

Abstract
The cannabinoid receptor type 2 (CB2) is a G protein-coupled seven transmembrane receptor that transmits endogenous cannabinoid signaling. The role of CB2 in the pathogenesis of kidney injury and fibrosis remains poorly understood. Here we demonstrate that CB2 was induced, predominantly in kidney tubular epithelium, in various models of kidney disease induced by unilateral ureteral obstruction, adriamycin or ischemia/reperfusion injury. In vitro, forced expression of CB2 or treatment with a CB2 agonist was sufficient to trigger matrix gene expression, whereas knockdown of CB2 by siRNA abolished transforming growth factor-β1-induced signaling and fibrogenic responses in kidney tubular cells. CB2 also mediated fibroblasts and macrophage activation in vitro. Mice with genetic ablation of CB2 were protected against kidney injury after ureteral obstruction, validating a pathogenic role of CB2 in renal fibrosis in vivo. By using in silico screening and medicinal chemistry modifications, we discovered a novel compound, XL-001, that bound to CB2 with high affinity and selectivity, and acted as an inverse agonist. Incubation with XL-001 inhibited in a dose-dependent fashion the fibrogenic response induced by CB2 overexpression, CB2 agonist or transforming growth factor-β1. In vivo, intraperitoneal injections of XL-100 ameliorated kidney injury, fibrosis and inflammation in both the obstruction and ischemia/reperfusion models. Delayed administration of XL-001 was also effective in ameliorating kidney fibrosis and inflammation. Thus, CB2 is a pathogenic mediator in kidney fibrosis and targeted inhibition with the novel inverse agonist XL-001 may provide a strategy in the fight against fibrotic kidney diseases.
Keywords: CB2, cannabinoid, renal fibrosis, inverse agonist, chronic kidney disease
Introduction
Kidney fibrosis is a hallmark and major outcome of virtually all kinds of progressive chronic kidney disease (CKD).1–3 Numerous epidemiological studies have shown that the prevalence of CKD in the general population is high and reaches to 13% in some countries.4–6 In this context, CKD is increasingly becoming a public health problem worldwide.7 Despite its high prevalence and severe morbidity and mortality, there is currently no effective therapy that can completely halt or reverse renal fibrogenesis and the progression of CKD.8 This enormous unmet medical need calls for more studies and better understanding of the patho-mechanism of kidney fibrosis, ultimately enabling one to identify new and effective targets for a therapeutic intervention of CKD.
The endocannabinoid system is an essential signaling scheme that regulates a diverse array of biological processes including memory, appetite, energy metabolism, and immunity.9–11 Endocannabinoids, such as anandamide (AEA) and 2-arachidonoyl glycerol (2-AG), are endogenous lipid ligands, which can bind to two G-protein-coupled seven transmembrane cannabinoid receptors, namely CB1 and CB2.12 Ligand-CB1 or -CB2 receptor engagement triggers a cascade of intracellular signal activation, leading to specific gene expression and various cellular responses. Earlier studies demonstrate that CB1 receptor is mainly expressed in the central and peripheral nervous systems, whereas CB2 is primarily produced by cells of hematopoietic origin including monocytes/macrophages.13, 14 However, recent findings point out that both CB1 and CB2 are widely expressed in a variety of organs, particularly in the pathologic conditions.15, 16 Such a wide expression pattern of CB receptors suggests a much broader spectrum of their biologic activities than originally thought. Indeed, both CB1 and CB2 have been implicated in regulating injury repair, inflammation and fibrosis in several organs including liver, lung and kidney after various insults.17–19
In the kidney, both CB1 and CB2 receptors are expressed and may play a role in the pathogenesis of kidney disorders.20, 21 Studies show that the abuse of synthetic cannabinoids is linked to acute kidney injury, as well as tubulointerstitial lesions.22, 23 Targeted inhibition of CB1 is known to protect podocyte integrity and reduce kidney fibrosis after chronic injury.19, 24 While the role of CB1 activation in mediating kidney injury is well established,16, 19, 25, 26 the function of renal CB2 is inconsistent and controversial.27, 28 Some studies show that CB2 expression is down-regulated in the kidney of diabetic nephropathy,29 whereas others demonstrate an induction of CB2 mRNA and protein after unilateral ureteral obstruction (UUO).27 Activation of CB2 by agonists has been shown to protect against albuminuria in diabetic nephropathy and alleviates renal fibrosis.19, 26 However, CB2 activation also augments immune cell influx, aggravates inflammation and modulates α-smooth muscle actin (α-SMA) positive myofibroblasts in fibrotic repair.30, 31 Hence, the potential role of CB2 in renal fibrosis remains to be defined by using more comprehensive approaches.
In this study, we have systematically investigated the role of CB2 in renal fibrogenesis by using in vitro and in vivo models, and by taking both genetic and pharmacologic approaches. More importantly, we have discovered a novel, specific and highly selective CB2 inverse agonist, which blocked CB2 actions. Our results indicate that CB2 activation plays an important role in mediating kidney fibrosis and inflammation, and identify this pathway as a potential therapeutic target.
Results
CB2 is upregulated in various models of CKD
We first examined the expression and regulation of CB2 in the pathogenesis of kidney fibrosis. To this end, we used three mouse models of CKD induced by UUO, adriamycin (ADR) or ischemia-reperfusion injury (IRI), respectively. These models are widely utilized and represent different etiologies leading to renal fibrosis.32–34 As shown in Figure 1, a and b, quantitative real-time RT-PCR (qRT-PCR) revealed that both CB1 and CB2 mRNA were significantly upregulated in the obstructed kidneys at 7 days after UUO, compared to sham controls. We next sought to determine the cellular source of CB2 protein expression in fibrotic kidneys by using immunohistochemical staining. As shown in Figure 1c, whereas CB2 protein was hardly detectable in sham control kidneys, it was markedly upregulated in the obstructed kidneys after UUO. The expression of CB2 was predominantly in renal tubular epithelium (Figure 1c, arrow). Notably, some interstitial cells were also stained positively for CB2 after obstructive injury (Figure 1c, arrowhead). To quantitatively determine the relative abundance of CB2 protein, we carried out Western blot analyses of whole kidney lysates. As shown in Figure 1, d and e, CB2 protein was induced about 8-folds in the obstructed kidneys at 7 days after UUO, compared with sham controls. Similar results were obtained when renal CB2 levels were examined at 3 weeks after ADR injection (Figure 1, f and g), or at 11 days after unilateral IRI (Figure 1, h and i). These data indicate that CB2 induction is a common pathologic feature in the fibrotic kidneys after various injuries.
Figure 1.

Renal expression of CB2 is induced in various models of CKD. (a, b) Quantitative real-time RT-PCR (qRT-PCR) showed the relative abundances of CB1 (a) and CB2 (b) mRNA in sham and UUO kidneys at 7 days after UUO. *P < 0.05 versus sham controls (n = 5 to 6). (c) Immunohistochemical staining demonstrated CB2 protein expression and localization in the obstructed kidneys at 7 days after UUO. Paraffin-embedded kidney sections were stained with CB2 antibody. Boxed area was enlarged. Arrow indicates positive tubular staining. Arrowhead denotes interstitial cells stained positively for CB2. Scale bar, 50 μm. (d, e) Western blotting analyses showed the renal expressions of CB2 protein in the obstructed kidneys at 7 days after UUO. Representative Western blot (d) and quantitative data (e) are presented. Numbers (1 ~ 4) indicate each individual animal in a given group. Relative CB2 levels (sham controls = 1.0) were presented after normalization with actin. *P < 0.05 versus sham controls (n = 5 to 6). (f, g) Renal CB2 expression is induced in adriamycin (ADR) nephropathy. CB2 expression was assessed in the kidneys at 3 weeks after ADR injection. Representative Western blot (f) and quantitative data (g) are presented. Numbers (1 ~ 4) indicate each individual animal in a given group. Relative CB2 levels (controls = 1.0) were presented after normalization with actin. *P < 0.05 versus controls (n = 5 to 6). (h, i) Western blotting analyses demonstrated renal CB2 expression at 11 days after ischemia/reperfusion injury (IRI). Representative Western blot (h) and quantitative data (i) are presented. Numbers (1 ~ 4) indicate each individual animal in a given group. Relative CB2 levels (sham controls = 1.0) were presented after normalization with actin. *P < 0.05 versus controls (n = 5).
CB2 promotes fibrogenic responses in vitro
To investigate the functional role of CB2 in renal fibrosis, we employed a gain-of-function approach by forced expression of CB2 in kidney tubular epithelial cells. To this end, human proximal tubular epithelial cells (HKC-8) were transiently transfected with CB2 expression vector (pCMV-CB2) or empty vector (pcDNA3). As shown in Figure 2, a and b, overexpression of CB2 induced fibronectin expression in cultured HKC-8 cells, suggesting a fibrogenic action of the CB2 receptor in kidney cells.
Figure 2.

CB2 mediates TGF-β1-induced fibrogenic responses in vitro. (a, b) Over-expression of CB2 induced fibronectin expression in vitro. HKC-8 cells were transfected with CB2 expression vector (pCMV-CB2) or empty vector (pcDNA3) as indicated. Numbers (1 to 3) represent triplicate wells in a given group. Representative Western blot (a) and quantitative data (b) are presented. *P < 0.05 versus pcDNA3 group. (c, d) Western blotting analyses show that TGF-β1 dose-dependently induced CB2 expressions in cultured tubular epithelial cells in vitro. Human kidney proximal tubular cells (HKC-8) were incubated with different concentrations of TGF-β1 as indicated. Representative Western blot (c) and quantitative data (d) are presented. *P < 0.05 versus controls. (e–g) Knockdown of CB2 inhibited TGF-β1-induced fibrogenic responses in vitro. HKC-8 cells were transfected with CB2-specific siRNA or control siRNA, followed by incubation with TGF-β1 (2 ng/ml). Cell lysates were immunoblotted with antibodies against CB2, fibronectin, α-SMA and GAPDH, respectively. Representative Western blot (e) and quantitative data on the relative abundance of fibronectin (f) and α-SMA (g) proteins in different groups are presented. *P < 0.05 versus controls. †P < 0.05 versus control siRNA in the presence of TGF-β1. (h) Representative images show immunofluorescence staining of fibronectin in different groups. Arrow indicates positive staining. Scale bar, 50 μm. (i) Western blotting analyses show that TGF-β1-induced signal transduction was significantly attenuated by knockdown of CB2. Human kidney proximal tubular cells (HKC-8) were transfected with CB2 siRNA, then incubated with TGF-β1. Quantitative data (j through m) are presented. *P < 0.05 versus controls. †P < 0.05 versus control siRNA in the presence of TGF-β1.
We next examined the potential upstream regulator of CB2 in the fibrotic kidneys. As TGF-β1 is a well-characterized profibrotic cytokine that is induced in virtually all forms of CKD,35 we investigated whether CB2 expression is regulated by TGF-β1 using an in vitro system. As shown in Figure 2, c and d, TGF-β1 significantly induced CB2 expression in a dose-dependent manner, suggesting a potential role for TGF-β1 in mediating CB2 induction in fibrotic kidneys.
Given that TGF-β1 induces CB2 expression, we then explored whether CB2 is required for the profibrotic action of TGF-β1 in kidney epithelial cells. Therefore, we knocked down the expression of CB2 by using a small interfering RNA (siRNA) strategy. As illustrated in Figure 2, e through g, HKC-8 cells were transfected with control or CB2-specific siRNA in the presence of TGF-β1. Efficient knockdown of the CB2 expression by siRNA was confirmed in whole-cell lysates (Figure 2e). Notably, depletion of CB2 in HKC-8 cells inhibited the expression of fibronectin and α-smooth muscle actin (α-SMA) by TGF-β1 (Figure 2, e through g). Similar results were obtained when fibronectin deposition was assessed by immunofluorescence staining (Figure 2h).
We then examined the role of CB2 in TGF-β1 signaling. As shown in Figure 2, i through m, knockdown of CB2 hampered TGF-β1-mediated phosphorylation and activation of Smad3, extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) in HKC-8 cells. These results suggest CB2 is also required for TGF-β1 signaling.
CB2 mediates interstitial fibroblast and macrophage activation in vitro
To investigate the role of CB2 in renal interstitial cells, we employed an in vitro system using cultured normal rat kidney fibroblast cells (NRK-49F) and mouse macrophage cells (RAW264.7). As shown in Figure 3, a and b, the expression of CB2 was induced by TGF-β1 in NRK-49F cells. Furthermore, knockdown of CB2 by siRNA approach hindered TGF-β1-mediated fibronectin and collagen I expression (Figure 3, c through e), suggesting that CB2 plays a critical role in mediating fibroblast activation and matrix production.
Figure 3.

CB2 mediates the activations of fibroblasts and macrophages. (a, b) Western blotting analyses show that TGF-β1 induced CB2 expressions in fibroblasts in vitro. Rat normal kidney interstitial fibroblasts cells (NRK-49F) were incubated with TGF-β1 (2 ng/ml). Representative Western blot (a) and quantitative data (b) are presented. *P < 0.05 versus controls. (c–e) Knockdown of CB2 inhibited TGF-β1-induced fibrogenic responses in NRK-49F cells in vitro. NRK-49F cells were transfected with CB2-specific siRNA or control siRNA, followed by incubation with TGF-β1 (2 ng/ml). Cell lysates were immunoblotted with antibodies against fibronectin, collagen I and α-tubulin, respectively. Representative Western blot (c) and quantitative data on the relative abundance of fibronectin (d) and collagen I (e) proteins in different groups are presented. *P < 0.05 versus controls. †P < 0.05 versus control siRNA in the presence of TGF-β1. (f, g) Western blotting analyses show that lipopolysaccharide (LPS) induced CB2 expressions in macrophages in vitro. Mouse macrophage cell line (RAW264.7) was incubated with LPS (5 ng/ml). Representative Western blot (f) and quantitative data (g) are presented. *P < 0.05 versus controls. (h–j) Knockdown of CB2 inhibited LPS-induced M1-macrophage activation in vitro. RAW264.7 cells were transfected with CB2-specific siRNA or control siRNA, followed by incubation with LPS (5 ng/ml). Cell lysates were immunoblotted with antibodies against TNF-α, iNOS and GAPDH, respectively. Representative Western blot (h) and quantitative data on the relative abundance of TNF-α (i) and iNOS (j) proteins in different groups are presented. *P < 0.05 versus controls. †P < 0.05 versus control siRNA in the presence of LPS. (k–l) Knockdown of CB2 inhibited IL-4-induced M2-macrophage activation in vitro. RAW264.7 cells were transfected with CB2-specific siRNA or control siRNA, followed by incubation with IL-4 (20 ng/ml). Cell lysates were immunoblotted with antibodies against Mannose R (MR) and actin, respectively. Representative Western blot (k) and quantitative data on the relative abundance of MR (l) proteins in different groups are presented. *P < 0.05 versus controls. †P < 0.05 versus control siRNA in the presence of IL-4.
We then investigated the role of CB2 in macrophage activation in vitro. As shown in Figure 3, f and g, activation of macrophages by lipopolysaccharide (LPS) induced CB2 expression in RAW264.7 cells. Interestingly, deprivation of CB2 attenuated LPS-mediated induction of M1 macrophage markers tumor necrosis factor-α (TNF-α) and inducible nitric oxide synthase (iNOS) (Figure 3, h through j). Similarly, knockdown of CB2 also reduced interleukin-4 (IL-4)-induced M2 macrophage marker mannose R (MR) (Figure 3, k and l). These results suggest that CB2 is also instrumental in mediating both M1 and M2 macrophage activation.
Mice with CB2 deficiency are protected against renal fibrosis
To define the role of CB2 in renal fibrosis in vivo, we utilized CB2 null mice (CB2−/−) in which CB2 gene is globally disrupted. The CB2−/−mice displayed no overt abnormality in kidney morphology and function in normal physiological conditions, compared to wild-type controls (CB2+/+). Age- and sex-matched CB2−/− and CB+/+ mice in the same genetic background were subjected to UUO. At 7 days after UUO, renal fibrotic lesions were assessed. As shown in Figure 4, a and b, ablation of CB2 markedly reduced collagen accumulation and deposition in renal tubulointerstitium, as illustrated by Masson’s trichrome staining. To quantitatively determine fibrotic lesions in vivo, we further examined renal expression of fibronectin and α-SMA proteins by Western blot analyses of whole kidney lysates. As shown in Figure 4, c through e, and Supplementary Figure S1, ablation of CB2 significantly attenuated renal expression of fibronectin and α-SMA. Similar results were obtained when kidney sections were immunostained for fibronectin and α-SMA, respectively (Figure 4f), suggesting that CB2 signaling plays a critical role in promoting renal myofibroblast activation and interstitial matrix production after UUO.
Figure 4.

Mice with CB2 deficiency are protected against renal fibrosis after obstructive injury. (a) Representative micrographs show Masson’s trichrome staining (MTS) of the obstructed kidneys in CB2 null mice (CB2−/−) and wild-type controls (CB2+/+) at 7 days after UUO. Blue staining (arrow) indicates fibrotic collagen deposition. Scale bar, 50 μm. (b) Quantitative determination of the fibrotic area in CB2−/−null mice and CB2+/+ wild-type controls at 7 days after UUO. *P < 0.05 versus CB2+/+ group (n = 5 to 6). (c) Western blot analyses show that ablation of CB2 reduced renal expression of fibronectin and α-SMA after UUO. Numbers (1 to 4) represent different individual animals in a given group. (d, e) Graphic presentations of relative renal fibronectin (d) and α-SMA (e) protein levels in two groups as indicated. *P < 0.05 versus CB2+/+ group. (f) Immunostaining for renal fibronectin and α-SMA proteins in CB2+/+ and CB2+/+ mice as indicated. Frozen sections and paraffin sections were stained with different antibodies against fibronectin and α-SMA, respectively. Arrow indicates positive staining. Scale bar, 50 μm. (g) Western blot analyses show renal expression of β-catenin and its downstream MMP-7 after UUO. Numbers (1 to 4) represent different individual animals in a given group. (h, i) Graphic presentations of renal β-catenin (h) and MMP-7 (i) expressions in two groups as indicated. *P < 0.05 versus CB2+/+ mice. (j) Immunostaining for renal β-catenin proteins in CB2+/+ and CB2+/+ mice as indicated. Arrow indicates positive staining. Scale bar, 50 μm.
To investigate the potential mechanism by which CB2 promotes renal fibrosis, we then examined renal expression of β-catenin, a principal mediator of canonic Wnt signaling, as extensive studies indicate that hyperactive Wnt/β-catenin plays a crucial role in renal fibrogenesis.36, 37 As shown in Figure 4, g and h, as well as in Supplementary Figure S1, Western blotting analyses revealed that ablation of CB2 substantially inhibited renal expression of β-catenin protein. In agreement with this, CB2 deficiency also inhibited renal expression of matrix metalloproteinase-7 (MMP-7) (Figure 4, g and i), a direct downstream target of Wnt/β-catenin.38 Immunohistochemical staining also showed a reduced β-catenin protein in CB2−/− kidneys after UUO, compared to CB2+/+ controls (Figure 4j). Furthermore, ablation of CB2 also inhibited renal Smad3 and ERK activation after obstructive injury (Supplementary Figure S1).
XL-001 as a novel CB2 inverse agonist
Having established a crucial role of CB2 in renal fibrosis, we then sought to develop an inhibitor that can block both constitutive and stimulated CB2 activity. To identify novel CB2-selective ligands, an in silico screening was carried out (Figure 5a), and triaryl sulfonamide was discovered as a new scaffold possessing significant CB2 receptor affinity and selectivity.39 To get a more effective drug, we further carried out computational-guided lead optimization and medicinal chemistry synthesis and discovered a more potent compound, XL-001 (Figure 5, a and b), which exhibited the strong binding affinity to CB2 receptor with Ki of 0.5 nM (Figure 5c). This compound also exhibited the best CB2 selectivity over the CB1 receptor, with a selectivity index of 2594.39
Figure 5.

Discovery and characterization of compound XL-001, a novel inverse agonist of CB2. (a) Screening strategy for identification of XL-001. (b) The chemical structure of XL-001. (c) Binding affinity of XL-001 on the CB2 receptor (CB2 Ki = 0.5 nM). (d) Binding pose of XL-001 with the CB2 receptor. (e, f) The detailed 2D (e) and 3D (f) interactions between XL-001 and CB2. Hydrogen bonding interactions of XL-001 with Asn20 (2.20 Å) and Ser285 (3.67 Å) of CB2. The p-diehtylaminobenzene group formed strong hydrophobic interactions with Thr114 (3.74 Å), Trp194 (3.29 Å), and Trp258 (3.52 Å). Moreover, the p-methoxylbenzene group interacted strongly with Phe87 (3.46 Å) and Val113 (3.62 Å), while the p-methylbenzene group had a strong hydrophobic interaction with Phe183 (3.77 Å).
We then performed molecular docking study for exploring the interaction between XL-001 and CB2. Surflex-Dock GeomX (SFXC), a docking program in SYBYL was used to generate the detailed ligand-receptor interactions, in which the docking score was expressed as -log10 (Kd).40 The putative binding pocket of CB2 was predicted using Fast Connolly Type implemented in the MOLCAD module in SYBYL-X 1.3 (Figure 5d). The detailed 2D and 3D interactions between XL-001 and CB2 were presented in Figure 5, e and f, showing the docking score of 9.5, which is well correlated to its experimental binding affinity. The sulfonyl group in XL-001 formed a strong hydrogen bonding interaction with Asn20 (2.20Å) and Ser285 (3.67 Å), respectively. The p-diehtylaminobenzene group formed strong hydrophobic interactions with Thr114 (3.74 Å), Trp194 (3.29 Å), and Trp258 (3.52 Å). Moreover, the p-methoxylbenzene group interacted strongly with Phe87 (3.46 Å) and Val113 (3.62 Å), while the p-methylbenzene group had a strong hydrophobic interaction with Phe183 (3.77 Å). All these results suggest a strong binding affinity of XL-001 with the CB2 receptor.
XL-001 inhibits CB2-mediated fibrogenic response in vitro
We then tested the inhibitory effect of XL-001 on CB2-mediated fibrogenic response in vitro. In cultured HKC-8 cells, incubation with a specific CB2 agonist AM1241 markedly induced fibronectin and α-SMA expression (Figure 6a), underscoring that CB2 activation is sufficient to promote a fibrotic response. Interestingly, XL-001 antagonized the fibrogenic action of AM1241 and abolished fibronectin and α-SMA expression (Figure 6, a through c). Similarly, constitutive expression of CB2 alone in HKC-8 cells stimulated fibrogenic responses, and AM1241 further promoted fibronectin and α-SMA expression (Figure 6d). XL-001 not only completely abolished the effect of AM1241, but also obliterated fibronectin and α-SMA expression induced by CB2 overexpression alone (Figure 6, d through f), consistent with its capacity as an inverse agonist.
Figure 6.

XL-001 inhibits CB2-mediated fibrogenic responses in vitro. (a) Western blot analyses show that XL-001 inhibited CB2 agonist-induced fibronectin and α-SMA protein expression. HKC-8 cells were incubated with specific CB2 agonist (AM1241) in the absence or presence of XL-001 for 24 hours. (b, c) Graphic presentations of the relative abundances of fibronectin (b) and α-SMA (c) proteins in different groups as indicated. *P < 0.05 versus controls; †P < 0.05 versus AM1241 alone. (d–e) XL-001 acted as a CB2 inverse agonist. HKC-8 cells were transiently transfected with CB2 expression vector (pCMV-CB2), and then incubated with AM1241 or XL-001 as indicated. Representative Western blotting (d) and quantitative data on fibronectin (e) and α-SMA (f) are presented. *P < 0.05 versus controls; †P < 0.05 versus pCMV-CB2 alone. (g) XL-001 also dose-dependently inhibited TGF-β1-induced fibronectin and α-SMA protein expression. HKC-8 cells were incubated with TGF-β1 (2 ng/ml) in the absence or presence of XL-001 for 24 hours. (h, i) Graphic presentations of the relative abundances of fibronectin (h) and α-SMA (i) proteins in different groups as indicated. *P < 0.05 versus controls; †P < 0.05 versus TGF-β1 alone. (j) Representative micrographs show the immunofluorescence staining of fibronectin expressions in different groups as indicated. Scale bar, 50 μm.
XL-001 was also able to inhibit TGF-β1-induced fibronectin and α-SMA protein expression in HKC-8 cells in a dose-dependent manner (Figure 6, g through i), suggesting the potent ability of this compound in abolishing TGF-β1-triggered fibrogenic response. Similarly, XL-001 also completely inhibited fibronectin assembly and deposition in the extracellular space in vitro, as illustrated by immunofluorescence staining for fibronectin (Figure 6j). Furthermore, XL-001 inhibited the TGF-β1-triggered Smad3 and MAPK activation (Supplementary Figure S2). These data suggest that XL-001, a novel CB2 inverse agonist, is a potent inhibitor that represses matrix gene expression in response to both CB2 activation and TGF-β1 stimulation.
XL-001 ameliorates fibrosis in obstructive nephropathy in vivo
The ability of XL-001 in blocking CB2- and TGF-β1-mediated fibrogenic response in vitro prompted us to test its potency in ameliorating kidney fibrosis in vivo. To this end, groups of mice were subjected to UUO, and treated with different doses of XL-001 via daily intraperitoneal injection (Figure 7a). As shown in Figure 7b, Masson’s trichrome staining revealed that administration of XL-001 dose-dependently reduced renal fibrotic lesions at 7 days after UUO. Quantitative determination by computer-aided morphometric analyses showed that XL-001 at 20 mg/kg significantly reduced renal fibrosis (Figure 7c). Similarly, immunostaining for collagen I and α-SMA also indicated that XL-001 inhibited the induction of these proteins in the obstructed kidneys (Figure 7d, arrows). Western blot analyses of whole kidney lysates further confirmed that XL-001 reduced renal fibronectin and α-SMA expression after UUO (Figure 7, e and g). To confirm the impact of CB2 inhibition on tubular injury, we examined renal expression of kidney injury molecule-1 (Kim-1), a marker for tubular damage, by western blotting. As shown in Figure 7, e and f, XL-001 was able to inhibit renal Kim-1 expression after UUO, illustrating its ability to prevent tubular injury. Similar results were obtained after semi-quantification of tubular injury score in kidney sections (Supplementary Figure S3).
Figure 7.

XL-001 reduces renal fibrosis in obstructive nephropathy. (a) Experimental design. (b) Representative micrographs show that XL-001 reduced renal interstitial collagen deposition and fibrosis. Kidney sections were subjected to Masson's trichrome staining (MTS). Arrow indicates positive staining. Scale bar, 50 μm. XL (10), XL-001 (10 mg/kg body weight); XL (20), XL-001 (20 mg/kg body weight). (c) Graphic presentation of kidney fibrotic lesions in different groups after quantitative determination. *P < 0.05 versus sham controls; †P < 0.05 versus UUO alone. (d) Representative macrographs show collagen I and α-SMA protein expression in different groups as indicated. Arrows indicate positive staining. Scale bar, 50 μm. (e) Western blotting analyses show renal protein levels of fibronectin, α-SMA, β-catenin, PAI-1 and Snail1 in different groups. Numbers (1 to 3) represent different individual animals in a given group. (f–i) Graphic presentations of renal fibronectin (f), α-SMA (g), β-catenin (h), PAI-1 and Snail1 (i) proteins in different groups as indicated. †P < 0.05 versus UUO alone.
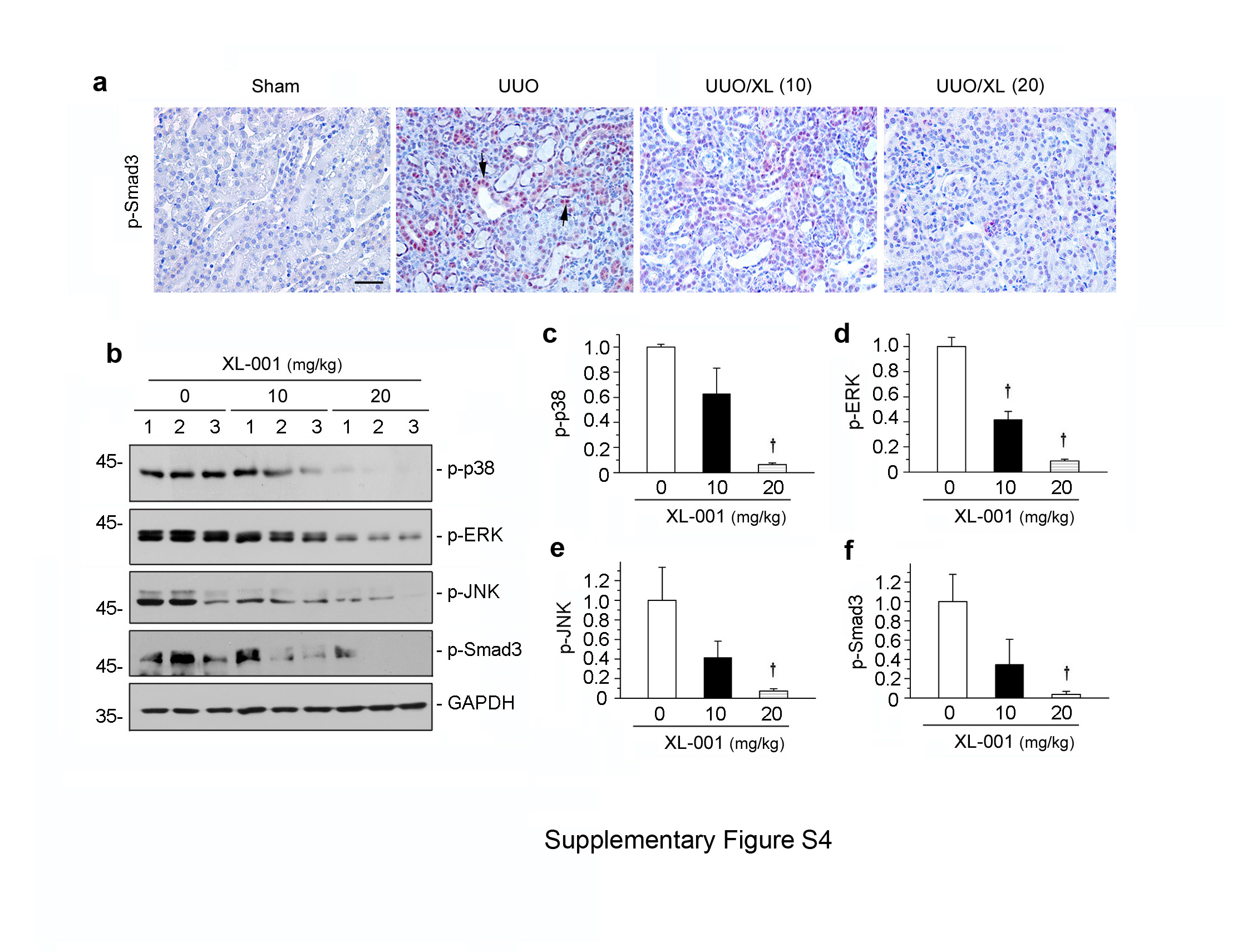
To further study the therapeutic action of XL-001, we examined its effect on renal expression of β-catenin and its downstream target genes. Consistent with the mitigation of renal fibrosis, XL-001 also markedly inhibited β-catenin expression and its downstream target genes such as plasminogen activator inhibitor-1 (PAI-1) and Snail1, as demonstrated by Western blot analyses of whole kidney lysates (Figure 7, e, h and i). We also examined TGF-β1 signaling in various groups. As shown in Supplementary Figure S4, treatment of XL-001 inhibited TGF-β-mediated Smad3, ERK, JNK and p38 MAPK activation in obstructive nephropathy after UUO.
XL-001 reduces renal inflammation in obstructive nephropathy
Renal infiltration of inflammatory cells is a major feature of kidney injury after UUO. Therefore, we also examined the effect of XL-001 on renal inflammation in obstructive nephropathy. As shown in Figure 8, a through d, major pro-inflammatory cytokines, including TNF-α, monocyte chemoattractant protein-1 (MCP-1), IL-6 and the regulated upon activation normal T cell expressed and secreted (RANTES) were all markedly induced in the obstructed kidneys after UUO. XL-001 suppressed renal mRNA expression of TNF-α, MCP-1 and IL-6 in a dose-dependent manner (Figure 8, a through c), whereas there was only a tendency of RANTES inhibition by XL-001 (Figure 8d). Consistent with this, XL-001 reduced renal infiltration of CD3+ T cells and MR+ macrophages in the obstructed kidneys after UUO (Figure 8e). We also investigated the expression and activation of p65 NF-κB, a key transcription factor involved in regulating tissue inflammation after injury. As shown in Figure 8, f through h, both phosphorylated active p65 and total p65 were inhibited by XL-001, suggesting an anti-inflammatory effect of XL-001 in vivo.
Figure 8.

XL-001 inhibits renal expression of pro-inflammatory cytokines and reduces renal inflammation after UUO. (a–d) qRT-PCR shows the relative abundances of renal TNF-α (a), MCP-1 (b), IL-6 (c), and RANTES (d) mRNA in different groups as indicated. *P < 0.05 versus sham controls. †P < 0.05 versus UUO alone (n = 5 to 6). The doses of XL-001 (0, 10, 20 mg/kg body weight) were indicated. (e) Representative micrographs show renal infiltration of CD3+ T cells and MR+ macrophages in different groups as indicated. Paraffin-embedded kidney sections were stained with antibodies against RANTES. Arrow indicates positive staining. Scale bar, 50 μm. (f) Western blot analyses show renal levels of the phosphorylated-p65 (p-p65) and total p65 in different groups as indicated. Numbers (1 to 3) represent different individual animals in a given group. (g, h) Graphic presentations show the relative protein levels of renal p-p65 (g) and total p65 (h) in different groups. †P < 0.05 versus UUO alone.
XL-001 attenuates renal fibrosis after ischemia/reperfusion injury
To confirm the therapeutic efficiency of XL-001, we carried out another model of kidney fibrosis induced by IRI.41 As shown in Figure 9a, XL-001 was administered through daily intraperitoneal injections starting at 4 days after unilateral IRI (UIRI), a time point beyond the initial acute kidney injury. The serum creatinine level was elevated at 11 days after UIRI compared to sham controls (Figure 9b). However, injections of XL-001 preserved renal function and decreased serum creatinine in these mice (Figure 9b). Similar results were obtained when urinary albumin levels were measured (Figure 9c). Masson’s trichrome staining (MTS) also showed that XL-001 in vivo ameliorated renal collagen deposition after UIRI (Figure 9d). We further carried out immunofluorescence and immunohistochemical staining for fibronectin and α-SMA, respectively. As shown in Figure 9e, XL-001 clearly inhibited the expression of fibronectin and α-SMA in kidneys at 11 days after UIRI. Similar results were obtained when whole kidney homogenates were immunoblotted with antibodies against fibronectin and α-SMA (Figure 9, f through h).
Figure 9.

XL-001 reduces renal fibrosis after ischemia-reperfusion injury. (a) Diagram shows the experimental design. Red and black arrows indicate the starting point of IRI and unilateral nephrectomy (UNx). Green bar and arrowheads indicate XL-001 treatment (20 mg/kg body weight). (b) Serum creatinine levels were measured at 11 days after IRI. *P < 0.05 versus sham controls. †P < 0.05 versus IRI alone (n = 5 to 6). (c) Urinary albumin levels in different groups at 11 days after IRI. Urinary albumin was expressed as milligrams per milligram creatinine. *P < 0.05 versus sham controls. †P < 0.05 versus IRI alone. (d) Representative micrographs show collagen deposition in different groups as indicated. Paraffin sections were subjected to Masson trichrome staining (MTS). Arrow indicates positive staining. Scale bar, 50 μm. (e) Representative micrographs show fibronectin and α-SMA expression in different groups as indicated. Kidney sections were stained with different antibodies against fibronectin and α-SMA, respectively. (f) Western blotting show that XL-001 inhibited fibronectin and α-SMA expression after IRI. Numbers (1 to 3) represent different individual animals in a given group. (g, h) Graphic presentations show the relative abundance of fibronectin (g) and α-SMA (g) protein expression in different groups as indicated. *P < 0.05 versus sham controls. †P < 0.05 versus IRI alone (n = 5 to 6). (i, j) Western blotting show that XL-001 inhibited renal β-catenin expression. Numbers (1 to 3) represent different individual animals in a given group. Representative Western blot (i) and quantitative data are presented. *P < 0.05 versus sham controls. †P < 0.05 versus IRI alone (n = 5 to 6). (k) Representative micrographs show renal β-catenin expression in different groups as indicated. Arrow indicates positive staining. Scale bar, 50 μm.
We then examined the expression of β-catenin in the kidneys after IRI. As shown in Figure 9, i and j, Western blotting and quantitation data indicated that β-catenin was induced in the kidneys after IRI and XL-001 significantly blocked IRI-induced β-catenin expression. Immunostaining also revealed that XL-001 reduced the expression of β-catenin protein in renal tubular epithelium (Figure 9k).
Delayed administration of XL-001 is effective in ameliorating renal fibrosis
To assess whether XL-001 is effective in ameliorating an established kidney injury and fibrosis, we treated UUO mice with XL-001 starting at 7 days after surgery, a time point when significant renal fibrosis is already established. The experimental scheme was shown in Figure 10a. As shown in Figure 10b, delayed administration of XL-001 at 20 mg/kg body weight was effective in ameliorating an established kidney injury and fibrosis. Similar results were obtained by western blot analysis for renal expression of fibronectin and α-SMA (Figure 10, c through e). Furthermore, we examined the expression of β-catenin and its downstream MMP-7 and PAI-1.38, 42 As shown in Figure 10, c, f through g, administration of XL-001 inhibited the induction of these fibrogenic genes. Delayed treatment with XL-001 also inhibited renal inflammation. As shown in Figure 10, i and j, renal expression of the phosphorylated-p65 was markedly reduced. Similarly, renal levels of TNF-α and RANTES mRNA were reduced (Figure 10, k and l), and renal infiltration MR+ macrophages was inhibited (Figure 10m). Together, these data suggest that delayed administration of XL-001 is also able to ameliorate an established kidney injury and fibrosis after UUO.
Figure 10.

XL-001 is effective in protecting against renal fibrosis and inflammation in an established obstructive nephropathy. (a) Experimental design. Red arrows indicate the time of UUO surgery. Green bars indicate XL-001 treatment. (b) Representative micrographs show kidney fibrotic lesions in different groups. Paraffin sections were used for Masson’s trichrome staining. Scale bar, 50 μm. (c) Western blot analyses show renal expression of fibronectin, α-SMA, β-catenin, PAI-1 and MMP-7 in different groups as indicated. Numbers (1 to 3) indicate individual animals in a given group. (d–h) Graphic presentations show the relative protein levels of fibronectin (d), α-SMA (e), β-catenin (f), PAI-1 (g) and MMP-7 (h) in different groups. *P < 0.05 versus controls; †P < 0.05 versus UUO alone (n = 5 to 6). (i) Western blotting analyses show p-p65 in different groups as indicated. Quantitative data (j) is presented. *P < 0.05 versus controls; †P < 0.05 versus UUO alone. (k, l) Graphic presentations show the relative levels of TNF-α (k) and RANTES (l) mRNA in different groups. (m) Representative micrographs show the immunofluorescence staining of mannose R expressions in the UUO kidneys after injections with vehicle or XL-001. Scale bar, 50 μm.
Discussion
The endocannabinoid system has been shown to contribute to the development of tissue fibrosis in different organs after injury.17–19, 43 In the kidney, increasing studies indicate the potential involvement of dysregulated CB receptors in the pathogenesis of experimental and human kidney disorders.19, 21, 43, 44 While CB1 is consistently upregulated in a wide variety of kidney injury models such as UUO and diabetic nephropathy,16, 21 data on the expression of the CB2 receptor in CKD are contradictory.19, 20, 27 This discrepancy warrants more comprehensive and systematic interrogation of CB2 regulation and function in the evolution of CKD. In contrast to some reports that CB2 is reno-protective,19, 27 we show in this study that CB2, similar to CB1, actually plays a detrimental role in kidney fibrogenesis. This conclusion is supported by several lines of evidence, including 1) CB2 upregulation in multiple models of CKD, 2) its ability to induce fibrotic responses in tubular cells and fibroblasts in vitro, 3) its ability to promote macrophage activation, 4) reduction of kidney fibrosis after injury in CB2−/− null mice, and 5) pharmacologic inhibition of kidney fibrosis in vivo. These studies establish that upregulation and/or activation of CB2 receptor play a critical role in promoting renal fibrogenesis. Our findings also illustrate that CB2 could be a novel target for a therapeutic intervention of a variety of CKD.
The CB2 expression was initially thought to be restricted to the immune system.45, 46 Subsequent studies reveal a widespread expression pattern in a variety of cells in the kidney including glomerular podocytes and tubular epithelial cells. Despite that downregulation of CB2 is reported in diabetic glomeruli and in the whole kidney of diet-induced obesity,29, 47 CB2 mRNA was actually upregulated after UUO, although its protein level was not assessed.19 Using three mouse models of kidney fibrosis induced by UUO, adriamycin or IRI, we demonstrated a consistent upregulation of CB2 protein in all models (Figure 1). Of interest, CB2 protein was primarily localized in the degenerated, dilated tubules after UUO, as well as in renal interstitial cells, presumably including fibroblasts and infiltrated inflammatory cells. Correspondingly, CB2 was induced in cultured tubular cells (HKC-8), interstitial fibroblasts (NRK-49F) and macrophages (RAW264.7) after TGF-β1 or LPS stimulation (Figures 2 and 3). These results indicate that CB2 upregulation is a common pathologic feature in diseased kidneys, regardless of the initial etiology.
In view of the CB2 induction after injury, it is imperative to illuminate the effect of CB2 activation on kidney cell biology. Using in vitro model, we showed that forced expression of CB2 alone was sufficient to induce matrix gene expression in HKC-8 cells (Figure 2), and so did CB2 activation by its agonist AM1241 (Figure 6). Of interest, constitutive expression of CB2 per se induces fibronectin, while addition of CB2 agonist further promotes its activity (Figure 6). Although HKC-8 cells were incubated in serum-free medium, we cannot exclude the possibility that tubular cells in culture may constitutively produce natural CB2 agonist. Nevertheless, these observations suggest a strong pro-fibrotic action of CB2, which could lead to an impaired tubular integrity.48–50 Consistent with this notion, we found that either siRNA-mediated knockdown or treatment with CB2 inverse agonist XL-001 blocks TGF-β1-mediated fibrogenic responses (Figures 2, 3 and 6), underscoring that CB2 is a downstream mediator of profibrotic TGF-β1 signaling. Furthermore, CB2 apparently is also instrumental in mediating macrophage activation (Figure 3). Taken together, these novel findings unambiguously demonstrate that CB2 is an important player in mediating kidney fibrotic response and inflammation after various injuries.
The strongest evidence for CB2 promoting kidney fibrosis in this study is the observation that genetic ablation of CB2 protects mice against the development of renal fibrosis after UUO (Figure 3). CB2 null mice have a normal phenotype under basal physiologic conditions. However, these mice are protected from developing fibrotic lesions after obstructive injury, a model with characteristic features of aggressive kidney fibrosis. These results are in harmony with in vitro observations (Figure 2), but are in sharp contrast to the previous report showing that CB2 is renal protective in UUO.19 In agreement with this genetic approach, pharmacologic inhibition with CB2 inverse agonist/antagonist XL-001 also reduce kidney injury and fibrotic lesion in both UUO and IRI models (Figures 7–9). At this stage, the mechanism underlying CB2 promoting fibrosis remains to be unraveled, but it could be related to the activation of β-catenin signaling (Figures 3, 7 and 9), a pathologic pathway that is well known to promote kidney fibrosis and CKD progression.33, 37 Indeed, genetic ablation or pharmacologic inhibition of CB2 receptor in vivo diminishes renal β-catenin accumulation and the expression of its downstream genes, suggesting a possible mechanistic connection.
Our data on the detrimental role of CB2 in kidney fibrosis are a direct contradiction to previous reports.19 The reason behind this discrepancy is unknown, but it may be related to the specificity and selectivity of the drugs used. It should be pointed out that the previous study on CB2 in UUO model was carried out only by using CB2 agonist and inhibitor.19 Given the high sequence homology between CB1 and CB2, there is a legitimate concern about the selectivity, as well as the off-target effect, of any inhibitors and agonists to these receptors. In contrast, the present study employed a combination of approaches including genetic knockout mice (Figure 4) and a compound (XL-001) with the highest selectivity over the CB1 receptor (Figure 5). It is worthwhile to note that our results are consistent with an earlier report that CB2 genetic deficiency decreases collagen deposition in atherosclerosis by slowing collagen synthesis and/or accelerating collagen degradation.51 Furthermore, over-expression or activation of the CB2 receptor in tubular epithelial cells, where CB2 is highly induced in vivo (Figure 1), is sufficient to induce fibronectin and α-SMA expression (Figure 6), providing an independent in vitro evidence for the profibrotic action of CB2. In addition, it has been shown that CB2 signaling is required for the formation of several T and B cell subsets,52 and therefore blockade of CB2 could alleviate the inflammatory response in the pathogenesis of CKD. In line with this view, we show here that CB2 is required for macrophage activation after LPS or IL-4 stimulation (Figure 3), and inhibition of CB2 signaling by XL-001 is accompanied by reduction of pro-inflammatory cytokine expression and macrophage and T cell infiltration (Figure 8). Taken together, our results on the role of CB2 in kidney fibrosis are quite consistent and cohesive, illustrated by both in vitro and in vivo evidence, and confirmed by genetic and pharmacologic approaches.
Another important finding in this study is the discovery of a novel CB2 inverse agonist XL-001. This compound was discovered through a high-throughput in silico screening and subsequent medicinal chemistry modifications.39 XL-001 exhibited the strongest binding affinity to the CB2 receptor, with the best selectivity over the CB1 receptor (more than 2500 folds). We show that it could block both constitutive and agonist-stimulated CB2 activity in kidney cells (Figure 6), thereby acting as an inverse agonist. Administration of XL-001 in the pre-clinical setting ameliorates kidney injury, inflammation and fibrosis in both UUO and IRI models, underscoring its bioavailability, safety and therapeutic efficacy in CKD. More importantly, XL-001 is still quite effective in mitigating renal fibrosis when kidney injury is already established (Figure 10), a situation closely imitating to clinical setting.
There are many questions that remain to be answered. One limitation of the present study is the lack of human relevance as no patient sample is used. In this regard, future studies on CB2 in CKD patients are warranted. Furthermore, it appears that CB2 is required for TGF-β1 signaling (Figure 2). However, the details of the interplay between CB2 and TGF-β receptor signaling remain to be delineated. Finally, we have shown an intimate connection between CB2 and β-catenin in diseased kidneys (Figures 4 and 9). Although the inhibition of β-catenin may simply reflect an improved pathology after genetic or pharmacologic ablation of CB2 in mice, possibility also exists that CB2 directly regulates β-catenin activation. Along this line, previous studies have indicated that stimulation of other G protein-coupled receptors leads to β-catenin activation by different mechanisms.53–56
In summary, our present study has provided convincing evidence for a detrimental role of CB2 in fibrotic CKD. Quite unexpectedly, we show that upregulation of CB2 protein is a common pathologic feature in CKD and CB2 activation is sufficient to trigger fibrogenic responses in kidney cells. We have also identified a novel, highly selective CB2 inverse agonist that can block its pro-fibrotic actions. Genetic ablation or pharmacologic inhibition of CB2 reduces renal fibrosis in two well-established animal models. Although more studies are needed, our studies demonstrate that the CB2 receptor is a novel therapeutic target for the treatment of fibrotic CKD.
Materials and Methods
Animal models
Mouse models of renal fibrosis were induced by UUO, UIRI and ADR, as described previously.33, 57 Briefly, male BALB/c mice weighing 22–24g were obtained from the Center of Experimental animal, Southern Medical University, Guangzhou, China. Under general anesthesia, UUO was carried out by double-ligating the left ureter using a 4-0 silk suture following a midline abdominal incision. Mice were randomly assigned to one of four groups (n = 5 to 6): 1) sham control, 2) UUO + vehicle control, 3) UUO + XL-001 (10 mg/kg body weight), and 4) UUO + XL-001 (20 mg/kg body weight). Another set of experiments was performed, in which XL-001 was given starting at 7 days after UUO via daily injection at 20 mg/kg body weight. Mice were sacrificed at 14 days after UUO (Figure 9a). XL-001 was dissolved in DMSO to a stock concentration of 10 mg/ml and stored at −20°C. Stock solutions were diluted in an 18:1:1 ratio of saline/emulphor-620/DMSO immediately prior to use. XL-001 was administered daily via intraperitoneal injection at the dosages of 10 to 20 mg/kg on the basis of pilot studies that have proven both efficacy and selectivity of these doses in mice (data not shown).
For UIRI model, mice were subjected to unilateral IRI by an established protocol as described previously.33 Briefly, left renal pedicles were clipped for 35 minutes using microaneurysm clamps (item no.18051–35; Fine Science Tools, Cambridge, UK) for IRI injury. After removal of the clamps, reperfusion of the kidneys was visually confirmed. During the ischemic period, body temperature was maintained between approximately 37°C and 38°C using a temperature-controlled heating system. After 10 days post-IRI, the intact right kidney was removed via a right flank incision. Mice were randomly assigned to one of three groups (n = 5 to 6): 1) sham control, 2) UIRI + vehicle control, 3) UIRI + XL-001 (20 mg/kg body weight). At 4 days after IRI, mice were subjected to daily intraperitoneal injections of XL-001 at 20 mg/kg body weight for 7 days. Mice were euthanized at 11 days post-IRI, respectively. Serum and kidney tissues were collected for various analyses.
For ADR mice, BALB/c mice were administered a single intravenous injection of ADR (doxorubicin hydrochloride; Sigma, St. Louis, MO) at 11 mg/kg body weight, and euthanized at 3 weeks after ADR injection. All animal experiments were approved by the Animal Ethic Committee at the Nanfang Hospital, Southern Medical University, and by the Institutional Animal Care and Use Committee at the University of Pittsburgh, respectively.
CB2 knockout mice and genotyping
Six- to 8-week-old breeding pairs of the CB2 null mice (CB2−/−) in C57BL/6J background were purchased from the Jackson Laboratory (stock #005786; The Jackson Laboratory, Bar Harbor, ME) and maintained in the University of Pittsburgh Medical Center animal facility under pathogen-free conditions. Age- and sex-matched CB2+/+ and CB2−/− mice on C57BL/6 background were subjected to UUO, respectively. CB2 receptor deficiency was confirmed by RT-PCR.
Urinary albumin and creatinine assay
Urine albumin was measured by using a mouse Albumin ELISA Quantitation kit, according to the manufacturer's protocol (Bethyl Laboratories, Inc., Montgomery, TX). Urinary and serum creatinine level were determined by use of a QuantiChrom creatinine assay kit (DICT-500, Bioassay Systems, Hayward, CA), according to the protocols specified by the manufacturer. Urinary albumin was normalized to urine creatinine and expressed as mg/mg urine creatinine.
Histology and immunohistochemical staining
Paraffin-embedded mouse kidney sections (3 μm thickness) were prepared by a routine procedure. Sections were stained with hematoxylin-eosin (HE), periodic acid-Schiff (PAS) reagent and Masson-Trichrome staining (MTS) for assessing collagen deposition and fibrotic lesions by standard procedures. Quantification of the fibrotic area was carried out by a computer-aided, point-counting technique.57 Immunohistochemical staining was performed using routine protocol. Antibodies used were as follows: rabbit polyclonal anti-CB2 (sc-10073; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-α-SMA (ab5694; Abcam, Cambridge, MA), rabbit polyclonal anti-Fsp1 (S100A4) (A5114; DAKO, Carpinteria, CA), rabbit polyclonal anti-β-catenin (ab15180; Abcam, Cambridge, MA), mouse monoclonal anti-RANTES (10R-R121A; Fitzgerald Industries International, Concord, MA), mouse monoclonal anti-CD3 (sc-20047; Santa Cruz Biotechnology, Santa Cruz, CA), and rabbit monoclonal anti-pSmad3 (9520; Cell Signaling Technologies).
Immunofluorescence staining
Kidney cryosections or cells cultured on coverslips were fixed with 3.7% paraformaldehyde for 15 min at room temperature. After blocking with 10% donkey serum for 30 min, the slides were immunostained with primary antibodies of rabbit polyclonal anti-fibronectin (F3648, Sigma, Darmstadt, Germany), rabbit polyclonal anti-collagen I (234167; EMD Millipore), and rabbit polyclonal anti-Mannose R (ab64693; Abcam, Cambridge, MA), mouse polyclonal anti-PAI-1 (sc-5297; Santa Cruz Biotechnology, Santa Cruz, CA). The slides were then stained with Cy3- or Cy2-conjugated secondary antibody (Jackson ImmunoResearch Laboratories), and mounted with Vectashield anti-fade mounting media by using DAPI to visualize the nuclei (Vector Laboratories, Burlingame, CA). Slides were viewed under a Nikon Eclipse E600 microscope.
Western blot analysis
Protein expression was analyzed by Western blot analysis.57 The primary antibodies used were as follows: anti-CB2 (ab45942; Abcam), anti-fibronectin (F3648; Sigma), anti-α-SMA (A2547; Sigma, St. Louis, MO), anti-β-catenin (610154; BD Transduction Laboratories), anti-MMP-7 (3801; Cell Signaling Technologies), anti-Snail1 (ab17732; Abcam), anti-PAI-1 (sc-5297; Santa Cruz Biotechnology), anti-phospho–NF-κB p65 (Ser536) (3036; Cell Signaling Technology), and anti-NF-κB p65 (3034; Cell Signaling Technologies), anti-pSmad3 (9520; Cell Signaling Technology), p-p38 (9211; Cell Signaling Technology), p-ERK (9101; Cell Signaling Technology), p-JNK (9251; Cell Signaling Technology), TNF-α (ab9739; Abcam, Cambridge, MA), iNOS (ab15323; Abcam, Cambridge, MA), and Mannose R (ab64693; Abcam, Cambridge, MA).
qRT-PCR
Total RNA isolation and quantitative, real-time RT-PCR (qRT-PCR) were carried out by a routine procedure.33 Briefly, first strand cDNA synthesis was carried out by using a Reverse Transcription System kit according to the instructions of the manufacturer (Promega, Madison, WI). Real-time RT-PCR was performed on ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). The PCR reaction mixture in a 25-μl volume contained 12.5 μl 2x SYBR Green PCR Master Mix (Applied Biosystems), 5 μl diluted RT product (1:10) and 0.5 μM sense and antisense primer sets. The sequences of the primer pairs used in RT-PCR or qRT- PCR were as follows: mouse CB1, 5’-TTCCACCGCAAAGATAGT-3’ and 5’-TGAAGGAGGCTGTAACCC-3’; mouse CB2, 5’-TATGCTGGTTCCCTGCACTG-3’ and 5’-GAGCGAATCTCTCCACTCCG-3’; mouse TNF-α, 5’-TCGTAGCAAACCACCAAGTG-3’ and 5’-CCTTGAAGAGAACCTGGGAG-3’ ; mouse MCP-1, 5’-CCCACTCACCTGCTGCTAC-3’ and 5’-TTCTTGGGGTCAGCACAGA-3’ ; mouse IL-6, 5’-CTTGGGACTGATGCTGGTG-3’ and 5’-TCCACGATTTCCCAGAGAAC-3’ ; mouse RANTES, 5’-TGCTGCTTTGCCTACCTCTC-3’ and 5’-TTGAACCCACTTCTTCTCTGG-3’; mouse β-actin, 5’-AGGCATCCTCACCCTGAAGTA-3’ and 5’-CACACGCAGCTCATTGTAGA-3’. PCR reaction was run by using standard conditions. Real-time PCR was performed using a Plantinum SYBR Green qPCR SuperMix-UDG kit (Invitrogen). After sequential incubations at 50°C for 2 min and 95°C for 10 min, respectively, the amplification protocol consisted of 50 cycles of denaturing at 95°C for 15 sec, annealing and extension at 60°C for 60 sec. The standard curve was made from series dilutions of template cDNA. The mRNA levels of various genes were calculated after normalizing with β-actin.
Identification and characterization of CB2 inverse agonist XL-001
N-(4-chlorophenethyl)-4-methyl-N-tosylbenzenesulfonamide was discovered as a novel CB2 ligand in our previous high-throughput screening research program.39 Our further chemical optimization and structure-activity relationship (SAR) studies offered us the best compound XL-001. Its chemical structure has been confirmed by NMR/ LC-MS and reported in our recent paper.39 Meanwhile, the binding affinity of XL-001 to CB1 and CB2 receptors were tested through radioligand competition binding assay, and the functional activity was measured by cAMP assay.
Cell culture and treatment
Human proximal tubular epithelial cells (HKC, clone-8) were provided by Dr. Lorraine C. Racusen (Johns Hopkins University, Baltimore, MD). Normal rat kidney interstitial fibroblast (NRK-49F) cell line and mouse macrophage cell line (RAW264.7) were obtained from American Type Culture Collection (ATCC, Manassas, VA), and cultured according to the instruction specified by the provider. HKC-8 cells were cultured according to the procedures described previously,57 and treated with human recombinant TGF-β1 (R&D Systems, Minneapolis, MN), specific CB2 agonist AM1241 (Cayman Chemical, Ann Arbor, MI), or CB2 inverse agonist XL-001, respectively. For some experiment, HKC cells were transfected with control vector (pcDNA3), CB2 expression vector (pCMV-CB2) or CB2-specific siRNA (Santa Cruz Biotechnology) by lipofectamine 2000 (Life Technologies, Carlsbad, CA), according to the manufacturer’s protocol. Transfected cells were then treated with AM1241 or XL-001 as indicated. NRK-49F cells and RAW264.7 cells were treated with TGF-β1, LPS or IL-4, as indicated. For some experiments, these cells were transfected with control or CB2-specific siRNA, followed by incubating with TGF-β1, LPS or IL-4, respectively. Whole-cell lysates were prepared and subjected to Western blot analyses.
Statistical analyses
All data examined were expressed as mean ± SEM. Statistical analysis of the data was carried out using SPSS 13.0 (SPSS Inc., Chicago, IL). Comparison between groups was made using one-way ANOVA followed by Student-Newman-Kuels test or Dunnett’s T3 procedure. P < 0.05 was considered significant.
Supplementary Material
Mice with CB2 deficiency are protected against TGF-β1 signaling and rnal fibrosis after obstructive injury. (a) Representative micrographs show renal expressions of p-Smad3 and α-SMA in different groups as indicated. Paraffin-embedded kidney sections were stained with antibodies against pSmad3 and α-SMA. Arrow indicates positive staining. Scale bar, 50 μm. (b) Western blot analyses show renal levels of the p-ERK, fibronectin, and β-catenin in different groups as indicated. Numbers (1 and 2) represent different individual animals in a given group. (c through e) Graphic presentations show the relative protein levels of p-ERK (c), fibronectin (d), and β-catenin (e) in different groups. *P < 0.05 versus sham controls. †P < 0.05 versus CB2+/+ UUO mice.
{kind=link}
XL-001 inhibits TGF-β1-induced signal transduction. (a) Western blotting analyses show that TGF-β1-induced signaling was significantly inhibited by XL-001. Human kidney proximal tubular cells (HKC-8) were pretreated with XL-001, then incubated with TGF-β1 (2 ng/ml) for 45 min. The expressions of pSmad3, p-ERK, p-p38 and p-JNK were analyzed by western blotting. Quantitative data (b through e) are presented. *P < 0.05 versus controls. †P < 0.05 versus TGF-β1 alone.
{kind=link}
Representative micrographs show kidney morphology after UUO in different groups as indicated. (a) Images of PAS staining are shown. Scale bar, 50 μm. XL (10), XL-001 (10 mg/kg body weight); XL (20), XL-001 (20 mg/kg body weight). (b) Quantitative analyses of injured tubules in three groups as indicated. Kidney sections were subjected to PAS staining. At least 20 randomly selected fields were evaluated under X400 magnification and results were averaged for each mouse. *P < 0.05 versus sham controls. †P < 0.05 versus UUO alone.
{kind=link}
XL-001 inhibits TGF-β1-induced signaling in vivo. (a) Representative micrographs show renal expressions of p-Smad3 in different groups as indicated. Paraffin-embedded kidney sections were stained with antibodies against p-Smad3. Arrow indicates positive staining. Scale bar, 50 μm. XL (10), XL-001 (10 mg/kg body weight); XL (20), XL-001 (20 mg/kg body weight). (b) Western blot analyses show renal levels of the p-p38, p-ERK, p-JNK and p-Smad3 in different groups as indicated. Numbers (1 to 3) indicate different individual animals in a given group. (c through f) Graphic presentations show the relative protein levels of p-p38 (c), p-ERK (d), p-JNK (e) and p-Smad3 (f) in different groups. †P < 0.05 versus UUO alone.
{kind=link}
Acknowledgments
This work was supported by National Natural Science Foundation of China Grant 81570620 and 81521003, and Guangdong Science Foundation grant 2014A030312014, and NIH/NIDA grant P30 DA035778A1.
Footnotes
Conflict of Interest
L.Z and P.Y are co-inventors of a patent involving XL-001 treatment for CKD.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–696. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeisberg M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010;21:1819–1834. doi: 10.1681/ASN.2010080793. [DOI] [PubMed] [Google Scholar]
- 3.Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest. 2014;124:2299–2306. doi: 10.1172/JCI72267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Nicola L, Minutolo R. Worldwide growing epidemic of CKD: fact or fiction? Kidney Int. 2016;90:482–484. doi: 10.1016/j.kint.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Zhang L, Wang F, Wang L, Wang W, Liu B, Liu J, Chen M, He Q, Liao Y, Yu X, Chen N, Zhang JE, Hu Z, Liu F, Hong D, Ma L, Liu H, Zhou X, Chen J, Pan L, Chen W, Wang W, Li X, Wang H. Prevalence of chronic kidney disease in China: a cross-sectional survey. Lancet. 2012;379:815–822. doi: 10.1016/S0140-6736(12)60033-6. [DOI] [PubMed] [Google Scholar]
- 6.Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, Van Lente F, Levey AS. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038–2047. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 7.Levin A, Tonelli M, Bonventre J, Coresh J, Donner JA, Fogo AB, Fox CS, Gansevoort RT, Heerspink HJL, Jardine M, Kasiske B, Kottgen A, Kretzler M, Levey AS, Luyckx VA, Mehta R, Moe O, Obrador G, Pannu N, Parikh CR, Perkovic V, Pollock C, Stenvinkel P, Tuttle KR, Wheeler DC, Eckardt KU participants ISNGKHS. Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet. 2017 doi: 10.1016/S0140-6736(17)30788-2. [DOI] [PubMed] [Google Scholar]
- 8.Breyer MD, Susztak K. The next generation of therapeutics for chronic kidney disease. Nat Rev Drug Discov. 2016;15:568–588. doi: 10.1038/nrd.2016.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshida R, Ohkuri T, Jyotaki M, Yasuo T, Horio N, Yasumatsu K, Sanematsu K, Shigemura N, Yamamoto T, Margolskee RF, Ninomiya Y. Endocannabinoids selectively enhance sweet taste. Proc Natl Acad Sci U S A. 2010;107:935–939. doi: 10.1073/pnas.0912048107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Busquets-Garcia A, Gomis-Gonzalez M, Srivastava RK, Cutando L, Ortega-Alvaro A, Ruehle S, Remmers F, Bindila L, Bellocchio L, Marsicano G, Lutz B, Maldonado R, Ozaita A. Peripheral and central CB1 cannabinoid receptors control stress-induced impairment of memory consolidation. Proc Natl Acad Sci U S A. 2016;113:9904–9909. doi: 10.1073/pnas.1525066113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu LY, Alexa K, Cortes M, Schatzman-Bone S, Kim AJ, Mukhopadhyay B, Cinar R, Kunos G, North TE, Goessling W. Cannabinoid receptor signaling regulates liver development and metabolism. Development. 2016;143:609–622. doi: 10.1242/dev.121731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gruden G, Barutta F, Kunos G, Pacher P. Role of the endocannabinoid system in diabetes and diabetic complications. Br J Pharmacol. 2016;173:1116–1127. doi: 10.1111/bph.13226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bahr BA, Karanian DA, Makanji SS, Makriyannis A. Targeting the endocannabinoid system in treating brain disorders. Expert Opin Investig Drugs. 2006;15:351–365. doi: 10.1517/13543784.15.4.351. [DOI] [PubMed] [Google Scholar]
- 14.Laprairie RB, Kelly ME, Denovan-Wright EM. The dynamic nature of type 1 cannabinoid receptor (CB(1) ) gene transcription. Br J Pharmacol. 2012;167:1583–1595. doi: 10.1111/j.1476-5381.2012.02175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai E, Zhang L, Ye L, Wan S, Feng L, Qi Q, Yao F, Li Z. Hepatic expression of cannabinoid receptors CB1 and CB2 correlate with fibrogenesis in patients with chronic hepatitis B. Int J Infect Dis. 2017 doi: 10.1016/j.ijid.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 16.Barutta F, Corbelli A, Mastrocola R, Gambino R, Di Marzo V, Pinach S, Rastaldi MP, Perin PC, Gruden G. Cannabinoid receptor 1 blockade ameliorates albuminuria in experimental diabetic nephropathy. Diabetes. 20103;59:1046–1054. doi: 10.2337/db09-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teixeira-Clerc F, Julien B, Grenard P, Tran Van Nhieu J, Deveaux V, Li L, Serriere-Lanneau V, Ledent C, Mallat A, Lotersztajn S. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med. 2006;12:671–676. doi: 10.1038/nm1421. [DOI] [PubMed] [Google Scholar]
- 18.Cinar R, Gochuico BR, Iyer MR, Jourdan T, Yokoyama T, Park JK, Coffey NJ, Pri-Chen H, Szanda G, Liu Z, Mackie K, Gahl WA, Kunos G. Cannabinoid CB1 receptor overactivity contributes to the pathogenesis of idiopathic pulmonary fibrosis. JCI Insight. 2017;2 doi: 10.1172/jci.insight.92281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lecru L, Desterke C, Grassin-Delyle S, Chatziantoniou C, Vandermeersch S, Devocelle A, Vernochet A, Ivanovski N, Ledent C, Ferlicot S, Dalia M, Said M, Beaudreuil S, Charpentier B, Vazquez A, Giron-Michel J, Azzarone B, Durrbach A, Francois H. Cannabinoid receptor 1 is a major mediator of renal fibrosis. Kidney Int. 2015;88:72–84. doi: 10.1038/ki.2015.63. [DOI] [PubMed] [Google Scholar]
- 20.Tam J. The emerging role of the endocannabinoid system in the pathogenesis and treatment of kidney diseases. J Basic Clin Physiol Pharmacol. 2016;27:267–276. doi: 10.1515/jbcpp-2015-0055. [DOI] [PubMed] [Google Scholar]
- 21.Hryciw DH, McAinch AJ. Cannabinoid receptors in the kidney. Curr Opin Nephrol Hypertens. 2016;25:459–464. doi: 10.1097/MNH.0000000000000249. [DOI] [PubMed] [Google Scholar]
- 22.Buser GL, Gerona RR, Horowitz BZ, Vian KP, Troxell ML, Hendrickson RG, Houghton DC, Rozansky D, Su SW, Leman RF. Acute kidney injury associated with smoking synthetic cannabinoid. Clin Toxicol (Phila) 2014;52:664–673. doi: 10.3109/15563650.2014.932365. [DOI] [PubMed] [Google Scholar]
- 23.Nanavati A, Herlitz LC. Tubulointerstitial Injury and Drugs of Abuse. Adv Chronic Kidney Dis. 2017;24:80–85. doi: 10.1053/j.ackd.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 24.Jourdan T, Szanda G, Rosenberg AZ, Tam J, Earley BJ, Godlewski G, Cinar R, Liu Z, Liu J, Ju C, Pacher P, Kunos G. Overactive cannabinoid 1 receptor in podocytes drives type 2 diabetic nephropathy. Proc Natl Acad Sci U S A. 2014;111:E5420–5428. doi: 10.1073/pnas.1419901111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janiak P, Poirier B, Bidouard JP, Cadrouvele C, Pierre F, Gouraud L, Barbosa I, Dedio J, Maffrand JP, Le Fur G, O'Connor S, Herbert JM. Blockade of cannabinoid CB1 receptors improves renal function, metabolic profile, and increased survival of obese Zucker rats. Kidney Int. 2007;72:1345–1357. doi: 10.1038/sj.ki.5002540. [DOI] [PubMed] [Google Scholar]
- 26.Barutta F, Grimaldi S, Gambino R, Vemuri K, Makriyannis A, Annaratone L, di Marzo V, Bruno G, Gruden G. Dual therapy targeting the endocannabinoid system prevents experimental diabetic nephropathy. Nephrol Dial Transplant. 2017 doi: 10.1093/ndt/gfx010. [DOI] [PubMed] [Google Scholar]
- 27.Barutta F, Grimaldi S, Franco I, Bellini S, Gambino R, Pinach S, Corbelli A, Bruno G, Rastaldi MP, Aveta T, Hirsch E, Di Marzo V, Gruden G. Deficiency of cannabinoid receptor of type 2 worsens renal functional and structural abnormalities in streptozotocin-induced diabetic mice. Kidney Int. 2014;86:979–990. doi: 10.1038/ki.2014.165. [DOI] [PubMed] [Google Scholar]
- 28.Jenkin KA, McAinch AJ, Briffa JF, Zhang Y, Kelly DJ, Pollock CA, Poronnik P, Hryciw DH. Cannabinoid receptor 2 expression in human proximal tubule cells is regulated by albumin independent of ERK1/2 signaling. Cell Physiol Biochem. 2013;32:1309–1319. doi: 10.1159/000354529. [DOI] [PubMed] [Google Scholar]
- 29.Barutta F, Piscitelli F, Pinach S, Bruno G, Gambino R, Rastaldi MP, Salvidio G, Di Marzo V, Cavallo Perin P, Gruden G. Protective role of cannabinoid receptor type 2 in a mouse model of diabetic nephropathy. Diabetes. 2011;60:2386–2396. doi: 10.2337/db10-1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frei RB, Luschnig P, Parzmair GP, Peinhaupt M, Schranz S, Fauland A, Wheelock CE, Heinemann A, Sturm EM. Cannabinoid receptor 2 augments eosinophil responsiveness and aggravates allergen-induced pulmonary inflammation in mice. Allergy. 2016;71:944–956. doi: 10.1111/all.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng JL, Yu TS, Li XN, Fan YY, Ma WX, Du Y, Zhao R, Guan DW. Cannabinoid receptor type 2 is time-dependently expressed during skin wound healing in mice. Int J Legal Med. 2012;126:807–814. doi: 10.1007/s00414-012-0741-3. [DOI] [PubMed] [Google Scholar]
- 32.Zhou D, Tian Y, Sun L, Zhou L, Xiao L, Tan RJ, Tian J, Fu H, Hou FF, Liu Y. Matrix Metalloproteinase-7 Is a Urinary Biomarker and Pathogenic Mediator of Kidney Fibrosis. J Am Soc Nephrol. 2017;28:598–611. doi: 10.1681/ASN.2016030354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao L, Zhou D, Tan RJ, Fu H, Zhou L, Hou FF, Liu Y. Sustained Activation of Wnt/beta-Catenin Signaling Drives AKI to CKD Progression. J Am Soc Nephrol. 2016;27:1727–1740. doi: 10.1681/ASN.2015040449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pippin JW, Brinkkoetter PT, Cormack-Aboud FC, Durvasula RV, Hauser PV, Kowalewska J, Krofft RD, Logar CM, Marshall CB, Ohse T, Shankland SJ. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol. 2009;296:F213–229. doi: 10.1152/ajprenal.90421.2008. [DOI] [PubMed] [Google Scholar]
- 35.Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 36.Tan RJ, Zhou D, Zhou LL, Liu YH. Wnt/beta-catenin signaling and kidney fibrosis. Kidney Int Suppl. 2014;4:84–90. doi: 10.1038/kisup.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol. 2009;20:765–776. doi: 10.1681/ASN.2008060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He W, Tan RJ, Li Y, Wang D, Nie J, Hou FF, Liu Y. Matrix metalloproteinase-7 as a surrogate marker predicts renal Wnt/beta-catenin activity in CKD. J Am Soc Nephrol. 2012;23:294–304. doi: 10.1681/ASN.2011050490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang P, Wang L, Feng R, Almehizia AA, Tong Q, Myint KZ, Ouyang Q, Alqarni MH, Wang L, Xie XQ. Novel triaryl sulfonamide derivatives as selective cannabinoid receptor 2 inverse agonists and osteoclast inhibitors: discovery, optimization, and biological evaluation. J Med Chem. 2013;56:2045–2058. doi: 10.1021/jm3017464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng Z, Alqarni MH, Yang P, Tong Q, Chowdhury A, Wang L, Xie XQ. Modeling, molecular dynamics simulation, and mutation validation for structure of cannabinoid receptor 2 based on known crystal structures of GPCRs. J Chem Inf Model. 2014;54:2483–2499. doi: 10.1021/ci5002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonventre JV, Basile D, Liu KD, McKay D, Molitoris BA, Nath KA, Nickolas TL, Okusa MD, Palevsky PM, Schnellmann R, Rys-Sikora K, Kimmel PL, Star RA Kidney Research National D. AKI: a path forward. Clin J Am Soc Nephrol. 2013;8:1606–1608. doi: 10.2215/CJN.06040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He W, Tan R, Dai C, Li Y, Wang D, Hao S, Kahn M, Liu Y. Plasminogen activator inhibitor-1 is a transcriptional target of the canonical pathway of Wnt/beta-catenin signaling. J Biol Chem. 2010;285:24665–24675. doi: 10.1074/jbc.M109.091256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jenkin KA, McAinch AJ, Grinfeld E, Hryciw DH. Role for cannabinoid receptors in human proximal tubular hypertrophy. Cell Physiol Biochem. 2010;26:879–886. doi: 10.1159/000323997. [DOI] [PubMed] [Google Scholar]
- 44.Larrinaga G, Varona A, Perez I, Sanz B, Ugalde A, Candenas ML, Pinto FM, Gil J, Lopez JI. Expression of cannabinoid receptors in human kidney. Histol Histopathol. 2010;25:1133–1138. doi: 10.14670/HH-25.1133. [DOI] [PubMed] [Google Scholar]
- 45.Chung YC, Shin WH, Baek JY, Cho EJ, Baik HH, Kim SR, Won SY, Jin BK. CB2 receptor activation prevents glial-derived neurotoxic mediator production, BBB leakage and peripheral immune cell infiltration and rescues dopamine neurons in the MPTP model of Parkinson's disease. Exp Mol Med. 2016;48:e205. doi: 10.1038/emm.2015.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benito C, Tolon RM, Pazos MR, Nunez E, Castillo AI, Romero J. Cannabinoid CB2 receptors in human brain inflammation. Br J Pharmacol. 2008;153:277–285. doi: 10.1038/sj.bjp.0707505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jenkin KA, O'Keefe L, Simcocks AC, Briffa JF, Mathai ML, McAinch AJ, Hryciw DH. Renal effects of chronic pharmacological manipulation of CB2 receptors in rats with diet-induced obesity. Br J Pharmacol. 2016;173:1128–1142. doi: 10.1111/bph.13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, Wu CC, Hagos Y, Burckhardt BC, Pentcheva-Hoang T, Nischal H, Allison JP, Zeisberg M, Kalluri R. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21:998–1009. doi: 10.1038/nm.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou D, Liu Y. Renal fibrosis in 2015: Understanding the mechanisms of kidney fibrosis. Nat Rev Nephrol. 2016;12:68–70. doi: 10.1038/nrneph.2015.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grande MT, Sanchez-Laorden B, Lopez-Blau C, De Frutos CA, Boutet A, Arevalo M, Rowe RG, Weiss SJ, Lopez-Novoa JM, Nieto MA. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 2015;21:989–997. doi: 10.1038/nm.3901. [DOI] [PubMed] [Google Scholar]
- 51.Netherland CD, Pickle TG, Bales A, Thewke DP. Cannabinoid receptor type 2 (CB2) deficiency alters atherosclerotic lesion formation in hyperlipidemic Ldlr-null mice. Atherosclerosis. 2010;213:102–108. doi: 10.1016/j.atherosclerosis.2010.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ziring D, Wei B, Velazquez P, Schrage M, Buckley NE, Braun J. Formation of B and T cell subsets require the cannabinoid receptor CB2. Immunogenetics. 2006;58:714–725. doi: 10.1007/s00251-006-0138-x. [DOI] [PubMed] [Google Scholar]
- 53.Posokhova E, Shukla A, Seaman S, Volate S, Hilton MB, Wu B, Morris H, Swing DA, Zhou M, Zudaire E, Rubin JS, St Croix B. GPR124 functions as a WNT7-specific coactivator of canonical beta-catenin signaling. Cell Rep. 2015;10:123–130. doi: 10.1016/j.celrep.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luo Y, Cai J, Xue H, Mattson MP, Rao MS. SDF1alpha/CXCR4 signaling stimulates beta-catenin transcriptional activity in rat neural progenitors. Neurosci Lett. 2006;398:291–295. doi: 10.1016/j.neulet.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 55.Nag JK, Rudina T, Maoz M, Grisaru-Granovsky S, Uziely B, Bar-Shavit R. Cancer driver G-protein coupled receptor (GPCR) induced beta-catenin nuclear localization: the transcriptional junction. Cancer Metastasis Rev. 2018;37:147–157. doi: 10.1007/s10555-017-9711-z. [DOI] [PubMed] [Google Scholar]
- 56.Trazzi S, Steger M, Mitrugno VM, Bartesaghi R, Ciani E. CB1 cannabinoid receptors increase neuronal precursor proliferation through AKT/glycogen synthase kinase-3beta/beta-catenin signaling. J Biol Chem. 2010;285:10098–10109. doi: 10.1074/jbc.M109.043711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou L, Li Y, Hao S, Zhou D, Tan R, Nie J, Hou FF, Kahn M, Liu Y. Multiple Genes of the Renin-Angiotensin System Are Novel Targets of Wnt/beta-Catenin Signaling. J Am Soc Nephrol. 2015;26:107–120. doi: 10.1681/ASN.2014010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mice with CB2 deficiency are protected against TGF-β1 signaling and rnal fibrosis after obstructive injury. (a) Representative micrographs show renal expressions of p-Smad3 and α-SMA in different groups as indicated. Paraffin-embedded kidney sections were stained with antibodies against pSmad3 and α-SMA. Arrow indicates positive staining. Scale bar, 50 μm. (b) Western blot analyses show renal levels of the p-ERK, fibronectin, and β-catenin in different groups as indicated. Numbers (1 and 2) represent different individual animals in a given group. (c through e) Graphic presentations show the relative protein levels of p-ERK (c), fibronectin (d), and β-catenin (e) in different groups. *P < 0.05 versus sham controls. †P < 0.05 versus CB2+/+ UUO mice.
XL-001 inhibits TGF-β1-induced signal transduction. (a) Western blotting analyses show that TGF-β1-induced signaling was significantly inhibited by XL-001. Human kidney proximal tubular cells (HKC-8) were pretreated with XL-001, then incubated with TGF-β1 (2 ng/ml) for 45 min. The expressions of pSmad3, p-ERK, p-p38 and p-JNK were analyzed by western blotting. Quantitative data (b through e) are presented. *P < 0.05 versus controls. †P < 0.05 versus TGF-β1 alone.
Representative micrographs show kidney morphology after UUO in different groups as indicated. (a) Images of PAS staining are shown. Scale bar, 50 μm. XL (10), XL-001 (10 mg/kg body weight); XL (20), XL-001 (20 mg/kg body weight). (b) Quantitative analyses of injured tubules in three groups as indicated. Kidney sections were subjected to PAS staining. At least 20 randomly selected fields were evaluated under X400 magnification and results were averaged for each mouse. *P < 0.05 versus sham controls. †P < 0.05 versus UUO alone.
XL-001 inhibits TGF-β1-induced signaling in vivo. (a) Representative micrographs show renal expressions of p-Smad3 in different groups as indicated. Paraffin-embedded kidney sections were stained with antibodies against p-Smad3. Arrow indicates positive staining. Scale bar, 50 μm. XL (10), XL-001 (10 mg/kg body weight); XL (20), XL-001 (20 mg/kg body weight). (b) Western blot analyses show renal levels of the p-p38, p-ERK, p-JNK and p-Smad3 in different groups as indicated. Numbers (1 to 3) indicate different individual animals in a given group. (c through f) Graphic presentations show the relative protein levels of p-p38 (c), p-ERK (d), p-JNK (e) and p-Smad3 (f) in different groups. †P < 0.05 versus UUO alone.