

Abstract
Background and Purpose
Mitochondria possess their own source of cAMP, that is, soluble adenylyl cyclase (sAC). Activation or expression of mitochondrial sAC promotes mitochondrial function. Oestrogen receptor signalling plays an essential role in the regulation of mitochondrial function. Here we aimed to determine whether 17β‐estradiol may affect mitochondrial cAMP signalling.
Experimental Approach
Expression of the intra‐mitochondrial proteins (Western blot), mitochondrial cAMP content (FRET‐based live imaging and MS assay), mitochondrial membrane potential and cytochrome oxidase activity were analysed in H9C2 and C2C12 cells.
Key Results
A 24 h treatment with 17β‐estradiol significantly reduced the basal level of mitochondrial cAMP, without affecting the intra‐mitochondrial content of sAC, phosphodiesterase 2 (PDE2) or PKA and the activity of the intra‐mitochondrial sAC. The effect of 17β‐estradiol on mitochondrial cAMP was prevented by inhibition of a cGMP‐activated PDE2 or soluble guanylyl cyclase (sGC), suggesting a role of NO signalling. Indeed, 17β‐estradiol raised cellular levels of cGMP and the intra‐mitochondrial expression of the catalytic subunit β of sGC was found. The 17β‐estradiol‐induced reduction of the mitochondrial cAMP level was accompanied by decreased cytochrome oxidase activity and mitochondrial membrane potential in a PDE2‐dependent manner.
Conclusions and Implications
17β‐estradiol reduced the basal level of mitochondrial cAMP content and cytochrome oxidase activity in a sAC‐independent but in a PDE2‐dependent manner. The results suggest a role of 17β‐estradiol‐induced activation of NO signalling in the regulation of mitochondrial cAMP content. Our study adds a new aspect to the complex action of oestrogens on mitochondrial biology, that is relevant to hormone replacement therapy.
Abbreviations
- CFP
cyan fluorescence protein
- EPAC
exchange protein directly activated by cAMP
- ER(s)
oestrogen receptor(s)
- Estradiol
17β‐estradiol
- GFP
green fluorescence protein
- HSP60
heat shock protein 60
- PPT
propylpyrazoletriol
- sAC
soluble AC
- sGC
soluble GC
- TIM23
translocase of inner mitochondrial membrane 23
- tmAC
transmembrane adenylyl cyclase
- TOM40
translocase of outer mitochondrial membrane 40
- YFP
yellow fluorescence protein
Introduction
There is increasing evidence for an evolutionary conserved role of mitochondrial http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2352/http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=284 signalling in controlling mitochondrial function in mammals (Acin‐Perez et al., 2009b; Acin‐Perez et al., 2011; Hebert‐Chatelain et al., 2016) and yeast (Hess et al., 2014). Although the plasmalemmal http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=257 were initially assumed to be a source of mitochondrial cAMP (DiPilato et al., 2004), recent studies (Acin‐Perez et al., 2009b; Di Benedetto et al., 2013) have shown that cytosolic cAMP does not permeate the mitochondrial membrane and thus a mitochondria‐localized cAMP source is required. Type 10 soluble AC (sAC), in addition to several other subcellular compartments, is localized in the mitochondrial matrix (Huttemann et al., 2007; Acin‐Perez et al., 2009b; De Rasmo et al., 2012; Valsecchi et al., 2013; Hebert‐Chatelain et al., 2016). The cAMP produced in mitochondria promotes cytochrome oxidase activity via PKA‐dependent phosphorylation of the cytochrome oxidase subunit IV (Acin‐Perez et al., 2009b; Valsecchi et al., 2013). Furthermore, intra‐mitochondrial cAMP prevents the digestion of nuclear‐encoded subunits of complex I by mitochondrial proteases and supports its http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4487‐ubiquinone oxidoreductase activity (De Rasmo et al., 2015). The activation of the mitochondrial sAC promotes http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1713 synthesis and attenuates oxygen radical production (Acin‐Perez et al., 2009a; Acin‐Perez et al., 2009b; Di Benedetto et al., 2013; Wang et al., 2016). In contrast, the suppression of sAC expression or activity jeopardizes mitochondrial function (Acin‐Perez et al., 2009b; Hebert‐Chatelain et al., 2016). Of note, advanced heart failure in rats is associated with significant down‐regulation of mitochondrial sAC that leads to the increased sensitivity to Ca2+ stress of isolated mitochondria (Wang et al., 2016), demonstrating a role of mitochondrial cAMP signalling in diseases.
Overall, the intra‐mitochondrial cAMP signalling supports mitochondrial function. Strategies directed towards the stimulation of the intra‐mitochondrial cAMP signalling may contribute to the prevention of myocardial pathologies accompanied by mitochondrial dysfunction, for example, ischaemic heart diseases or heart failure. However, tools or treatments affecting mitochondrial cAMP signalling are currently missing.
The female hormone oestrogen plays an essential role in the regulation of mitochondrial function by promoting the expression of several mitochondrial proteins, for example, pyruvate dehydrogenase and subunits of complexes I, IV and V of the mitochondrial respiratory chain (Hsieh et al., 2006; Chen et al., 2009; Rettberg et al., 2014). Whether oestrogen may affect mitochondrial, that is, sAC‐dependent, cAMP signalling remains unknown and was the aim of the present study. Using female rodent cardiac (H9C2) and skeletal muscle (C2C12) cell lines, we found that a 24 h treatment with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1013 (estradiol) significantly reduced mitochondrial cAMP content, decreased cytochrome oxidase activity and led to partial mitochondrial depolarization in a PDE2‐dependent manner. The results suggest that http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2509‐http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2347 signalling was involved in the effects of estradiol in these cells.
Methods
Cell culture and treatments
Cardiac rat embryonic myoblasts (H9C2, ATCC CRL‐1446) and mouse myoblasts (C2C12, ATCC CRL‐1772) were purchased from the American Type Culture Collection. Cells were expanded and frozen in aliquots within 4 weeks of purchase. Cells were cultured in Gibco DMEM (Thermo Fisher Scientific, Berlin, Germany) supplemented with 10% FBS, L‐glutamine (2 mmol·L−1), sodium pyruvate (1 mmol·L−1) and antibiotics (100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin) in a 5% CO2 incubator at 37°C. For the experiments, cells were cultured in phenol red‐free, starvation DMEM supplemented with 2.5% charcoal‐stripped FBS (CS‐FBS) (Biochrom, Berlin, Germany) for 24 h before treatment.
Analyses of the basal cytosolic ATP and mitochondrial morphology did not reveal any marked alteration induced by 48 h starvation in DMEM supplemented with 2.5% CS‐FBS (Supporting Information Figure S4).
Cells were treated with water‐soluble 17β‐estradiol (Sigma, Germany), propylpyrazoletriol (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=2819; Tocris, Germany), KB5 (Karo Pharma, Sweden), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=1014 (Tocris) or corresponding vehicle (dextrin, ethanol, DMSO) for additional 24 h in DMEM supplemented with 2.5% CS‐FBS. Treatments with 40 μmol·L−1 http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5190 (Sigma), 200 μmol·L−1 http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=388 (Sigma), 100 nmol·L−1 http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5147 (Cayman, USA), 100 μmol·L−1 http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=10023 (kindly provided by Dr J. Buck, Cornell University, NY, USA), 200 μmol·L−1 http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5172 (BioLog, Germany), 10 μmol·L−1 http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6554 (Tocris), 100 μmol·L−1 NOC‐12 (Calbiochem, Germany) or 300 μmol·L−1 http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5213 (Santa Cruz Biotechnology, TX, USA) were performed as indicated.
Luciferase assay
H9C2 cells were seeded in 12‐well sterile plastic culture plates at a density of 25 × 103 cells per well in DMEM supplemented with 10% FBS, followed by 48 h in phenol red‐free starvation DMEM supplemented with 2.5% CS‐FBS. During the initial 24 h of starvation, H9C2 cells were transfected with a firefly luciferase reporter construct containing three copies of oestrogen response element (3xERE http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=626, Addgene, Germany) (500 ng per well) and Renilla luciferase reporter plasmid phRL‐TK (10 ng per well) (Promega, Germany). Cells were additionally co‐transfected with http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=620 or http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=621 vectors (500 ng per well) (a kind gift from Dr P. Chambon, INSERM, France) or corresponding empty vector (pSG5, pcDNA3.1 Promega), using FuGENE®‐HD Transfection Reagent (Promega) with a 3:1 FuGENE to DNA ratio according to the manufacturer's instructions. After overnight incubation, cells were treated with estradiol (30 nmol·L−1) or vehicles for 24 h. Firefly and Renilla luciferase activities were measured using the Dual‐Glow™‐Luciferase Assay System (Promega) according to the manufacturer's instructions. To determine the ER activity, firefly luciferase activity was normalized to Renilla luciferase activity.
Subcellular fractionation
Cells were lysed by mechanical homogenization (eight strokes) with Teflon Potter Elvehjem homogenizer (Roth, Germany) in mitochondria isolation buffer (1 mmol·L−1 Tris‐MOPS, 10 mol·L−1 EGTA‐Tris, 200 mmol·L−1 sucrose, pH = 7.4) as previously described (Frezza et al., 2007). The efficiency of cell lysis (>60%) was confirmed by Trypan blue staining. Lysed cells were centrifuged at 800× g for 15 min (4°C), and the supernatant was further centrifuged at 12 000× g for 15 min (4°C). The pellet was defined as the crude mitochondrial fraction and the supernatant as the enriched cytosolic, mitochondria‐free fraction. The purity of the enriched cytosolic fraction was confirmed by the Western blot analysis of cytochrome oxidase.
To obtain intra‐mitochondrial proteins, the crude mitochondrial fraction was washed twice with mitochondria isolation buffer and incubated in the buffer for 15 min with 10 μg·mL−1 trypsin (Biochrom) on ice followed by centrifugation at 12 000× g for 15 min at 4°C. The purity of the mitochondrial fraction was verified by Western blot analysis for TATA binding protein (TBP, nucleus marker) and protein disulfide isomerase (PDI, endoplasmic reticulum marker). The efficiency of trypsin treatment was confirmed by the absence of TOM40 (outer mitochondrial membrane protein), whereas the integrity of mitochondria was evaluated based on the presence of HSP60 (matrix protein) applying Western blot analysis (Figure 1A).
Figure 1.
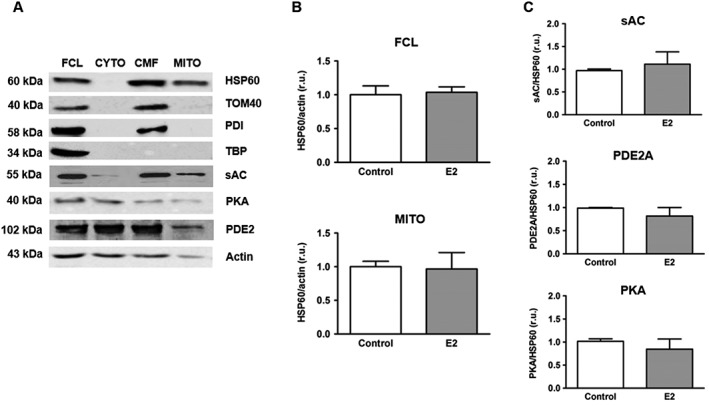
Effect of estradiol on the expression of sAC, PDE2 and PKA in mitochondria. (A) Western blot analysis of HSP60, TOM40, PDI, TBP, sAC, PKA catalytic subunit α (PKA) and PDE2 performed in full cell lysate (FCL), enriched cytosolic (CYTO), crude (CMF) and purified (MITO) mitochondrial fractions of H9C2 cells. The proteins investigated were used as the following markers: HSP60 – mitochondrial matrix, TOM40 – outer mitochondrial membrane, PDI – endoplasmic reticulum, TBP – nucleus. Data are representative of three independent experiments with similar results. (B) Statistical analysis of HSP60 expression in FCL and in purified mitochondrial fraction (MITO) of either control H9C2 cells or cells treated with 30 nmol·L−1 estradiol (E2) for 24 h. Values are means ± SEM, n = 8. (C) Statistical analysis of sAC, PDE2 and PKA expression examined by Western blots, in purified mitochondrial fraction of either control H9C2 cells or cells treated with 30 nmol·L−1 estradiol (E2) for 24 h. Values are means ± SEM, n = 5–6. All data were normalized to the corresponding control and expressed in relative units (r.u.).
Western blotting
Cells were lysed in a Laemmli buffer containing 2% SDS, 10% glycerol, 50 mmol·L−1 1,4‐dithiothreitol, 0.1% bromphenol blue and 62.5 mmol·L−1 Tris–HCl (pH = 6.8). Equal amounts of total proteins were separated on SDS‐polyacrylamide gels and transferred to a nitrocellulose membrane. The membranes were immunoblotted overnight with the following primary antibodies: sAC (clones R21, CEP Biotech, NY, USA), PDE2 (Proteintech, USA), PKA catalytic subunit α (Abcam, Germany), soluble GC (sGC) (Cayman), cytochrome oxidase (subunit IV), HSP60 and PDI (Enzo Life Science, Germany), TIM23 (BD Bioscience, Germany), actin (Millipore, Germany), cytochrome oxidase (Cell Signalling, Germany), ERβ (Acris, Germany), ERα (G20), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=221 (N‐15), TOM40 and TBP (Santa Cruz, Germany). After incubation with the secondary antibodies, specific bands were visualized by chemiluminescence using an ECL kit (Promega). Equal sample loading was confirmed by stripping the membranes with Restore Western Blot Stripping Buffer followed by treatment antibodies against actin and HSP60.
FRET‐based analyses
For FRET‐based live imaging analyses, cells were transfected with plasmids encoding the corresponding sensors. Transfections were performed at 50–60% confluence by electroporation with the Amaxa™ Nucleofector™ II device (Lonza, Switzerland) applying the T‐20 programme. To transfect 106 cells, 9 μg of total cDNA, diluted in 100 μL of electroporation buffer (5 mmol·L−1 KCl, 15 mmol·L−1 MgCl2, 50 mmol·L−1 mannitol, 120·mmol L−1 Na2HPO4/NaH2PO4, pH = 7.2), was used.
Before electroporation, cells were cultured in DMEM supplemented with 10% FBS. After electroporation, cells were seeded on 24 mm diameter glass coverslips and cultured in 10% FBS for 24 h, followed by 24 h of starvation in DMEM supplemented with 2.5% CS‐FBS and 24 h treatments in similar medium. Live imaging experiments were performed on the third day after the transfection.
For cAMP analysis, cells were transfected with http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=259‐based cAMP‐sensor without (H30) or with a mitochondrial targeted sequence (4mtH30) (Di Benedetto et al., 2013). The 4mtH30 sensor derives from the cytosolic H30 sensor, which was tested for cGMP sensitivity upon cell stimulation with the NO donor, nitroprusside. The FRET‐signal of H30 was not affected by nitroprusside leading to the conclusion that cGMP does not detectably affect the conformation of the sensor (Ponsioen et al., 2004).
During the experiments, cells were maintained at room temperature in HEPES‐buffered Ringer‐modified saline (110 mmol·L−1 NaCl, 5 mmol·L−1 KCl, 1 mmol·L−1 Na3PO4, 1 mmol·L−1 MgSO4, 20 mmol·L−1 HEPES, 2 mmol·L−1 CaCl2 and 5 mmol·L−1 glucose). For treatment with bicarbonate, a buffer containing 100 mmol·L−1 NaHCO3, 20 mmol·L−1 HEPES, 10 mmol·L−1 NaCl, 5 mmol·L−1 KCL, 1 mmol·L−1 Na3PO4, 1 mmol·L−1 MgSO4, 2 mmol·L−1 CaCl2 and 5 mmol·L−1 glucose was added to the chamber at a proportion of 1:1. cAMP probes were excited at 430 nm, and emission light was acquired at 470 nm for cyan fluorescence protein (CFP) and 530 nm for yellow fluorescence protein (YFP) channel. Images were acquired with an inverted microscope (oil immersion objective 40×, Zeiss, Germany) and an imaging system (Visitron, Germany) every 6 s. The analysis of the FRET signal was performed with VisiView software (Visitron). Emission signals obtained in the cell‐free region (background) were subtracted from the corresponding emission signals obtained within the region of interest and presented as a CFP/YFP ratio.
For analysis of mitochondrial pH, cells were transfected with mitochondrial SypHer (9 μg of total cDNA per 106 cells) by electroporation, as described above. Experiments were performed in HEPES‐buffered Ringer‐modified saline (2.5 mmol·L−1 KCL, 500 μmol·L−1 NaH2PO4, 500 μmol·L−1 MgSO4, 10 mmol·L−1 HEPES, 125 mmol·L−1 NaCl, 2 mmol·L−1 CaCl2 and 5 mmol·L−1 glucose, pH set to 7.4). For pH calibration, the buffer containing 20 mmol·L−1 NaCl, 125 mmol·L−1 KCl, 0.5 mmol·L−1 MgCl2, 0.2 mmol·L−1 EGTA and 20 mmol·L−1 Tris (for pH range 8–8.5) or 20 mmol·L−1 HEPES (for pH range 7–7.5) supplemented with 10 μmol·L−1 nigericin (Sigma) and 1 μmol L−1 valinomycin (Sigma) was used. SypHer was excited at 430 and 480 nm, and emission was acquired at 535 nm. Fluorescence ratios (480/430) were calculated by VisiView and analysed in Excel and GraphPad Prism 5.
For analysis of cytosolic ATP, cells were transfected with FRET probe AT1.03 (9 μg of total cDNA per 106 cells) by electroporation followed by FRET‐based live imaging, as described above.
Mitochondrial co‐localization of the EPAC‐based cAMP‐sensor and mitochondrial morphology imaging
H9C2 cells were transfected with 4mtH30 sensor and seeded on glass coverslips; 48 h after the transfection, cells were loaded with 50 nmol·L−1 MitoTracker Deep Red (Thermo Fisher Scientific, Germany) in DMEM medium with 0.5% of FBS for 20 min at 37°C. Subsequently, cells were washed with growth medium for additional 20 min. Coverslips were mounted into a chamber, and cells were bathed in DMEM medium with 0.5% FBS at 30°C and in a gas controlled incubator at 5% CO2. Images were acquired with 63× oil‐immersion objective using a Nikon spinning disc confocal microscope CSU‐X and EMCCD camera iXon3 DU‐888 Ultra.
To perform a qualitative analysis of mitochondrial morphology, H9C2 cells were stained with 200 nmol·L−1 MitoTracker Green (Thermo Fisher Scientific) in DMEM medium with 2.5 or 10% FBS for 20 min at 37°C followed by 20 min washing period. Similar technique and handling were applied as described above for MitoTracker Deep Red.
Analysis of mitochondrial membrane potential (ΔΨm)
ΔΨm was monitored with the fluorescence dye JC‐1 (Thermo Fisher Scientific). For this purpose, H9C2 cells were loaded with JC‐1 (5 μmol·L−1) at 37°C for 20 min followed by washing for an additional 20 min. The loaded cells were excited at 490 nm, and the emitted fluorescence was collected at 535 and 590 nm. ΔΨm was presented as the fluorescence ratio (590 nm/535 nm) after subtraction of the ratio obtained from completely depolarized mitochondria achieved by treatment with 500 nmol·L−1 carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone.
cGMP analysis
The analysis of total cellular cGMP content was performed using a cGMP elisa kit (Enzo). The measured absorbance at 405 nm was used to calculate the concentration of cGMP by applying a calibration curve. According to the manufacturer, the sensitivity of the assay is 0.025 pmol·L−1 cGMP. The cross reactivity for a number of related compounds tested by the manufacturer was less than 0.001% for GMP, GTP, cAMP, AMP, ATP cyclic UMP and CTP.
Enzymic assays
Mitochondria isolated from H9C2 cells (as previously described) were used for analysis of cytochrome oxidase activity as previously described (Medja et al., 2009). Briefly, oxidation rate of 100 μmol·L−1 reduced cytochrome oxidase was analysed in 50 mmol·L−1 potassium phosphate buffer with pH 7.0 at 37°C and defined as changes in absorbance at 550 nm. cytochrome oxidase activity was normalized to subunit IV of cytochrome oxidase expression defined by Western blot.
MS analysis of mitochondrial cAMP
The MS measurement of cAMP content in crude mitochondrial fraction was performed as previously described (Hartwig et al., 2014). Briefly, the crude mitochondrial fraction was isolated as described above, washed twice with mitochondria isolation buffer and incubated in the buffer with corresponding compounds at 37°C for 15 min. After treatment, mitochondria were pelleted and stored at −80°C.
Data and statistical analysis
Data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). The data are given as mean ± SEM. Group sizes (n) for all experiment are provided and refer to independent single measurements. Data subjected to statistical analysis have n of at least five per group except for some experiments requiring a large amount of material, for example, analyses performed in isolated mitochondria. When applicable, the values from treatment groups were normalized to the corresponding control values. GraphPad Prism 5 (San Diego, USA) was employed for data analysis. For comparison of two groups, the unpaired t‐test was used. Statistical significance was accepted when P < 0.05.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c)
Results
Effect of estradiol on expression of sAC, PDE2 and PKA in mitochondria
Because estradiol promotes expression of many mitochondrial proteins (Klinge, 2008), we first examined the effects of estradiol on the expression of two major proteins involved in cAMP synthesis and degradation in mitochondria, that is, sAC and PDE2 (Acin‐Perez et al., 2011). H9C2 cells were treated with 30 nmol·L−1 estradiol for 24 h. These cells displayed a marked expression of three oestrogen receptors (ERα, ERβ and GPR30) (Supporting Information Figure S1A, B). Furthermore, a luciferase‐based reporter assay confirmed the efficacy of oestrogen treatment to activate the ERα and ERβ (Supporting Information Figure S1D). Additionally, estradiol treatment promoted http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514 phosphorylation and cell proliferation (Supporting Information Figure S1C, E), typical markers of estradiol action in various cell types (Alvaro et al., 2002; Keshamouni et al., 2002).
To obtain a purified mitochondrial fraction enriched with matrix proteins, the crude mitochondrial fraction was shortly treated with trypsin. This treatment led to both the cleavage of proteins localized at the outer mitochondrial membrane and the exclusion of contamination with endoplasmic reticulum (Figure 1A). The protease resistance of matrix proteins was confirmed by the presence of HSP60. As expression of HSP60 was not affected by estradiol treatment in either full cell lysate or mitochondria (Figure 1B), we used this intra‐mitochondrial protein as a loading control for further analyses.
Estradiol treatment for 24 h had no effect on the expression of mitochondrial sAC and PDE2 (Figure 1C), a major cAMP degrading enzyme in the mitochondrial matrix (Acin‐Perez et al., 2011). As PKA can be a key downstream target of intra‐mitochondrial cAMP signalling and directly affects the activity of oxidative phosphorylation, due to phosphorylation of cytochrome oxidase subunits I and IV (Acin‐Perez et al., 2009b) we assessed PKA expression inside the mitochondria. Western blot analysis for the PKA catalytic subunit α revealed no effect of estradiol treatment on the amount of this protein in mitochondria (Figure 1C).
Effect of estradiol on the activity of mitochondrial sAC and basal cAMP
Activity of sAC is regulated by divalent cations (Ca2+, Mg2+) and bicarbonate (Geng et al., 2005; Zippin et al., 2013). The mitochondrial concentration of these ions depends on the expression and activity of several intra‐mitochondrial proteins, for example, carbonic anhydrases, mitochondrial Ca2+ uniporter and Na+/Ca2+ exchanger, whose expression or activity may be affected by estradiol (Rettberg et al., 2014; Kim et al., 2017), and as a result, the sAC activity may be altered. To test whether treatment with estradiol influences sAC activity, transfection of cells with a mitochondria‐targeted EPAC‐based cAMP‐sensor (4mtH30) was performed. The cAMP level was assessed by measuring the 470/535 nm emission ratio (emission for CFP and YFP respectively, see Methods section) upon excitation at 430 nm. When cells transiently expressing 4mtH30 were examined, the probe was mainly localized within mitochondria, that is, the CFP fluorescence signal obtained in mitochondria‐free regions was less than 2% of the signal obtained from mitochondria‐rich regions (Supporting Information Figure S2A). Furthermore, the merge image of EPAC‐based cAMP sensor and MitoTracker Deep Red demonstrated that 4mtH30 was mainly co‐localized with mitochondria (Supporting Information Figure S2B).
To confirm the mitochondrial cAMP‐sensor's sensitivity to cAMP, cells were treated with the selective EPAC agonist 8‐pCPT‐2‐O‐Me‐cAMP, which led to a marked increase of the CFP/YFP ratio in mitochondria (Supporting Information Figure S2C). Similarly, the activation of sAC with bicarbonate led to a significant rise of the CFP/YFP ratio, which was prevented by treatment with LRE1 (Supporting Information Figure S2D), a novel selective sAC inhibitor (Ramos‐Espiritu et al., 2016).
To demonstrate that elevation of mitochondrial cAMP under bicarbonate treatment is not due to the import of cytosolic cAMP, which can be synthesized by sAC localized in cytosol, a cAMP elevation test was performed. Here, treatment with forskolin, an activator of plasmalemmal AC, did not affect the FRET ratio in mitochondria, that is, in cells expressing a mitochondria‐targeted 4mtH30 probe. By contrast, in cells expressing a non‐targeted H30 probe, the forskolin treatment led to a marked rise of the FRET‐ratio in the cytosol (Supporting Information Figure S2E). These results further confirm the previous finding that the inner mitochondrial membrane is impermeable to cytosolic cAMP and that mitochondria contain sAC as their own source of cAMP (Di Benedetto et al., 2013; Lefkimmiatis et al., 2013).
Applying a treatment with 50 mmol·L−1 bicarbonate to examine the stimulated sAC activity, we found no effect of estradiol pretreatment in H9C2 cells (Figure 2A, B). Similarly, pretreatment with estradiol did not affect the stimulated sAC activity in C2C12 skeletal muscle cells (Figure 2D, E). Taken together, these data are in accordance with the finding that estradiol does not influence the expression of mitochondrial sAC.
Figure 2.
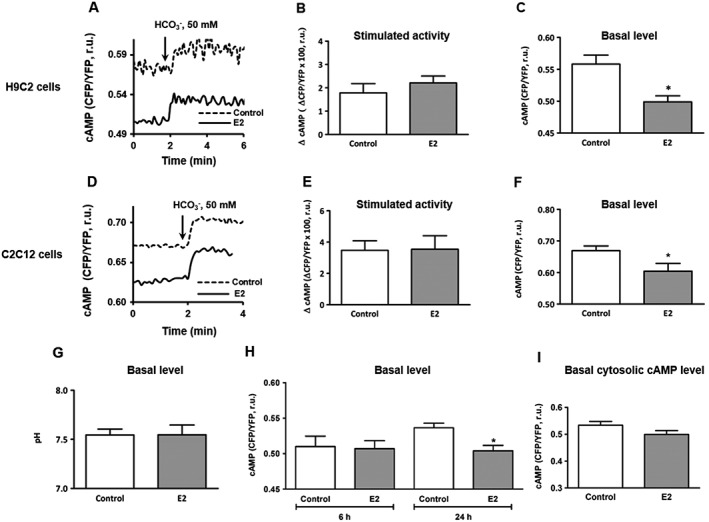
FRET‐based analysis of intra‐mitochondrial cAMP measured in cells transfected with 4mtH30 sensor. Representative kinetics of mitochondrial cAMP level (A, D) followed by statistical analysis of the maximal cAMP elevation (stimulated activity, ΔcAMP) induced by 50 mmol·L−1 bicarbonate (B, E) and basal cAMP level (C, F) performed in H9C2 and C2C12 cells, either pretreated with 30 nmol·L−1 estradiol (E2) or vehicle (control) for 24 h. Values are means ± SEM, n = 5–6. *P < 0.05,significantly different from control. (G) Statistical analysis of mitochondrial pH under basal conditions measured in H9C2 cells pretreated with 30 nmol·L−1 estradiol (E2) or vehicle (control) for 24 h. Values are means ± SEM, n = 5. (H) Statistical analysis of the basal cAMP level measured in H9C2 cells, either pretreated with 30 nmol·L−1 estradiol (E2) or vehicle (control) for 6 or 24 h. Values are means ± SEM, n = 6. *P < 0.05, significantly different from control. (I) Statistical analysis of the basal cytosolic cAMP level measured in H9C2 cells transfected with H30 cytosolic cAMP sensor and either pretreated with 30 nmol·L−1 estradiol (E2) or vehicle (control) for 24 h. Values are means ± SEM, n = 8.
It is noteworthy that estradiol treatment significantly reduced the basal level of mitochondrial cAMP in both cell types (Figure 2C, F). cAMP analysis with green fluorescence protein (GFP)‐derived probes is technically difficult, as pH affects the fluorescence intensity and most GFP mutants are sensitive to pH changes in the physiological range (Llopis et al., 1998; Abad et al., 2004). Therefore, we tested for an effect of estradiol treatment on mitochondrial pH under basal conditions, using transfection with mitochondria‐targeted pH sensor SypHer in H9C2 cells (Poburko et al., 2011). Treatment with estradiol for 24 h did not affect the basal pH value in the mitochondrial matrix (Figure 2G).
To assess the effects of estradiol treatment on mitochondrial cAMP at earlier time points, which would be an indication for non‐genomic actions, treatments with estradiol for 6 and 24 h were compared in H9C2 cells. There was no effect of estradiol on the mitochondrial cAMP after 6 h treatment (Figure 2H). Interestingly, while affecting the basal mitochondrial cAMP level, 24 h treatment with estradiol did not affect the basal cytosolic cAMP concentration (Figure 2I). These data argue for an estradiol‐induced reduction of the basal cAMP level in mitochondria only.
To strengthen the finding, treatment with ER‐specific agonists was performed. Interestingly, only the ERα agonist PPT simulated the effects of estradiol (Supporting Information Figure S3B). We further examined whether an estradiol‐induced reduction in mitochondrial cAMP concentration may be due to reduced basal sAC activity. To test this hypothesis, the basal sAC activity was indirectly analysed, using IBMX, a non‐selective inhibitor of PDEs. Such treatment gradually increased the FRET ratio, indicating an accumulation of cAMP in the mitochondrial matrix due to mitochondrial sAC activity (Figure 3A). Pretreatment of H9C2 or C2C12 cells with estradiol for 24 h did not significantly change this response to IBMX (Figure 3B, C).
Figure 3.
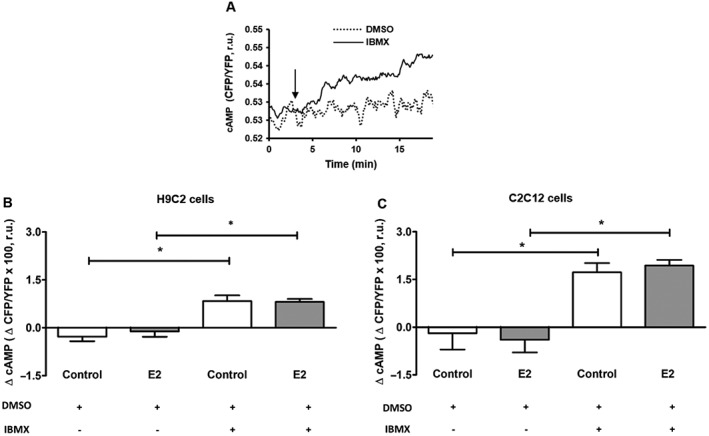
FRET‐based indirect assay of the basal mitochondrial sAC activity performed in cells transfected with 4mtH30 sensor. (A) Representative kinetics of mitochondrial cAMP level observed in H9C2 cells treated with 200 μmol·L−1 IBMX or vehicle (DMSO). Data are representative of four independent experiments with similar results. (B, C) Statistical analysis of cAMP changes (ΔcAMP) within 15 min after treatment with IBMX or DMSO performed in H9C2 or C2C12 cells pretreated with either 30 nmol·L−1 estradiol (E2) or vehicle (control). Values are means ± SEM, n = 5. *P < 0.05, significantly different from corresponding group under treatment with DMSO only.
Role of mitochondrial PDE2 in estradiol‐induced reduction of mitochondrial cAMP
To further explore the mechanisms responsible for the reduction of mitochondrial cAMP level through estradiol, the contribution of PDE2, a major cAMP degrading enzyme found in the mitochondrial matrix (Acin‐Perez et al., 2011), was assessed. For this purpose, cells were treated for 15 min before and during the experiment, either with the non‐selective PDE inhibitor IBMX (200 μmol·L−1) or with the PDE2 specific inhibitor Bay 60‐7550 (100 nmol·L−1). Treatment of H9C2 cells with both PDE inhibitors abolished the estradiol‐induced reduction of cAMP (Figure 4A, C). Similar effects were found in C2C12 cells (Figure 4B, D). To substantiate the findings and exclude the potential effects of PDE2 inhibition in the cytosol, isolated mitochondria were incubated with Bay 60‐7550, followed by mitochondrial cAMP analysis by MS. In agreement with the FRET‐based data, estradiol reduced the basal mitochondrial cAMP level (control 1.04 ± 0.03 and estradiol, 0.67 ± 0.10 nmol·μg−1 of protein; n = 4; incomplete data, not subject to statistical analysis). This effect was attenuated by treatment with Bay 60‐7550: control 1.07 ± 0.04 and estradiol, 0.88 ± 0.11 nmol·μg−1 of proteins; n = 3 (incomplete data, not subject to statistical analysis). Altogether, the findings obtained with different techniques suggest that PDE2 was significantly involved in the estradiol‐induced reduction of mitochondrial cAMP.
Figure 4.
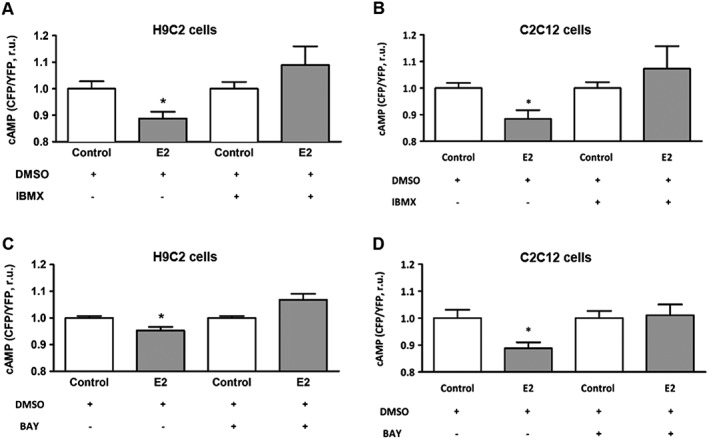
Inhibition of PDEs abolished estradiol effect on the basal mitochondrial cAMP level measured in cells transfected with 4mtH30 sensor. FRET‐based analyses of basal mitochondrial cAMP concentration performed in H9C2 and C2C12 cells pretreated with either 30 nmol·L−1 estradiol (E2) or vehicle (control) for 24 h, and additionally treated with IBMX (200 μmol·L−1) (A, B) or Bay 60‐7550 (100 nmol·L−1) (C, D) 15 min before and during cAMP measurement. Values are means ± SEM, n = 5 for H9C2 cells and n = 5–6 for C2C12 cells. *P < 0.05, significantly different from corresponding control.
Role of NO‐cGMP signalling in estradiol‐induced reduction of mitochondrial cAMP
PDE2 is a cGMP‐activated PDE (Martinez et al., 2002). Increasing evidence suggests that ER stimulation promotes NO‐cGMP signalling (Marathe et al., 2012) and, therefore, may lead to the activation of mitochondrial PDE2. To test this hypothesis, cellular cGMP analysis was performed. Treatment of H9C2 cells with the NO donor (NOC‐12, 100 μmol·L−1) for 30 min significantly increased total cellular cGMP level (Figure 5A). Similar to NO donor, 24 h treatment with estradiol led to a significant rise in cellular cGMP (Figure 5B). Thus, in agreement with previous reports, in our model, estradiol leads to the activation of the NO system, which, in turn, may promote sGC activity in mitochondria as the mitochondrial membrane is permeable to NO.
Figure 5.
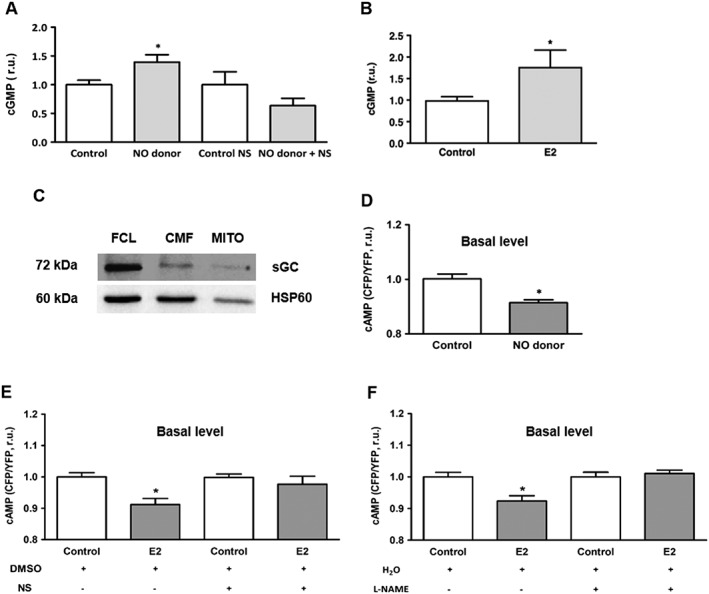
Role of NO‐cGMP signalling in estradiol‐induced reduction of mitochondrial cAMP level. (A, B) Statistical analysis of total cellular cGMP concentration in H9C2 cells pretreated with the NO donor (NOC‐12, 100 μmol·L−1, 30 min) or estradiol (E2, 30 nmol·L−1, 24 h). Additional treatment with the sGC inhibitor NS‐2880 (10 μmol·L−1, 30 min) or vehicle (DMSO) was performed to show the efficacy of sGC inhibitor (A). Values are means ± SEM, n = 5–7 for (A) and n = 8 for (B). *P < 0.05 significantly different from corresponding control. (C) Western blot analysis of the β catalytic subunit of sGC performed in full cell lysate (FCL), crude (CMF) and purified (MITO) mitochondrial fractions of H9C2 cells. Data are representative of three independent experiments with similar results. (D) Statistical analyses of the basal mitochondrial cAMP level in H9C2 cells transfected with 4mtH30 sensor and pretreated with NO donor (NOC‐12, 100 μmol·L−1) or vehicle (control) for 30 min. Values are means ± SEM, n = 7. *P < 0.05, significantly different from control. (E) FRET‐based analysis of basal cAMP level performed in H9C2 cells transfected with 4mtH30 sensor and either pretreated with 30 nmol·L−1 estradiol (E2) or vehicle (control) for 24 h and additionally treated for 30 min before and during cAMP measurement with the sGC inhibitor NS‐2880 (10 μmol·L−1) or vehicle (DMSO). Values are means ± SEM, n = 5–6. *P < 0.05, significantly different from control. All data were normalized to the corresponding control and expressed in relative units (r.u.). (F) FRET‐based analysis of basal cAMP level performed in H9C2 cells transfected with 4mtH30 sensor and either pretreated with estradiol (E2, 30 nmol·L−1) or vehicle (control) for 24 h and additionally treated for 30 min before and during cAMP measurement with the NOS inhibitor L‐NAME (300 μmol·L−1) or vehicle (H2O). Values are means ± SEM, n = 6–7. *P < 0.05, significantly different from control. All data were normalized to the corresponding control and expressed in relative units (r.u.).
To our knowledge, there are no data demonstrating the expression of sGC in the mitochondrial matrix, though indirect evidence has been provided (Kimura and Murad, 1974; Nakazawa et al., 1976; Acin‐Perez et al., 2011). To investigate whether H9C2 cells express sGC in mitochondria, we used the fractionation technique described above (Figure 1A). Western blot analysis revealed the presence of the sGC catalytic subunit β in mitochondria (Figure 5C). To confirm the involvement of NO‐sGC signalling in the estradiol‐induced reduction of intra‐mitochondrial cAMP, first, a FRET‐based analysis of mitochondrial cAMP was performed in H9C2 cells pretreated with the NO donor. Similar to the estradiol treatment, pre‐incubation of cells with NO donor NOC‐12 significantly reduced the basal mitochondrial cAMP level (Figure 5D). Second, the estradiol‐induced reduction of mitochondrial cAMP content was prevented by sGC inhibition with NS‐2880 (10 μmol·L−1, 30 min) (Figure 5E) indicating the causal role of this cyclase. Third, the effect of estradiol was abolished by treatment with the NOS inhibitor L‐NAME (Figure 5F). Altogether, these data suggest that NO‐cGMP signalling plays a role in the estradiol‐induced decrease of intra‐mitochondrial cAMP.
Estradiol suppresses cytochrome oxidase activity and reduces mitochondrial membrane potential
Mounting evidence emphasizes the importance of mitochondrial cAMP for mitochondrial function (Acin‐Perez et al., 2009b; Hess et al., 2014; De Rasmo et al., 2015; Hebert‐Chatelain et al., 2016). Particularly, the alteration of mitochondrial cAMP is directly translated to the cytochrome oxidase activity (Acin‐Perez et al., 2009b). Thus, we investigated whether estradiol‐induced reduction of mitochondrial cAMP concentration may affect cytochrome oxidase activity. For this purpose, we analysed cytochrome oxidase enzymic activity in mitochondria isolated from H9C2 cells. As expected, estradiol treatment led to a significant reduction of cytochrome oxidase activity (Figure 6A). Interestingly, although estradiol reduced cytochrome oxidase activity, it also increased its expression (Figure 6B), which may, at least in part, compensate the negative effect of estradiol on mitochondrial respiratory chain activity.
Figure 6.
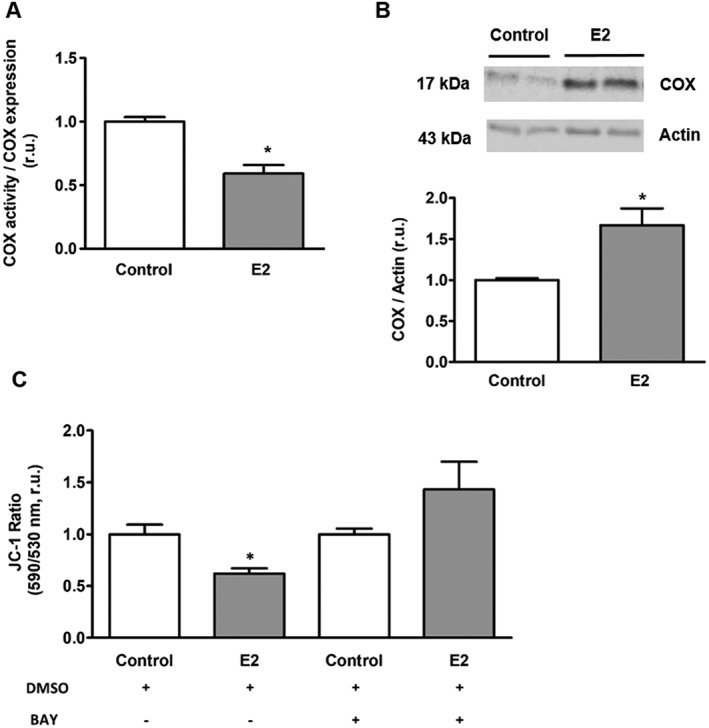
Estradiol effect on cytochrome oxidase (COX) activity and mitochondrial membrane potential. (A) Statistical analysis of cytochrome oxidase activity in mitochondria isolated from H9C2 cells pretreated with either 30 nmol·L−1 estradiol (E2) or vehicle (Control) for 24 h. Values are means ± SEM, n = 7. *P < 0.05, significantly different from control. (B) Western blot and statistical analyses of cytochrome oxidase expression performed in H9C2 cells after treatment with either 30 nmol·L−1 estradiol (E2) or vehicle (Control) for 24 h. Values are means ± SEM, n = 6. *P < 0.05, significantly different from control. (C) Statistical analysis of the basal mitochondrial membrane potential in H9C2 cells treated with either 30 nmol·L−1 estradiol (E2) or vehicle (Control) for 24 h. Cells were treated for 15 min before and during analysis with either Bay 60‐7550 (100 nmol·L−1) or vehicle (DMSO). Values are means ± SEM, n = 7 for DMSO and n = 6–7 for BAY treated groups. *P < 0.05, significantly different from control . All data were normalized to the corresponding control and expressed in relative units (r.u.).
Mitochondrial membrane potential is the main driving force for mitochondrial ATPase activity and, thereby, for mitochondrial ATP synthesis. As the activity of the mitochondrial electron transport chain is crucial for the development of the mitochondrial membrane potential, we aimed to investigate whether estradiol may also affect mitochondrial membrane potential by applying the ratiometric sensor JC‐1. The analysis revealed a significant reduction of mitochondrial membrane potential in cells treated with estradiol in a PDE2‐dependent manner (Figure 6C).
Discussion
The aim of this study was to determine whether estradiol could affect mitochondrial cAMP signalling in muscle cells. The main findings are as follows: (i) treatment with estradiol did not affect the intra‐mitochondrial content of sAC, PDE2 or PKA catalytic subunit α; (ii) analysis of the mitochondrial sAC activity, basal and stimulated did not reveal any significant effect of estradiol treatment; (iii) by contrast, estradiol significantly reduced the basal mitochondrial cAMP level and cytochrome oxidase activity in a PDE2‐dependent manner. Together, these results suggest that NO‐cGMP signalling serves as a mechanistic link between estradiol and mitochondrial PDE2 (Figure 7).
Figure 7.
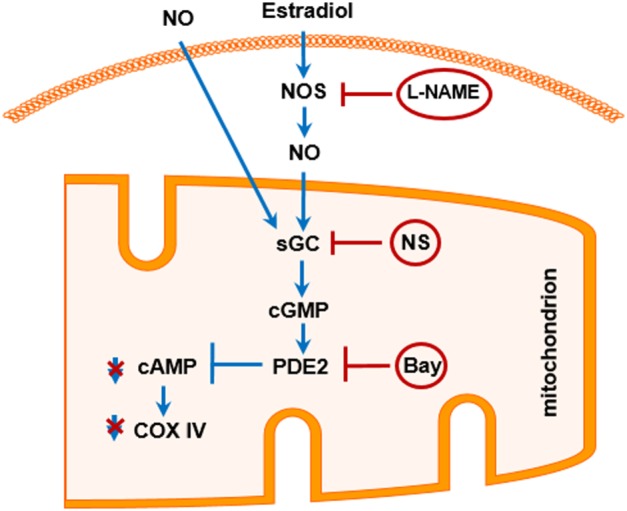
Schematic model of the proposed cellular mechanisms involved in the regulation of mitochondrial cAMP by estradiol. Activation of the cellular NO signalling either by estradiol or by NO donor stimulates mitochondrial sGC followed by activation of PDE2 and degradation of mitochondrial cAMP. Reduction of mitochondrial cAMP concentration results in the reduced cytochrome oxidase (COX IV) activity. Inhibition of NOS with L‐NAME, sGC with NS‐2880 (NS) or PDE2 with Bay 60‐7550 (Bay) prevents these effects of estradiol.
The functional significance of sAC in many cellular function has been demonstrated (Acin‐Perez et al., 2009b; Appukuttan et al., 2012; Di Benedetto et al., 2013; Appukuttan et al., 2014; Chagtoo et al., 2018; see also Pozdniakova and Ladilov, 2018). A convincing body of evidence supports the functional significance of the mitochondrial cAMP signalling in eukaryotes (Acin‐Perez et al., 2009b; Acin‐Perez et al., 2011; De Rasmo et al., 2012; Hess et al., 2014; Hebert‐Chatelain et al., 2016). Moreover, recent data suggest that mitochondria possess their own source of cAMP, that is, matrix‐localized sAC, thus being independent of the cytosolic variations in cAMP concentration (Di Benedetto et al., 2013; Lefkimmiatis et al., 2013). In accordance with this, the present study demonstrated that the activation of plasmalemmal AC with forskolin caused the rapid elevation of cytosolic cAMP concentration but did not affect the cAMP concentration in mitochondria. Of note, both we and other groups (Di Benedetto et al., 2013; Lefkimmiatis et al., 2013; Mukherjee et al., 2016; Wang et al., 2016) have observed a rapid increase in intra‐mitochondrial cAMP following sAC stimulation with bicarbonate.
The promotion of mitochondrial cAMP signalling, for example, through overexpression of sAC, improves cytochrome oxidase activity and ATP synthesis by mitochondria (Acin‐Perez et al., 2009b). Currently, there are no pharmacological or molecular tools that can be applied in vivo to promote mitochondrial cAMP signalling. In this study, we investigated the hypothesis that estradiol may have an effect on this pathway. Estradiol positively regulated cAMP signalling in a variety of cell types, for example, in rat cardiomyocytes (Buitrago et al., 2000) or coronary artery smooth muscle cells (Yu et al., 2014), through non‐genomic action of plasmalemmal ER on the tmAC activity. Similarly, the limited data demonstrating a genomic action of estradiol on cAMP signalling support its positive regulatory effect (Asano et al., 2005; Ismail et al., 2015).
On the other hand, positive effects of estradiol on the expression and activity of numerous mitochondrial proteins have also been reported (Chen et al., 2009; Lagranha et al., 2010; Galmes‐Pascual et al., 2017). Whether and how estradiol may affect mitochondrial cAMP signalling remain unknown. In the present study, expression analysis of three main members of the mitochondrial cAMP signalling, that is, sAC, PDE2 and PKA, revealed no effect of estradiol on the content of these proteins in the mitochondrial matrix.
Aside from expression, estrogens may also modulate the activity of mitochondrial enzymes via various post‐translational modifications (Lagranha et al., 2010; Luo et al., 2016) and, thereby, affect mitochondrial cAMP signalling. Applying mitochondria‐targeted expression of EPAC‐based cAMP sensor, we found that estradiol significantly reduced cAMP level in the mitochondrial matrix. This effect of estradiol on intra‐mitochondrial cAMP is not due to sensor mistargeting or other artefacts. First, our analyses confirmed the proper localization of the 4mtH30 sensor inside the mitochondria (Supporting Information Figure S2). Thus, the signal obtained from mitochondria‐targeted cAMP sensor has an intra‐mitochondrial origin. Second, the known effect of pH on GFP‐derived sensors is unlikely to have contributed to the effect of estradiol on the basal intra‐mitochondrial cAMP level, as intra‐mitochondrial pH analyses revealed no effect of estradiol (Figure 2G). Third, the estradiol effect on the mitochondrial cAMP concentration was confirmed by the MS analysis of cAMP in isolated mitochondria.
To understand the underlying cellular mechanisms responsible for the estradiol‐induced reduction of the intra‐mitochondrial cAMP level, the role of mitochondrial cAMP generating (sAC) and degrading (PDE2) enzymes was investigated. Our results revealed that the estradiol effect on the basal intra‐mitochondrial cAMP level was not due to the alteration in basal cAMP synthesis by sAC but rather due to an enhanced cAMP degradation by mitochondrial PDE2. Indeed, in our model, inhibition of PDE2 abolished estradiol‐induced reduction in mitochondrial cAMP. Although from the live imaging experiments we cannot completely exclude a possible contribution of cytosolic PDE2 inhibition, the treatment of isolated mitochondria with PDE2 inhibitor led to a similar result, as estimated by MS analysis. Thus, mitochondrial PDE2 is responsible for the estradiol effect on the mitochondrial cAMP concentration.
PDE2 is a cGMP‐activated PDE isoform and may be activated by estradiol via up‐regulation of NO signalling. Indeed, (i) estradiol pretreatment, similar to treatment with a NO donor, elevates total cellular cGMP content, and (ii) inhibition of sGC or NOS prevented effect of estradiol on mitochondrial cAMP. In line with our findings, numerous reports demonstrated estradiol‐induced up‐regulation of eNOS and iNOS in various cell types (MacRitchie et al., 1997; Jayachandran et al., 2001), including cardiomyocytes (Nuedling et al., 1999), accompanied by increased NO synthesis (for a review, see also Ling et al., 2006). Therefore, it seems likely that NOS is a target of estradiol leading to up‐regulation of NO signalling.
The NO‐induced generation of cGMP in cardiac mitochondria has already been suggested (Seya et al., 2007). It is important to note that this study was performed in isolated mitochondria, thus excluding a possible import of cytosolic cGMP. Whether cellular NO elevation may affect mitochondrial cAMP signalling had not been investigated before. To address this issue, we, first, confirmed the presence of sGC in the mitochondrial matrix. Second, the treatment of cells with a NO donor led to a significant reduction in the mitochondrial cAMP level. Third, the inhibition of sGC prevented the effects of estradiol on mitochondrial cAMP, confirming the causal role of sGC. In agreement with our data, an earlier study of Acin‐Perez and colleagues demonstrated a cGMP‐induced stimulation of cAMP‐degrading activity of PDE2 in mitochondria isolated from brain and liver (Acin‐Perez et al., 2011). Thus, the present study, together with previous reports, suggests that the sGC‐PDE2 axis is present in the mitochondrial matrix and responds to cellular NO elevation leading to a reduction of mitochondrial cAMP.
In addition to the cGMP synthesis in the mitochondrial matrix, cytosolic cGMP may be imported. Though there are no studies investigating this issue, previous reports demonstrated that the mitochondrial inner membrane is not permeable for cAMP (Di Benedetto et al., 2013; Lefkimmiatis et al., 2013), which has similar size and charge. Thus, it seems unlikely that cGMP penetrates the inner mitochondrial membrane.
In order to understand the functional significance of the estradiol‐induced reduction in mitochondrial cAMP concentration, the cytochrome oxidase activity was examined. Previous reports suggest that intra‐mitochondrial cAMP plays a role in the regulation of cytochrome oxidase activity (Acin‐Perez et al., 2009b; Acin‐Perez et al., 2011). In agreement with these reports, a reduction in mitochondrial cAMP was accompanied by the reduced cytochrome oxidase activity in our experiment. Of note, estradiol increased cytochrome oxidase expression in our study, which is in agreement with a known genomic effect of estradiol on mitochondrial biogenesis (Chen et al., 2009). Thus, oestrogen has two apparently opposing effects on mitochondrial function: increasing respiratory capacity by increasing cytochrome oxidase expression and at the same time, limiting maximal cytochrome oxidase activity.
The negative effect of estradiol on cytochrome oxidase activity was associated with a partial mitochondrial depolarization in the present study. Though this effect may be due to reduced cytochrome oxidase activity, other factors may also affect mitochondrial membrane potential, for example, up‐regulated expression of mitochondrial uncoupling proteins. However, several studies have suggested a down‐regulation of these proteins by estradiol in muscle cells (Alexanderson et al., 2009; Nagai et al., 2016).
Although our results present a discrepancy with the majority of findings that suggest a stimulatory effect of estradiol on mitochondrial function (Chen et al., 2009; Liu et al., 2017), the known multiple effects of estradiol on mitochondria may compensate the observed suppression of cytochrome oxidase activity. Indeed, in line with previous reports, we found a significant up‐regulation of cytochrome oxidase expression under estradiol treatment. Whether the estradiol‐induced down‐regulation of mitochondrial cAMP and cytochrome oxidase activity may have positive or negative effects in different physiological and pathological situations needs further investigation. One may suppose that similar to the allosteric ATP‐inhibition of cytochrome oxidase (Ramzan et al., 2010), estradiol‐induced inhibition of cytochrome oxidase may limit free radical generation by reducing membrane potential.
The intriguing finding of the study is that oestrogen indirectly affects mitochondrial cAMP and cytochrome oxidase activity via activation of NO signalling unveiling a complex action of oestrogen on mitochondria. This finding may, therefore, contribute to explain negative or even harmful effects (Lobo, 2017; Azam et al., 2018) of hormone replacement therapy.
In summary, the present study reports that estradiol did not affect the expression or activity of mitochondrial sAC, whereas it markedly reduced the mitochondrial cAMP concentration and cytochrome oxidase activity and induced partial mitochondrial depolarization. These results suggest that the effects of estradiol are mechanistically linked to the activation of mitochondrial sGC‐PDE2 axis due to the stimulatory action on the cellular NO signalling.
Author contributions
S.P. and Y.L. conceived and designed the study, performed data analysis and wrote the manuscript. S.P., M.G.‐M., G.G., G.d.B. and Y.L. performed the experiments and procedures; V.R.‐Z. generated research funds and coordinated the project. All authors commented on the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 (A, B) Western blot expression analysis of estrogen receptor (ER) alpha, beta and GRP30 in H9C2 cells. The uterus was used as a positive control for estrogen receptors alpha and beta, while MCF7 cells were used as a positive control for GRP30. Data are representative of three independent experiments with similar results. (C) Western bot analysis of phosphorylated ERK1/2 (pERK) performed with H9C2 cells after treatment with 30 nmol·L−1 estradiol (E2) or vehicle (Control) for 24 h. Data are representative of three independent experiments with similar results. (D) Analysis of estradiol‐induced activation of estrogen receptors performed in H9C2 cells overexpressing estrogen receptor alpha or beta. Cells were treated with 30 nmol·L−1 estradiol (E2) or vehicle (Control) for 24 h. Values are means ± SEM. n = 8. *P < 0.05 vs. corresponding Control. (E) Analysis of cell number (normalized to Control) after treatment with nmol·L−1 estradiol (E2) or vehicle (Control) for 24 h. Values are means ± SEM. n = 6. *P < 0.05 vs Control.
Figure S2 Characterization of mitochondria‐targeted cAMP probe expressed in H9C2 cells. (A) Statistical analysis of CFP fluorescence obtained in H9C2 cells transfected with mitochondria‐targeted cAMP probe 4mtH30 from mitochondria‐free, i.e., cytosolic, or mitochondria‐rich regions. Values are means ± SEM. n = 4. (B) Representative fluorescent images of H9C2 cells transfected with EPAC‐based mitochondria‐targeted cAMP probe 4mtH30 (upper panel, green) and stained with MitoTracker deep Red (middle panel, red). The merge image (lower panel) demonstrates a high level of 4mtH30 and MitoTracker deep Red co‐localization (yellow). (C) Representative kinetics of mitochondrial cAMP level observed in H9C2 cells after treatment with either 200 nmol·L−1 EPAC activator (8‐pCPT2’‐O‐Me‐cAMP) or vehicle (Control). Values are means ± SEM. n = 4. *P < 0.05. Arrow indicates the start of treatment. (D) Representative kinetics of mitochondrial cAMP level observed in H9C2 cells under treatment with 50 nmol·L−1 bicarbonate. Cells were pretreated for 30 min with either 100 nmol·L−1 LRE1 (a sAC inhibitor) or vehicle (DMSO). Values are means ± SEM. n = 4. *P < 0.05 (E) Representative kinetics of cytosolic (transfection with non‐targeted H30 probe) or mitochondrial (transfection with mitochondria‐targeted 4mtH30 probe) cAMP observed in a H9C2 cell under treatment with forskolin (40 nmol·L−1) and IBMX (200 nmol·L−1). Units are presented in % and were calculated as (R‐Rmin)/(Rmax‐Rmin)*100%, where R = CFP/YFP, Rmin = R before treatment, Rmax = maximal R after treatment with forskolin and IBMX. Values are means from three experiments.
Figure S3 Effect of ER agonists on mitochondrial cAMP. Statistical analysis of mitochondrial cAMP elevation (Stimulated activity) induced by 50 nmol·L−1 bicarbonate (A) and basal cAMP level (B) were measured in H9C2 cells transfected with 4mtH30 sensor and either pretreated with 1 nmol·L−1 estrogen receptor alpha agonist (PPT) or vehicle (Control) for 24 h. Values are means ± SEM. n = 7. *P < 0.05. Statistical analysis of mitochondrial cAMP elevation (Stimulated activity) induced by 50 nmol·L−1 bicarbonate (C) and basal cAMP level (D) measured in H9C2 cells, either pretreated with 10 nmol·L−1 estrogen receptor beta agonist (KB5) or vehicle (Control) for 24 h. Values are means ± SEM. n = 10. Statistical analysis of mitochondrial cAMP elevation (Stimulated activity) induced by 50 nmol·L−1 bicarbonate (E) and basal cAMP level (F) measured in H9C2 cells, either pretreated with 10 nmol·L−1 GRP30 agonist (G‐1) or vehicle (Control) for 24 h. Values are means ± SEM. n = 10.
Figure S4 Effect of FBS reduction on cellular ATP and mitochondrial morphology. (A) FRET‐based assay of the basal cytosolic ATP measured in H9C2 cells transfected with ATP sensor. Cell were kept for 48 h.
Acknowledgements
We would like to express our gratitude to K. Lefkimmiatis, V. Jayarajan, E. Dworatzek, N. Haritonow and A. Kuehne for their assistance with theoretical and technical components of this research. We thank Pierre Chambon (Institute of Genetics, Molecular and Cellular Biology, CNRS/INSERM, College de France, Illkirch Cedex, France) for providing us with ERα and ERβ plasmids (HEGO vector). We also express our gratitude to Karo bio (Sweden) for providing us with KB5.This study was supported by the European Union (Radox Grant FP7‐PEOPLE‐2012‐ITN), by DZHK (German Centre for Cardiovascular Research) partner site Berlin (Grants B14‐003 and 81Z2100201), by Margarete‐Ammon‐Stiftung and was a part of S. Pozdniakova's thesis project submitted in fulfilment of the requirements for the degree of Doctor of Philosophy at the Freie Universität Berlin (Germany).
Pozdniakova, S. , Guitart‐Mampel, M. , Garrabou, G. , Di Benedetto, G. , Ladilov, Y. , and Regitz‐Zagrosek, V. (2018) 17β‐Estradiol reduces mitochondrial cAMP content and cytochrome oxidase activity in a phosphodiesterase 2‐dependent manner. British Journal of Pharmacology, 175: 3876–3890. 10.1111/bph.14455.
References
- Abad MF, Di Benedetto G, Magalhaes PJ, Filippin L, Pozzan T (2004). Mitochondrial pH monitored by a new engineered green fluorescent protein mutant. J Biol Chem 279: 11521–11529. [DOI] [PubMed] [Google Scholar]
- Acin‐Perez R, Salazar E, Brosel S, Yang H, Schon EA, Manfredi G (2009a). Modulation of mitochondrial protein phosphorylation by soluble adenylyl cyclase ameliorates cytochrome oxidase defects. EMBO Mol Med 1: 392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acin‐Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G (2009b). Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab 9: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acin‐Perez R, Russwurm M, Gunnewig K, Gertz M, Zoidl G, Ramos L et al (2011). A phosphodiesterase 2A isoform localized to mitochondria regulates respiration. J Biol Chem 286: 30423–30432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexanderson C, Eriksson E, Stener‐Victorin E, Lonn M, Holmang A (2009). Early postnatal oestradiol exposure causes insulin resistance and signs of inflammation in circulation and skeletal muscle. J Endocrinol 201: 49–58. [DOI] [PubMed] [Google Scholar]
- Alvaro D, Onori P, Metalli VD, Svegliati‐Baroni G, Folli F, Franchitto A et al (2002). Intracellular pathways mediating estrogen‐induced cholangiocyte proliferation in the rat. Hepatology 36: 297–304. [DOI] [PubMed] [Google Scholar]
- Appukuttan A, Kasseckert SA, Micoogullari M, Flacke JP, Kumar S, Woste A et al (2012). Type 10 adenylyl cyclase mediates mitochondrial bax translocation and apoptosis of adult rat cardiomyocytes under simulated ischaemia/reperfusion. Cardiovasc Res 93: 340–349. [DOI] [PubMed] [Google Scholar]
- Appukuttan A, Flacke JP, Flacke H, Posadowsky A, Reusch HP, Ladilov Y (2014). Inhibition of soluble adenylyl cyclase increases the radiosensitivity of prostate cancer cells. Biochim Biophys Acta 1842: 2656–2663. [DOI] [PubMed] [Google Scholar]
- Asano K, Okawa T, Matsuoka I, Suzuki Y, Sato A (2005). Effects of sex steroids on expression of adenylyl cyclase messenger RNA in rat uterus. J Endocrinol Invest 28: 357–362. [DOI] [PubMed] [Google Scholar]
- Azam S, Lange T, Huynh S, Aro AR, Von Euler‐Chelpin M, Vejborg I et al (2018). Hormone replacement therapy, mammographic density, and breast cancer risk: a cohort study. Cancer Causes Control 29: 495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buitrago C, Massheimer V, De Boland AR (2000). Acute modulation of Ca2+ influx on rat heart by 17β‐estradiol. Cell Signal 12: 47–52. [DOI] [PubMed] [Google Scholar]
- Chagtoo M, George N, Pathak N, Tiwari S, Godbole MM, Ladilov Y (2018). Inhibition of intracellular type 10 adenylyl cyclase protects cortical neurons against reperfusion‐induced mitochondrial injury and apoptosis. Mol Neurobiol 55: 2471–2482. [DOI] [PubMed] [Google Scholar]
- Chen JQ, Cammarata PR, Baines CP, Yager JD (2009). Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim Biophys Acta 1793: 1540–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Brit J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rasmo D, Signorile A, Larizza M, Pacelli C, Cocco T, Papa S (2012). Activation of the cAMP cascade in human fibroblast cultures rescues the activity of oxidatively damaged complex I. Free Radic Biol Med 52: 757–764. [DOI] [PubMed] [Google Scholar]
- De Rasmo D, Signorile A, Santeramo A, Larizza M, Lattanzio P, Capitanio G et al (2015). Intramitochondrial adenylyl cyclase controls the turnover of nuclear‐encoded subunits and activity of mammalian complex I of the respiratory chain. Biochim Biophys Acta 1853: 183–191. [DOI] [PubMed] [Google Scholar]
- Di Benedetto G, Scalzotto E, Mongillo M, Pozzan T (2013). Mitochondrial Ca(2)(+) uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab 17: 965–975. [DOI] [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, Zhang J (2004). Fluorescent indicators of cAMP And Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A 101: 16513–16518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L (2007). Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc 2: 287–295. [DOI] [PubMed] [Google Scholar]
- Galmes‐Pascual BM, Nadal‐Casellas A, Bauza‐Thorbrugge M, Sbert‐Roig M, Garcia‐Palmer FJ, Proenza AM et al (2017). 17β‐Estradiol improves hepatic mitochondrial biogenesis and function through PGC1B. J Endocrinol 232: 297–308. [DOI] [PubMed] [Google Scholar]
- Geng W, Wang Z, Zhang J, Reed BY, Pak CY, Moe OW (2005). Cloning and characterization of the human soluble adenylyl cyclase. Am J Physiol Cell Physiol 288: C1305–C1316. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS guide to pharmacology in 2018: updates and expansion to encompass the new guide to immunopharmacology. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig C, Bahre H, Wolter S, Beckert U, Kaever V, Seifert R (2014). cAMP, cGMP, cCMP and cUMP concentrations across the tree of life: high cCMP and cUMP levels in astrocytes. Neurosci Lett 579: 183–187. [DOI] [PubMed] [Google Scholar]
- Hebert‐Chatelain E, Desprez T, Serrat R, Bellocchio L, Soria‐Gomez E, Busquets‐Garcia A et al (2016). A cannabinoid link between mitochondria and memory. Nature 539: 555–559. [DOI] [PubMed] [Google Scholar]
- Hess KC, Liu J, Manfredi G, Muhlschlegel FA, Buck J, Levin LR et al (2014). A mitochondrial CO2‐adenylyl cyclase‐cAMP signalosome controls yeast normoxic cytochrome c oxidase activity. FASEB J 28: 4369–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YC, Yu HP, Suzuki T, Choudhry MA, Schwacha MG, Bland KI et al (2006). Upregulation of mitochondrial respiratory complex IV by estrogen receptor‐β is critical for inhibiting mitochondrial apoptotic signaling and restoring cardiac functions following trauma‐hemorrhage. J Mol Cell Cardiol 41: 511–521. [DOI] [PubMed] [Google Scholar]
- Huttemann M, Lee I, Samavati L, Yu H, Doan JW (2007). Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim Biophys Acta 1773: 1701–1720. [DOI] [PubMed] [Google Scholar]
- Ismail N, Giribabu N, Muniandy S, Salleh N (2015). Estrogen and progesterone differentially regulate the levels of cystic fibrosis transmembrane regulator (CFTR), adenylate cyclase (AC), and cyclic adenosine mono‐phosphate (cAMP) in the rat cervix. Mol Reprod Dev 82: 463–474. [DOI] [PubMed] [Google Scholar]
- Jayachandran M, Hayashi T, Sumi D, Iguchi A, Miller VM (2001). Temporal effects of 17β‐estradiol on caveolin‐1 mRNA and protein in bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol 281: H1327–H1333. [DOI] [PubMed] [Google Scholar]
- Keshamouni VG, Mattingly RR, Reddy KB (2002). Mechanism of 17‐β‐estradiol‐induced Erk1/2 activation in breast cancer cells. A role for HER2 and PKC‐δ. J Biol Chem 277: 22558–22565. [DOI] [PubMed] [Google Scholar]
- Kim K, Lee D, Ahn C, Kang HY, An BS, Seong YH et al (2017). Effects of estrogen on esophageal function through regulation of Ca2+‐related proteins. J Gastroenterol 52: 929–939. [DOI] [PubMed] [Google Scholar]
- Kimura H, Murad F (1974). Evidence for two different forms of guanylate cyclase in rat heart. J Biol Chem 249: 6910–6916. [PubMed] [Google Scholar]
- Klinge CM (2008). Estrogenic control of mitochondrial function and biogenesis. J Cell Biochem 105: 1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E (2010). Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res 106: 1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkimmiatis K, Leronni D, Hofer AM (2013). The inner and outer compartments of mitochondria are sites of distinct cAMP/PKA signaling dynamics. J Cell Biol 202: 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S, Komesaroff P, Sudhir K (2006). Cellular mechanisms underlying the cardiovascular actions of oestrogens. Clin Sci (Lond) 111: 107–118. [DOI] [PubMed] [Google Scholar]
- Liu A, Philip J, Vinnakota KC, Van Den Bergh F, Tabima DM, Hacker T et al (2017). Estrogen maintains mitochondrial content and function in the right ventricle of rats with pulmonary hypertension. Physiol Rep 5: e13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llopis J, Mccaffery JM, Miyawaki A, Farquhar MG, Tsien RY (1998). Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc Natl Acad Sci U S A 95: 6803–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo RA (2017). Hormone‐replacement therapy: current thinking. Nat Rev Endocrinol 13: 220–231. [DOI] [PubMed] [Google Scholar]
- Lu B, Lee J, Nie X, Li M, Morozov YI, Venkatesh S et al (2013). Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Mol Cell 49: 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo T, Liu H, Kim JK (2016). Estrogen protects the female heart from ischemia/reperfusion injury through manganese superoxide dismutase phosphorylation by mitochondrial P38β at threonine 79 and serine 106. Plos One 11: e0167761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRitchie AN, Jun SS, Chen Z, German Z, Yuhanna IS, Sherman TS et al (1997). Estrogen upregulates endothelial nitric oxide synthase gene expression in fetal pulmonary artery endothelium. Circ Res 81: 355–362. [DOI] [PubMed] [Google Scholar]
- Marathe N, Rangaswami H, Zhuang S, Boss GR, Pilz RB (2012). Pro‐survival effects of 17β‐estradiol on osteocytes are mediated by nitric oxide/cGMP via differential actions of cGMP‐dependent protein kinases I and II. J Biol Chem 287: 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez SE, Wu AY, Glavas NA, Tang XB, Turley S, Hol WG et al (2002). The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc Natl Acad Sci U S A 99: 13260–13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medja F, Allouche S, Frachon P, Jardel C, Malgat M, Mousson De Camaret B et al (2009). Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 9: 331–339. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Jansen V, Jikeli JF, Hamzeh H, Alvarez L, Dombrowski M et al (2016). A novel biosensor to study cAMP dynamics in cilia and flagella. Elife 5: e14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai S, Ikeda K, Horie‐Inoue K, Shiba S, Nagasawa S, Takeda S et al (2016). Estrogen modulates exercise endurance along with mitochondrial uncoupling protein 3 downregulation in skeletal muscle of female mice. Biochem Biophys Res Commun 480: 758–764. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, Sano M, Saito T (1976). Subcellular distribution and properties of guanylate cyclase in rat cerebellum. Biochim Biophys Acta 444: 563–570. [DOI] [PubMed] [Google Scholar]
- Nuedling S, Kahlert S, Loebbert K, Doevendans PA, Meyer R, Vetter H et al (1999). 17β‐Estradiol Stimulates Expression Of Endothelial And Inducible No Synthase In Rat Myocardium In‐Vitro And In‐Vivo. Cardiovasc Res 43: 666–674. [DOI] [PubMed] [Google Scholar]
- Poburko D, Santo‐Domingo J, Demaurex N (2011). Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J Biol Chem 286: 11672–11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponsioen B, Zhao J, Riedl J, Zwartkruis F, Van Der Krogt G, Zaccolo M et al (2004). Detecting cAMP‐induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep 5: 1176–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozdniakova S, Ladilov Y (2018). Functional significance of the Adcy10‐dependent intracellular cAMP compartments. J Cardiovasc Dev Dis 5: E29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos‐Espiritu L, Kleinboelting S, Navarrete FA, Alvau A, Visconti PE, Valsecchi F et al (2016). Discovery of LRE1 as a specific and allosteric inhibitor of soluble adenylyl cyclase. Nat Chem Biol 12: 838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramzan R, Staniek K, Kadenbach B, Vogt S (2010). Mitochondrial respiration and membrane potential are regulated by the allosteric ATP‐inhibition of cytochrome c oxidase. Biochim Biophys Acta 1797: 1672–1680. [DOI] [PubMed] [Google Scholar]
- Rettberg JR, Yao J, Brinton RD (2014). Estrogen: a master regulator of bioenergetic systems in the brain and body. Front Neuroendocrinol 35: 8–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardanelli AM, Signorile A, Nuzzi R, Rasmo DD, Technikova‐Dobrova Z, Drahota Z et al (2006). Occurrence of A‐kinase anchor protein and associated cAMP‐dependent protein kinase in the inner compartment of mammalian mitochondria. FEBS Lett 580: 5690–5696. [DOI] [PubMed] [Google Scholar]
- Seya K, Motomura S, Furukawa K (2007). Cardiac mitochondrial cGMP stimulates cytochrome c release. Clin Sci (Lond) 112: 113–121. [DOI] [PubMed] [Google Scholar]
- Valsecchi F, Ramos‐Espiritu LS, Buck J, Levin LR, Manfredi G (2013). cAMP and mitochondria. Physiology (Bethesda) 28: 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu D, Varin A, Nicolas V, Courilleau D, Mateo P et al (2016). A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death. Cell Death Dis 7: e2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Li F, Klussmann E, Stallone JN, Han G (2014). G protein‐coupled estrogen receptor 1 mediates relaxation of coronary arteries via cAMP/PKA‐dependent activation of MLCP. Am J Physiol Endocrinol Metab 307: E398–E407. [DOI] [PubMed] [Google Scholar]
- Zippin JH, Chen Y, Straub SG, Hess KC, Diaz A, Lee D et al (2013). CO2/HCO3(−)‐ and calcium‐regulated soluble adenylyl cyclase as a physiological ATP sensor. J Biol Chem 288: 33283–33291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A, B) Western blot expression analysis of estrogen receptor (ER) alpha, beta and GRP30 in H9C2 cells. The uterus was used as a positive control for estrogen receptors alpha and beta, while MCF7 cells were used as a positive control for GRP30. Data are representative of three independent experiments with similar results. (C) Western bot analysis of phosphorylated ERK1/2 (pERK) performed with H9C2 cells after treatment with 30 nmol·L−1 estradiol (E2) or vehicle (Control) for 24 h. Data are representative of three independent experiments with similar results. (D) Analysis of estradiol‐induced activation of estrogen receptors performed in H9C2 cells overexpressing estrogen receptor alpha or beta. Cells were treated with 30 nmol·L−1 estradiol (E2) or vehicle (Control) for 24 h. Values are means ± SEM. n = 8. *P < 0.05 vs. corresponding Control. (E) Analysis of cell number (normalized to Control) after treatment with nmol·L−1 estradiol (E2) or vehicle (Control) for 24 h. Values are means ± SEM. n = 6. *P < 0.05 vs Control.
Figure S2 Characterization of mitochondria‐targeted cAMP probe expressed in H9C2 cells. (A) Statistical analysis of CFP fluorescence obtained in H9C2 cells transfected with mitochondria‐targeted cAMP probe 4mtH30 from mitochondria‐free, i.e., cytosolic, or mitochondria‐rich regions. Values are means ± SEM. n = 4. (B) Representative fluorescent images of H9C2 cells transfected with EPAC‐based mitochondria‐targeted cAMP probe 4mtH30 (upper panel, green) and stained with MitoTracker deep Red (middle panel, red). The merge image (lower panel) demonstrates a high level of 4mtH30 and MitoTracker deep Red co‐localization (yellow). (C) Representative kinetics of mitochondrial cAMP level observed in H9C2 cells after treatment with either 200 nmol·L−1 EPAC activator (8‐pCPT2’‐O‐Me‐cAMP) or vehicle (Control). Values are means ± SEM. n = 4. *P < 0.05. Arrow indicates the start of treatment. (D) Representative kinetics of mitochondrial cAMP level observed in H9C2 cells under treatment with 50 nmol·L−1 bicarbonate. Cells were pretreated for 30 min with either 100 nmol·L−1 LRE1 (a sAC inhibitor) or vehicle (DMSO). Values are means ± SEM. n = 4. *P < 0.05 (E) Representative kinetics of cytosolic (transfection with non‐targeted H30 probe) or mitochondrial (transfection with mitochondria‐targeted 4mtH30 probe) cAMP observed in a H9C2 cell under treatment with forskolin (40 nmol·L−1) and IBMX (200 nmol·L−1). Units are presented in % and were calculated as (R‐Rmin)/(Rmax‐Rmin)*100%, where R = CFP/YFP, Rmin = R before treatment, Rmax = maximal R after treatment with forskolin and IBMX. Values are means from three experiments.
Figure S3 Effect of ER agonists on mitochondrial cAMP. Statistical analysis of mitochondrial cAMP elevation (Stimulated activity) induced by 50 nmol·L−1 bicarbonate (A) and basal cAMP level (B) were measured in H9C2 cells transfected with 4mtH30 sensor and either pretreated with 1 nmol·L−1 estrogen receptor alpha agonist (PPT) or vehicle (Control) for 24 h. Values are means ± SEM. n = 7. *P < 0.05. Statistical analysis of mitochondrial cAMP elevation (Stimulated activity) induced by 50 nmol·L−1 bicarbonate (C) and basal cAMP level (D) measured in H9C2 cells, either pretreated with 10 nmol·L−1 estrogen receptor beta agonist (KB5) or vehicle (Control) for 24 h. Values are means ± SEM. n = 10. Statistical analysis of mitochondrial cAMP elevation (Stimulated activity) induced by 50 nmol·L−1 bicarbonate (E) and basal cAMP level (F) measured in H9C2 cells, either pretreated with 10 nmol·L−1 GRP30 agonist (G‐1) or vehicle (Control) for 24 h. Values are means ± SEM. n = 10.
Figure S4 Effect of FBS reduction on cellular ATP and mitochondrial morphology. (A) FRET‐based assay of the basal cytosolic ATP measured in H9C2 cells transfected with ATP sensor. Cell were kept for 48 h.