

Abstract
Many patients with chronic psychotic disorders including schizophrenia (SZ) maintain meaningful levels of plasticity (i.e., capacity for change) within neurocognition-relevant brain mechanisms, as evidenced by gains in neurocognition and function after interventions such as targeted cognitive training. However, like many clinical features of these disorders, therapeutic responses in SZ are heterogeneous, and prospectively identifying treatment-sensitive individuals and individualized treatment modalities remains an unmet challenge. We propose that available plasticity in neurocognition-relevant brain mechanisms in individual SZ patients can be detected by gains in laboratory measures of early auditory information processing (EAIP) and auditory learning after a single challenge-dose of a pharmacologic agent; here, we present supportive data for this strategy with the non-competitive NMDA antagonist, memantine, and the psychostimulant, amphetamine. We describe a novel therapeutic model where this “challenge dose” strategy is used to prospectively identify a sensitive cohort of patients, and in these patients, a therapeutic response is elicited by pairing drug-enhanced EAIP and auditory learning with auditory-based targeted cognitive training.
Keywords: amphetamine, auditory steady state response, memantine, mismatch negativity, neurocognition, prepulse inhibition, schizophrenia
1.1 Introduction
Schizophrenia (SZ) is a severe brain disorder affecting 1% of the world population. Its cost to society is well documented (Rice 2009), as are stories of lifelong suffering among SZ patients and their families. Almost 60 years after the introduction of drugs designed to target its symptoms, antipsychotics (APs) are at best modestly effective, and the neurobiological targets of these medications are not firmly anchored in a mechanistic understanding of the biology of this disorder.
While APs blunt severe acute psychotic symptoms, they may not have a meaningful impact on real-life function (Keefe et al. 2007, 2016; Leucht et al. 2009; Lieberman et al. 2005). Evidence that daily function in SZ is closely linked to neurocognition (Green 1996) has stimulated efforts to develop procognitive agents as adjuncts to APs; these efforts have largely yielded negative results (cf. Barch 2010, 2011; Buchanan et al. 2007; Goff et al. 1996, 1999, 2007, 2008; Green 2007). Importantly, procognitive trials generally suffer from two important weaknesses. First, they are not conducted in the context of cognitive interventions (cf. Barch 2010). Simply adding a putative procognitive drug to a daily AP regimen may not provide a sensitive test of its activity: drugs that enhance specific domains of neurocognition, e.g. working memory, might not yield clinical benefits unless paired with interventions that access those domains, i.e. utilize/place demands on working memory. This is precisely the rationale for the use of pro-extinction drugs to enhance clinical benefits of cognitive and behavioral interventions for anxiety disorders (Choi et al. 2010; Norberg et al. 2008; Ressler et al. 2004). Second, SZ is heterogeneous, and pro-cognitive trials in SZ suffer from the absence of biomarkers that identify “sensitive” clinical subgroups.
A key consideration in the development of pro-cognitive agents for SZ is the degree to which neural function underlying neurocognition retains its plasticity (i.e. capacity for change) in this disorder, and may therefore be a rational target for therapeutics. If plasticity of cognition-relevant circuitry is limited due, for example, to inherent flaws in circuit connectivity resulting from significant errors in neuronal migration or synaptic connectivity, this might argue against a viable therapeutic target. No drug will likely unscramble circuit design flaws imparted two decades earlier, which form the foundation of neurocognitive impairments. If, on the other hand, intact plasticity can be identified prospectively, this might indicate sensitivity for positive change, given an appropriate intervention. Because SZ is heterogeneous in presentation and presumably in its underlying pathophysiology, it is very likely that the amount of retained meaningful plasticity will differ greatly across patients, and across brain circuitries that are impacted by their illnesses. Thus, a means to prospectively identify retained plasticity (“room to move”) among individual SZ patients, within cognition-relevant brain mechanisms, could serve as a critically important biomarker for stratifying patients into groups that are more vs. less likely to show neurocognitive gains in response to a therapeutic intervention. Implicit in this model is that the therapeutic target would be healthy brain mechanisms that have retained a degree of plasticity; by contrast, attempts to “correct” brain function that is disrupted based on presumed neurodevelopmental pathology in schizophrenia have met with little success, despite efforts spanning almost 60 years.
1.2 Using Drugs to Identify Plasticity in Cognition-relevant Brain Substrates in Schizophrenia
One way to identify intact plasticity within cognition-relevant brain mechanisms is to “challenge” those mechanisms pharmacologically, while monitoring informative laboratory measures of cognition-relevant brain events. The use of a drug challenge to identify enriched, sensitive subgroups of patients parallels the common use of a “test dose” to predict clinical benefit from interventions ranging from hormones (Biller 2007) to anti-Parkinsonian therapies (Hughes et al. 1990) to bronchodilators (Fruchter and Yigla 2009); it is an established way to acutely probe the brain for healthy biological mechanisms that might be leveraged in the service of therapeutics.
Which laboratory measures might be most informative for identifying plasticity within cognition-relevant brain mechanisms? One suggestion came from our studies of neurophysiological endophenotypes, conducted by the Consortium on the Genetics of Schizophrenia (COGS). Using structural equation modeling (SEM) in 1415 SZ patients, the COGS group (Thomas et al. 2017) reported that measures of early auditory information processing (EAIP) had a direct (mediating) effect on cognition (p<0.001), that cognition had a direct effect on negative symptoms (p<0.001), and that both cognition (p<0.001) and negative symptoms (p<0.001) had direct effects on functional outcome. Overall, EAIP had a fully mediated effect on functional outcome, engaging general rather than modality (auditory)-specific cognition. One measure of EAIP in this study was mismatch negativity (MMN), a phenomenon described elsewhere in this Special Issue as the negative event-related potential (ERP) that is automatically elicited in response to a deviant sound within the context of repetitive, identical sounds. Explicitly, this model predicts that a 1 μV change in the MMN EAIP response complex will result in improvements of d=0.78 for cognition and d=0.28 for psychosocial functioning. While the time-course for such cognitive and functional changes in relation to increased EAIP is not known (see below, Fig. 4), these findings nonetheless suggest that interventions that reliably enhance measures of EAIP in SZ patients would be rational targets for therapeutic development.
Figure 4.

A model for how drug-induced gains in EAIP and “intermediate steps” (“Auditory Fidelity”) will enhance the therapeutic impact of TCT. We know that drugs can enhance EAIP (Light et al. 2017; Swerdlow et al. 2016), and that EAIP mediates neurocognition and function (Thomas et al. 2017). This “bottom-up” model predicts that increased auditory processing fidelity and auditory learning can be intermediate steps between drug-induced gains in EAIP, and augmented therapeutic effects of TCT. Each step in this model has a time constant, “τ”. For example we know that for memantine, τ1 ≈ 4h; we predict a tight physiological “coupling” of EAIP and auditory fidelity, i.e. a brief τ2. TCT enhances neurocognition and function after ≈ 40h of training, but we expect τ3 and τ4 to be shorter than this, if drug-enhanced auditory fidelity and learning accelerate and amplify the benefits of TCT.
To determine whether measures of EAIP retained plasticity in SZ patients, we also examined changes in EAIP after an acute drug challenge. Memantine (MEM) is an uncompetitive NMDA receptor antagonist with low-affinity but rapid blocking and unblocking ability. It has little impact on basal NMDA transmission; this distinguishes it mechanistically from other NMDA antagonists (Lipton 2006). It has positive effects on cognitive measures in both healthy animals and a range of human and animal models for dementia, depression, ischemia and neuroinflammation (Kim et al. 2010; Ma et al. 2015). MEM enhances hippocampal long-term potentiation (LTP), and reverses an experimentally-induced loss of LTP (Ma et al. 2015); it also alters excitation/inhibition (E/I) dynamics in frontal circuitry implicated in models of SZ neuropathology (Smith et al. 2011) and associated with MMN and cognitive deficits in SZ (Rowland et al. 2015). Of most relevance to the present topic, acute MEM increased MMN in healthy humans (Korostenskaja et al. 2007); in this particular study, MMN amplitude to frequency deviants increased 0.91 μV – enough to produce large effect-size increases in cognition in these healthy subjects. Such drug effects in intact / healthy brains support the therapeutic model proposed here, in which drugs target healthy rather than pathological brain circuitry.
2.1 Memantine and Early Auditory Information Processing
We studied the acute effects of MEM on measures of EAIP in chronic, antipsychotic (AP)-medicated SZ patients. In addition to MMN, we measured prepulse inhibition of acoustic startle (PPI) and the auditory steady state response (ASSR). These three measures were chosen because they: 1) are neurophysiological measures of EAIP, i.e. of the brain’s automatic response to a simple sensory event proximal to, or independent of, a point at which it engages conscious or volitional processing; 2) are reliable, objective and quantitative; 3) consistently detect EAIP deficits in SZ patients; 4) reflect “automatic” vs. volitional processes and are relatively insensitive to motivational or effort-based artifact; 5) are suited to repeated testing in a cross-over design without significant order or “carry-over” effects, and 6) are each regulated by NMDA mechanisms, with at least some evidence for enhanced performance associated with NMDA blockade (Korostenskaja et al. 2007; Swerdlow et al. 2009; Hiyoshi et al. 2014). The study (Light et al. 2017; Swerdlow et al. 2016) used a double-blind, placebo-controlled crossover comparison of placebo vs. MEM (10 and 20 mg po) in both SZ patients and healthy subjects (HS). Test days were separate by about 1 week. Details of the methods can be found in the original data reports (Light et al. 2017; Swerdlow et al. 2016).
In brief, MEM significantly enhanced performance in each of these 3 measures of EAIP (Fig. 2). For PPI (Fig. 2A), the effects were more robust among patients than HS; for MMN (Fig. 2B), they were somewhat more robust among HS than patients, and for ASSR (Figs. 2C, D), the effects were roughly comparable across groups. Compared to published findings of a 0.91 μV increase in MMN after 30 mg of MEM in HS (Korostenskaja et al. 2007), we detected a maximum increase of about 1.1 μV MMN after 20 mg of MEM in HS, but only a maximum increase of about 0.4 μV among SZ patients. Nonetheless, for each of the 3 EAIP measures, MEM “improved” performance levels, i.e. moved them in a direction associated with less pathology. Importantly, while in this group of patients there were significant deficits in MMN and ASSR, their PPI was quantitatively intact (consistent with the fact that all were AP-medicated, and almost all were taking 2nd-generation AP’s, which are known to normalize PPI (cf. Swerdlow et al. 2008)). Thus, MEM’s effects were not dependent on deficits in EAIP measures, and were not impacted by AP medication, also consistent with the possibility that MEM was acting on intact mechanisms that were performing at “normal levels”.
Figure 2.

Effects of memantine on A: PPI; B: MMN; C, D: ASSR in HS and SZ patients. SZ patients and HS (n’s=42 & 42) were tested after placebo or memantine (20 mg p.o. shown here). Memantine significantly enhanced PPI (Swerdlow et al. 2016) (A; p<0.04 for 10–120 ms; p<0.01 for 60 ms), MMN (Swerdlow et al. 2016) (B; p<0.014; Duration; Pitch; Combined) and ASSR (Light et al. 2017) (C: Evoked Power, 40 Hz *p<0.025; D: Gamma Phase Locking (Coherence), *p<0.002). Figure modified and reproduced from Swerdlow et al. (2016) and Light et al. (2017).
The only robust predictor of MEM “sensitivity” in these EAIP measures was patient age, which significantly predicted more sensitivity to the PPI-enhancing effects of MEM (p=0.005), but also significantly predicted less sensitivity to the MMN- (p<0.025) and ASSR-enhancing (coherence: p<0.005; power: p<0.0015) effects of MEM. While illness chronicity is often difficult to disentangle from subject age, each of the 4 relationships noted above were weakened when illness duration was substituted for age, such that only the negative relationships with MEM sensitivity on ASSR measures (p<0.045 and p<0.02) remained statistically significant.
Most importantly, this study revealed that, among EAIP measures thought to mediate cognition and function in SZ patients, significant plasticity (i.e. capacity for change) could be identified via an acute “challenge” with MEM. Clearly, not every patient exhibited this evidence of plasticity (Figure 3), consistent with the heterogeneous neurobiology of this disorder. When identifying “sensitive” subgroups (Figure 1), such heterogeneity is expected, and criteria can be tested and empirically validated to identify the magnitude of MEM-stimulated plasticity that predicts a sensitive treatment subgroup (Figure 3).
Figure 3.
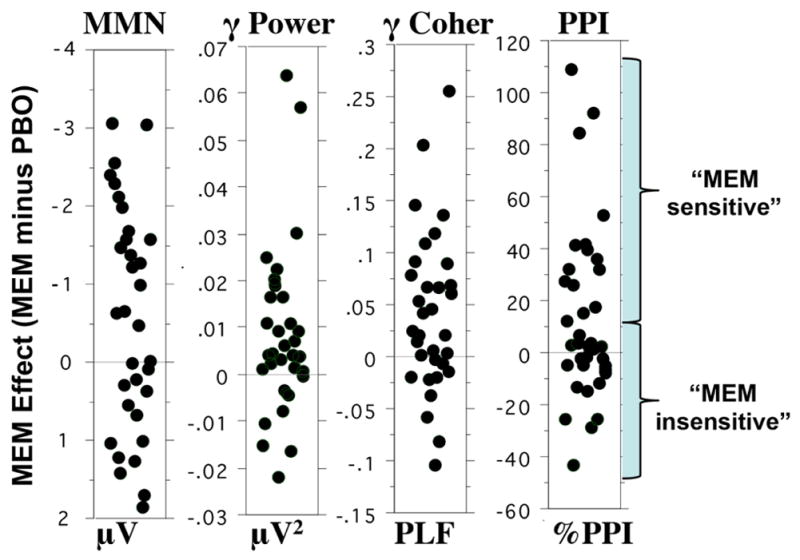
Distribution of mean values of memantine sensitivity (response after memantine minus response after placebo) for SZ subjects in the measures shown in Figure 2: MMN and ASSR (Gamma Band Power and Coherence) and PPI (Swerdlow et al. 2016; Light et al. 2017). Units are shown below each distribution. As a group, patients exhibited a significant increase in each EAIP measure after memantine, there was clear response heterogeneity. We propose to use this hetereogeneous EAIP response to acute drug challenge as evidence for available neuroplasticity within neurocognition-relevant brain mechanisms (EAIP), and hence as a biomarker to predict therapeutic sensitivity (example shown for PPI, based on a median split of MEM sensitivity).
Figure 1.

Schematic showing the idealized impact of adding a biomarker to a heterogeneous population defined by a clinical phenotype to enhance the likelihood of treatment response. = treatment-sensitive patient.
The expectation based on these findings is that, over time, in a sensitive subgroup identified based on the magnitude of increased EAIP after acute MEM challenge, MEM (or a mechanistically similar compound) should facilitate gains in neurocognition and function in AP-medicated schizophrenia patients. However, simply based on these findings, we absolutely would not expect severely ill schizophrenia patients to have an “awakening” of enhanced neurocognitive function based on EAIP gains after 1 pill of MEM. Nonetheless, we might reasonably expect that by positively engaging existing plasticity within systems responsible for EAIP, MEM will enhance the ability of these systems to benefit from exercises designed to train auditory-based cognitive processes. This (as of yet, untested) hypothesis is further discussed below.
2.2 Amphetamine and Auditory Learning in Targeted Cognitive Training
Because AP treatment largely fails to enhance function in SZ patients, alternative, non-pharmacologic treatments for SZ have been the focus of intensive investigation. Cognitive, behavioral and social therapies modestly reduce symptoms and improve function in SZ (Demily and Frank, 2008; Klingberg et al. 2009; Lecardeur et al. 2009; McGurk et al. 2007; Medalia and Choi 2009; Tai and Turkington 2009), with sustained benefits in many cases lasting years (Eack et al. 2010; Granholm et al. 2007; McGurk et al. 2009). In Targeted Cognitive Training (TCT), SZ patients train on auditory processing tasks concomitant with domain-specific attention and working-memory operations; this “bottom-up” approach seeks to improve speed and accuracy of auditory information processing, to generate “upstream” gains in cognition and function. Immediately after 50h of TCT, SZ patients show large effect-size gains in global cognition (Fisher et al. 2009), but durable changes for some patients may require extended training (Fisher et al. 2010), functional gains are modest, and individual responses are quite variable: 45% of SZ patients fail to benefit (d ≤ 0.2) (Murthy et al. 2012) even after 100h of TCT (Fisher et al. 2010). Given this rate of “non-response” and modest functional gains, the costs and logistical impediments associated with getting severely ill SZ patients to complete 100h of TCT can be prohibitive.
We, and others, have asked whether drugs can enhance the impact of cognitive therapies in SZ. Strategies for the Pharmacologic Augmentation of Cognitive Therapy (PACT) (Swerdlow 2011) in SZ would not involve the development of “new antipsychotics”: while suppressing active psychosis with APs can generally benefit a cognitive intervention, it might be possible to identify drugs that more specifically, and perhaps synergistically, enhance the clinical benefits of cognitive therapies. The goal of such interventions would be to enhance basic neural mechanisms that mediate cognitive and functional gains resulting from cognitive therapies.
Recent findings from our group (Perez et al. 2017) suggest that EAIP measures predict the amount of auditory learning by patients within a TCT session; it thus seems parsimonious to speculate that interventions that enhance EAIP should increase or accelerate learning, and hence clinical gains, from TCT. In a study of severely ill SZ patients, training-induced gains in auditory processing speed (APS) in the “sound Sweeps” auditory training paradigm – one component of the larger TCT suite – were significantly predicted by baseline (pre-training) MMN amplitude (r = 0.49; p<0.01).
The fact that patients are capable of perceptual “learning” as demonstrated by their enhanced APS in this Sound Sweeps paradigm by itself demonstrates some degree of plasticity; this plasticity is not limited to early sensory mechanisms, since therapeutic benefits of TCT generalize to several cognitive domains. Nonetheless, as noted above, clinical gains from TCT alone are only modest, and in a recent study, we asked whether APS learning could be enhanced pharmacologically. If so, this might demonstrate a mechanism by which plasticity could be potentiated in SZ patients, in a manner that – in theory – should increase or accelerate the clinical gains from TCT.
Because APS learning can be detected in a single session, it might be possible to detect the sensitivity to pharmacologically-enhanced TCT using a drug-challenge design, similar to that described above for MEM. We tested the effects of the psychostimulant, amphetamine (AMPH; 10 mg po), on APS learning in the Posit Science “Sound Sweeps” session, in both SZ patients and HS. Briefly, participants were presented with pairs of frequency-modulated sound “sweeps” and indicated whether they perceived each sweep as becoming higher or lower in pitch (Fisher et al. 2009). As in other studies of TCT, training was continuously adaptive: sweep duration, frequency range, and interstimulus interval (ISI) become shorter after correct responses, but longer after incorrect responses. To progress through training levels, participants must discriminate progressively shorter sound sweeps. Before and after each training session, participants completed a “baseline” APS assessment, which were used to generate standardized APS improvement scores for all participants. Detailed methods are found in (Swerdlow et al. 2017).
APS learning was significantly enhanced by AMPH vs. placebo, particularly (but not exclusively) in patients. Among patients, AMPH-induced increases in APS learning were positively associated with AMPH-enhanced Attention / Vigilance Domain scores on the MATRICS Cognitive Consensus Battery (r=0.34, p<0.05) (Swerdlow et al. 2017). Presumably, auditory learning mechanisms in SZ patients retain adequate plasticity such that stimulation of AMPH-sensitive mechanisms – neurochemical, neurocognitive (e.g. attention / vigilance), or both - are able to potentiate this learning. By comparing baseline (no pill) APS learning to post-pill APS learning, it was possible to demonstrate that patients retained their “extra” learning – the gains experienced during AMPH dosing – at least one week later. In other words, this AMPH-enhanced learning is not strictly “state-dependent”. Neither age nor illness chronicity were associated with AMPH-enhanced learning in these patients. Interestingly, the magnitude of the AMPH-enhanced learning was significantly correlated with measures of baseline EAIP (i.e., P3a latency; r=−0.43, p<0.05); AMPH effects on EAIP were not measured in this study.
3.1 Discussion
The observations that early auditory information processing (EAIP): 1) mediates cognition and function in SZ, and 2) retains significant plasticity as demonstrated by increases after acute pharmacologic challenge, and 3) that this plasticity can take the form of enhanced and retained learning, has several implications for SZ therapeutics.
The most straightforward predictions from these observations are that EAIP is a rational “target” for SZ medications, that EAIP appear pharmacologically sensitive, and that under the right conditions, drug-enhanced EAIP might be expected to be associated with improved cognitive, clinical and functional outcome. However, the most important of these predictions – that drug-enhanced EAIP might, under the right conditions, have a positive therapeutic impact, has not yet been tested. In fact, the only evidence to-date from the studies described above is that - despite gains in EAIP after one pill of MEM in SZ patients - measures 240 minutes post-pill detected a non-specific reduction in MATRICS Comprehensive Cognitive Battery (MCCB) performance among HS across all neurocognitive domains (Bhakta et al. 2016). The disconnect between acute changes in EAIP, and those of MCCB performance, might suggest: 1) changes in EAIP are orthogonal to neurocognition (not consistent with Thomas et al. (2017)); 2) “off-target” effects of 20 mg of MEM (4X larger than a typical starting dose) are independent of EAIP mechanisms, similar to off-target effects after initial exposure to other psychotherapeutic agents (Amado-Boccara et al. 1992); 3) insensitivity of the MCCB to procognitive changes after acute drug challenges (consistent with its insensitivity to acute pro-attentional effects of amphetamine; Chou et al., 2013; Swerdlow et al. 2017; and counter to the intended purpose for developing the MCCB); or 4) there are intermediate steps in information processing, between enhanced EAIP, and “downstream” gains in more complex cognitive domains. In fact, the strong mediating effect of EAIP on cognition and function in 1400 SZ patients was detected at a single point in time, typically decades after the onset of illness. Thus, the relationships represented in these mediating effects evolved over long periods of time; these relationships (e.g. 1 μV increase in EAIP translating to a large (d=0.78) gain in cognition) do not in any way suggest that a change in EAIP will lead to an instantaneous improvement in cognition, i.e. an “awakening”.
Conceivably, gains at many intermediate steps between enhanced EAIP and neurocognition–such as gains in the fidelity of an auditory signal, or gains in the amount of information capacity of that auditory signal – might occur with their own time constants (Figure 4); ultimately, when this enhanced processing capacity is matched with inputs that put demands on that capacity – such as might occur over 30–40 hours of TCT - these gains could augment the impact of TCT on neurocognition and function (Figure 4). This model, in which immediate changes in EAIP trigger a sequence of time-linked changes leading to an enhanced sensitivity to cognitive training, is both speculative and testable. No study has yet conducted the critical test to determine whether sensitivity to the acute EAIP-enhancing effects of MEM predict sensitivity to MEM-enhanced TCT performance or therapeutic impact. However, the evidence that EAIP predicts TCT gains, and that 30–40 h of TCT leads to cognitive and functional gains, provides a logical framework for such speculation.
Two critical points deserve clarification. First, the plasticity (capacity for change) demonstrated by an acute drug response is likely to be mechanistically distinct from the type of “neuroplasticity” often described in learning paradigms such as TCT. There is no presumption (and in fact, we would view it as very unlikely) that the mechanism underlying acute MEM-induced gains in EAIP measures reflected LTP or other lasting shifts in cell function that are hypothesized to regulate TCT and its impact on neurocognition. Second, none of our findings to date suggest that gains in EAIP after acute MEM challenge reflect “compensatory” changes in schizophrenia patients. In fact, to a greater or lesser degree, each of these changes is also detected in healthy subjects. For this reason, our working (though not tested) hypothesis is that the gains in EAIP after MEM challenge in schizophrenia patients reflect the action of MEM on residual healthy brain mechanisms. Whether these apparent “intact” brain mechanisms, identified via MEM challenge, can be harnessed in the service of therapeutic gains is another question awaiting experimental evidence.
In contrast to the use of a MEM challenge to detect “room to move” in measures of EAIP, our findings with AMPH-enhanced APS learning reflect a different strategy for demonstrating pharmacologically-accessible therapeutic “plasticity” in schizophrenia patients. In this case, AMPH challenge is used to assess gains in the specific behavioral process (learning to detect auditory frequency modulation) presumed to be the basis for at least some of the therapeutic impact of TCT. This approach avoids any inference related to the potential functional impact of increased drug-enhanced EAIP measures, but does assume that enhanced “learning” within a TCT module will ultimately translate to greater therapeutic impact. This assumption is currently being tested.
Adding drugs to already complex regimens of medications in chronic psychosis patients is never without risks, and such risks might be of particular concern when the added drugs produce neurochemical changes – e.g. NMDA blockade (memantine) and enhanced dopaminergic release (amphetamine) – that are associated mechanistically with models for the genesis or exacerbation of psychosis. However, in AP-medicated SZ patients (total n>1000), published findings of clinical trials with memantine augmentation report excellent safety (cf. Kishi et al. 2017; Zheng et al. 2017). Psychostimulant use in AP-medicated SZ patients has been less studied; our studies with a single dose of amphetamine detected no adverse consequences, consistent with a literature documenting the safety, and symptomatic and neurocognitive benefits, of amphetamine in antipsychotic-mediated schizophrenia patients (cf. Swerdlow et al. 2017). For example, in a controlled trial (Lasser et al. 2013), AMPH (5.9–20.8 mg/d) was administered daily for up to 14 weeks to antipsychotic-medicated schizophrenia patients, without increased adverse events either during treatment or upon withdrawal, and with evidence for some clinical gains. We also reported that SZ patients were less sensitive to autonomic activation after amphetamine, compared to HS (Swerdlow et al. 2017), consistent with findings from controlled trials (e.g. Modell & Hussar 1965). Using cross-species models, we reported (Swerdlow et al. 2017) that the effects of amphetamine on sensorimotor gating in AP-medicated SZ patients were best explained by increased prefrontal D1 activation together with subcortical D2 blockade, a profile that might be expected to maximize cognitive gains and minimize clinical risk among these patients.
Perhaps most importantly, the model proposed here to leverage drug-enhanced auditory plasticity in SZ patients would involve a very limited exposure to drug, since it would only be used during the course of TCT or other cognitive interventions. For example, in the case of amphetamine – which enhances TCT learning after a single pill – this might limit total drug exposure to 30–40 pills (1 per TCT session) over a 10-week period (approximately 15–50% of the total exposure documented to be safe by Lasser et al. (2013)). Nonetheless, it remains clear that drugs can have a propensity for precipitating psychosis in vulnerable individuals, and that any PACT strategy must be pursued weighing both the risks and benefits of added drug exposure even in the context of careful oversight and clinical monitoring.
We provide two examples of acute drug-enhanced auditory processing, with MEM (MEM-enhanced EAIP) and AMPH (AMPH-enhanced auditory frequency modulation learning). It is reasonable to consider whether other drugs might acutely enhance early auditory processing or learning, and thereby be candidate “pro-TCT” agents. To our knowledge, this possibility has not been tested with “sound sweeps” learning; in terms of EAIP measures, both MMN and ASSR are relatively insensitive to acute drug effects (with MEM being an exception to this rule, in both healthy subjects and schizophrenia patients (Korostenskaja et al., 2007; Swerdlow et al., 2016)). By contrast, PPI has been shown to be enhanced in healthy subjects by a number of drugs with different mechanisms, including atypical antipsychotics (Swerdlow et al., 2006; Vollenweider et al., 2006), amantadine (Swerdlow et al. 2002), ketamine (Duncan et al., 2001), nicotine (Hong et al., 2008) and MDMA (Vollenweider et al., 1999). We have reported that AMPH acutely enhances PPI in some subgroups of healthy individuals (Talledo et al. 2009) and in schizophrenia patients (Swerdlow et al. 2018). Clearly, the ability of a drug to enhance PPI in healthy subjects, absent other information, is not a sufficient basis to propose its utility in a PACT strategy.
In summary, we present evidence for the importance of EAIP in the mediation of cognition and function in SZ patients, and for available plasticity within EAIP systems to serve as a potential therapeutic target for pro-cognitive and TCT-enhancing interventions. The fact that substantial plasticity in EAIP in SZ patients is both experimentally demonstrable and augmented by available and well-tested drugs offers significant promise for new intervention models in SZ.
Acknowledgments
Role of the Funding Source: Supported by NIMH grants R01-MH059803 & R01-MH094320, and by a Distinguished Investigator Award from the Behavior & Brain Research Foundation (NRS) and by the Veteran’s Administration VISN 22 Mental Illness Research, Education, and Clinical Center (GAL). Dr. Bhakta was supported by 1KL2TR001444.
We thank Ms. Maria Bongiovanni for her expert assistance with manuscript preparation.
Footnotes
Contributors: Dr. Swerdlow wrote the first draft of the manuscript; Drs. Light and Bhakta made edits to this manuscript; all authors approved the final manuscript.
Conflict of Interest: Dr. Light has received consulting compensation from Astellas, Boehringer Ingelheim, Merck, Lundbeck, Neuroverse, NeuroSig and Takeda. Drs. Swerdlow and Bhakta report no consulting compensation.
Funding and Disclosure
Supported by R01-MH059803 & R01-MH094320, and by a Distinguished Investigator Award from the Behavior & Brain Research Foundation (NRS); SB was supported by 1KL2TR001444. GAL has served as a consultant for Astellas, Boehringer Ingelheim, Heptares, Merck, Lundbeck, Neuroverse, NeuroSig and Takeda.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amado-Boccara I, Gougoulis N, Poirier LM, Galinowski A, Loo H. Effects of antidepressants on cognitive functions: A review. Neurosci Biobehav Rev. 1992;19(3):479–93. doi: 10.1016/0149-7634(94)00068-c. [DOI] [PubMed] [Google Scholar]
- Barch DM. Pharmacological strategies for enhancing cognition in schizophrenia. Curr Top Behav Neurosci. 2010;4:43–96. doi: 10.1007/7854_2010_39. [DOI] [PubMed] [Google Scholar]
- Barch DM. There are currently no proven pharmacological or psychological treatments for the core cognitive deficits of schizophrenia. Biol Psychiatry. 2011;69:6S. [Google Scholar]
- Bhakta SG, Chou HH, Rana B, Talledo JA, Balvaneda B, Gaddis L, Light GA, Swerdlow NR. Effects of acute memantine administration on MATRICS Consensus Cognitive Battery performance in psychosis: Testing an experimental medicine strategy. Psychopharmacology (Berl) 2016;233:2399–2410. doi: 10.1007/s00213-016-4291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan RW, Javitt DC, Marder SR, Schooler NR, Gold JM, McMahon RP, Heresco-Levy U, Carpenter WT. The Cognitive and Negative Symptoms in Schizophrenia Trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psychiatry. 2007;164:1593–1602. doi: 10.1176/appi.ajp.2007.06081358. [DOI] [PubMed] [Google Scholar]
- Choi DC, Rothbaum BO, Gerardi M, Ressler KJ. Pharmacological enhancement of behavioral therapy: focus on posttraumatic stress disorder. Curr Top Behav Neurosci. 2010;2:279–299. doi: 10.1007/7854_2009_10. [DOI] [PubMed] [Google Scholar]
- Chou HH, Talledo J, Lamb S, Thompson W, Swerdlow NR. Amphetamine effects on MATRICS Consensus Cognitive Battery performance in healthy adults. Psychopharmacology. 2013;227:165–176. doi: 10.1007/s00213-012-2948-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demily C, Franck N. Cognitive remediation: a promising tool for the treatment of schizophrenia. Expert Rev Neurother. 2008;8:1029–1036. doi: 10.1586/14737175.8.7.1029. [DOI] [PubMed] [Google Scholar]
- Duncan EJ, Madonick SH, Parwani A, Angrist B, Rajan R, Chakravorty S, Efferen TR, Szilagyi S, Stephanides M, Chappell PB, Gonzenbach S, Ko GN, Rotrosen JP. Clinical and sensorimotor gating effects of ketamine in normals. Neuropsychopharmacology. 2001;25:72–83. doi: 10.1016/S0893-133X(00)00240-2. [DOI] [PubMed] [Google Scholar]
- Eack SM, Hogarty GE, Cho RY, Prasad KM, Greenwald DP, Hogarty SS, Keshavan MS. Neuroprotective effects of cognitive enhancement therapy against gray matter loss in early schizophrenia: results from a 2-year randomized controlled trial. Arch Gen Psychiatry. 2010;67:674–682. doi: 10.1001/archgenpsychiatry.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M, Holland C, Merzenich MM, Vinogradov S. Using neuroplasticity-based auditory training to improve verbal memory in schizophrenia. Am J Psychiatry. 2009;166:805–811. doi: 10.1176/appi.ajp.2009.08050757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M, Holland C, Subramaniam K, Vinogradov S. Neuroplasticity-based cognitive training in schizophrenia: an interim report on the effects 6 months later. Schizophr Bull. 2010;36:869–74. doi: 10.1093/schbul/sbn170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DC, Cather C, Gottlieb JD, Evins AE, Walsh J, Raeke L, Otto MW, Schoenfeld D, Green MF. Once-weekly D-cycloserine effects on negative symptoms and cognition in schizophrenia: an exploratory study. Schizophr Res. 2008;106:320–327. doi: 10.1016/j.schres.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DC, Keefe R, Citrome L, Davy K, Krystal JH, Large C, Thompson TR, Volavka J, Webster EL. Lamotrigine as add-on therapy in schizophrenia: results of 2 placebo-controlled trials. J Clin Psychopharmacol. 2007;27:582–589. doi: 10.1097/jcp.0b013e31815abf34. [DOI] [PubMed] [Google Scholar]
- Goff DC, Tsai G, Levitt J, Amico E, Manoach D, Schoenfeld DA, Hayden DL, McCarley R, Coyle JT. A placebo-controlled trial of D-cycloserine added to conventional neuroleptics in patients with schizophrenia. Arch Gen Psychiatry. 1999;56:21–27. doi: 10.1001/archpsyc.56.1.21. [DOI] [PubMed] [Google Scholar]
- Goff DC, Tsai G, Manoach DS, Flood J, Darby DG, Coyle JT. D-cycloserine added to clozapine for patients with schizophrenia. Am J Psychiatry. 1996;153:1628–1630. doi: 10.1176/ajp.153.12.1628. [DOI] [PubMed] [Google Scholar]
- Granholm E, McQuaid JR, McClure FS, Link PC, Perivoliotis D, Gottlieb JD, Patterson TL, Jeste DV. Randomized controlled trial of cognitive behavioral social skills training for older people with schizophrenia: 12-month follow-up. J Clin Psychiatry. 2007;68:730–737. doi: 10.4088/jcp.v68n0510. [DOI] [PubMed] [Google Scholar]
- Green MF. Cognition, drug treatment, and functional outcome in schizophrenia: A tale of two transitions. Am J Psychiatry. 2007;164:992–994. doi: 10.1176/ajp.2007.164.7.992. [DOI] [PubMed] [Google Scholar]
- Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 1996;153:321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- Hiyoshi T, Kambe D, Karasawa JI, Chaki S. Differential effects of NMDA receptor antagonists at lower and higher doses on basal gamma band oscillation power in rat cortical electroencephalograms. Neuropharmacology. 2014;85:384–396. doi: 10.1016/j.neuropharm.2014.05.037. [DOI] [PubMed] [Google Scholar]
- Hong LE, Wonodi I, Lewis J, Thaker GK. Nicotine effect on prepulse inhibition and prepulse facilitation in schizophrenia patients. Neuropsychopharmacology. 2008;33:2167–2174. doi: 10.1038/sj.npp.1301601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe RS, Bilder RM, Davis SM, Harvey PD, Palmer BW, Gold JM, Meltzer HY, Green MF, Capuano G, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Davis CE, Hsiao JK, Lieberman JA CATIE Investigators; Neurocognitive Working Group. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch Gen Psychiatry. 2007;64:633–647. doi: 10.1001/archpsyc.64.6.633. [DOI] [PubMed] [Google Scholar]
- Keefe RS, Haig GM, Marder SR, Harvey PD, Dunayevich E, Medalia A, Davidson M, Lombardo I, Bowie CR, Buchanan RW, Bugarski-Kirola D, Carpenter WT, Csernansky JT, Dago PL, Durand DM, Frese FJ, Goff DC, Gold JM, Hooker CI, Kopelowicz A, Loebel A, McGurk SR, Opler LA, Pinkham AE, Stern RG. Report on ISCTM Consensus Meeting on Clinical Assessment of Response to Treatment of Cognitive Impairment in Schizophrenia. Schizophr Bull. 2016;42:19–33. doi: 10.1093/schbul/sbv111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YW, Shin JC, An YS. Changes in cerebral glucose metabolism in patients with posttraumatic cognitive impairment after memantine therapy: a preliminary study. Ann Nucl Med. 2010;24:363–369. doi: 10.1007/s12149-010-0360-3. [DOI] [PubMed] [Google Scholar]
- Kishi T, Matsuda Y, Iwata N. Memantine add-on to antipsychotic treatment for residual negative and cognitive symptoms of schizophrenia: a meta-analysis. Psychopharmacology (Berl) 2017 doi: 10.1007/s00213-017-4616-7. in press. [DOI] [PubMed] [Google Scholar]
- Klingberg S, Wittorf A, Herrlich J, Wiedemann G, Meisner C, Buchkremer G, Frommann N, Wölwer W. Cognitive behavioural treatment of negative symptoms in schizophrenia patients: study design of the TONES study, feasibility and safety of treatment. Eur Arch Psychiatry Clin Neurosci. 2009;259:S149–S154. doi: 10.1007/s00406-009-0047-8. [DOI] [PubMed] [Google Scholar]
- Korostenskaja M, Nikulin VV, Kicić D, Nikulina AV, Kähkönen S. Effects of NMDA receptor antagonist memantine on mismatch negativity. Brain Res Bull. 2007;72:275–283. doi: 10.1016/j.brainresbull.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Lasser RA, Dirks B, Nasrallah H, Kirsch C, Gao J, Pucci ML, Knesevich MA, Lindenmayer JP. Adjunctive lisdexamfetamine dimesylate therapy in adult outpatients with predominant negative symptoms of schizophrenia: open-label and randomized-withdrawal phases. Neuropsychopharmacology. 2013;38:2140–9. doi: 10.1038/npp.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecardeur L, Stip E, Giguere M, Blouin G, Rodriguez JP, Champagne-Lavau M. Effects of cognitive remediation therapies on psychotic symptoms and cognitive complaints in patients with schizophrenia and related disorders: a randomized study. Schizophr Res. 2009;111:153–158. doi: 10.1016/j.schres.2009.03.029. [DOI] [PubMed] [Google Scholar]
- Leucht S, Arbter D, Engel RR, Kissling W, Davis JM. How effective are second-generation antipsychotic drugs? A meta-analysis of placebo-controlled trials. Mol Psychiatry. 2009;14:429–447. doi: 10.1038/sj.mp.4002136. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Light GA, Zhang W, Joshi YB, Bhakta S, Talledo JA, Swerdlow NR. Single-Dose memantine improves cortical oscillatory response dynamics in patients with schizophrenia. Neuropsychopharmacology. 2017 doi: 10.1038/npp.2017.81. Epub ahead of print, Apr. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- Ma J, Mufti A, Stan Leung L. Effects of memantine on hippocampal long-term potentiation, gamma activity, and sensorimotor gating in freely moving rats. Neurobiol Aging. 2015;36(9):2544–2554. doi: 10.1016/j.neurobiolaging.2015.05.017. [DOI] [PubMed] [Google Scholar]
- McGurk SR, Mueser KT, DeRosa TJ, Wolfe R. Work, recovery, and comorbidity in schizophrenia: a randomized controlled trial of cognitive remediation. Schizophr Bull. 2009;35:319–35. doi: 10.1093/schbul/sbn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk SR, Twamley EW, Sitzer DI, McHugo GJ, Mueser KT. A meta-analysis of cognitive remediation in schizophrenia. Am J Psychiatry. 2007;164:1791–802. doi: 10.1176/appi.ajp.2007.07060906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medalia A, Choi J. Cognitive remediation in schizophrenia. Neuropsychol Rev. 2009;19:353–64. doi: 10.1007/s11065-009-9097-y. [DOI] [PubMed] [Google Scholar]
- Modell W, Hussar AE. Failure of dextroamphetamine sulfate to influence eating and sleeping patterns in obese schizophrenic patients: Clinical and pharmacological significance. JAMA. 1965;193:275–8. doi: 10.1001/jama.1965.03090040019006. [DOI] [PubMed] [Google Scholar]
- Murthy NV, Mahncke H, Wexler BE, Maruff P, Inamdar A, Zucchetto M, Lund J, Shabbir S, Shergill S, Keshavan M, Kapur S, Laruelle M, Alexander R. Computerized cognitive remediation training for schizophrenia: an open label, multi-site, multinational methodology study. Schizophr Res. 2012;139:87–91. doi: 10.1016/j.schres.2012.01.042. [DOI] [PubMed] [Google Scholar]
- Norberg MM, Krystal JH, Tolin DF. A meta-analysis of D-cycloserine and the facilitation of fear extinction and exposure therapy. Biol Psychiatry. 2008;63:1118–1126. doi: 10.1016/j.biopsych.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Perez V, Tarasenko M, Miyakoshi M, Pianka S, Makeig S, Braff D, Swerdlow NR, Light G. Mismatch Negativity is a Sensitive and Predictive Biomarker of Perceptual Learning During Auditory Cognitive Training in Schizophrenia. Neuropsychopharmacology. 2017;42:2206–2213. doi: 10.1038/npp.2017.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K, Zimand E, Hodges L, Davis M. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- Rice DP. The economic impact of schizophrenia. J Clin Psychiatry. 2009;60(Suppl 1):4–6. discussion 28–30. [PubMed] [Google Scholar]
- Rowland LM, Summerfelt A, Wijtenburg SA, Du X, Chiapelli JJ, Krishna N, West J, Muellerklein F, Kochunov P, Hong LE. Frontal glutamate and γ-aminobutyric acid levels and their associations with mismatch negativity and digit sequencing task performance in schizophrenia. JAMA Psychiatry. 2016;73:166–174. doi: 10.1001/jamapsychiatry.2015.2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR. Are we studying and treating schizophrenia correctly? Schizophr Res. 2011;130:1–10. doi: 10.1016/j.schres.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR. Beyond antipsychotics: Pharmacologically-augmented cognitive therapies (PACTs) for schizophrenia. Neuropsychopharmacology. 2011;37:310–311. doi: 10.1038/npp.2011.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Bhakta S, Chou HH, Talledo JA, Balvaneda B, Light GA. Memantine Effects On Sensorimotor Gating and Mismatch Negativity in Patients with Chronic Psychosis. Neuropsychopharmacology. 2016;41:419–430. doi: 10.1038/npp.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Bhakta SG, Talledo JA, Franz DM, Hughes EL, Rana BK, Light GA. Effects of amphetamine on sensorimotor gating and neurocognition in antipsychotic-medicated schizophrenia patients. Neuropsychopharmacology. 2017 doi: 10.1038/npp.2017.285. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Bhakta S, Thomas M, Joshi Y, Zhang W, Talledo J, Light G. Leveraging auditory information processing neuroplasticity towards therapeutic development in schizophrenia. Biol Psychiatry. 2017;81:S175. [Google Scholar]
- Swerdlow NR, Bhakta SG, Talledo J, Franz DM, Hughes EL, Rana B, Light GA. Effects of Amphetamine on Sensorimotor Gating and Neurocognition in Antipsychotic-Medicated Schizophrenia Patients. Neuropsychopharmacology. 2018;43:708–717. doi: 10.1038/npp.2017.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Stephany N, Shoemaker JM, Ross L, Wasserman LC, Talledo J, Auerbach PP. Effects of amantadine and bromocriptine on startle and sensorimotor gating: parametric studies and cross-species comparisons. Psychopharmacology. 2002;164:82–92. doi: 10.1007/s00213-002-1172-5. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Talledo J, Sutherland AN, Nagy D, Shoemaker JM. Antipsychotic effects on prepulse inhibition in normal ‘low gating’ humans and rats. Neuropsychopharmacology. 2006;31:2011–2021. doi: 10.1038/sj.npp.1301043. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Tarasenko M, Bhakta SG, Talledo J, Alvarez AI, Hughes EL, Rana B, Vinogradov S, Light GA. Amphetamine enhances gains in auditory discrimination training in adult schizophrenia patients. Schizophrenia Bulletin. 2017;43:872–880. doi: 10.1093/schbul/sbw148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, van Bergeijk DP, Bergsma F, Weber E, Talledo J. The effects of memantine on prepulse inhibition. Neuropsychopharmacology. 2009;34:1854–64. doi: 10.1038/npp.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology. 2008;199:331–88. doi: 10.1007/s00213-008-1072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai S, Turkington D. The evolution of cognitive behavior therapy for schizophrenia: current practice and recent developments. Schizophr Bull. 2009;35:865–73. doi: 10.1093/schbul/sbp080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talledo JA, Sutherland Owens AN, Schortinghuis T, Swerdlow NR. Amphetamine effects on startle gating in normal women and female rats. Psychopharmacology. 2009;204:165–75. doi: 10.1007/s00213-008-1446-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas ML, Green MF, Hellemann G, Sugar CA, Tarasenko M, Calkins ME, Greenwood TA, Gur RE, Gur RC, Lazzeroni LC, Nuechterlein KH, Radant AD, Seidman LJ, Shiluk AL, Siever LJ, Silverman JM, Sprock J, Stone WS, Swerdlow NR, Tsuang DW, Tsuang MT, Turetsky BI, Braff DL, Light GA. Modeling the Cascade of Deficits from Early Auditory Information Processing to Psychosocial Functioning in Schizophrenia. JAMA Psychiatry. 2017;74:37–46. doi: 10.1001/jamapsychiatry.2016.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollenweider FX, Barro M, Csomor PA, Feldon J. Clozapine enhances prepulse inhibition in healthy humans with low but not with high prepulse inhibition levels. Biol Psychiatry. 2006;60:597–603. doi: 10.1016/j.biopsych.2006.03.058. [DOI] [PubMed] [Google Scholar]
- Vollenweider FX, Remensberger S, Hell D, Geyer MA. Opposite effects of 3,4-methylenedioxymethamphetamine (MDMA) on sensorimotor gating in rats versus healthy humans. Psychopharmacology. 1999;143:365–372. doi: 10.1007/s002130050960. [DOI] [PubMed] [Google Scholar]
- Zheng W, Li XH, Yang XH, Cai DB, Ungvari GS, Ng CH, Wang SB, Wang YY, Ning YP, Xiang YT. Adjunctive memantine for schizophrenia: a meta-analysis of randomized, double-blind, placebo-controlled trials. Psychol Med. 2017;22:1–10. doi: 10.1017/S0033291717001271. [DOI] [PubMed] [Google Scholar]