

Abstract
The acute promyelocytic leukemia (APL) has been treated with all-trans retinoic acid (RA) for decades. While RA has largely been ineffective in non-APL AML subtypes, co-treatments combining RA and other agents are currently in clinical trials. Using the RA-responsive non-APL AML cell line HL-60, we tested the efficacy of the Src family kinase (SFK) inhibitor bosutinib on RA-induced differentiation. HL-60 has been recently shown to bear fidelity to a subtype of AML that respond to RA. We found that co-treatment with RA and bosutinib enhanced differentiation evidenced by increased CD11b expression, G1/G0 cell cycle arrest, and respiratory burst. Expression of the SFK members Fgr and Lyn was enhanced, while SFK activation was inhibited. Phosphorylation of several sites of c-Raf was increased and expression of AhR and p85 PI3K was enhanced. Expression of c-Cbl and mTOR was decreased. Our study suggests that SFK inhibition enhances RA-induced differentiation and may have therapeutic value in non-APL AML.
Keywords: Retinoic acid, leukemia, SFK inhibitors
Introduction
Acute myeloid leukemia (AML), the most common form of acute leukemia in adults, is characterized by defective differentiation and excessive accumulation of proliferatively active progenitor cells in bone marrow and blood [1,2]. The acute promyelocytic leukemia (APL) subtype of AML is characterized by a t(15,17) cytogenetic marker resulting in the PML-RARα fusion protein seminal to a block of leukocyte differentiation at the promyelocyte stage and accounts for approximately 10–15% of all AML cases [3,4]. Once considered one of the most lethal forms of acute leukemia, the advent of all trans-retinoic acid (RA) and arsenic trioxide therapy has revolutionized treatment of APL [4]. Now, APL is characterized by complete remission rates of 90% and cure rates of around 80% [5].
While RA is effective in treating APL, it is not effective in treating other subtypes of AML. Moreover, many APL patients who initially respond to RA treatment relapse; subsequent RA treatment is ineffective due to RA-resistance [6,7]. It is therefore of great interest to combine RA with other agents, such as other differentiation-inducing compounds or kinase inhibitors, in order to promote RA-induced differentiation of APL as well as non-APL AML [8,9].
Src family kinases (SFKs) are a group of enzymes that are important in leukemia cell proliferation, survival, adhesion, and differentiation [2]. SFKs have been shown to positively regulate MAPK signaling, cell proliferation, and contribute to cell transformation [10].
SFKs are overexpressed in many cancer types and are typically associated with acute and chronic myeloid malignancies and metastasis [11]. Lyn has been found to be the primary active SFK expressed in AML cells [12,13]. However, in the RA-responsive non-APL AML cell line, patient derived HL-60 cells, expression of both Lyn and Fgr, the only SFKs we detected in these cells, are upregulated following RA treatment leading to differentiation [14–16]. SFK inhibition has been effective in slowing leukemic cell growth [17]. It is therefore of interest to determine the impact of SFK expression and activity on RA-induced differentiation therapy.
SFK inhibitors PP2 and dasatinib have been reported to enhance RA-induced differentiation [13,14,18,19]. However, some reports demonstrate that SFKs themselves positively regulate RA-induced differentiation [15,16].
It is ergo unclear what role SFKs themselves have in regulating RA-induced differentiation. One focus of interest is the impact of SFK inhibitors on the mitogen-activated protein kinase (MAPK) pathway, where different signaling/phosphorylation signatures on the same targeted signaling molecules can cause different outcomes. Defining these signatures is important to understanding the differential signaling attributes that might be exploited for therapeutic intervention. RA has been found to elicit MAPK pathway activation necessary for HL-60 cell differentiation and growth arrest [20]. PP2 and dasatinib co-treatments with RA were found to affect the rapidly accelerated fibrosarcoma (Raf)/mitogen-activated protein kinase kinase (MEK)/extracellular-signal-regulated kinase (ERK) axis by upregulating c-Raf pS259 while not impacting MEK or ERK expression or phosphorylation [14]. The results are somewhat counter-intuitive, but suggest signaling events that are of importance to inducing differentiation of the leukemic cells. They motivate interest in signaling that drives differentiation and in particular, they motivate targeting SFKs to probe for signaling attributes driving differentiation.
Bosutinib is a second-generation SFK inhibitor that has been used clinically to treat phases of intolerant or resistant Philadelphia chromosome (t(19,21)/Bcr-Abl) – positive chronic myeloid leukemias (CML) that do not harbor the T315I or V299LABL kinase domain mutations [21]. Bosutinib treatment has been studied extensively in CML, but its effects on AML have not been well characterized. Additionally, although the effects of SFK inhibition on APL and AML have been widely reported using PP2, the drug is for research use only; dasatinib has also been studied, but has a worse toxicity profile than that of bosutinib [22]. Studies to characterize the effects of bosutinib are therefore warranted; bosutinib’s clinical usage to treat CML and benign toxicity profile make it an attractive candidate for combination treatment of AML [21,23]. We recently reported that certain AML primary cells responded favorably following co-treatment with RA and bosutinib and that the HL-60 cell line may represent an RA-responsive non-APL AML subtype [9]. Significantly, bosutinib is already in clinical use for CML and this potential off-label application to differentiation therapy could accelerate its deployment in AML.
In the present study, we examined the effects of RA/bosutinib co-treatments on HL-60 cells to determine the effects of bosutinib on HL-60 differentiation and the MAPK and mammalian target of rapamycin (mTOR) pathways. We found that RA/bosutinib enhanced RA-induced expression of the differentiation marker CD11b at 48 and 72 h and G1/G0 arrest at 48 h. Induced oxidative metabolism and associated marker p47phox, a component of the NADPH oxidative metabolism machine, were also enhanced with RA/bosutinib compared to RA. With RA/bosutinib treatment, SFK members Fgr and Lyn saw an increase in expression, while pan-SFK activation decreased. Bosutinib also enhanced RA-induced c-Raf phosphorylation at S259, S621, and the C-terminal domain and decreased expression of c-Cbl and mTOR compared to RA alone. Bosutinib enhanced RA-induced expression of AhR and p85 PI3K. Hence bosutinib affected several known key signaling molecule regulators of RA-induced leukemic cell differentiation. Combined RA/bosutinib therapy may thus be useful in differentiation therapy for AML.
Materials and methods
Cell culture and treatments
Reagents, unless specified otherwise, were purchased from commercial suppliers in the highest purity available. HL-60 human myeloblastic leukemia cells derived from the original patient isolates, was a generous gift of Dr. Robert Gallagher, which were maintained in his laboratory in Roswell Park Memorial Institute medium (RPMI) 1640 supplemented with 5% heat inactivated fetal bovine serum (GE Healthcare, Chicago, IL) and 1 × antibiotic/antimycotic (Thermo Fisher Scientific, Waltham, MA) in a 5% carbon dioxide humidified atmosphere at 37 °C. The cells used were certified as mycoplasma free HL-60 by Bio-Synthesis, Lewisville, TX, in August 2017. Viability was monitored by 0.2% Trypan Blue (Invitrogen, Carlsbad, CA) exclusion and routinely exceeded 95%. Experimental cultures were initiated at a density of 0.1 × 106 cells/mL.
There were three treatment regimens studied: (1) untreated, (2) RA, and (3) RA/bosutinib. All-trans retinoic acid (RA) (Sigma, St. Louis, MO) was added from a 5 mM stock solution in 100% ethanol to a final concentration of 1 μM in culture. Bosutinib (Sigma) was used from a stock of 5 mM in Dimethyl sulfoxide (DMSO; Sigma) to make the final concentrations in culture indicated.
Flow cytometric phenotypic analysis
Immunostaining for CD11b and CD38 was performed as previously described and was analyzed using a Becton Dickinson LSR II flow cytometer (Franklin Lakes, NJ). Gating was set to exclude 95% of the untreated wild-type HL-60 samples. Propidium iodide (PI) cell cycle analysis was performed as previously described [24,25].
Respiratory burst quantification
Respiratory burst quantification was performed as previously described [25] and analyzed using a Becton Dickinson LSR II flow cytometer. Gating was set to exclude 95% of the DMSO-treated samples. The shift in fluorescence intensity in response to 12-O-tetradecanoylphorbol-13-acetate (TPA) was used to determine the percent cells with the capability to generate inducible oxidative metabolites.
Antibodies
CD38 and CD11b for flow cytometry were from Becton Dickinson (Franklin Lakes, NJ). Lyn, Fgr, pY416-SFK, AhR, p47phox, mTOR, c-Raf pS259, c-Raf pS621, c-Raf pS289/296/301(c-Raf pC-terminal domain), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), horseradish peroxidase anti-mouse, and anti-rabbit antibodies were from Cell Signaling (Danvers, MA). Total c-Raf was from Becton Dickinson. c-Cbl (C-15) and AhR (H211) were from Santa Cruz Biotechnology (Santa Cruz, CA).
Western blot analysis
Cells were pelleted, washed twice with PBS, and lysed with ice cold mammalian protein extraction reagent (Pierce, Rockford, IL) with protease and phosphatase inhibitor cocktails (Sigma, St. Louis, MO). Samples were incubated overnight at −80 °C and debris was pelleted. Protein concentration was determined using the Pierce BCA Protein Assay(Thermo Fisher Scientific Co., Waltham, MA) according to the manufacturer’s protocol. Lysate was subjected to standard sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)., using 25 μg of lysate per lane under denaturing conditions. Membranes were blocked with 5% dry nonfat milk in phosphate buffer saline (PBS) and were probed with antibodies described above. Enhanced chemiluminescence reagent (GE Healthcare, Pittsburg, PA) was used for detection. Films were scanned and bands of interest were quantified using ImageJ (NIH, Bethesda, Maryland).
Statistical analysis
Statistics were analyzed using Microsoft Excel. Means of treatment groups of interest were compared using two-tailed paired-sample t-tests. The data represent the means of three repeats ± standard error of the mean (SEM). A p value of <.05 was considered significant.
Results
RA/bosutinib enhances CD11b and G1/G0 arrest
We first determined the effect of bosutinib on RA-induced differentiation in HL-60 cells by comparing differentiation markers of cells treated with RA alone or in combination with bosutinib (RA/B) over a 72 h treatment period: we assessed CD38 and CD11b expression, G1/G0 cell cycle arrest, and population growth. We chose to use 0.25 μM bosutinib based on an initial dose-response experiment, included in the supplementary information (Supplementary Figures S1 and S2). It is the lowest dose that yields significant increases compared to control; i.e. the lowest observable effect level (LOEL). A previous study also found that this dosage of bosutinib inhibited cancer cell migration and invasion [26]. The dose chosen elicited no detectable toxicity or significant phenotypic shift by itself.
We measured expression of two cell surface markers, CD38 and CD11b, using flow cytometry. CD38 expression was nearly 100% in both RA and RA/bosutinib treated cells at 24, 48, and 72 h, respectively (Figure (A)). CD38 mean expression per cell also yielded no difference between RA and RA/bosutinib at the three time points (Figure 1(B)). We measured CD11b, a differentiation marker of RA-induced differentiation, at 48 and 72 h. RA/bosutinib significantly increases CD11b expression at 48and 72 h (Figure (C)). Mean expression per cell of CD11b also showed a significant increase with RA/bosutinib treatment at 48 h (Figure (D)).
Figure 1.
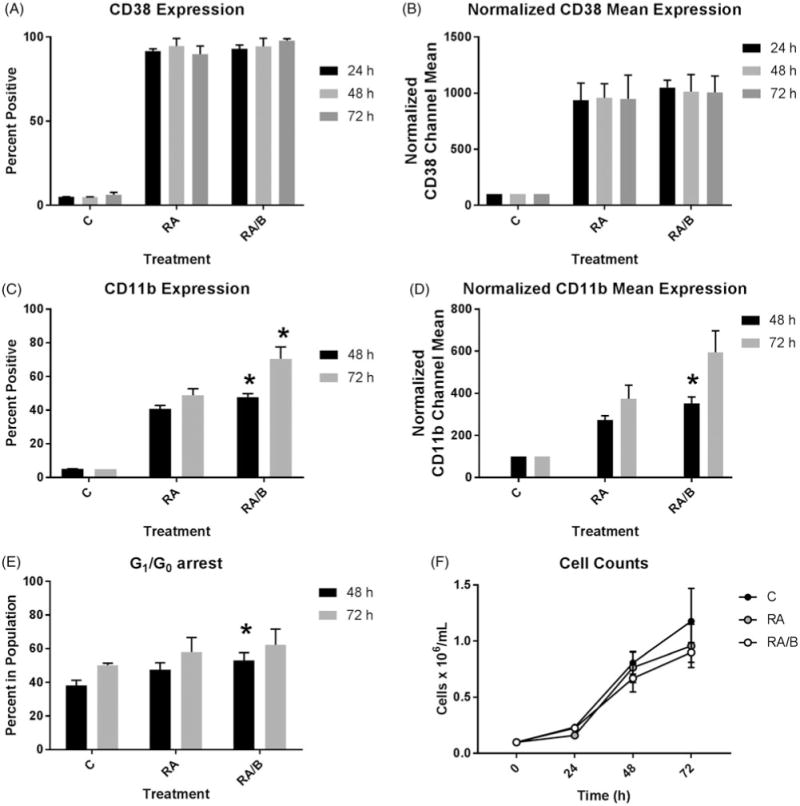
Phenotypic analysis of HL-60 cells treated with RA/bosutinib. (A) HL-60 cells were cultured in the presence of 1 μM RA or 1 μM RA and 0.25 μM bosutinib as indicated. CD38 expression was assessed by flow cytometry following 24, 48, and 72 h treatment periods. Gating to discriminate positive cells was set to exclude 95% of untreated controls (n = 3). Error bars indicate SEM. (B) Normalized means of CD38 expression per cell at 24, 48, or 72 h (n = 3). (C) CD11b expression was assessed by flow cytometry at 48 and 72 h (n = 3). *p < .05 comparing RA-treated samples to RA/bosutinib-treated samples. Two-tailed paired-sample t-tests were used to determine significance. (D) Normalized means of CD11b expression per cell at 48and 72 h (n = 3). *p < .05 comparing RA-treated samples to RA/bosutinib-treated samples. (E) Cell cycle distribution showing the percentage of cells in G1/G0 was analyzed using flow cytometry with propidium iodide staining at 24, 48, and 72 h (n = 4). *p < .05 comparing RA-treated samples to RA/bosutinib-treated samples. (F) Cell counts were taken at 24, 48, and 72 h using a hemocytometer and 0.2% Trypan Blue exclusion staining (n = 3).
At 48 h but not 72 h, RA/bosutinib showed significant enhancement of G1/G0 arrest compared to RA alone (Figure (E)). Addition of bosutinib to the cells did not result in any apparent toxicity compared to RA, as both RA/bosutinib and RA treatments yield similar cell growth patterns over the 72 h treatment period (Figure (F)). Both RA and RA/bosutinib induce growth retardation compared to untreated cells (Figure 1(F)).
RA/bosutinib increases RA-induced respiratory burst activity
To determine the effects of RA/bosutinib on a functional differentiation markerand respiratory burst, we measured inducible reactive oxygen species production (ROS) by flow cytometry at 72 h. RA caused an increase in ROS compared to untreated cells and RA/bosutinib treated cells show a significant further increase in oxidative metabolism compared to RA alone (Figure 2(A)).
Figure 2.
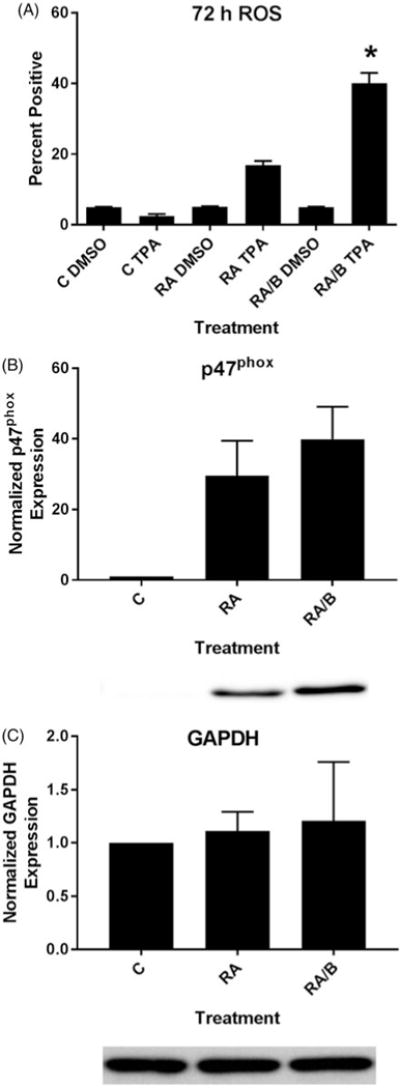
HL-60 cells treated with RA/bosutinib displayed enhanced respiratory burst and p47phox expression. (A) HL-60 cells were cultured in the presence of 1 μM RA or 1 μM RA and 0.25 μM bosutinib as indicated. Respiratory burst was analyzed by measuring inducible reactive oxygen species (ROS) production by flow cytometry using the 2′,7′-dichlorofluorescein (DCF) assay. Gates to determine percent increase of expression with treatment were set to exclude 95% of the DMSO-treated control population for each culture condition; TPA-treated samples show induced ROS (n = 3). Error bars indicate SEM. *p < .05 comparing RA-treated samples to RA/bosutinib-treated samples. Two-tailed paired-sample t-tests were used to determine significance. (B) HL-60 cells were cultured for 48 h in the presence of 1 μM RA or 1 μM RA and 0.25 μM B as indicated and whole cell lysate was collected. Twenty five microgram of lysate per lane was run. Western blots of PAGE-resolved lysates were probed for p47phox (n = 3). Films were scanned and bands of interest were quantified using NIH ImageJ. Error bars indicate SEM. A representative blot, cropped to show only the band of interest, is included. (C) Western blots of GAPDH were used as loading controls following the procedure described above.
To corroborate the ROS assay, we also measured expression of a subunit of the NADPH oxidase complex that produces the respiratory burst, p47phox [27,28]. After a 48 h treatment period, we collected cell lysate and performed western blots for p47phox (Figure 2(B)). Expression was greater in both RA and RA/bosutinib compared to untreated cells and RA/bosutinib further enhanced p47phox levels compared to RA alone (Figure 2(B)). GAPDH was used as a loading control (Figure 2(C)).
RA/bosutinib increases RA-induced SFK expression, but decreases SFK activation
As bosutinib is an SFK inhibitor and the SFK members Fgr and Lyn have been shown to be upregulated by RA treatment in HL-60 cells [3,26,28], we assessed levels and phosphorylation of these members by Western blot. Since Fgr and Lyn are the primary SFK members that are upregulated (i.e. Fgr, Lyn) or activated (i.e. Lyn) in response to RA treatment in AML, we did not probe for other members [12–16]. We collected lysate following a 48 h treatment period. Both RA and RA/bosutinib increased Fgr and Lyn levels compared to untreated cells and RA/bosutinib induced further increases of Fgr and Lyn levels compared to RA alone (Figure 3(A,B)).
Figure 3.
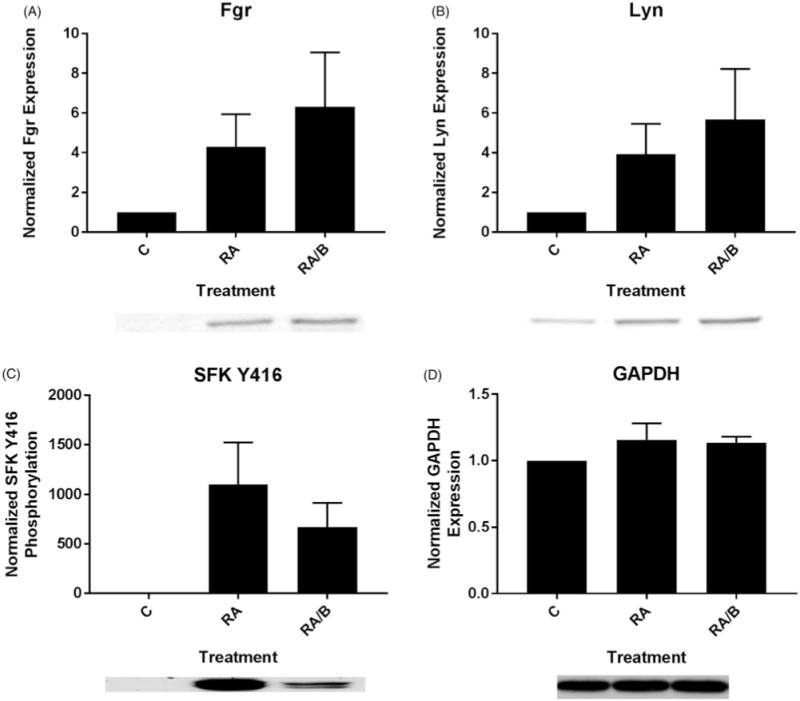
Bosutinib enhances RA-induced SFK expression and diminishes SFK phosphorylation. (A) HL-60 cells were cultured for 48 h in the presence of 1 μM RA or 1 μM RA and 0.25 μM bosutinib (B) as indicated and whole cell lysate was collected. Twenty five microgram of lysate per lane was run. Western blots of PAGE-resolved lysates were probed for Fgr (n = 3). Films were scanned and bands of interest were quantified using NIH ImageJ. Error bars indicate SEM. A representative blot, cropped to show only the band of interest, is included. (B) Western blots of Lyn following the procedure described above. (C) Western blots of phosphorylated pan-Y416 SFK following the procedure described above. (D) Western blots of GAPDH were used as loading controls following the procedure described above.
We measured SFK activation using a pan-SFK anti-body that detects active site (Y416) phosphorylation in all family members, including Fgr and Lyn. While treatment with RA and RA/bosutinib both caused increases in SFK phosphorylation compared to untreated cells, treatment with RA/bosutinib decreased SFK phosphorylation compared to RA alone (Figure 3(A–C)). GAPDH was used as a loading control (Figure 3(D)).
RA/bosutinib augments RA-induced c-Raf phosphorylation
We also examined phosphorylation of several sites of c-Raf previously shown to drive RA-induced differentiation[29]. Since SFKs can regulate MAPK signaling, we analyzed the effect of bosutinib on RA-induced c-Raf activation. We evaluated the phosphorylation status of c-Raf regulatory phosphorylation’s at S259, S621 and the c-Raf C-terminal domain by western blot following a 48 h treatment period. RA increases the amount of c-Raf pS259, c-Raf pS621, and c-Raf pC-terminal domain. We observed modest enhancements of levels of each of these in cells treated with RA/bosutinib compared to RA alone (Figure 4(A–C)). Total c-Raf levels were modestly increased with both RA and RA/bosutinib treatment (Figure 4(D)). GAPDH was used as a loading control (Figure 4(E)).
Figure 4.
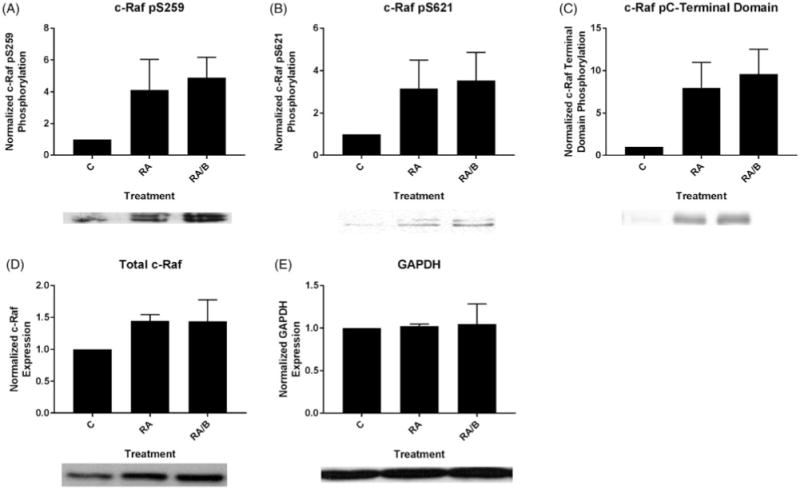
Bosutinib enhances levels of RA-induced phosphorylated c-Raf. (A) HL-60 cells were cultured for 48 h in the presence of 1 μM RA or 1 μM RA and 0.25 μM bosutinib as indicated and whole cell lysate was collected. Twenty five microgram of lysate per lane was run. Western blots of PAGE-resolved lysates were probed for c-Raf pS259 (n = 3). Films were scanned and bands of interest were quantified using NIH ImageJ. Error bars indicate SEM. A representative blot, cropped to show only the band of interest, is included. (B) Western blots of c-Raf pS621 following the procedure described above. (C) Western blots of c-Raf pC-terminal domain following the procedure described above. (D) Western blots of total c-Raf following the procedure described above. (E) Western blots of GAPDH were used as loading controls following the procedure described above.
RA/bosutinib inhibits c-Cbl and mTOR expression, and enhances AhR and p85 PI3K expression
We next assessed levels of several proteins associated with RA-induced differentiation, AhR, c-Cbl, and p85 PI3K, as well as one known to be aberrantly activated in AML, mTOR [8,24,30,31]. After a 48 h treatment period, we collected cell lysate and analyzed expression of these proteins via Western blot. C-Cbl expression was upregulated in cells treated with RA and RA/bosutinib, but RA/bosutinib inhibited c-Cbl expression compared to cells treated with RA alone (Figure 5(A)). mTOR expression followed a similar pattern (Figure 5(B)). AhR and p85 PI3K levels, however, were further upregulated in RA/bosutinib-treated cells compared to RA-treated cells (Figure 5(C, D)). GAPDH was used as a loading control (Figure 5(E)).
Figure 5.
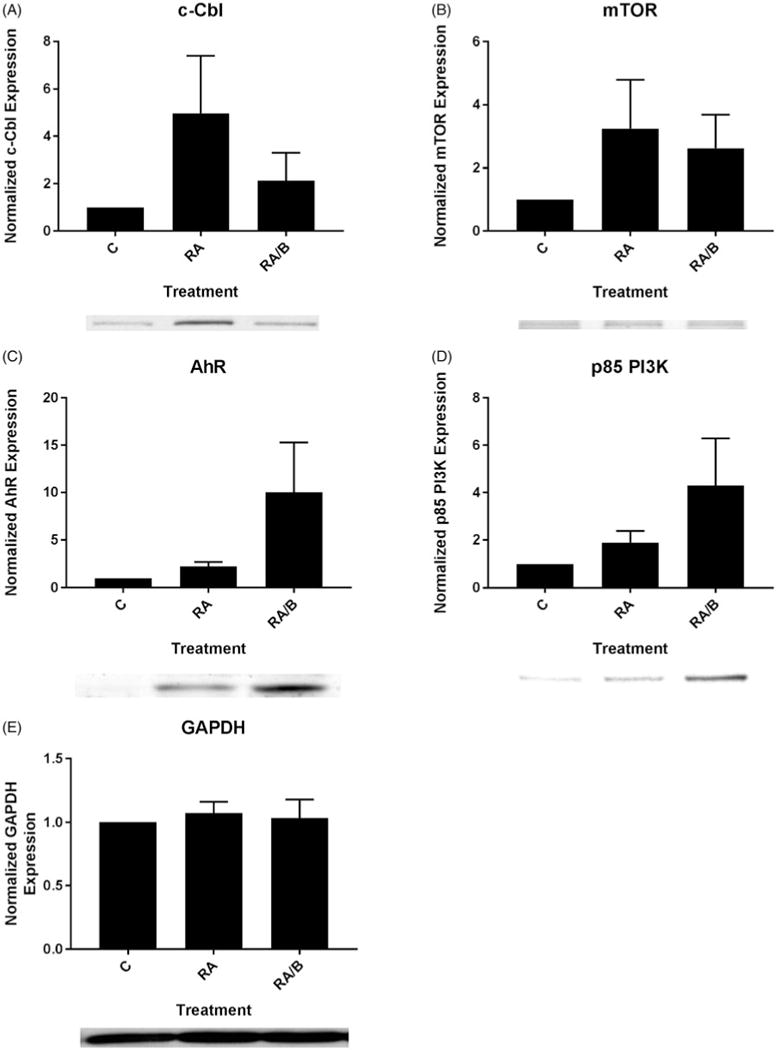
The effect of bosutinib on c-Cbl, mTOR, AhR, and p85 PI3K. (A) HL-60 cells were cultured for 48 h in the presence of 1 μM RA or 1 μM RA and 0.25 μM bosutinib (B) as indicated and whole cell lysate was collected. Twenty five microgram of lysate per lane was run. Western blots of PAGE-resolved lysates were probed for c-Cbl (n = 3). Films were scanned and bands of interest were quantified using NIH ImageJ. Error bars indicate SEM. A representative blot, cropped to show only the band of interest, is included. (B) Western blots of mTOR following the procedure described above. (C) Western blots of AhR following the procedure described above. (D) Western blots of p85 PI3K following the procedure described above. (E) Western blots of GAPDH were used as loading controls following the procedure described above.
Discussion
We sought to provide an initial characterization of the effects of treatment with bosutinib on RA-induced differentiation of HL-60 cells in order to explore its potential therapeutic value in non-APL AML. We recently reported that the HL-60 cell line bears fidelity to a previously undefined RA-responsive, non-APL subtype of AML [9]. Here, we observed that RA/bosutinib treatment enhanced several myeloid lineage differentiation markers compared with RA treatment alone: CD11b expression, G1/G0 cell cycle arrest, and respiratory burst, a functional marker of mature myeloid series cells [28]. Expression of CD38, an early marker of RA-induced differentiation, was not affected by the addition of bosutinib. Recent reports from our laboratory, however, suggest that CD38 may not be necessary for the differentiation process, although it can enhance it [9,21,32].
Since addition of bosutinib enhanced phenotypic and functional markers of RA-induced differentiation, we explored its effects on activation of the key signaling protein c-Raf. Cells treated with RA/bosutinib displayed increased phosphorylated c-Raf levels. The particular phosphorylation sites assayed, S259, S621, and the C-terminal domain, are all associated with active c-Raf in HL-60 cells and thus drive RA-induced differentiation [29]. The Western blotting results are consistent with our phenotypic results and previous findings that RA co-treatments with PP2 or dasatinib also increased c-Raf phosphorylation, namely c-Raf pS259 and c-Raf pS621, in HL-60 cells [14]. These findings are congruous with the notion that c-Raf is a downstream target of SFK inhibitors like bosutinib.
Given that bosutinib is a SFK inhibitor, we assessed its effects on the two prominent SFK members expressed in RA-treated HL-60 cells, Fgr and Lyn, the primary active SFK expressed in AML cells [12–16]. As anticipated, we saw that RA/bosutinib treatment decreased levels of phosphorylated Y416, a mark of activated SFKs. We did, however, observe increased levels of Fgr and Lyn in RA/bosutinib compared to RA alone; increase in expression could perhaps be an attempt to compensate for the inhibited activity of the proteins. These results are consistent with previous findings with RA/dasatinib co-treatments [14]. Dasatinib, like bosutinib, is a second generation SFK inhibitor that is used clinically to treat CML. Bosutinib, however, may be more interesting as a potential therapeutic intervention in AML because it has a lower toxicity profile compared to dasatinib; it is used in patients with CML who have shown intolerance to dasatinib [22].
c-Cbl interacts with CD38 to promote RA-induced differentiation and G1/G0 arrest of HL-60 cells [30]. As previously reported, c-Cbl expression is upregulated by RA treatment; RA/bosutinib treatment partially inhibits RA-induced upregulation. Since c-Cbl expression has been shown to drive RA-induced differentiation, this is surprising, but it is possible that bosutinib drives RA-induced differentiation via other pathways such as c-Raf activation.
The mTOR/p70S6K/4EBP1 pathway, a driver of cellular anabolism/metabolism, is constitutively activated in AML cells, leading to increased cell proliferation and inhibition of differentiation [26]. mTOR is thought to be downstream of the pathways implicated above to regulate differentiation. Dasatinib inactivates the mTOR pathway, correlating with an improvement in cell survival in the AML-derived cell line PVTL-1 [33]. Lyn is hypothesized to serve as the intermediary; Lyn stimulates mTOR expression, and is inhibited by dasatinib [33]. Lending credence to this, silencing Lyn was found to inhibit the mTOR pathway in primary AML cells, and PP2 mimics rapamycin, a selective inhibitor of mTOR [12].
To our knowledge, the effects of SFK inhibitors on the mTOR pathway in APL are unknown. It was hence of interest to determine if bosutinib has an inhibitory effect on mTOR in an RA-responsive AML cell line as found in other forms of AML. Both RA and RA/bosutinib induced greater mTOR expression compared to untreated cells, however, RA/bosutinib decreased mTOR expression compared to RA (Figure 5(B)). It appears that a decrease in the activated SFKs yields an inhibitory effect on the mTOR pathway in RA-treated cells. This inhibitory effect on the mTOR pathway suggests part of the process by which bosutinib enhances differentiation compared to RA alone.
We note that MAPK signaling pathway activation is promoting differentiation and cell cycle arrest in RA-treated HL-60 cells, yet it is also thought to drive mTOR and, consequentially, cellular anabolism to promote proliferation. Hence, RA appears to be redirecting these pathways to support differentiation instead of proliferation. The mechanism of this is a profound but yet unresolved question in leukemic cell differentiation.
AhR is known to drive differentiation and is known to be linked to phosphorylated c-Raf in an RA-activated signalsome [19]. We found that RA treatment increased AhR levels, which is consistent with previous studies (Figure 5(C)) [3,34,35]. Co-treatment with bosutinib further increased AhR expression, consistent with the increase in phosphorylated c-Raf expression in RA/bosutinib treated samples (Figures 3 and 5(C)).
p85 PI3K activity is upregulated during granulocytic maturation [6]. It was therefore of interest to determine the effect of bosutinib on p85 PI3K expression; the protein exhibited an expression pattern similar to that of AhR, as bosutinib caused an increase in RA-induced upregulation of p85 PI3K expression. Interestingly, while both CD38 and c-Cbl are known to interact with p85 PI3K [6], RA/bosutinib treatment did not affect the RA-induced expression of these proteins in the same fashion.
Bosutinib, like other SFK inhibitors, has promising effects on enhancing RA-induced differentiation in HL-60 cells. These effects include modulation of the SFK members Fgr and Lyn and impacts on the MAPK and mTOR pathways. Further studies may elucidate the effects of RA/bosutinib co-treatments on RA-resistant APL cell lines or other myeloid leukemias. Bosutinib is already in clinical trials as a cytotoxic chemotherapeutic agent, but our results suggest that it can also be potentially useful as an agent used in combination therapy with RA for differentiation therapy of AML.
Supplementary Material
Acknowledgments
This work was supported by grant R01 CA152870 from the National Institutes of Health (AY) and by the Center on the Physics of Cancer Metabolism through Award Number 1U54CA210184-01 from the National Cancer Institute.
Footnotes
Supplemental data for this article can be accessed here.
Disclaimers
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Potential conflict of interest: Disclosure forms provided by the authors are available with the full text of this article online at https://doi.org/10.1080/10428194.2018.1452213.
References
- 1.Hu S, Ueda M, Stetson L, et al. A novel glycogen synthase kinase-3 inhibitor optimized for acute myeloid leukemia differentiation activity. Mol Cancer Ther. 2016;15:1485–1494. doi: 10.1158/1535-7163.MCT-15-0566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iriyama N, Yuan B, Hatta Y, et al. Lyn, a tyrosine kinase closely linked to the differentiation status of primary acute myeloid leukemia blasts, associates with negative regulation of all-trans retinoic acid (ATRA) and dihydroxyvitamin D3 (VD3)-induced HL-60 cells differentiation. Cancer Cell Int. 2016;16:37. doi: 10.1186/s12935-016-0314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ibabao CN, Bunaciu RP, Schaefer DM, et al. The AhR agonist VAF347 augments retinoic acid-induced differentiation in leukemia cells. FEBS Open Bio. 2015;5:308–318. doi: 10.1016/j.fob.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watts JM, Tallman MS. Acute promyelocytic leukemia: what is the new standard of care? Blood Rev. 2014;28:205–212. doi: 10.1016/j.blre.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Coombs CC, Tavakkoli M, Tallman MS. Acute promyelocytic leukemia: where did we start, where are we now, and the future. Blood Cancer J. 2015;5:e304. doi: 10.1038/bcj.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warrell RP., Jr Retinoid resistance in acute promyelocytic leukemia: new mechanisms, strategies, and implications. Blood. 1993;82:1949–1953. [PubMed] [Google Scholar]
- 7.Freemantle SJ, Spinella MJ, Dmitrovsky E. Retinoids in cancer therapy and chemoprevention: promise meets resistance. Oncogene. 2003;22:7305–7315. doi: 10.1038/sj.onc.1206936. [DOI] [PubMed] [Google Scholar]
- 8.Jensen HA, Bunaciu RP, Varner JD, et al. GW5074 and PP2 kinase inhibitors implicate nontraditional c-Raf and Lyn function as drivers of retinoic acid-induced maturation. Cellr Signal. 2015;27:1666–1675. doi: 10.1016/j.cellsig.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bunaciu RP, MacDonald RJ, Gao F, et al. Potential for subsets of wt-NPM1 primary AML blasts to respond to retinoic acid treatment. Oncotarget. 2018;9:4134–4149. doi: 10.18632/oncotarget.23642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim MP, Park SI, Kopetz S, et al. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res. 2009;335:249–259. doi: 10.1007/s00441-008-0682-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Logue JS, Morrison DK. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 2012;26:641–650. doi: 10.1101/gad.186965.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dos Santos C, Demur C, Bardet V, et al. A critical role for Lyn in acute myeloid leukemia. Blood. 2008;111:2269–2279. doi: 10.1182/blood-2007-04-082099. [DOI] [PubMed] [Google Scholar]
- 13.Kropf PL, Wang L, Zang Y, et al. Dasatinib promotes ATRA-induced differentiation of AML cells. Leukemia. 2010;24:663–665. doi: 10.1038/leu.2009.267. [DOI] [PubMed] [Google Scholar]
- 14.Congleton J, MacDonald R, Yen A. Src inhibitors, PP2 and dasatinib, increase retinoic acid-induced association of Lyn and c-Raf (S259) and enhance MAPK-dependent differentiation of myeloid leukemia cells. Leukemia. 2012;26:1180–1188. doi: 10.1038/leu.2011.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katagiri K, Yokoyama KK, Yamamoto T, et al. Lyn and Fgr protein-tyrosine kinases prevent apoptosis during retinoic acid-induced granulocytic differentiation of HL-60 cells. J Biol Chem. 1996;271:11557–11562. doi: 10.1074/jbc.271.19.11557. [DOI] [PubMed] [Google Scholar]
- 16.Notario V, Gutkind JS, Imaizumi M, et al. Expression of the fgr protooncogene product as a function of myelomonocytic cell maturation. J Cell Biol. 1989;109:3129–3136. doi: 10.1083/jcb.109.6.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerrouahen BS, Futami M, Vaklavas C, et al. Dasatinib inhibits the growth of molecularly heterogeneous myeloid leukemias. Clin Cancer Res. 2010;16:1149–1158. doi: 10.1158/1078-0432.CCR-09-2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miranda MB, Redner RL, Johnson DE. Inhibition of Src family kinases enhances retinoic acid induced gene expression and myeloid differentiation. Mol Cancer Ther. 2007;6:3081–3090. doi: 10.1158/1535-7163.MCT-07-0514. [DOI] [PubMed] [Google Scholar]
- 19.Jensen HA, Styskal LE, Tasseff R, et al. The Src-family kinase inhibitor PP2 rescues inducible differentiation events in emergent retinoic acid-resistant myeloblastic leukemia cells. PloS One. 2013;8:e58621. doi: 10.1371/journal.pone.0058621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yen A, Roberson MS, Varvayanis S, et al. Retinoic acid induced mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase-dependent MAP kinase activation needed to elicit HL-60 cell differentiation and growth arrest. Cancer Res. 1998;58:3163–3172. [PubMed] [Google Scholar]
- 21.Abbas R, Hsyu PH. Clinical pharmacokinetics and pharmacodynamics of bosutinib. Clin Pharmacokinet. 2016;55:1191–1204. doi: 10.1007/s40262-016-0391-6. [DOI] [PubMed] [Google Scholar]
- 22.Kantarjian HM, Cortes JE, Kim DW, et al. Bosutinib safety and management of toxicity in leukemia patients with resistance or intolerance to imatinib and other tyrosine kinase inhibitors. Blood. 2014;123:1309–1318. doi: 10.1182/blood-2013-07-513937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cortes JE, Kantarjian HM, Brummendorf TH, et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011;118:4567–4576. doi: 10.1182/blood-2011-05-355594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacDonald RJ, Shrimp JH, Jiang H, et al. Probing the requirement for CD38 in retinoic acid-induced HL-60 cell differentiation with a small molecule dimerizer and genetic knockout. Sci Rep. 2017;7:17406. doi: 10.1038/s41598-017-17720-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bunaciu RP, Yen A. Activation of the aryl hydrocarbon receptor AhR Promotes retinoic acid-induced differentiation of myeloblastic leukemia cells by restricting expression of the stem cell transcription factor Oct4. Cancer Res. 2011;71:2371–2380. doi: 10.1158/0008-5472.CAN-10-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vultur A, Buettner R, Kowolik C, et al. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol Cancer Ther. 2008;7:1185–1194. doi: 10.1158/1535-7163.MCT-08-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace AS, Supnick HT, Bunaciu RP, et al. RRD-251 enhances all-trans retinoic acid (RA)-induced differentiation of HL-60 myeloblastic leukemia cells. Oncotarget. 2016;7:46401–46418. doi: 10.18632/oncotarget.10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jensen HA, Bunaciu RP, Ibabao CN, et al. Retinoic acid therapy resistance progresses from unilineage to bilineage in HL-60 leukemic blasts. PloS One. 2014;9:e98929. doi: 10.1371/journal.pone.0098929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yen A, Williams M, Platko JD, et al. Expression of activated RAF accelerates cell differentiation and RB protein down-regulation but not hypophosphorylation. Eur J Cell Biol. 1994;65:103–113. [PubMed] [Google Scholar]
- 30.Shen M, Yen A. c-Cbl interacts with CD38 and promotes retinoic acid-induced differentiation and G0 arrest of human myeloblastic leukemia cells. Cancer Res. 2008;68:8761–8769. doi: 10.1158/0008-5472.CAN-08-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Congleton J, Shen M, MacDonald R, et al. Phosphorylation of c-Cbl and p85 PI3K driven by all-trans retinoic acid and CD38 depends on Lyn kinase activity. Cell Signal. 2014;26:1589–1597. doi: 10.1016/j.cellsig.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamkin TJ, Chin V, Varvayanis S, et al. Retinoic acid-induced CD38 expression in HL-60 myeloblastic leukemia cells regulates cell differentiation or viability depending on expression levels. J Cell Biochem. 2006;97:1328–1338. doi: 10.1002/jcb.20745. [DOI] [PubMed] [Google Scholar]
- 33.Nagao T, Kurosu T, Umezawa Y, et al. Proliferation and survival signaling from both Jak2-V617F and Lyn involving GSK3 and mTOR/p70S6K/4EBP1 in PVTL-1 cell line newly established from acute myeloid leukemia transformed from polycythemia vera. PLoS One. 2014;9:e84746. doi: 10.1371/journal.pone.0084746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raffetto JD, Vasquez R, Goodwin DG, et al. Mitogen-activated protein kinase pathway regulates cell proliferation in venous ulcer fibroblasts. Vasc Endovascular Surg. 2006;40:59–66. doi: 10.1177/153857440604000108. [DOI] [PubMed] [Google Scholar]
- 35.Bunaciu RP, Yen A. 6-Formylindolo (3,2-b)carbazole (FICZ) enhances retinoic acid (RA)-induced differentiation of HL-60 myeloblastic leukemia cells. Mol Cancer. 2013;12:39. doi: 10.1186/1476-4598-12-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.