TCEP) to elute presulfides from the beads selectively before subjected to MS analysis [58].
It was reported that tag-switch assay could selectively detect protein S-sulfhydrated residues. Firstly, methylsulfonyl benzothiazole, −SH block agent, reacted with both –SH and –S-SH to form –S-BT and –S-S-BT. Secondly, –S-S-BT could respond to a biotin-linked cyanoacetate (CN-biotin) to form stable thioether linkages. In the contrast, −S-BT could not be sensitive to CN-biotin. Biotinylated protein could be pulled down by streptavidin agarose beads and then detected by Western blot or MS [59].
Dóka et al. developed the protein persulfide detection protocol (ProPerDP), which could detect S-sulfhydrated protein easily and reliably. In this protocol, both –SH and –S-SH were alkylated by the biotin-labeled alkylating agent, EZ-Link Iodoacetyl-PEG2-Biotin. And then biotinylated proteins were pulled down by streptavidin magnetic beads. Dithiothreitol or TCEP was used to cleave the persulfidated proteins off the beads. Either Western blot or MS was used to detect protein S-sulfhydration, which was dependent on the composition and concentration of samples [60].
2.2.4. Role of H2S in Maintenance of Vascular Tone
Studies have shown that H2S is a vasorelaxant mediator. The plasma level of H2S in hypertension rats was lower than that in normal rats. After injection of H2S, the blood pressure was obviously reduced [61]. H2S could cause a concentration-dependent relaxation from preconstricted aortic rings. The cyclic guanosine monophosphate (cGMP) level was increased by H2S or overexpression of CSE in vasorelaxant process [62]. H2S also directly opened the ATP-sensitive K+ channel (KATP channel) for physiological relaxation [25]. H2S induced intracellular acidification via activation of Cl−/HCO3− exchanger, which is partially responsible for H2S-mediated vasorelaxation [63]. Metabolic inhibition is also required for the vasorelaxant effects of H2S [64]. Nox4 is a positive transcriptional regulator of CSE in endothelial cells and propose that it may in turn contribute to the regulation of vascular tone via the modulation of H2S production [65]. H2S at low concentrations opened potassuim channels after smooth muscle calcium activated. It also may involve another mechanism, for example, mitochondrial complex I and III led to uncoupling of force, and promoted vasodilation [66].
2.2.5. Role of H2S in Heart Protection
What is more, H2S plays an important role to protect against heart failure. The growing evidence indicated that H2S levels were decreased in the heart failure of mice [67]. ROS accumulation is a major factor to lead to heart failure. Wu et al. reported that Sirt1 was regulated by H2S to reduce ROS for cardiovascular protection [68]. H2S also can increase Trx1 to protect against ischemic-induced heart failure [69]. Renin release was inhibited by H2S to prevent heart failure [70]. H2S may stimulate angiogenesis to regulate cardiac remodeling [71]. H2S is a critical regulator of cardiac mitochondrial content and it can promote mitochondrial biogenesis through an AMPK peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) signaling pathway [56]. H2S reduced recruitment of CD11b+Gr-1 cells in mice and has helpful effects on cardiac remolding after myocardial infarction (MI) [72]. H2S increased proteasomal activity and function n an Nrf2-dependent manner during the development of heart failure [73]. GYY4137, H2S donor, prevented cardiac dysfunction and adverse remodeling through promoting early postischemic endogenous natriuretic peptide activity [74]. H2S suppressed endoplasmic reticulum stress stemming from high-fat diet-induced cardiac dysfunction [75] (Figure 3).
Figure 3.

Cardioprotective role of H2S. [68]: Wu et al.; [69]: Nicholson et al.; [70]: Liu et al.; [56]: Shimizu et al.; [73]: Shimizu et al.; [74]: Lilyanna et al.; [75]: Barr et al.
3. Physical Properties, Biosynthesis, and Reactivity of NO
3.1. Physical Properties of NO
Due to the importance of NO in physiological regulation, it was selected as the gaseous transmitter molecule of the year 1992 and the Nobel Prize in Physiology or Medicine was awarded in 1998. NO is a simple molecule with oxygen and nitrogen, which is uncharged. Based on this characteristic, NO penetrates into the membrane freely, which is independent on membrane receptors. NO is also a radical molecule with an unpaired electron. Therefore, NO has a short half-life from 2–30 seconds and it is very reactive to deliver the signal [76].
Although it is difficult to exactly detect NO due to its lability, there are several methods developed to overcome this defect. NO is electrochemical reactive, and electrochemical detection is developed to measure its concentration from cultured cells and rat hearts [77, 78]. Oxyhemoglobin (HbO2) can react with NO to yield methemoglobin (metHb) and nitrate (NO3−), and metHb is measured by spectrophotometric analysis [79]. Nitrogen dioxide (NO2) is generated from the reaction of NO with ozone (O3), and the light from NO2 can be detected, because the excited state of NO2 returns to the ground state. NO concentration can also be detected by chemiluminescent methods [80]. Based on the green fluorescent protein, a biosensor is used to measure the level of NO in cells [81].
3.2. Biosynthesis and Reactivity of NO
NO is enzymatically synthesized by the NO synthase (NOS) family of proteins [82]. There are three distinct isoforms of NOS proteins, neuronal NOS (nNOS) (encoded by NOS1), endothelial inducible NOS (iNOS) (encoded by NOS2), and NOS (eNOS) (encoded by NOS3). These three NOSs are oxidoreductase homodimer enzymes including two domains, an amino-terminal oxygenase domain and a reductase domain. The amino-terminal domain contains three binding sites for a ferric haem cluster, cofactor tetrahydrobiopterin (BH4) and substrate L-arginine. The reductase domain includes three binding sites for flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), and the electron donor nicotinamide adenine dinucleotide phosphate (NADPH). These two domains are linked by a sequence that binds calcium-complexed calmodulin [83]. Upon NOS activation, FAD and FMS transfer electrons from NADPH to heme. Reductase domains of monomers bind calmodulin and then boost the transfer of electrons. The electrons promote binding of O2 to the ferrous through reduced haem iron. L-Arginine binds the ferrous form to generate L-citrulline and NO [84–86] (Figure 4).
Figure 4.
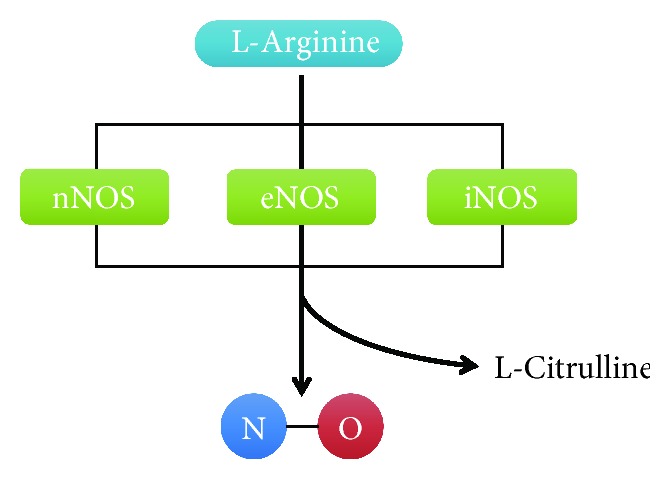
Biosynthesis of NO. L-Arginine is catalyzed by three different enzymes, nNOS, eNOS, and iNOS, to produce NO.
nNOS is the first NOS to be cloned and is mainly expressed in the sarcoplasmic reticulum (SR) of cardiac myocytes [87], in autonomic cardiac neurons and ganglia [88], and within vascular smooth muscle cells (VSMCs) [89, 90]. eNOS is highly expressed not only in the endothelial cells but also in cardiac myocytes [91, 92] and platelets [93]. iNOS can be found in a lot of cell types, such as leukocytes, endothelial cells, VSMCs, cardiac myocytes, nerve cells, and fibroblasts [94, 95]. Increased intracellular Ca2+ levels could promote eNOS and nNOS to produce NO. Unlike eNOS and nNOS, NO generation from iNOS is calcium independent. Inflammatory stimuli easily induce the expression of iNOS, such as cytokine (TNF-α or IFN-γ), bacterial proteins/peptides, or lipopolysaccharide (LPS). The dimeric enzyme catalytic activity of iNOS is much higher than those of nNOS and eNOS, when NOSs are assembled. iNOS maintains larger quantity of NO until exhaustion of substrate and cofactors or enzyme degradation; therefore, elevated iNOS expression is mostly associated with pathological stress [2, 84, 96, 97]. In addition, NO3− and nitrite (NO2−), which exist in diet, generate NO through the nonenzymatical pathway. Therefore, NO can be supplied through daily diet [98]. S-Nitroso-L-glutathione (GSNO) is another form of NO storage. GSNO releases NO through catalyzing enzymes, such as GSH peroxidase and thioredoxin reductase [99, 100].
3.2.1. Role of NO in Angiogenesis
NO has an important role for angiogenesis. Vascular endothelial growth factor (VEGF) upregulated eNOS expression and increased NO release to stimulate angiogenesis [101]. VEGF-induced cell proliferation was attenuated by NG-nitro-L-arginine methyl ester (L-NAME) [102, 103]. X-Box binding protein 1 (XBP1) stimulated endothelial cell migration via regulating eNOS expression [104]. Cavin-2 promoted the generation of NO in endothelial cells by controlling activity of eNOS and then stimulated endothelial cell angiogenesis [105]. C-reactive protein (CRP) quenched the production of NO through posttranscriptional effect on eNOS mRNA stability and then inhibited angiogenesis [106]. Phosphorylation of eNOS and NO production were mediated by Akt and then regulated angiogenesis [107] (Figure 5).
Figure 5.
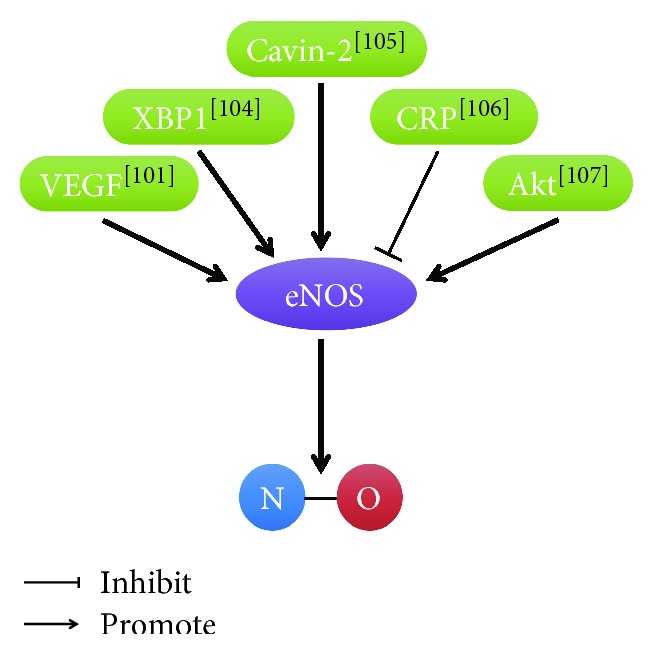
Regulatory role of NO in endothelial cell angiogenesis. [101]: Fukumura et al.; [104]: Yang et al.; [105]: Boopathy et al.; [106]: Verma et al.; [107]: Dimmeler et al.
3.2.2. Proteins S-Nitrosylation by NO
S-nitrosylation is one main way of NO to mediate protein activity. NO can S-nitrosylate G protein-coupled receptor kinases (GRKs) to suppress their activity and block phosphorylation [108]. p65 subunit of NF-κB is S-nitrosylated at cys38 to protect against inflammation [109]. S-nitrosylated arginase1 contributed to endothelial dysfunction in the aging cardiovascular system [110]. NO S-nitrosylated N-ethylmaleimide-sensitive factor (NSF) and then inhibited exocytosis of Weibel-Palade bodies [111]. Using transgenic mice to titrate the levels of S-nitrosylation protein, Irie et al. uncovered major roles for protein S-nitrosylation generally and for phospholamban (PLN) and cardiac troponin C (cTnC) S-nitrosylation in particular, in β-AR-dependent regulation of Ca2+ homeostasis [112]. Dynamic S-nitrosylation/denitrosylation of β-arrestin 2 regulated stimulus-induced GPCR trafficking [113]. nNOS deficiency impaired ryanodine receptor (RyR) S-nitrosylation and led to altered Ca2+ homeostasis [114]. S-nitrosylation of native transient receptor potential channel 5 (TrpC5) at cys553 and nearby cys558 upon G protein-coupled ATP receptor stimulation elicited entry of Ca2+ into endothelial cells [115]. S-nitrosylation at the cys215 residue of PTP1B protected against H2O2-induced irreversible oxidation [116]. S-nitrosylation of GAPDH triggered binding to Siah1, an E3 ubiquitin ligase, nuclear translocation, and cell apoptosis [117] (Table 2).
Table 2.
Selected protein targets of NO.
3.2.3. Role of NO in Maintenance of Vascular Tone
The primary function of NO is identified as endothelial-derived relaxation factor (EDRF) [118, 119]. Endothelial cells can produce small quantities of NO to stimulate vascular smooth muscle relaxation. Due to short half-life, vasoconstriction does not happen unless persistent NO has been generated. NOS inhibitors are used to investigate the physiological roles of NO in biological systems, such as L-N-mono-methyl-arginine (L-NMMA) and L-NAME. When inhibitors are added, NO is attenuated and blood pressure increases. Increase of blood flow enhances NO production and causes vessel relaxation. So, NO also is an endogenous mediator of blood flow [2, 120]. NO regulated adrenomedullin-induced vasorelaxation via the cGMP pathway [121]. NO stimulated Ca2+-dependent K+ channel activity and opened Ca2+-dependent K+ channels to induce relaxation of vascular smooth muscle cells [122]. NO deficit increased vascular tone in the contribution of Cav3.1 and Cav3.2 T-type calcium channels through regulating the bioavailability of ROS produced by NADPH oxidase [123].
3.2.4. Role of NO in Heart Protection
Congestive heart failure results in cardiovascular dysfunction and diminishes vascular NO production. Targeted overexpression of the eNOS gene within the vascular endothelium in mice attenuated both cardiac and pulmonary dysfunction and dramatically improves survival during severe congestive heart failure [124]. During ischemia/reperfusion, the more serious heart functions were found in eNOS-deficient mice compared with wild-type mice [125]. NO is an important modulator of left ventricular (LV) remodeling after myocardial infarction (MI). Cardiomyocyte-restricted overexpression of eNOS limited LV dysfunction and remodeling after MI [126]. NO stimulated PKG activity and opened the KATP channel to induce ROS generation in cardiomyocytes [127]. Nitrite increased NO levels and prevented the progression of hypertrophy and heart failure via cGMP/GS3Kβ signaling [128]. The coronary arteries from the heart failure rats exhibited reduced NO bioavailability, whereas the MI rats exhibited increased NO bioavailability because of the increased eNOS/nNOS/PI3K/Akt pathway and a reduction in ROS generation [129] (Figure 6).
Figure 6.
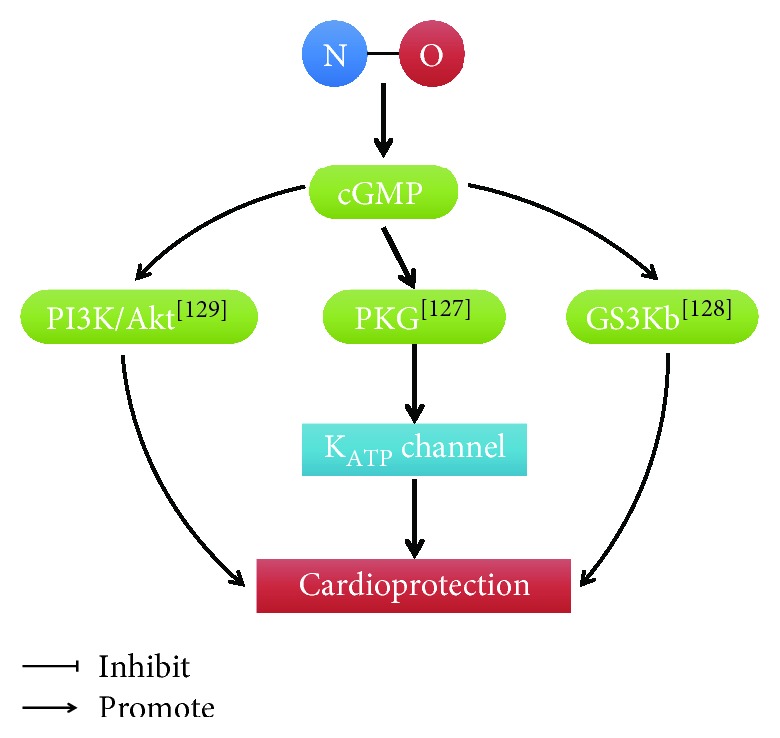
Cardioprotective role of NO. [127]: Xu et al.; [128]: Bhushan et al.; [129]: Couto et al.
4. Cross-Talk between H2S and NO
In the past several years, attention has been given to gas cross-talk. Growing evidence has shown that these two gasotransmitters interact with each other's biosynthesis and physiological response in many ways. However, there is still no clarity about the nature of the interaction [7, 130]. H2S and NO can affect not only the generation of each other through enzymatic expression and activity but also the further downstream signaling pathway [131–133]. The next part will provide current understanding of the interactions and mechanisms between H2S and NO in the cardiovascular system.
4.1. Biosynthesis of H2S and NO Interaction
The activity of CBS was suppressed by NO through binding to the enzyme. A five-coordinate ferrous nitrosyl species was formed and ligands are lost [134]. H2S directly inhibited the activity of recombinant eNOS to cause the increase of aortic contractility. However, the natural mechanism is not clear [132]. In the model of myocarditis, there were high iNOS mRNA and protein expression. H2S therapy inhibited iNOS overexpression to limit inflammatory cell infiltration, suppress cardiac edema, and attenuate myocardial lesions [135]. These two gasotransmitters inhibit each other's production; however, it has been shown that NO and H2S promote their respective synthesis. NO obviously increased CSE expression and H2S generation from vascular tissues. The dose-dependent relaxation curve of H2S was shifted to the right by L-NAME [25]. Likely, H2S enhanced iNOS expression and NO production through the IL-1β-induced NF-κB signaling pathway [136]. In endothelial cells, H2S increased NO generation twofold from eNOS and eNOS was activated at Ser 1177 through the Akt pathway [137]. It was also found that H2S obviously increased calcium concentrations and activated eNOS at phosphoserine residue 1179. A calcium chelator abolished H2S-induced NO synthesis in endothelial cells. So, eNOS activation and NO generation were regulated by H2S through calcium release [138]. Altaany et al. reported that although H2S stimulated eNOS activation, the expression of eNOS is slightly affected by H2S in endothelial cells. NO production was inhibited by CSE knockdown, whereas CSE overexpression enhanced NO generation [139]. H2S concentrations were decreased in heart failure. H2S therapy regulated eNOS and increased NO production to protect against heart failure [67]. In isoproterenol-induced myocardial injury, H2S played a cardioprotective role but this effect was abrogated when NOS was inhibited [140]. In CSE KO mice, H2S reduction caused eNOS dysfunction, limitation of NO production, and elevated oxidative stress. When using exogenous H2S therapy, eNOS was activated and NO levels were increased and oxidative stress was obviously inhibited. However, H2S did not diminish oxidative stress injury in eNOS phosphomutant mice. Therefore, H2S-mediated cytoprotection was closely correlated to eNOS activation and NO generation [141] (Figure 7).
Figure 7.

The interactions between H2S and NO on biosynthesis and chemical reaction. [25]: Zhao et al.; [67]: Kondo et al.; [132]: Kubo et al.; [135]: Hua et al.; [136]: Jeong et al.; [137]: Predmore et al.; [138]: Kida et al.; [139]: Altaany et al.; [140]: Sojitra et al.; [141]: King et al.; [145]: Eberhardt et al.; [146]: Whiteman et al.; [147]: Yong et al.; [148]: Ali et al.; [149]: Yong et al.; [150]: Filipovic et al.; [151]: Ondrias et al.; [152]: Cortese-Krott et al.; [158]: Wang et al.; [165]: Geng et al.
4.2. Posttranslational Modifications of H2S and NO Interaction
S-sulfhydration and S-nitrosylation are important mechanisms for H2S and NO, respectively, to modify target protein. H2S could enhance eNOS activity by S-sulfhydration. There are both monomeric eNOS and dimeric eNOS in cells, but NO is produced only by eNOS dimers. H2S promoted eNOS dimer formation to increase NO generation. Cys443 in eNOS was not only a S-sulfhydration site but also a S-nitrosylation site. S-sulfhydration of eNOS was not affected by NO, whereas H2S inhibited the S-nitrosylation of eNOS [142]. In chronic tissue ischemia, H2S increased NO generation through eNOS activity and nitrite reduction mechanism [143]. Cys215 in PTP1B was also both S-nitrosylated and s-sulfhydrated. S-nitrosylation of PTP1B could increase its activity, but s-sulfhydration of PTP1B could prevent its activity [55, 116]. Similarly, GAPDH was also modified by both S-nitrosylation and s-sulfhydration at cys150. NO inhibited the activity of GAPDH, while H2S increased the activity of GAPDH [45, 117]. Intriguingly, the p65 subunit of NF-κB at cys38 was both S-nitrosylated and s-sulfhydrated, and either NO or H2S could increase NF-κB activity [47, 109]. NaHS treatment on reperfusion increases S-nitrosylation to a level comparable to that with S-nitroso-N-acetylpenicillamine (SNAP) treatment. In addition, there was an additive increase in S-nitrosylation but not S-sulfhydration when SNAP and NaHS were added together at reperfusion. Thus, part of the benefit of sodium hydrosulfide (NaHS) is an increase in S-nitrosylation and the magnitude of the protective effect is related to the magnitude of the increase in S-nitrosylation [144]. Taken together, S-sulfhydration and S-nitrosylation at the same cys residues of target protein dynamics balance and compete with each other to maintain normal function of the protein (Figure 8).
Figure 8.

The interactions between H2S and NO on the same protein targets. [45]: Mustafa et al.; [47]: Sen et al.; [55]: Krishnan et al.; [109]: Kelleher et al.; [117]: Hara et al.; [142]: Altaany et al.; [143]: Bir et al.
4.3. Biochemistry of H2S and NO Interaction
Although above evidences indicate that these two gases affect each other's synthesis, other researchers suggest that H2S and NO chemical interact to from a novel molecule. Nitroxyl (HNO) is a sibling of NO that releases calcitonin gene-related peptide (CGRP) to play a cardioprotective role. Eberhardt et al. showed that H2S interacted with NO to form HNO. NO and H2S converged at transient receptor potential channel A1 (TRPA1). TRPA1 was activated by HNO through the formation of disulfide bonds and CGRP was released to regulate vascular tone [145]. It also has been shown that a novel nitrosothiol was produced by a direct interaction between NO and H2S, which was detected by a combination of analysis methods. This result was also identified in the LPS-treated liver. The nitrosothiol did not increase cGMP concentration without Cu2+ in RAW264.7 cells [146]. H2S and NO play a key role to regulate heart function. The extent of myocyte contraction was reduced by NO, while H2S also had small effect to stimulate heart contractility. However, H2S + NO did not play an inotropic role in the presence of thiols. This result suggested that H2S may interact with NO to generate a new molecule, which was very sensitive to thiols. This new thiol-sensitive molecule has not been identified, but it exerts significant regulatory role in the heart. Further work needs to be done to explain this question and offer new therapeutic application [147]. Ali et al. reported that H2S and NO might interact together to produce an unidentified nitrosothiol, which inhibited vasorelaxant potential of NO not only in vitro but also in vivo. The authors also proposed that, although H2S had vasorelaxant activity, the crucial function of H2S was to regulate local levels of NO [148]. A thiol-sensitive molecule was formed by the chemical interaction between H2S and NO, which had positive inotropic and lusitropic effects [149]. S-Nitrosothiols could interact with H2S to produce thionitrous acid (HSNO), which provided NO+, NO, and NO− to play a physiological role [150]. Ondrias et al. reported that nitrosothiols reacted with H2S to release NO to exert biological functions [151]. When nitrosothiol interacted with sulfide in excess, SSNO− was obtained, which was very stable at physiological pH and produced polysulfides and NO [152] (Figure 7).
4.4. Role of the Interaction of H2S and NO in Angiogenesis
Accumulating evidences report that H2S and NO play an important role in endothelial cell angiogenesis. However, the mechanisms of interaction between H2S and NO are still exclusive. We found that H2S and NO converged at the same downstream molecular target, Sirt1. Sirt1 activation increased the VEGF level and cGMP concentration. Evoked by the increase in cGMP levels, cGMP/PKG and downstream molecules, including p38 and ERK, were activated to participate in the regulation of angiogenesis [153]. Coletta et al. also reported that both NO and H2S could increase intracellular cGMP. H2S decreased cGMP degradation by preventing phosphodiesterase type 5 (PDE5), while NO activated sGC to produce cGMP [154]. As mentioned previously, H2S stimulated angiogenesis via Akt phosphorylation. eNOS activity was induced by the increase of Akt phosphorylation [107]. It predicted that H2S activated Akt and increases eNOS phosphorylation at its activating site Ser1177 [154] (Figure 9).
Figure 9.
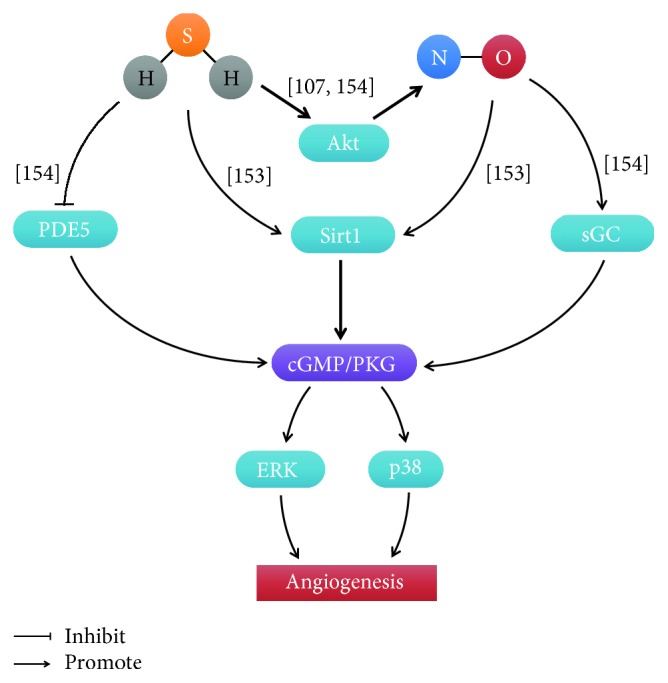
The interactions between H2S and NO in endothelial cell angiogenesis. [107]: Dimmeler et al.; [153]: Hu et al.; [154]: Coletta et al.
4.5. Role of the Interaction between H2S and NO in Vascular Tone
Several studies have shown that the interactions between H2S and NO maintain vascular tone. Hosoki et al. firstly found that H2S induced much stronger vascular relaxation in the presence of NO [155]. The vascular denervation did not affect H2S-induced vasorelaxation; however, the vasorelaxant effect of H2S was inhibited in the absence of endothelium. When the nitric oxide synthase was blocked, H2S-induced vasorelaxation was attenuated [25]. We also found that H2S and NO generation from ZYZ-803, a novel H2S- and NO-conjugated donor, cooperatively regulated vascular tone through the cGMP pathway. Either blocking CSE and/or eNOS activity, or uncontaining endothelium could prevent ZYZ-803-induced vasorelaxation [156]. In line with these results, Coletta et al. reported that H2S and NO are mutually dependent to regulate endothelium-dependent vasorelaxation [154]. The data above showed that H2S have a vasoregulatory role in a NO-dependent manner. However, the regulatory role of H2S and NO in vasorelaxation was differently found by other labs. 1H-oxadiazolo-quinoxalin-1-one (ODQ) and NS-2028 were cGMP inhibitors, which suppressed SNP-induced relaxation. But ODQ and NS-2028 could promote H2S-induced vasorelaxation. Moreover, the vasorelaxant potent of SNP was reduced when aortic tissues were pretreated with H2S [157]. Similarly, Whiteman et al. [146] and Ali et al. [148] found that the vasorelaxant effect of NO was inhibited by H2S. Wang et al. reported that PAG inhibited the relaxant effect of NO [158].
4.6. Role of the Interaction of H2S and NO in Heart Protection
Both H2S and NO have a cardioprotective role in the heart. H2S protected against ischemic injury via increasing NO release and adding L-NAME attenuated the cardioprotective of H2S [159]. Sojitra et al. also found that H2S alleviated isoproterenol-induced cardiomyopathy through elevating myocardial and serum NO levels and inhibition of NOS activity abrogated the cardioprotective role of H2S [140]. H2S postconditioning conferred the protective effects against ischemia-reperfusion injury through the activation of eNOS pathways [160]. Similarly, CSE KO mice exhibited reduced levels of NO and reduced NO synthesis via eNOS, which increased oxidative stress and an exacerbated response to myocardial ischemia/reperfusion injury [141]. We also found that H2S and NO cooperatively attenuated left ventricular remodeling and dysfunction during the development of heart failure through the VEGF/cGMP pathway [161]. A novel H2S donor, SG-1002, prevented the transition from compensated to decompensated heart failure in part via upregulation of eNOS and increased nitric oxide bioavailability [67]. Bibli et al. reported that H2S preserved eNOS activity via inhibiting proline-rich tyrosine kinase 2 (PYK2) in H9c2 cells under oxidative stress [162]. Sodium nitrite (NaNO2) significantly improves LV function in ischemia-induced chronic heart failure via increasing H2S bioavailability, Nrf2 activation, and antioxidant defenses [163]. GYY4137, a slow-releasing H2S donor, protected the heart against lethal reperfusion injury through activation of the PI3K/Akt pathway, with partial dependency on NO [164]. However, the other labs reported some conflicting results. Kubo et al. showed that the activity of eNOS was inhibited by H2S in rat and mouse aortic rings [132]. In addition, Geng et al. found that both exogenous and endogenous H2S reduced NO generation and prevented eNOS activity and transcription [165]. This is possible that differences of H2S concentration and experimental model cause the conflicting result and the interaction between H2S and NO in heart protection is still needed to study (Figure 10).
Figure 10.
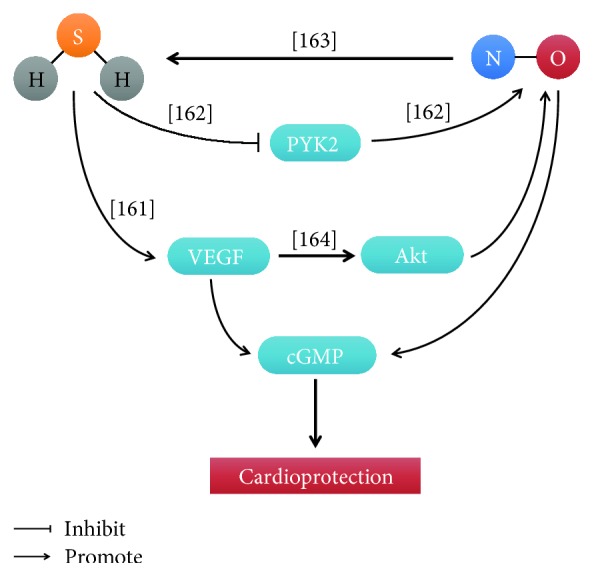
The interactions between H2S and NO in cardioprotection. [161]: Wu et al.; [162]: Bibli et al.; [163]: Donnarumma et al.; [164]: Karwi et al.
5. Conclusion
In this review, we have summarized the biological functions of H2S and NO and described the interactions between these two gases in the cardiovascular system. As gasotransmitters, there are some similar functions between H2S and NO. They do not only have similar biological reactivity but also have similar biological effects. H2S and NO interact with each other's synthesizing enzymes and affect their generation. Moreover, H2S and NO directly produce a new unidentified compound by chemical interaction. Both of them are endothelial-derived relaxation factors to regulate vascular tone. They also stimulate endothelia cell angiogenesis and protect against heart injury. In addition, H2S and NO regulate target protein activity through S-sulfhydration and S-nitrosylation at special cysteine residue to exert biological effects.
It is beyond debate that either H2S or NO plays a critical role in regulation of the mammalian cardiovascular system. Although accumulating evidence has suggested that H2S and NO interact with each other in the cardiovascular system, the natural precise mechanism of the interactions remains unclear. Both H2S and NO work on each other's physiological generation and response in the cardiovascular system; however, these conclusions sometimes appear inconsistent. This may be caused by different gasotransmitter donors and levels, different experimental models and parameters, and so on. Some groups have found that H2S and NO chemically produced a novel compound, like HNO. But another group also showed that H2S and NO interaction generated other chemical species. This will be needed to develop novel methods or instruments to measure these unknown species. Moreover, H2S and NO can modify the same target protein even at the same cysteine residue, but the competitive mechanism is still covered and how the posttranslational modifications affect target protein activity. Although there is plenty of information pointing towards a physiological regulation of H2S and NO, much work needs to be done to investigate the cross-talk between H2S and NO. A deeper understanding of the interactions will cause the development of novel therapeutic strategies for cardiovascular diseases.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (no. 81703499), Shanghai Chenguang Program (17CG13), Shanghai Sailing Program (no. 16YF1410400), Shanghai Municipal Commission of Health and Family Planning (no. ZHYY-ZXYJHZX-201618), and Beijing Medical Award Foundation (YJHYXKYJJ-152).
Abbreviations
- 3-MST:
3-Mercaptopyruvate sulfurtransferase
- AMPK:
5′ adenosine monophosphate-activated protein kinase
- AOAA:
Aminooxyacetate
- BCA:
β-Cyano-L-alanine
- BH4:
Tetrahydrobiopterin
- CAT:
Cysteine aminotransferase
- CBS:
Cystathionine β-synthase
- cGMP:
Cyclic guanosine monophosphate
- CGRP:
Calcitonin gene-related peptide
- CN:
Cyanoacetate
- CNS:
Central nervous system
- CO:
Carbon monoxide
- CRP:
C-reactive protein
- CSE:
Cystathionine γ-lyase
- cTnC:
Cardiac troponin C
- CVS:
Cardiovascular system
- EDRF:
Endothelial-derived relaxation factor
- eNOS:
Endothelial NOS
- FAD:
Flavin adenine dinucleotide
- FMN:
Flavin mononucleotide
- GAPDH:
Glyceraldehyde-3-phosphate dehydrogenase
- GRKs:
G protein-coupled receptor kinases
- GSNO:
S-Nitroso-L-glutathione
- H2S:
Hydrogen sulfide
- HA:
Hydroxylamine
- HbO2:
Oxyhemoglobin
- HNO:
Nitroxyl
- HS−:
Hydrogen sulfide ions
- IHD:
Ischemic heart disease
- iNOS:
Inducible NOS
- KATP channel:
ATP-sensitive K+ channel
- KLF5:
Krüppel-like factor 5
- L-NAME:
L-NG-Nitroarginine methyl ester
- L-NMMA:
L-N-Mono-methyl-arginine
- LPS:
Lipopolysaccharide
- LV:
Left ventricular
- MAPK:
Mitogen-activated protein kinase
- MEK1:
Mitogen-activated protein kinase kinase 1
- metHb:
Methemoglobin
- MI:
Myocardial infarction
- MMTS:
S-Methyl methanethiosulfonate
- MS:
Mass spectrometry
- mTOR:
Mammalian target of rapamycin
- NADPH:
Nicotinamide adenine dinucleotide phosphate
- NaHS:
Sodium hydrosulfide
- NaNO2:
Sodium nitrite
- nNOS:
Neuronal NOS
- NO:
Nitric oxide
- NO2:
Nitrogen dioxide
- NO2−:
Nitrite
- NO3−:
Nitrate
- NOS:
NO synthase
- NSF:
N-Ethylmaleimide-sensitive factor
- ODQ:
1H-Oxadiazolo-quinoxalin-1-one
- O3:
Ozone
- PAG:
D,L-Propargylglycine
- PARP-1:
Poly[ADP-ribose] polymerase 1
- PDE5:
Phosphodiesterase type 5
- PGC-1α:
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PLN:
Phospholamban
- PP2A:
Protein phosphatase 2A
- PTPs:
Protein tyrosine phosphatases
- PYK2:
Proline-rich tyrosine kinase 2
- ROS:
Reactive oxygen species
- RyRs:
Ryanodine receptors
- S2−:
Sulfide ions
- SNAP:
S-Nitroso-N-acetylpenicillamine
- Sp1:
Specificity protein 1
- SR:
Sarcoplasmic reticulum
- STAT3:
Signal transducer and activator of transcription 3
- TCEP:
Tris(2-carboxyethyl)phosphine
- TRPA1:
Transient receptor potential channel A1
- TrpC5:
Transient receptor potential channel 5
- VEGF:
Vascular endothelial growth factor
- VEGFR2:
Vascular endothelial growth factor receptor 2
- VSMCs:
Vascular smooth muscle cells
- XBP1:
X-Box binding protein 1.
Contributor Information
Dan Wu, Email: wu_dan@tongji.edu.cn.
Deqiu Zhu, Email: zdq_0726@163.com.
Conflicts of Interest
None of the authors has any conflict of interests to disclose.
Authors' Contributions
Qingxun Hu, Dan Wu, and Deqiu Zhu designed the subject content of the review article. Qingxun Hu and Dan Wu conducted the initial search of literature, drafted the manuscript, and prepared the figures and tables. Deqiu Zhu gave some critical suggestions and had primary responsibility of the final content. All authors had read and approved the final manuscript. Qingxun Hu and Dan Wu contributed equally to this work.
References
- 1.Gewaltig M. T., Kojda G. Vasoprotection by nitric oxide: mechanisms and therapeutic potential. Cardiovascular Research. 2002;55(2):250–260. doi: 10.1016/S0008-6363(02)00327-9. [DOI] [PubMed] [Google Scholar]
- 2.Papapetropoulos A., Rudic R. D., Sessa W. C. Molecular control of nitric oxide synthases in the cardiovascular system. Cardiovascular Research. 1999;43(3):509–520. doi: 10.1016/S0008-6363(99)00161-3. [DOI] [PubMed] [Google Scholar]
- 3.Wang R. The gasotransmitter role of hydrogen sulfide. Antioxidants & Redox Signaling. 2003;5(4):493–501. doi: 10.1089/152308603768295249. [DOI] [PubMed] [Google Scholar]
- 4.Wang R. Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? The FASEB Journal. 2002;16(13):1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 5.Li L., Hsu A., Moore P. K. Actions and interactions of nitric oxide, carbon monoxide and hydrogen sulphide in the cardiovascular system and in inflammation — a tale of three gases! Pharmacology & Therapeutics. 2009;123(3):386–400. doi: 10.1016/j.pharmthera.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Whiteman M., Moore P. K. Hydrogen sulfide and the vasculature: a novel vasculoprotective entity and regulator of nitric oxide bioavailability? Journal of Cellular and Molecular Medicine. 2009;13(3):488–507. doi: 10.1111/j.1582-4934.2009.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lo Faro M. L., Fox B., Whatmore J. L., Winyard P. G., Whiteman M. Hydrogen sulfide and nitric oxide interactions in inflammation. Nitric Oxide. 2014;41:38–47. doi: 10.1016/j.niox.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 8.Szabo C. Gaseotransmitters: new frontiers for translational science. Science Translational Medicine. 2010;2(59, article 59ps54) doi: 10.1126/scitranslmed.3000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pace N. R. A molecular view of microbial diversity and the biosphere. Science. 1997;276(5313):734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- 10.Sugahara S., Suzuki M., Kamiya H., et al. Colorimetric determination of sulfide in microsamples. Analytical Sciences. 2016;32(10):1129–1131. doi: 10.2116/analsci.32.1129. [DOI] [PubMed] [Google Scholar]
- 11.Qian Y., Karpus J., Kabil O., et al. Selective fluorescent probes for live-cell monitoring of sulphide. Nature Communications. 2011;2(1):p. 495. doi: 10.1038/ncomms1506. [DOI] [PubMed] [Google Scholar]
- 12.Tan B., Jin S., Sun J., et al. New method for quantification of gasotransmitter hydrogen sulfide in biological matrices by LC-MS/MS. Scientific Reports. 2017;7(1, article 46278) doi: 10.1038/srep46278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mok Y. Y. P., Mohammed Atan M. S. B., Ping C. Y., et al. Role of hydrogen sulphide in haemorrhagic shock in the rat: protective effect of inhibitors of hydrogen sulphide biosynthesis. British Journal of Pharmacology. 2004;143(7):881–889. doi: 10.1038/sj.bjp.0706014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doeller J. E., Isbell T. S., Benavides G., et al. Polarographic measurement of hydrogen sulfide production and consumption by mammalian tissues. Analytical Biochemistry. 2005;341(1):40–51. doi: 10.1016/j.ab.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 15.Ishigami M., Hiraki K., Umemura K., Ogasawara Y., Ishii K., Kimura H. A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxidants & Redox Signaling. 2009;11(2):205–214. doi: 10.1089/ars.2008.2132. [DOI] [PubMed] [Google Scholar]
- 16.Ubuka T. Assay methods and biological roles of labile sulfur in animal tissues. Journal of Chromatography B. 2002;781(1-2):227–249. doi: 10.1016/S1570-0232(02)00623-2. [DOI] [PubMed] [Google Scholar]
- 17.Mason J., Cardin C. J., Dennehy A. The role of sulphide and sulphide oxidation in the copper molybdenum antagonism in rats and guinea pigs. Research in Veterinary Science. 1978;24(1):104–108. [PubMed] [Google Scholar]
- 18.Richardson C. J., Magee E. A. M., Cummings J. H. A new method for the determination of sulphide in gastrointestinal contents and whole blood by microdistillation and ion chromatography. Clinica Chimica Acta. 2000;293(1-2):115–125. doi: 10.1016/S0009-8981(99)00245-4. [DOI] [PubMed] [Google Scholar]
- 19.Abe K., Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. The Journal of Neuroscience. 1996;16(3):1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furne J., Saeed A., Levitt M. D. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2008;295(5):R1479–R1485. doi: 10.1152/ajpregu.90566.2008. [DOI] [PubMed] [Google Scholar]
- 21.Erickson P. F., Maxwell I. H., Su L. J., Baumann M., Glode L. M. Sequence of cDNA for rat cystathionine γ-lyase and comparison of deduced amino acid sequence with related Escherichia coli enzymes. Biochemical Journal. 1990;269(2):335–340. doi: 10.1042/bj2690335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stipanuk M. H., Beck P. W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochemical Journal. 1982;206(2):267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishii I., Akahoshi N., Yu X. N., et al. Murine cystathionine γ-lyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochemical Journal. 2004;381(1):113–123. doi: 10.1042/BJ20040243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geng B., Yang J., Qi Y., et al. H2S generated by heart in rat and its effects on cardiac function. Biochemical and Biophysical Research Communications. 2004;313(2):362–368. doi: 10.1016/j.bbrc.2003.11.130. [DOI] [PubMed] [Google Scholar]
- 25.Zhao W., Zhang J., Lu Y., Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. The EMBO Journal. 2001;20(21):6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang G., Wu L., Jiang B., et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science. 2008;322(5901):587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beltowski J., Jamroz-Wisniewska A. Hydrogen sulfide and endothelium-dependent vasorelaxation. Molecules. 2014;19(12):21183–21199. doi: 10.3390/molecules191221183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shibuya N., Tanaka M., Yoshida M., et al. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxidants & Redox Signaling. 2009;11(4):703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- 29.Lavu M., Bhushan S., Lefer D. J. Hydrogen sulfide-mediated cardioprotection: mechanisms and therapeutic potential. Clinical Science. 2011;120(6):219–229. doi: 10.1042/CS20100462. [DOI] [PubMed] [Google Scholar]
- 30.Kabil O., Banerjee R. Enzymology of H2S biogenesis, decay and signaling. Antioxidants & Redox Signaling. 2014;20(5):770–782. doi: 10.1089/ars.2013.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kimura H. Hydrogen sulfide: its production, release and functions. Amino Acids. 2011;41(1):113–121. doi: 10.1007/s00726-010-0510-x. [DOI] [PubMed] [Google Scholar]
- 32.Kimura Y., Goto Y. I., Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxidants & Redox Signaling. 2010;12(1):1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- 33.Szabo C. Hydrogen sulphide and its therapeutic potential. Nature Reviews Drug Discovery. 2007;6(11):917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 34.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiological Reviews. 2012;92(2):791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 35.Whiteman M., Le Trionnaire S., Chopra M., Fox B., Whatmore J. Emerging role of hydrogen sulfide in health and disease: critical appraisal of biomarkers and pharmacological tools. Clinical Science. 2011;121(11):459–488. doi: 10.1042/CS20110267. [DOI] [PubMed] [Google Scholar]
- 36.Sun Q., Collins R., Huang S., et al. Structural basis for the inhibition mechanism of human cystathionine γ-lyase, an enzyme responsible for the production of H2S. Journal of Biological Chemistry. 2009;284(5):3076–3085. doi: 10.1074/jbc.M805459200. [DOI] [PubMed] [Google Scholar]
- 37.Asimakopoulou A., Panopoulos P., Chasapis C. T., et al. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE) British Journal of Pharmacology. 2013;169(4):922–932. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Laan A. M., Piek J. J., van Royen N. Targeting angiogenesis to restore the microcirculation after reperfused MI. Nature Reviews Cardiology. 2009;6(8):515–523. doi: 10.1038/nrcardio.2009.103. [DOI] [PubMed] [Google Scholar]
- 39.Cai W., Wang M., Moore P., Jin H., Yao T., Zhu Y. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovascular Research. 2007;76(1):29–40. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- 40.Papapetropoulos A., Pyriochou A., Altaany Z., et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(51):21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tao B. B., Liu S. Y., Zhang C. C., et al. VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045–Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxidants & Redox Signaling. 2013;19(5):448–464. doi: 10.1089/ars.2012.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kan J., Guo W., Huang C., Bao G., Zhu Y., Zhu Y. Z. S-propargyl-cysteine, a novel water-soluble modulator of endogenous hydrogen sulfide, promotes angiogenesis through activation of signal transducer and activator of transcription 3. Antioxidants & Redox Signaling. 2014;20(15):2303–2316. doi: 10.1089/ars.2013.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou Y., Li X. H., Zhang C. C., et al. Hydrogen sulfide promotes angiogenesis by downregulating miR-640 via the VEGFR2/mTOR pathway. American Journal of Physiology-Cell Physiology. 2016;310(4):C305–C317. doi: 10.1152/ajpcell.00230.2015. [DOI] [PubMed] [Google Scholar]
- 44.van den Born J. C., Mencke R., Conroy S., Zeebregts C. J., van Goor H., Hillebrands J. L. Cystathionine γ-lyase is expressed in human atherosclerotic plaque microvessels and is involved in micro-angiogenesis. Scientific Reports. 2016;6(1, article 34608) doi: 10.1038/srep34608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mustafa A. K., Gadalla M. M., Sen N., et al. H2S signals through protein S-sulfhydration. Science Signaling. 2009;2(96, article ra72) doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang G., Zhao K., Ju Y., et al. Hydrogen sulfide protects against cellular senescence via S-Sulfhydration of Keap 1 and activation of Nrf 2. Antioxidants & Redox Signaling. 2013;18(15):1906–1919. doi: 10.1089/ars.2012.4645. [DOI] [PubMed] [Google Scholar]
- 47.Sen N., Paul B. D., Gadalla M. M., et al. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Molecular Cell. 2012;45(1):13–24. doi: 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheung S. H., Lau J. Y. W. Hydrogen sulfide mediates athero-protection against oxidative stress via S-sulfhydration. PLoS One. 2018;13(3, article e0194176) doi: 10.1371/journal.pone.0194176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Du C., Lin X., Xu W., et al. Sulfhydrated sirtuin-1 increasing its deacetylation activity is an essential epigenetics mechanism of anti-atherogenesis by hydrogen sulfide. Antioxidants & Redox Signaling. 2018 doi: 10.1089/ars.2017.7195. [DOI] [PubMed] [Google Scholar]
- 50.Modis K., Ju Y., Ahmad A., et al. S-sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacological Research. 2016;113(Part A):116–124. doi: 10.1016/j.phrs.2016.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meng G., Xiao Y., Ma Y., et al. hydrogen sulfide regulates Krüppel‐like factor 5 transcription activity via specificity protein 1 S‐sulfhydration at Cys664 to prevent myocardial hypertrophy. Journal of the American Heart Association. 2016;5(9, article e004160) doi: 10.1161/JAHA.116.004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saha S., Chakraborty P. K., Xiong X., et al. Cystathionine β-synthase regulates endothelial function via protein S-sulfhydration. The FASEB Journal. 2016;30(1):441–456. doi: 10.1096/fj.15-278648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao K., Ju Y., Li S., Altaany Z., Wang R., Yang G. S‐sulfhydration of MEK1 leads to PARP‐1 activation and DNA damage repair. EMBO Reports. 2014;15(7):792–800. doi: 10.1002/embr.201338213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie Z. Z., Shi M. M., Xie L., et al. Sulfhydration of p66Shc at cysteine59 mediates the antioxidant effect of hydrogen sulfide. Antioxidants & Redox Signaling. 2014;21(18):2531–2542. doi: 10.1089/ars.2013.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krishnan N., Fu C., Pappin D. J., Tonks N. K. H2S-induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Science Signaling. 2011;4(203, article ra86) doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimizu Y., Polavarapu R., Eskla K. L., et al. Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. Journal of Molecular and Cellular Cardiology. 2018;116:29–40. doi: 10.1016/j.yjmcc.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pan J., Carroll K. S. Persulfide reactivity in the detection of protein S-sulfhydration. ACS Chemical Biology. 2013;8(6):1110–1116. doi: 10.1021/cb4001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Longen S., Richter F., Köhler Y., Wittig I., Beck K. F., Pfeilschifter J. Quantitative persulfide site identification (qPerS-SID) reveals protein targets of H2S releasing donors in mammalian cells. Scientific Reports. 2016;6(1, article 29808) doi: 10.1038/srep29808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang D., Macinkovic I., Devarie-Baez N. O., et al. Detection of protein S-sulfhydration by a tag-switch technique. Angewandte Chemie International Edition. 2014;53(2):575–581. doi: 10.1002/anie.201305876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dóka E., Pader I., Bíró A., et al. A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Science Advances. 2016;2(1, article e1500968) doi: 10.1126/sciadv.1500968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan H., Du J., Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochemical and Biophysical Research Communications. 2004;313(1):22–27. doi: 10.1016/j.bbrc.2003.11.081. [DOI] [PubMed] [Google Scholar]
- 62.Bucci M., Papapetropoulos A., Vellecco V., et al. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(10):1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- 63.Lee S. W., Cheng Y., Moore P. K., Bian J. S. Hydrogen sulphide regulates intracellular pH in vascular smooth muscle cells. Biochemical and Biophysical Research Communications. 2007;358(4):1142–1147. doi: 10.1016/j.bbrc.2007.05.063. [DOI] [PubMed] [Google Scholar]
- 64.Kiss L., Deitch E. A., Szabo C. Hydrogen sulfide decreases adenosine triphosphate levels in aortic rings and leads to vasorelaxation via metabolic inhibition. Life Sciences. 2008;83(17-18):589–594. doi: 10.1016/j.lfs.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mistry R. K., Murray T. V. A., Prysyazhna O., et al. Transcriptional regulation of cystathionine-γ-Lyase in endothelial cells by NADPH oxidase 4-dependent signaling. Journal of Biological Chemistry. 2016;291(4):1774–1788. doi: 10.1074/jbc.M115.685578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hedegaard E. R., Gouliaev A., Winther A. K., et al. Involvement of potassium channels and calcium-independent mechanisms in hydrogen sulfide–induced relaxation of rat mesenteric small arteries. The Journal of Pharmacology and Experimental Therapeutics. 2016;356(1):53–63. doi: 10.1124/jpet.115.227017. [DOI] [PubMed] [Google Scholar]
- 67.Kondo K., Bhushan S., King A. L., et al. H2S protects against pressure overload–induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127(10):1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu D., Hu Q., Liu X., Pan L., Xiong Q., Zhu Y. Z. Hydrogen sulfide protects against apoptosis under oxidative stress through SIRT1 pathway in H9c2 cardiomyocytes. Nitric Oxide. 2015;46:204–212. doi: 10.1016/j.niox.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 69.Nicholson C. K., Lambert J. P., Molkentin J. D., Sadoshima J., Calvert J. W. Thioredoxin 1 is essential for sodium sulfide–mediated cardioprotection in the setting of heart failure. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(4):744–751. doi: 10.1161/ATVBAHA.112.300484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu Y. H., Lu M., Xie Z. Z., et al. Hydrogen sulfide prevents heart failure development via inhibition of renin release from mast cells in isoproterenol-treated rats. Antioxidants & Redox Signaling. 2014;20(5):759–769. doi: 10.1089/ars.2012.4888. [DOI] [PubMed] [Google Scholar]
- 71.Polhemus D. J., Kondo K., Bhushan S., et al. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circulation: Heart Failure. 2013;6(5):1077–1086. doi: 10.1161/CIRCHEARTFAILURE.113.000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu T., Li H., Wu B., et al. Hydrogen sulfide reduces recruitment of CD11b+DG Gr-1+DG cells in mice with myocardial infarction. Cell Transplantation. 2017;26(5):753–764. doi: 10.3727/096368917X695029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shimizu Y., Nicholson C. K., Lambert J. P., et al. Sodium sulfide attenuates ischemic-induced heart failure by enhancing proteasomal function in an Nrf2-dependent manner. Circulation: Heart Failure. 2016;9(4, article e002368) doi: 10.1161/CIRCHEARTFAILURE.115.002368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lilyanna S., Peh M. T., Liew O. W., et al. GYY4137 attenuates remodeling, preserves cardiac function and modulates the natriuretic peptide response to ischemia. Journal of Molecular and Cellular Cardiology. 2015;87:27–37. doi: 10.1016/j.yjmcc.2015.07.028. [DOI] [PubMed] [Google Scholar]
- 75.Barr L. A., Shimizu Y., Lambert J. P., Nicholson C. K., Calvert J. W. Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide. 2015;46:145–156. doi: 10.1016/j.niox.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lowenstein C. J., Dinerman J. L., Snyder S. H. Nitric oxide: a physiologic messenger. Annals of Internal Medicine. 1994;120(3):227–237. doi: 10.7326/0003-4819-120-3-199402010-00009. [DOI] [PubMed] [Google Scholar]
- 77.Tsukahara H., Gordienko D. V., Goligorsky M. S. Continuous monitoring of nitric oxide release from human umbilical vein endothelial cells. Biochemical and Biophysical Research Communications. 1993;193(2):722–729. doi: 10.1006/bbrc.1993.1685. [DOI] [PubMed] [Google Scholar]
- 78.Engelman D. T., Watanabe M., Engelman R. M., et al. Constitutive nitric-oxide release is impaired after ischemia and reperfusion. Journal of Thoracic and Cardiovascular Surgery. 1995;110(4):1047–1053. doi: 10.1016/S0022-5223(05)80173-4. [DOI] [PubMed] [Google Scholar]
- 79.Tarpey M. M., Fridovich I. Methods of detection of vascular reactive species: nitric oxide, superoxide, hydrogen peroxide, and peroxynitrite. Circulation Research. 2001;89(3):224–236. doi: 10.1161/hh1501.094365. [DOI] [PubMed] [Google Scholar]
- 80.Braman R. S., Hendrix S. A. Nanogram nitrite and nitrate determination in environmental and biological materials by vanadium(III) reduction with chemiluminescence detection. Analytical Chemistry. 1989;61(24):2715–2718. doi: 10.1021/ac00199a007. [DOI] [PubMed] [Google Scholar]
- 81.Sato M., Hida N., Umezawa Y. Imaging the nanomolar range of nitric oxide with an amplifier-coupled fluorescent indicator in living cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(41):14515–14520. doi: 10.1073/pnas.0505136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yin S., Fuangthong M., Laratta W. P., Shapleigh J. P. Use of a green fluorescent protein-based reporter fusion for detection of nitric oxide produced by denitrifiers. Applied and Environmental Microbiology. 2003;69(7):3938–3944. doi: 10.1128/AEM.69.7.3938-3944.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Farah C., Michel L. Y. M., Balligand J. L. Nitric oxide signalling in cardiovascular health and disease. Nature Reviews Cardiology. 2018;15(5):292–316. doi: 10.1038/nrcardio.2017.224. [DOI] [PubMed] [Google Scholar]
- 84.Alderton W. K., Cooper C. E., Knowles R. G. Nitric oxide synthases: structure, function and inhibition. Biochemical Journal. 2001;357(3):593–615. doi: 10.1042/bj3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abu-Soud H. M., Stuehr D. J. Nitric oxide synthases reveal a role for calmodulin in controlling electron transfer. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(22):10769–10772. doi: 10.1073/pnas.90.22.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stuehr D., Pou S., Rosen G. M. Oxygen reduction by nitric-oxide synthases. Journal of Biological Chemistry. 2001;276(18):14533–14536. doi: 10.1074/jbc.R100011200. [DOI] [PubMed] [Google Scholar]
- 87.Xu K. Y., Huso D. L., Dawson T. M., Bredt D. S., Becker L. C. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(2):657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Choate J. K., Danson E. J. F., Morris J. F., Paterson D. J. Peripheral vagal control of heart rate is impaired in neuronal NOS knockout mice. American Journal of Physiology-Heart and Circulatory Physiology. 2001;281(6):H2310–H2317. doi: 10.1152/ajpheart.2001.281.6.H2310. [DOI] [PubMed] [Google Scholar]
- 89.Schwarz P. M., Kleinert H., Forstermann U. Potential functional significance of brain-type and muscle-type nitric oxide synthase I expressed in adventitia and media of rat aorta. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19(11):2584–2590. doi: 10.1161/01.ATV.19.11.2584. [DOI] [PubMed] [Google Scholar]
- 90.Piech A., Dessy C., Havaux X., Feron O., Balligand J. L. Differential regulation of nitric oxide synthases and their allosteric regulators in heart and vessels of hypertensive rats. Cardiovascular Research. 2003;57(2):456–467. doi: 10.1016/S0008-6363(02)00676-4. [DOI] [PubMed] [Google Scholar]
- 91.Balligand J. L., Kobzik L., Han X., et al. Nitric oxide-dependent parasympathetic signaling is due to activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes. Journal of Biological Chemistry. 1995;270(24):14582–14586. doi: 10.1074/jbc.270.24.14582. [DOI] [PubMed] [Google Scholar]
- 92.Petroff M. G. V., Kim S. H., Pepe S., et al. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nature Cell Biology. 2001;3(10):867–873. doi: 10.1038/ncb1001-867. [DOI] [PubMed] [Google Scholar]
- 93.Wallerath T., Gath I., Aulitzky W., Pollock J., Kleinert H., Förstermann U. Identification of the NO synthase isoforms expressed in human neutrophil granulocytes, megakaryocytes and platelets. Thrombosis and Haemostasis. 1997;77(1):163–167. doi: 10.1055/s-0038-1655925. [DOI] [PubMed] [Google Scholar]
- 94.Wilcox J. N., Subramanian R. R., Sundell C. L., et al. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17(11):2479–2488. doi: 10.1161/01.ATV.17.11.2479. [DOI] [PubMed] [Google Scholar]
- 95.Balligand J. L., Ungureanu-Longrois D., Simmons W. W., et al. Cytokine-inducible nitric oxide synthase (iNOS) expression in cardiac myocytes. Characterization and regulation of iNOS expression and detection of iNOS activity in single cardiac myocytes in vitro. Journal of Biological Chemistry. 1994;269(44):27580–27588. [PubMed] [Google Scholar]
- 96.Liu V., Huang P. Cardiovascular roles of nitric oxide: a review of insights from nitric oxide synthase gene disrupted mice. Cardiovascular Research. 2008;77(1):19–29. doi: 10.1016/j.cardiores.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sessa W. C. The nitric oxide synthase family of proteins. Journal of Vascular Research. 1994;31(3):131–143. doi: 10.1159/000159039. [DOI] [PubMed] [Google Scholar]
- 98.Benjamin N., O'Driscoll F., Dougall H., et al. Stomach NO synthesis. Nature. 1994;368(6471):p. 502. doi: 10.1038/368502a0. [DOI] [PubMed] [Google Scholar]
- 99.Hou Y., Guo Z., Li J., Wang P. G. Seleno compounds and glutathione peroxidase catalyzed decomposition of S-nitrosothiols. Biochemical and Biophysical Research Communications. 1996;228(1):88–93. doi: 10.1006/bbrc.1996.1620. [DOI] [PubMed] [Google Scholar]
- 100.Nikitovic D., Holmgren A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. Journal of Biological Chemistry. 1996;271(32):19180–19185. doi: 10.1074/jbc.271.32.19180. [DOI] [PubMed] [Google Scholar]
- 101.Fukumura D., Gohongi T., Kadambi A., et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(5):2604–2609. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Babaei S., Teichert-Kuliszewska K., Monge J. C., Mohamed F., Bendeck M. P., Stewart D. J. Role of nitric oxide in the angiogenic response in vitro to basic fibroblast growth factor. Circulation Research. 1998;82(9):1007–1015. doi: 10.1161/01.RES.82.9.1007. [DOI] [PubMed] [Google Scholar]
- 103.Papapetropoulos A., Garcia-Cardena G., Madri J. A., Sessa W. C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. The Journal of Clinical Investigation. 1997;100(12):3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang J., Xu J., Danniel M., et al. The interaction between XBP1 and eNOS contributes to endothelial cell migration. Experimental Cell Research. 2018;363(2):262–270. doi: 10.1016/j.yexcr.2018.01.016. [DOI] [PubMed] [Google Scholar]
- 105.Boopathy G. T. K., Kulkarni M., Ho S. Y., et al. Cavin-2 regulates the activity and stability of endothelial nitric-oxide synthase (eNOS) in angiogenesis. Journal of Biological Chemistry. 2017;292(43):17760–17776. doi: 10.1074/jbc.M117.794743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Verma S., Wang C. H., Li S. H., et al. A self-fulfilling prophecy: C-reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation. 2002;106(8):913–919. doi: 10.1161/01.CIR.0000029802.88087.5E. [DOI] [PubMed] [Google Scholar]
- 107.Dimmeler S., Fleming I., Fisslthaler B., Hermann C., Busse R., Zeiher A. M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 108.Whalen E. J., Foster M. W., Matsumoto A., et al. Regulation of β-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129(3):511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 109.Kelleher Z. T., Matsumoto A., Stamler J. S., Marshall H. E. NOS2 regulation of NF-κB by S-nitrosylation of p65. Journal of Biological Chemistry. 2007;282(42):30667–30672. doi: 10.1074/jbc.M705929200. [DOI] [PubMed] [Google Scholar]
- 110.Santhanam L., Lim H. K., Lim H. K., et al. Inducible NO synthase–dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circulation Research. 2007;101(7):692–702. doi: 10.1161/CIRCRESAHA.107.157727. [DOI] [PubMed] [Google Scholar]
- 111.Matsushita K., Morrell C. N., Cambien B., et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115(2):139–150. doi: 10.1016/S0092-8674(03)00803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Irie T., Sips P. Y., Kai S., et al. S-Nitrosylation of calcium-handling proteins in cardiac adrenergic signaling and hypertrophy. Circulation Research. 2015;117(9):793–803. doi: 10.1161/CIRCRESAHA.115.307157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ozawa K., Whalen E. J., Nelson C. D., et al. S-nitrosylation of β-arrestin regulates β-adrenergic receptor trafficking. Molecular Cell. 2008;31(3):395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gonzalez D. R., Beigi F., Treuer A. V., Hare J. M. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(51):20612–20617. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yoshida T., Inoue R., Morii T., et al. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nature Chemical Biology. 2006;2(11):596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- 116.Chen Y. Y., Chu H. M., Pan K. T., et al. Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. Journal of Biological Chemistry. 2008;283(50):35265–35272. doi: 10.1074/jbc.M805287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hara M. R., Agrawal N., Kim S. F., et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nature Cell Biology. 2005;7(7):665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 118.Ignarro L. J., Buga G. M., Wood K. S., Byrns R. E., Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(24):9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Palmer R. M. J., Ferrige A. G., Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327(6122):524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 120.Dusting G. J. Nitric oxide in cardiovascular disorders. Journal of Vascular Research. 1995;32(3):143–161. doi: 10.1159/000159089. [DOI] [PubMed] [Google Scholar]
- 121.Hayakawa H., Hirata Y., Kakoki M., et al. Role of nitric oxide–cGMP pathway in adrenomedullin-induced vasodilation in the rat. Hypertension. 1999;33(2):689–693. doi: 10.1161/01.HYP.33.2.689. [DOI] [PubMed] [Google Scholar]
- 122.Mistry D. K., Garland C. J. Nitric oxide (NO)-induced activation of large conductance Ca2+-dependent K+ channels (BKCa) in smooth muscle cells isolated from the rat mesenteric artery. British Journal of Pharmacology. 1998;124(6):1131–1140. doi: 10.1038/sj.bjp.0701940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Howitt L., Kuo I. Y., Ellis A., et al. Chronic deficit in nitric oxide elicits oxidative stress and augments T-type calcium-channel contribution to vascular tone of rodent arteries and arterioles. Cardiovascular Research. 2013;98(3):449–457. doi: 10.1093/cvr/cvt043. [DOI] [PubMed] [Google Scholar]
- 124.Jones S. P., Greer J. J. M., van Haperen R., Duncker D. J., de Crom R., Lefer D. J. Endothelial nitric oxide synthase overexpression attenuates congestive heart failure in mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(8):4891–4896. doi: 10.1073/pnas.0837428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jones S. P., Girod W. G., Palazzo A. J., et al. Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. American Journal of Physiology-Heart and Circulatory Physiology. 1999;276(5):H1567–H1573. doi: 10.1152/ajpheart.1999.276.5.H1567. [DOI] [PubMed] [Google Scholar]
- 126.Janssens S., Pokreisz P., Schoonjans L., et al. Cardiomyocyte-specific overexpression of nitric oxide synthase 3 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circulation Research. 2004;94(9):1256–1262. doi: 10.1161/01.RES.0000126497.38281.23. [DOI] [PubMed] [Google Scholar]
- 127.Xu Z., Ji X., Boysen P. G. Exogenous nitric oxide generates ROS and induces cardioprotection: involvement of PKG, mitochondrial KATP channels, and ERK. American Journal of Physiology-Heart and Circulatory Physiology. 2004;286(4):H1433–H1440. doi: 10.1152/ajpheart.00882.2003. [DOI] [PubMed] [Google Scholar]
- 128.Bhushan S., Kondo K., Polhemus D. J., et al. Nitrite therapy improves left ventricular function during heart failure via restoration of nitric oxide–mediated cytoprotective signaling. Circulation Research. 2014;114(8):1281–1291. doi: 10.1161/CIRCRESAHA.114.301475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Couto G. K., Britto L. R. G., Mill J. G., Rossoni L. V. Enhanced nitric oxide bioavailability in coronary arteries prevents the onset of heart failure in rats with myocardial infarction. Journal of Molecular and Cellular Cardiology. 2015;86:110–120. doi: 10.1016/j.yjmcc.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 130.Pae H. O., Lee Y. C., Jo E. K., Chung H. T. Subtle interplay of endogenous bioactive gases (NO, CO and H2S) in inflammation. Archives of Pharmacal Research. 2009;32(8):1155–1162. doi: 10.1007/s12272-009-1806-9. [DOI] [PubMed] [Google Scholar]
- 131.Bertova A., Cacanyiova S., Kristek F., Krizanova O., Ondrias K., Tomaskova Z. The hypothesis of the main role of H2S in coupled sulphide-nitroso signalling pathway. General Physiology and Biophysics. 2010;29(4):402–410. doi: 10.4149/gpb_2010_04_402. [DOI] [PubMed] [Google Scholar]
- 132.Kubo S., Doe I., Kurokawa Y., Nishikawa H., Kawabata A. Direct inhibition of endothelial nitric oxide synthase by hydrogen sulfide: contribution to dual modulation of vascular tension. Toxicology. 2007;232(1-2):138–146. doi: 10.1016/j.tox.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 133.Grossi L. Hydrogen sulfide induces nitric oxide release from nitrite. Bioorganic & Medicinal Chemistry Letters. 2009;19(21):6092–6094. doi: 10.1016/j.bmcl.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 134.Taoka S., Banerjee R. Characterization of NO binding to human cystathionine β-synthase:: possible implications of the effects of CO and NO binding to the human enzyme. Journal of Inorganic Biochemistry. 2001;87(4):245–251. doi: 10.1016/S0162-0134(01)00335-X. [DOI] [PubMed] [Google Scholar]
- 135.Hua W., Chen Q., Gong F., Xie C., Zhou S., Gao L. Cardioprotection of H2S by downregulating iNOS and upregulating HO-1 expression in mice with CVB3-induced myocarditis. Life Sciences. 2013;93(24):949–954. doi: 10.1016/j.lfs.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 136.Jeong S. O., Pae H. O., Oh G. S., et al. Hydrogen sulfide potentiates interleukin-1β-induced nitric oxide production via enhancement of extracellular signal-regulated kinase activation in rat vascular smooth muscle cells. Biochemical and Biophysical Research Communications. 2006;345(3):938–944. doi: 10.1016/j.bbrc.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 137.Predmore B. L., Julian D., Cardounel A. J. Hydrogen sulfide increases nitric oxide production from endothelial cells by an Akt-dependent mechanism. Frontiers in Physiology. 2011;2:p. 104. doi: 10.3389/fphys.2011.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kida M., Sugiyama T., Yoshimoto T., Ogawa Y. Hydrogen sulfide increases nitric oxide production with calcium-dependent activation of endothelial nitric oxide synthase in endothelial cells. European Journal of Pharmaceutical Sciences. 2013;48(1-2):211–215. doi: 10.1016/j.ejps.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 139.Altaany Z., Yang G., Wang R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. Journal of Cellular and Molecular Medicine. 2013;17(7):879–888. doi: 10.1111/jcmm.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sojitra B., Bulani Y., Putcha U. K., et al. Nitric oxide synthase inhibition abrogates hydrogen sulfide-induced cardioprotection in mice. Molecular and Cellular Biochemistry. 2012;360(1-2):61–69. doi: 10.1007/s11010-011-1044-6. [DOI] [PubMed] [Google Scholar]
- 141.King A. L., Polhemus D. J., Bhushan S., et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(8):3182–3187. doi: 10.1073/pnas.1321871111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Altaany Z., Ju Y., Yang G., Wang R. The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Science Signaling. 2014;7(342, article ra87) doi: 10.1126/scisignal.2005478. [DOI] [PubMed] [Google Scholar]
- 143.Bir S. C., Kolluru G. K., McCarthy P., et al. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia‐inducible factor‐1α and vascular endothelial growth factor–dependent angiogenesis. Journal of the American Heart Association. 2012;1(5, article e004093) doi: 10.1161/JAHA.112.004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sun J., Aponte A. M., Menazza S., Gucek M., Steenbergen C., Murphy E. Additive cardioprotection by pharmacological postconditioning with hydrogen sulfide and nitric oxide donors in mouse heart: S-sulfhydration vs. S-nitrosylation. Cardiovascular Research. 2016;110(1):96–106. doi: 10.1093/cvr/cvw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Eberhardt M., Dux M., Namer B., et al. H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO–TRPA1–CGRP signalling pathway. Nature Communications. 2014;5(1) doi: 10.1038/ncomms5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Whiteman M., Li L., Kostetski I., et al. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochemical and Biophysical Research Communications. 2006;343(1):303–310. doi: 10.1016/j.bbrc.2006.02.154. [DOI] [PubMed] [Google Scholar]
- 147.Yong Q. C., Cheong J. L., Hua F., et al. Regulation of heart function by endogenous gaseous mediators-crosstalk between nitric oxide and hydrogen sulfide. Antioxidants & Redox Signaling. 2011;14(11):2081–2091. doi: 10.1089/ars.2010.3572. [DOI] [PubMed] [Google Scholar]
- 148.Ali M. Y., Ping C. Y., Mok Y. Y., et al. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? British Journal of Pharmacology. 2006;149(6):625–634. doi: 10.1038/sj.bjp.0706906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yong Q. C., Hu L. F., Wang S., Huang D., Bian J. S. Hydrogen sulfide interacts with nitric oxide in the heart: possible involvement of nitroxyl. Cardiovascular Research. 2010;88(3):482–491. doi: 10.1093/cvr/cvq248. [DOI] [PubMed] [Google Scholar]
- 150.Filipovic M. R., Miljkovic J. L., Nauser T., et al. Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols. Journal of the American Chemical Society. 2012;134(29):12016–12027. doi: 10.1021/ja3009693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ondrias K., Stasko A., Cacanyiova S., et al. H2S and HS− donor NaHS releases nitric oxide from nitrosothiols, metal nitrosyl complex, brain homogenate and murine L1210 leukaemia cells. Pflügers Archiv. 2008;457(2):271–279. doi: 10.1007/s00424-008-0519-0. [DOI] [PubMed] [Google Scholar]
- 152.Cortese-Krott M. M., Fernandez B. O., Santos J. L. T., et al. Nitrosopersulfide (SSNO−) accounts for sustained NO bioactivity of S-nitrosothiols following reaction with sulfid. Redox Biology. 2014;2:234–244. doi: 10.1016/j.redox.2013.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hu Q., Wu D., Ma F., et al. Novel angiogenic activity and molecular mechanisms of ZYZ-803, a slow-releasing hydrogen sulfide–nitric oxide hybrid molecule. Antioxidants & Redox Signaling. 2016;25(8):498–514. doi: 10.1089/ars.2015.6607. [DOI] [PubMed] [Google Scholar]
- 154.Coletta C., Papapetropoulos A., Erdelyi K., et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(23):9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hosoki R., Matsuki N., Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochemical and Biophysical Research Communications. 1997;237(3):527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 156.Wu D., Hu Q., Ma F., Zhu Y. Z. Vasorelaxant effect of a new hydrogen sulfide-nitric oxide conjugated donor in isolated rat aortic rings through cGMP pathway. Oxidative Medicine and Cellular Longevity. 2016;2016:10. doi: 10.1155/2016/7075682.7075682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tang G., Yang G., Jiang B., Ju Y., Wu L., Wang R. H2S is an endothelium-derived hyperpolarizing factor. Antioxidants & Redox Signaling. 2013;19(14):1634–1646. doi: 10.1089/ars.2012.4805. [DOI] [PubMed] [Google Scholar]
- 158.Wang Y. F., Mainali P., Tang C. S., et al. Effects of nitric oxide and hydrogen sulfide on the relaxation of pulmonary arteries in rats. Chinese Medical Journal. 2008;121(5):420–423. [PubMed] [Google Scholar]
- 159.Pan T. T., Feng Z. N., Lee S. W., Moore P. K., Bian J. S. Endogenous hydrogen sulfide contributes to the cardioprotection by metabolic inhibition preconditioning in the rat ventricular myocytes. Journal of Molecular and Cellular Cardiology. 2006;40(1):119–130. doi: 10.1016/j.yjmcc.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 160.Yong Q. C., Lee S. W., Foo C. S., Neo K. L., Chen X., Bian J. S. Endogenous hydrogen sulphide mediates the cardioprotection induced by ischemic postconditioning. American Journal of Physiology-Heart and Circulatory Physiology. 2008;295(3):H1330–H1340. doi: 10.1152/ajpheart.00244.2008. [DOI] [PubMed] [Google Scholar]
- 161.Wu D., Hu Q., Xiong Y., Zhu D., Mao Y., Zhu Y. Z. Novel H2S-NO hybrid molecule (ZYZ-803) promoted synergistic effects against heart failure. Redox Biology. 2018;15:243–252. doi: 10.1016/j.redox.2017.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bibli S. I., Szabo C., Chatzianastasiou A., et al. Hydrogen sulfide preserves endothelial nitric oxide synthase function by inhibiting proline-rich kinase 2: implications for cardiomyocyte survival and cardioprotection. Molecular Pharmacology. 2017;92(6):718–730. doi: 10.1124/mol.117.109645. [DOI] [PubMed] [Google Scholar]
- 163.Donnarumma E., Bhushan S., Bradley J. M., et al. Nitrite therapy ameliorates myocardial dysfunction via H2S and nuclear factor‐erythroid 2‐related factor 2 (Nrf2)‐dependent signaling in chronic heart failure. Journal of the American Heart Association. 2016;5(8, article e003551) doi: 10.1161/JAHA.116.003551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Karwi Q. G., Whiteman M., Wood M. E., Torregrossa R., Baxter G. F. Pharmacological postconditioning against myocardial infarction with a slow-releasing hydrogen sulfide donor, GYY4137. Pharmacological Research. 2016;111:442–451. doi: 10.1016/j.phrs.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 165.Geng B., Cui Y., Zhao J., et al. Hydrogen sulfide downregulates the aortic L-arginine/nitric oxide pathway in rats. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2007;293(4):R1608–R1618. doi: 10.1152/ajpregu.00207.2006. [DOI] [PubMed] [Google Scholar]