

Abstract
Background and Purpose
Pulmonary arterial hypertension (PAH) is a life‐threatening disease that leads to progressive pulmonary hypertension, right heart failure and death. Parenteral prostaglandins (PGs), including treprostinil, a prostacyclin analogue, represent the most effective medical treatment for severe PAH. We investigated the effect of treprostinil on established severe PAH and underlying mechanisms using the rat SU5416 (SU, a VEGF receptor‐2 inhibitor)‐chronic hypoxia (Hx) model of PAH.
Experimental Approach
Male Sprague Dawley rats were injected with SU (20 mg·kg−1, s.c.) followed by 3 weeks of Hx (10% O2) to induce severe PAH. Four weeks post‐SU injection, baseline right ventricular (RV) systolic pressure (RVSP) was measured, and the rats were randomized to receive vehicle or treprostinil treatment (Trep‐100: 100 ng·kg−1·min−1 or Trep‐810: 810 ng·kg−1·min−1). Following 3 weeks of treatment, haemodynamic and echocardiographic assessments were performed, and tissue samples were collected for protein expression and histological analysis.
Key Results
At week 7, no difference in RVSP or RV hypertrophy was observed between vehicle and Trep‐100; however, Trep‐810 significantly reduced RVSP and RV hypertrophy. Trep‐810 treatment significantly improved cardiac structure and function. Further, a short‐term infusion of treprostinil in rats with established PAH at 4 weeks post‐SU produced an acute, dose‐dependent reduction in RVSP consistent with a vasodilator effect. However, chronic Trep‐810 treatment did not alter media wall thickness, degree of vascular occlusion or total vessel count in the lungs.
Conclusions and Implications
Treprostinil exerts therapeutic benefits in PAH through decreased vascular resistance and improved cardiac structure and function; however, treprostinil treatment does not have direct impact vascular remodelling.
Abbreviations
- BMPR2
bone morphogenic protein receptor 2
- CO
cardiac output
- HR
heart rate
- Hx
chronic hypoxia
- LV
left ventricle
- MCT
monocrotaline
- OCT
optimum cutting temperature formulation
- PA
pulmonary artery
- PAAT
pulmonary artery acceleration time
- PAH
pulmonary arterial hypertension;
- PASMC
pulmonary artery smooth‐muscle cell
- PET
pulmonary ejection time
- PVR
pulmonary vascular resistance
- RV
right ventricle
- RVID‐d/LVID‐d
RV chamber size expressed as the ratio of end‐diastolic RV to LV internal diameter
- RVSP
right ventricular systolic pressure
- S
septum
- SMAD
homologues of the Drosophila protein, mothers against decapentaplegic (Mad) and the Caenorhabditis elegans protein Sma
- SU5416
1,3‐dihydro‐3‐[(3,5‐dimethyl‐1H‐pyrrol‐2‐yl)methylene]‐2H‐indol‐2‐one
- SUHx
SU5416 with chronic hypoxia
- SV
stroke volume
- TAPSE
tricuspid annulus planar systolic excursion
- Trep‐100
100 ng·kg−1·min−1 treprostinil
- Trep‐810
810 ng·kg−1·min−1 treprostinil
Introduction
Pulmonary arterial hypertension (PAH) is a progressive disease of the lung vascular system that leads to increased pulmonary vascular resistance (PVR), right heart failure and death. PAH remains an incurable disease despite recent advances that have led to approval of multiple drug therapies, which have improved the quality of life of PAH patients (Lau et al., 2017). Among these therapies, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1915 analogues have become the cornerstone of the pharmacotherapy for PAH patients (Farber and Gin‐Sing 2016). Continuous i.v. infusion of a prostacyclin analogue, epoprostenol, was the first PAH‐specific drug therapy to be introduced (Sitbon and Vonk Noordegraaf 2017). Newer prostacyclin analogues, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1895 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5820, have better stability profiles, making it feasible to deliver them via continuous s.c. or i.v. infusion. Prostacyclin therapy improves the functional capacity and survival of PAH patients (Simonneau et al., 2002; Lau et al., 2017).
The development of effective drug therapies for PAH has been hampered by the lack of animal models that faithfully reproduce the salient pathophysiological features of this disease. Most therapies to date have been developed using animal models of PAH that fail to reproduce complex arterial occlusive remodelling such as plexiform lesions, which are the hallmark features of the human disease (Ryan et al., 2011). While all the animal models of PAH have limitations, the rat http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5056 (SU; http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1813&familyId=324&familyType=CATALYTICRECEPTOR inhibitor) and chronic hypoxia (SUHx) model is currently well accepted. The SUHx model better reproduces the relevant pathological features of human PAH (Taraseviciene‐Stewart et al., 2001; Abe et al., 2010); however, far less is known about the effects of currently available PAH therapies in this model. Unlike in the monocrotaline (MCT) model (Schermuly et al., 2005; Yigitaslan and Sirmagul 2012), chronic treatment with inhaled iloprost did not show improved pulmonary haemodynamics, pulmonary vascular remodelling or cardiac hypertrophy in the SUHx model (Gomez‐Arroyo et al., 2015). Furthermore, the study showed direct effects of iloprost on the heart that lead to improved cardiac function in the SUHx model (Gomez‐Arroyo et al., 2015). Another study using the SUHx model showed that acute i.v. infusion of iloprost decreased right ventricular systolic pressure (RVSP) and lead to a trend towards increased cardiac output (Oka et al., 2007). Similar to iloprost, treprostinil has been shown to prevent and reverse pulmonary hypertension in the MCT and chronic hypoxia model (Nikam et al., 2010; Yang et al., 2010; Zhou et al., 2013). The protective effects of treprostinil in the MCT model included reduced muscularization of small pulmonary arteries, improved pulmonary haemodynamics and improved cardiac function (Yang et al., 2010; Zhou et al., 2013). However, the efficacy of treprostinil has not yet been established in the SUHx model. Therefore, in the present study, we explored the effects of treprostinil treatment on haemodynamic parameters, vascular remodelling and right ventricular function in this model of severe PAH.
Methods
All study protocols were approved by the Animal Care Committee (University of Ottawa, Ontario, Canada) and conducted according to the guidelines from the Canadian Council for the Care of on Animal Care. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010).
Rat SU5416 chronic hypoxia (SUHx) model of PAH
Male Sprague Dawley (Harlan Laboratories, Indianapolis, IN, USA) rats weighing 150–200 g were used for this study. PAH was induced by a single s.c. injection of SU5416 (SU: Tocris, Bristol, UK) in 0.5% carboxymethyl cellulose followed by 3 weeks normobaric chronic hypoxia (Hx, 9–10% O2) using the A‐Chamber ProOx 110 system (BioSpherix Ltd., NY, USA), as previously described (Taraseviciene‐Stewart et al., 2001; Jiang et al., 2016). Following completion of hypoxia treatment, the rats were housed under normoxic condition for an additional 4 weeks (Figure 1A).
Figure 1.

(A) Schematic demonstrating the protocol for treprostinil treatment. Rats were injected with SU (20 mg·kg−1, s.c.) and subjected to 3 weeks of normobaric Hx (10% O2) followed by additional 4 weeks of normoxia exposure. At 4 weeks post‐SU, baseline RVSP was measured and a s.c. osmotic pump for continuous delivery of treprostinil was installed. Echocardiography and RVSP measurement was performed at the end the study (7 weeks post‐SU). (B) Bar graph showing treprostinil plasma concentration in Trep‐100 (n = 12) or Trep‐810 (n = 9) treated rats.
Measurement of RVSP and RV hypertrophy
RVSP was measured using high‐fidelity pressure catheters (Transonic‐Scisense Inc., ON, Canada) at 4 weeks post‐SU (baseline) and at 7 weeks post‐SU (end study) (Figure 1A). For RV catheterization, rats were anaesthetized by an i.p. injection of xylazine (7 mg·kg−1) and ketamine (35 mg·kg−1). The pressure catheter was inserted into the right jugular vein and advanced through the superior vena cava and right atrium into the RV. Haemodynamic parameters were recorded and analysed using the LabScribe3 software (iWorx, Dover, NH, USA). At 7 weeks, after data acquisition, animals were euthanized by exsanguination under anaesthesia. The heart was excised, and the ventricles were dissected from the atria, the aorta and the pulmonary trunk. The RV and left ventricle (LV) and septum (S) were separated, and RV hypertrophy was calculated by measuring the ratio of RV weight to LV plus septum weight (RV/LV + S, Fulton index). The operators acquiring the RVSP and RV hypertrophy data were blinded to the treatment allocation.
Treprostinil treatment
Treprostinil was kindly provided by United Therapeutics Corp. (NC, USA). Treprostinil was suspended in water for injection and dissolved by addition of 1 N sodium hydroxide; pH was adjusted using 1 N and 0.01 N HCl between pH 6.7 and 6.9. Concentration of stock treprostinil was calculated based on the dose of treprostinil and the average weight of the animals at the time of treatment initiation (4 weeks post‐SU). Vehicle was prepared by the same method except without treprostinil. Treprostinil and vehicle solutions were filtered through a 0.22 μ filter and loaded into the ALZET osmotic minipumps 2ML4 (DURECT Corporation, Cupertino, CA, USA) under sterile conditions. The osmotic minipumps were primed for at least 60 min in sterile water before implantation. The pumps were then implanted s.c. into the SUHx rats at 4 weeks post‐SU, immediately after baseline RVSP measurement (Figure 1A). The rats were randomized to receive treprostinil (Trep‐100: 100 ng·kg−1·min−1, n = 14; Trep‐810: 810 ng·kg−1·min−1, n = 9) or vehicle (Veh‐100: vehicle for 100 ng·kg−1·min−1, n = 13; Veh‐810: vehicle for 810 ng·kg−1·min−1, n = 8) treatment before RVSP measurement.
Non‐invasive assessment by echocardiography
Echocardiography was performed at the end of the study using the Vevo2100 ultrasonography system (VisualSonics, ON, Canada). Pulmonary artery acceleration time (PAAT), pulmonary ejection time (PET), pulmonary artery (PA) diameter, tricuspid annulus planar systolic excursion (TAPSE), RV chamber size expressed as the ratio of end‐diastolic RV to LV internal diameter (RVID‐d/LVID‐d), RV anterior wall (RVAW) thickness, stroke volume (SV) and cardiac output (CO) were measured as described previously (Urboniene et al., 2010; Jiang et al., 2016). The operators acquiring and analysing the echocardiographic images were blinded to the treatment allocation.
Lung histological measurements
The left lobe of the lung was inflated via the trachea with 50:50 optimum cutting temperature formulation (OCT)/saline solution (Tissue‐Tek OCT; Qiagen, Mississauga, ON, Canada) and then removed. The left lobe was then cut into thick cross sections and fixed in 4% paraformaldehyde for 24 h, rinsed and washed in PBS for 8 h and stored in 70% ethanol until the day of paraffin embedding. Tissue blocks were sectioned (5 μm thickness) with a microtome (Leica Microsystems, Concord, ON, Canada), placed onto poly‐L‐lysine‐coated slides, dried at 37°C for 16 h and then dewaxed and rehydrated through graded alcohols. For microscopy and quantitative morphometry of the lung, haematoxylin and eosin staining was performed with standard protocols. Images were acquired by Panoramic DESK (3DHISTECH, Hungary) scanscope using Panoramic Scanner and analysed using Panoramic Viewer (3DHISTECH, Hungary). Ten random high power fields (10× magnification) for each rat were analysed for media wall thickness, total vessel count and vascular occlusion. Media wall thickness as a % of external diameter was estimated as described previously (Ogura et al., 2013). The % medial wall thickness = ((distance between the internal and external lamina × 2)/external diameter) × 100. For total vessel count, all the vessels were counted from the 10 random fields. The numbers of completely, partially and non‐occlusive distal arterioles (<100 μm) were quantified from the random fields.
Western blotting
Right lung was collected at the end of study, flash‐frozen in liquid nitrogen and stored at −80°C until further processing. Lung lysates were prepared in CelLytic™ MT Cell Lysis Reagent (Sigma, ON, Canada) containing cOmplete™ protease inhibitor cocktail (Sigma, ON, Canada) and PhosSTOP™ (Sigma, ON, Canada) and using the TissueLyser (Qiagen, ON, Canada) two cycles of 25 Hz for 3 min. The tissue lysate was then centrifuged at 12 000× g for 10 min, and the supernatant was collected. Protein concentration of the protein extract was determined colorimetrically by the DC Protein Assay Kit (Bio‐Rad, ON, Canada), using BSA as standard. SDS‐PAGE of lung protein extract (50 μg) was performed with NuPAGE® Novex® 4–12% Bis‐Tris Protein Gels (ThermoFisher Scientific, ON, Canada). Following transfer of the separated proteins to nitrocellulose membranes (NOVEX iBLOT Gel Transfer Stacks, ThermoFisher Scientific), blots were blocked with 2% BSA in PBS‐T (PBS containing 0.1% Tween 20, pH 7.4). Blots were then incubated with primary antibodies to http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1794 (BMPR2) (BD Pharmingen, Cat# 612292), phospho‐http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=303 (Cell Signalling Technologies, Cat# 13820), VEGFR2 (Cell Signalling Technologies, Cat# 2479S), cleaved http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1619 (Cell Signalling Technologies, Cat# 9661S) or β‐actin (ThermoFisher Scientific, Cat# A5441) for overnight at 4°C. Then the blots were washed, three times for 15 min, with PBS‐T and incubated with appropriate IRDye® anti‐rabbit or anti‐mouse secondary antibodies (LI‐COR Biotechnology, NE, USA) in 2% BSA/PBS‐T. Further, the blots were washed, three times for 15 min, with PBS‐T and imaged with Odyssey® imaging system (LI‐COR Biotechnology). The blots were quantified using the Image Studio™ Software (LI‐COR Biotechnology) and expressed as a percentage of control to reduce the variation between blots. Equal numbers of samples from each group were used per blot to reduce sampling bias.
Caspase 3/7 activity assay
Caspase 3/7 activity in the lung lysates was assessed using Apo‐ONE® Homogeneous Caspase‐3/7 Assay (Promega Corp., WI, USA) according to manufacturer's protocol with slight modifications. Briefly, lung lysate were diluted to 1 μg·μL−1 with CelLytic™ MT Cell Lysis Reagent. Then, 50 μL of diluted reagent (substrate and buffer combined) was added directly to 50 μL samples and incubated at 25°C for 2.5 h. Fluorescence was measured every 30 min using excitation wavelength of 480 nm and emission wavelength of 520 nm. Caspase activity was calculated using gain of fluorescence between 30 min intervals. Amount of metabolized substrate was determined from standard curve of Rhodamin 110.
Plasma treprostinil measurement
Analysis of plasma treprostinil was performed by Tendam Labs (Durham, NC, USA). For the quantitative determination of treprostinil in rat plasma, a method was validated over the concentration range of 0.500 to 500 ng·mL−1. An aliquot (25 μL) of rat plasma was added to 225 μL of methanol containing 10.0 ng·mL−1 of the internal standard, treprostinil‐d4, in a 96‐well analysis plate. The plate was then capped, vortex‐mixed and centrifuged. An aliquot (50 μL) of the supernatant and 200 μL of reagent grade water were then transferred to a clean 96‐well analysis plate. From each well, a 10.0 μL aliquot of the extract was injected onto an ultra‐performance liquid chromatographic system equipped with a triple quadrupole tandem mass spectrometer (AB/MDS Sciex API‐5000) detector operated in negative TurboIonSpray® mode. Separation of treprostinil from extracted matrix materials was accomplished using a Waters Acquity BEH C18 (2.1 × 100 mm, 1.7 μm) column operated at 65°C. Mobile phase A consisted of 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in acetonitrile at a total flow rate of 0.775 mL·min−1. Calibration standards, prepared in rat plasma from 0.500 to 500 ng·mL−1, were used to construct standard curves for treprostinil. Linear‐weighted (1/concentration2) regression analysis of peak area ratio versus theoretical concentration was used to produce calibration curves.
Acute treprostinil treatment in anaesthetized SUHx rats
At 4 weeks, SUHx rats were anaesthetized by an i.p. injection of xylazine (7 mg·kg−1) and ketamine (35 mg·kg−1), and 50% dose of ketamine/xylazine was administered every 10 min until the end of the study. Rats were catheterized for RVSP measurement as described above. For measurement of systolic BP (SBP), the left carotid artery was cannulated and a catheter was placed in the aorta. Left jugular vein was cannulated for continuous i.v. infusion of treprostinil. Following catheterization, RVSP and SBP were recorded for 10 min to establish a stable baseline reading. Treprostinil stock solution (1 mg·mL−1) was prepared as described above. At the end of baseline, treprostinil (810 or 3000 ng·kg−1·min−1, i.v.) or vehicle was administered for 30 min by syringe pump. The injection rate was kept constant at 10 μL·min−1 and a working stock solution of treprostinil was prepared, based on animal weight, by dilution in saline (Baxter, ON, Canada). Following 30 min of acute treprostinil or vehicle administration, the infusion was discontinued, and pressures were monitored for an additional 15 min.
Statistical analysis
Data are represented as mean ± SEM unless otherwise stated. Statistical analysis was performed using GraphPad Prism 7.0 (GraphPad Software, Inc., CA, USA). For statistical comparisons, Student's t‐test or one‐way ANOVA (>2 groups) were performed followed by Tukey multiple comparison test with significance level of P < 0.05.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Results
Treprostinil plasma concentration with continuous s.c. delivery in rat SUHx model
We tested the effects of low‐dose (100 ng·kg−1·min−1; Trep‐100) and high‐dose (810 ng·kg−1·min−1; Trep‐810) treprostinil on established PAH using the rat SUHx model of severe PAH. The low and high doses of treprostinil were selected based on the previous studies that investigated the effects of treprostinil in pulmonary artery banding model and that demonstrated reversal of established PAH in MCT model (Yang et al., 2010; Zhou et al., 2013; Axelgaard et al., 2017). To confirm a clinically relevant plasma concentration of treprostinil was achieved in our study, we analysed the plasma levels of treprostinil in naïve, vehicle, Trep‐100 and Trep‐810‐treated rats. Both treatments achieved measurable increases in plasma concentrations; however, this was trivial for Trep‐100 (Figure 1B) whereas Trep‐810 resulted in higher plasma levels of drug, comparable to therapeutic plasma levels reported in clinical studies (McSwain et al., 2008; Kumar et al., 2016).
Treprostinil improves pulmonary haemodynamics and RV hypertrophy in rat SUHx model with established PAH
At the time of randomization (4 weeks post‐SU), there was no difference in baseline RVSP between vehicle and treprostinil‐treated groups (Figure 2A and Supporting Information Figure S1A). In contrast to previous reports using the MCT model, no effect of 3 weeks treatment of Trep‐100 (low‐dose) was observed on RVSP or RV hypertrophy in the SUHx model (Supporting Information Figure S1B–D). Notably, the higher dose of treprostinil (Trep‐810) significantly lowered RVSP and RV hypertrophy compared to the vehicle controls (Figure 2B,C). Trep‐810 prevented the increase in RVSP observed in the vehicle treatment group but did not significantly reduce the established PAH from baseline levels (Figure 2D). For Trep‐100‐treated rats, we observed no effect on echocardiographic parameters of haemodynamic function (PAAT, PET and PAAT/PET) (Supporting Information Figure S2A–C) compared to vehicle control, which is consistent with the lack of change in RVSP (Supporting Information Figure S1D). Surprisingly, no significant effects of Trep‐810 treatment were observed on echocardiographic parameters of haemodynamic function (PAAT/PET or PAT) (Figure 3A,B); however, a significant increase in PET with treprostinil treatment (Figure 3C) was observed. Further, to better understand the changes in pulmonary haemodynamics with treprostinil treatment, we estimated PVR using the ratio of RVSP/CO as a surrogate measure for PVR (Urboniene et al., 2010). Compared to the naïve controls, we observed significant increase in RVSP/CO in vehicle‐treated SUHx rats that was reduced with Trep‐810 treatment (Figure 3D) highlighting the improvement in PVR with Trep‐810. However, this effect on PVR was not evident in Trep‐100 treated rats (Supporting Information Figure S2D). There was a small but significant decrease in PA diameter in the vehicle‐treated SUHx rats (Figure 3E), compared to sham rats, despite the higher arterial pressure. Interestingly, treatment with Trep‐810 resulted in a modest but significant increase in PA diameter compared to the vehicle treated SUHx rats (Figure 3E), despite a lower internal pressures.
Figure 2.
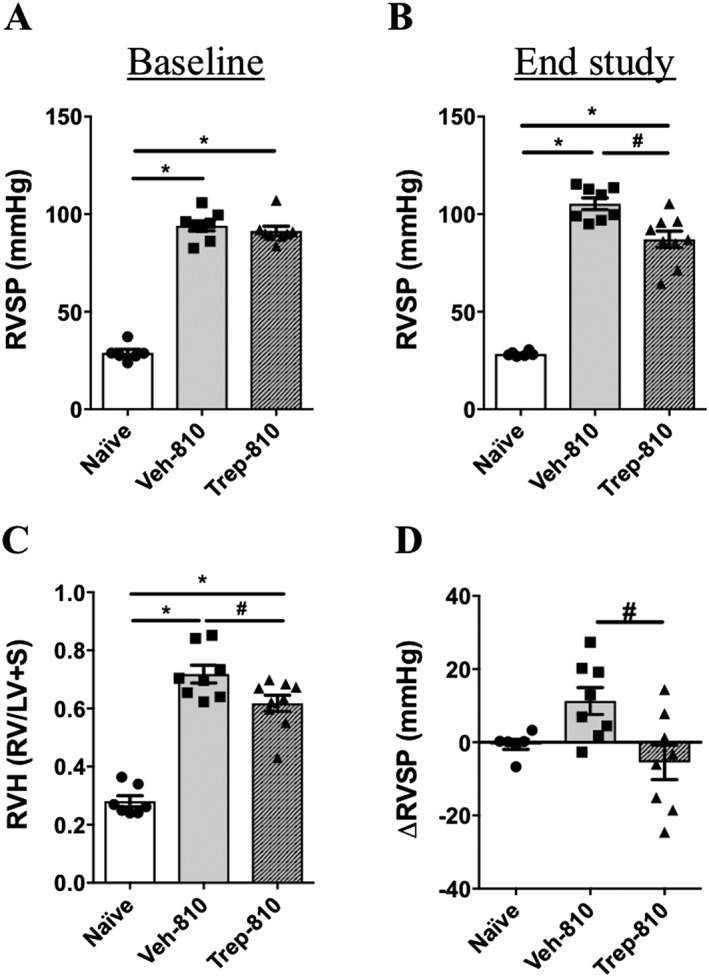
Treprostinil (Trep‐810) improves pulmonary haemodynamics and RV hypertrophy in the rat SUHx model with established PAH. (A) Baseline RVSP (week‐4); (B) RVSP at end study (week‐7); (C) RV hypertrophy (Fulton Index, RV/LV + S); and (D) change in RVSP from baseline (4‐week) to end of study (7‐week). *P < 0.05 compared to naïve and # P < 0.05 compared to Veh‐810. Naïve (n = 6), Veh‐810 (n = 8) and Trep‐810 (n = 8).
Figure 3.

Echocardiographic parameters to estimate pulmonary haemodynamics in SUHx rats treated with Trep‐810. Bar graphs showing (A) PAAT/PET, (B) PAAT, (C) PET, (D) RVSP/CO as a surrogate marker for PVR and (E) PA diameter. *P < 0.05 compared to naïve and # P < 0.05 compared to Veh‐810. Naïve (n = 5), Veh‐810 (n = 8) and Trep‐810 (n = 9).
Trep‐810 improves RV function and structure in SUHx model of PAH
Earlier studies have reported direct beneficial effects of iloprost on RV in rat SUHx model (Gomez‐Arroyo et al., 2015); therefore, we investigated the effect of treprostinil on cardiac structure and function using echocardiography. Decrease in CO and SV in vehicle‐treated SUHx rats was observed, and both were improved with Trep‐810 treatment (Figure 4A,B). Trep‐810 did not have any effect on heart rate (HR) (Figure 4C). We observed decrease in TAPSE in vehicle‐treated SUHx rats compared to naïve rats and treprostinil treatment did not improve TAPSE in SUHx rats (Figure 4D). Furthermore, SUHx treatment resulted in significant increase in RV chamber size (RVIDd/LVIDd) compared to naïve rats and Trep‐810 treatment lowered RV chamber size compared to vehicle controls (Figure 4E). We also observed reduction in RVAW thickness (Figure 4F,G) with Trep‐810 compared to vehicle controls that was consistent with reduction in RV hypertrophy (Figure 2C). Together, improvements in cardiac structure and function were seen in treprostinil‐treated SUHx rats; however, cardiac contractility was not improved. Consistent with no change in haemodynamic parameters, no significant effect of Trep‐100 was observed on RV structure (RV hypertrophy and RVIDd/LVIDd) or RV function (CO, SV and HR) (Supporting Information Figures S1C and S2E–H).
Figure 4.
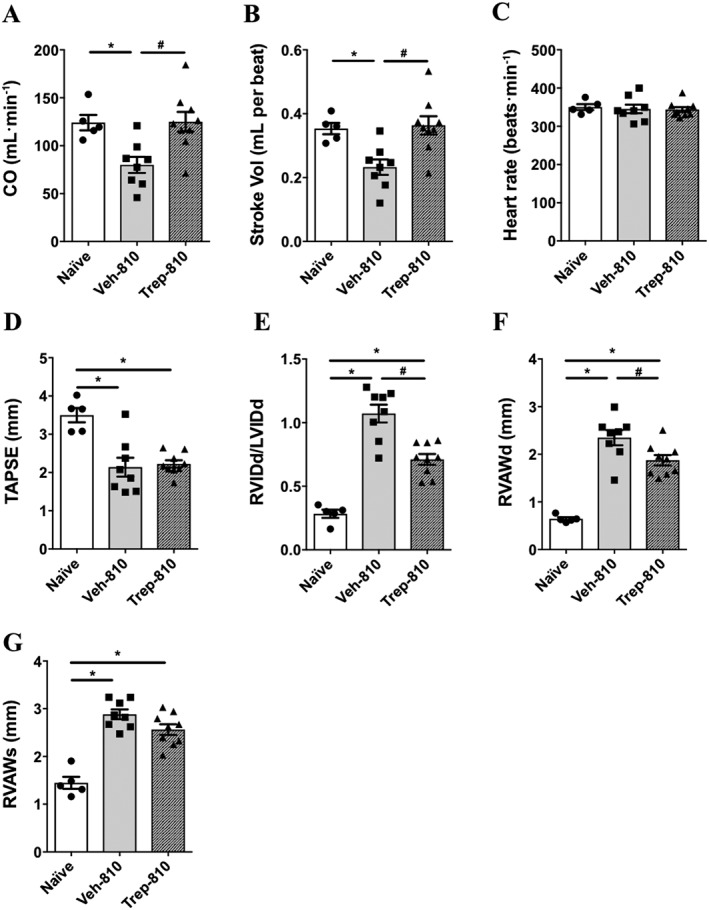
Echocardiographic parameters as an estimate of RV function and structure in SUHx rats treated with Trep‐810. Bar graphs showing (A) CO, (B) SV, (C) HR, (D) TAPSE, (E) RVIDd/LVIDd, (F) diastolic right ventricular anterior wall thickness (RVAWd) and (G) systolic right ventricular anterior wall thickness (RVAWs). *P < 0.05 compared to naïve and # P < 0.05 compared to Veh‐810. Naïve (n = 5), Veh‐810 (n = 8) and Trep‐810 (n = 9).
Treprostinil treatment does not alter vascular remodelling in SUHx model
Further, we investigated the effects of Trep‐810 on vascular remodelling in SUHx model. Compared to naïve controls, significantly greater vessel wall thickness was observed in the lungs of vehicle‐treated SUHx rats across different size blood vessels (Figure 5A). Trep‐810 had no effect on vessel wall thickness of different size vessels compared to vehicle‐treated controls (Figure 5A). Interestingly, we observed significant decrease in total vessel count in the lungs of vehicle‐treated SUHx rats compared to naïve controls and Trep‐810 did not have any effect on total vessel counts compared to vehicle controls (Figure 5B,C). We also studied vessel occlusion in response to SUHx and Trep‐810. Vehicle control lungs had significantly lower non‐occluded and higher partially or completely occluded vessels compared to naïve rats (Figure 5D–G). Trep‐810 did not have significant effects on vessel occlusion compared to vehicle controls (Figure 5D–G). These data indicate that the beneficial effects of treprostinil against SUHx induced severe PAH are independent of vascular remodelling changes in this model.
Figure 5.
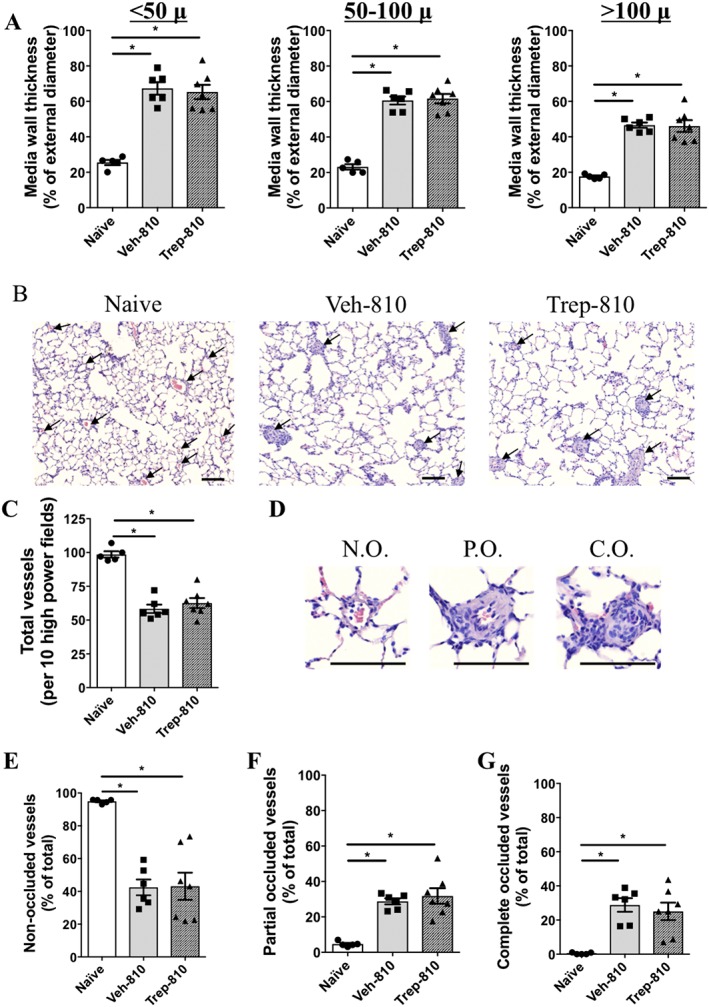
Pulmonary vascular remodelling in naïve or SUHx rats treated with vehicle or Trep‐810. (A) Bar graphs showing media wall thickness in different size blood vessels in naïve, Veh‐810 or Trep‐810‐treated rat lungs. (B) Representative images of haematoxylin and eosin staining of lungs from naïve, Veh‐810 or Trep‐810 rats. (C) Bar graph demonstrating total vessel count in naïve, Veh‐810 or Trep‐810‐treated rat lungs. (D) Representative images of non‐occluded (N.O.), partially occluded (P.O.) and completely occluded (C.O.) vessels. Bar graphs showing quantification of (E) non‐occluded, (F) partially occluded and (G) completely occluded vessels in naïve, Veh‐810 or Trep‐810‐treated rat lungs. *P < 0.05 compared to naïve and # P < 0.05 compared to Veh‐810. Scale bar = 50 μ. Naïve (n = 5), Veh‐810 (n = 6) and Trep‐810 (n = 7).
Protective effects of treprostinil are not associated with BMPR2, VEGFR‐2 and cleaved caspase‐3 expression in the lungs
Previous studies have demonstrated a direct effect of prostacyclin analogues on BMPR2 signalling. Iloprost and treprostinil both have been shown to increase phospho‐SMAD1/5 expression in vitro and in vivo (Yang et al., 2010). Hence, we explored expression of BMPR2 and its downstream target phospho‐SMAD1/5/9. No difference in BMPR2 or phospho‐SMAD1/5/9 expression was observed among naïve, vehicle control and Trep‐810 (Figure 6A,B). Notwithstanding the lack of a significant difference between the groups, levels of BMPR2 varied somewhat in the Veh‐810 group, such that lower BMPR2 levels correlated with a higher RVSP but that this correlation was not present in the Trep‐810 group (Figure 6C) suggesting that the improvement in RVSP with treprostinil treatment was independent of changes in BMPR2 expression in the lungs. Previous studies have shown that SU induces endothelial apoptosis that leads to increase in cleaved caspase‐3 expression and activity in the lungs. Therefore, we examined the expression and activity of cleaved caspase‐3 in the lungs of vehicle or Trep‐810‐treated rats. Interestingly, we observed trend towards higher cleaved caspase‐3 expression and activity in vehicle‐treated SUHx rat lungs compared to naïve controls, even at 7 weeks post‐SU (Figure 6D,E). Trep‐810 treatment had no significant effect on cleaved caspase‐3 expression or activity compared to vehicle controls (Figure 6D,E). We also examined the changes in VEGFR‐2 expression and observed lower VEGFR‐2 in vehicle treated SUHx rat lungs compared to naïve controls and again Trep‐810 did not affect VEGFR‐2 expression in response to SUHx (Figure 6F). Together, these data indicate that the protective effects of treprostinil do not involve changes in cleaved caspase‐3, BMPR2 signalling or VEGFR‐2 in the lungs.
Figure 6.

Effect of Trep‐810 treatment on BMPR2, caspase and VEGFR‐2 signalling. Representative Western blot images and bar graphs showing (A) BMPR2 and (B) SMAD1/5/9 in whole lung homogenate of naïve, Veh‐810 or Trep‐810‐treated rats. (C) Dot plot showing correlation between RVSP and BMPR2 expression in the lungs of Veh‐810 or Trep‐810. (D) Bar graph representing cleaved caspase‐3/7 activity in whole lung homogenate of naïve, Veh‐810 or Trep‐810‐treated rats. (E) Representative Western blot images and bar graphs showing cleaved caspase‐3 expression in whole lung homogenate of naïve, Veh‐810 or Trep‐810‐treated rats. (F) Representative Western blot images and bar graphs showing VEGFR‐2 in whole lung homogenate of naïve, Veh‐810 or Trep‐810‐treated rats. *P < 0.05 compared to naïve and # P < 0.05 compared to Veh‐810; n = 6 per group.
Acute treprostinil treatment produces pulmonary vasodilatation in SUHx rats
To study the acute vasodilator effects of treprostinil in the SUHx rats, we investigated changes in RVSP following short‐term administration and withdrawal of treprostinil in anesthetized SUHx rats at 4 weeks post‐SU. Administration of treprostinil (810 ng·kg−1·min−1) resulted in an immediate reduction in RVSP, which was stable throughout the infusion and promptly returned to baseline following withdrawal of treprostinil (Figure 7A). Trep‐810 treatment had no effect on systemic BP (Figure 7B,C). Further, treatment with the higher dose of treprostinil (3000 ng·kg−1·min−1) produced a greater reduction in RVSP that was more sustained after discontinuation of the infusion (Figure 7A). The higher dose of treprostinil decreased systemic BP (systolic and diastolic) (Figure 7B,C). These data reveal an important element of vasoconstriction in the SUHx model and is consistent with vasodilatation being the primary mode of action of treprostinil in PAH.
Figure 7.

Acute vasodilator effects of treprostinil. Line graphs demonstrating acute effects of treprostinil (810 or 3000 ng·kg−1·min−1, i.v.) on (A) RVSP; (B) SBP and (C) DBP in anaesthetized SUHx rats at 4 weeks post‐SU; n = 3 per group.
Discussion
We report that high‐dose treprostinil (Trep‐810) improved pulmonary haemodynamics, cardiac structure and function in rats with established PAH in the SUHx model of severe PAH. However, treprostinil treatment did not reduce vascular remodelling as assessed by medial wall thickening and vascular occlusion. Treprostinil treatment also did not alter BMPR2 expression, cleaved caspase‐3 or VEGFR‐2 to exert the beneficial effects. Further, an acute infusion of treprostinil produced a substantial vasodilator response in SUHx model, suggesting that the effects of treprostinil were due primarily to pulmonary vasodilatation.
A deficiency of prostacyclin activity has long been identified as an important part of the pathobiology of PAH (Christman et al., 1992; Tuder et al., 1999), and prostacyclin analogues, including treprostinil, have been developed to target this deficit of prostacyclin in PAH. Treprostinil treatment has been shown to reduce pulmonary arterial pressure, decrease vascular resistance and improve cardiac index in PAH patients (Simonneau et al., 2002). Similarly, we observed a decrease in RVSP and PVR and improvement in cardiac output with treprostinil treatment in the SUHx rats with established PAH. Further, the moderate improvement observed in the haemodynamic parameters in the SUHx model is consistent with the effects of treprostinil in human PAH patients that showed moderate changes in mean pulmonary arterial pressures (Simonneau et al., 2002).
Previous studies using MCT model of PAH have shown improvement in RVSP, PVR and vascular remodelling with treprostinil treatment (Yang et al., 2010; Zhou et al., 2013). Interestingly, these effects in the rat MCT model were observed with lower doses of treprostinil that were based on average human dose used in clinics (45 ng·kg−1·min−1 or ~0.064 mg·kg−1·day−1; 0.1–0.3 mg·kg−1·day−1) (Yang et al., 2010; Zhou et al., 2013; LeVarge 2015). However, in our study, the Trep‐100 dose (100 ng·kg−1·min−1 or ~0.14 mg·kg−1·day−1) of treprostinil was found ineffective in the SUHx model. Notably, the plasma concentrations of treprostinil in the SUHx rats in this study were lower than the plasma concentrations reported in humans with equivalent doses of treprostinil (McSwain et al., 2008; Kumar et al., 2016). For example, 100 ng·kg−1·min−1 dose in human would result in ~14 ng·mL−1 plasma treprostinil (McSwain et al., 2008) compared to 1.19 ng·mL−1 observed in SUHx rats in this study. This highlights important differences in the pharmacokinetics of treprostinil between the two species, and these differences should be taken into consideration when extrapolating the doses between the species. The effectiveness of treprostinil, with possible lower plasma levels, in MCT model but not in SUHx model could be due to differences in the disease severity or in the prostacyclin signalling between the two animal models of PAH. Nevertheless, treprostinil, at higher dose, improved pulmonary haemodynamics and cardiac function in the rat SUHx model, which is consistent with the effects observed in the MCT model and PAH patients.
While treprostinil has been shown to be effective for treatment of PAH in patients as well as in animal models, the mechanism(s) remain unclear. The primary target of treprostinil appears to be the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=345&familyId=58&familyType=GPCR (PGI2 receptor) on vascular smooth‐muscle cells (Whittle et al., 2012). Activation of the IP receptor leads to adenylate cyclase activation, an increase in intracellular cAMP levels and smooth‐muscle relaxation producing vasodilatation. Treprostinil has been shown to have affinity for other http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=58 as well, including the DP1, EP2 and EP4 receptors (Whittle et al., 2012), and activation of these receptors could also produce vasodilatation. However, the contribution of an individual receptor in mediating the beneficial effects of treprostinil in PAH remains to be investigated. Irrespective of the receptor involved, vasodilatation appears to be the primary mode of action of treprostinil. In the present study, we observed a decrease in RVSP and increase in cardiac output in the absence of a reduction in medial thickness or an increase in arterial number. This is consistent with vasodilatation of resistance vessels in the pulmonary circulation in response to treprostinil, which confirmed the demonstration of important vasodilatation in response to acute treprostinil infusion in the SUHx rats with established PAH.
Apart from vasodilatation, treprostinil has been shown to suppress pulmonary artery smooth‐muscle cell (PASMC) proliferation. Clapp et al. reported anti‐proliferative properties of prostacyclin analogues on serum‐induced proliferation of normal PASMC (Clapp et al., 2002). Anti‐proliferative properties of prostacyclin on healthy and mutant PAMSCs, carrying a pathogenic nonsense mutation of the BMPR2 gene, have also been reported (Yang et al., 2010). Furthermore, animal studies have shown improvement in vascular remodelling with treprostinil treatment in MCT model of severe PAH (Yang et al., 2010). On the other hand, other groups have reported that treprostinil treatment improved clinical parameters and ameliorated symptoms of heart failure in the MCT model in the absence of beneficial effects on pulmonary arterial remodelling (van Albada et al., 2006). Moreover, autopsy studies have demonstrated that prostacyclin treatment does not reverse lung vascular remodelling in patients with PAH (Rich et al., 2010; Pogoriler et al., 2012). These results further support the claim that the 3 week treatment with treprostinil, in SUHx model, exerts beneficial effects through vasodilatation without affecting smooth‐muscle cell proliferation (at least in vivo) or vascular remodelling that is consistent with the effects of prostanoids in PAH patients (Rich et al., 2010; Pogoriler et al., 2012). Anti‐proliferative effects of treprostinil on the PASMC in vitro required much higher treprostinil concentrations (i.e. ~1 μM) than those achieved in the present study or during parenteral treprostinil therapy in PAH patients (~10 ng·mL−1 or ~25 nM), which could explain the lack of effect on vascular remodelling in this study as well as in PAH patents (Rich et al., 2010; Pogoriler et al., 2012).
Prostacyclin analogues have also been reported to affect cardiac function and RV remodelling. One year treatment of PAH patients with prostacyclin analogues improved echocardiographic parameters to estimate RV systolic pressure and assess RV structure (Tonelli et al., 2014). Further, in a patient treated with epoprostenol for 18 years, near normal cardiac output was observed, even in the presence of advances vascular remodelling (Rich et al., 2010). Gomez‐Arroyo et al. made similar observation in the SUHx model and reported improvement in cardiac output and exercise capacity with reduction in PVR and cardiac fibrosis in response to inhaled iloprost despite lack of reduction in pulmonary arterial pressure, vascular remodelling or RV hypertrophy (Gomez‐Arroyo et al., 2015). Further, Holmboe et al. demonstrated that iloprost has inotropic properties that directly improve RV function in MCT model of pulmonary hypertension as well as in pulmonary artery banding model (Holmboe et al., 2013), consistent with a direct effects of prostacyclin analogues on the RV. Interestingly, another study from Holmboe et al., using isolated heart perfusion system, demonstrated that iloprost, treprostinil and MRE‐269 improved RV function in the healthy rat hearts; however, the positive inotropic effects of these prostacyclin analogues were abolished in hypertrophied RV (Holmboe et al., 2017b). Axelgaard et al. has also demonstrated that the chronic treatment with treprostinil in a moderate or high dose does not reverse or attenuate RV hypertrophy or function in pulmonary artery banding model (Axelgaard et al., 2017) that is contrary to the effects of iloprost (Holmboe et al., 2013). Further, the lack of direct inotropic effect of PGs on isolated atrial strips from the normal and pressure‐overloaded human right hearts has been reported (Holmboe et al., 2017a). Overall, there is evidence to support the direct effects of some prostacyclin analogues on the heart; however, these effects may vary dependent on prostacyclin analogue as well as on condition of the RV. In the present study, treprostinil treatment increased stroke volume and cardiac output as well as decreased RV chamber size, wall thickness and Fulton index in the SUHx rats. However, we did not observe any effect of treprostinil treatment on TAPSE, a marker for RV contractility, indicating towards absence of positive inotropic effect of treprostinil in SUHx model. Therefore, we believe that the improvement in cardiac structure and function in response to treprostinil treatment in rat SUHx model was mostly driven by the improvement in PVR.
We also explored the effects of treprostinil on important pathways in the development and/or progression of PAH including the BMPR2 pathway. Treprostinil and iloprost have been demonstrated to have direct effect on BMPR2 signalling (Yang et al., 2010). Decrease in BMPR2 expression in the lungs of MCT‐treated rats was seen, and although that was not altered by treprostinil or iloprost, both analogues of prostacyclin did correct the phosphorylation of SMAD1/5 in vitro as well as in vivo (Yang et al., 2010). In the SUHx model, we did not observe a strong decrease in BMPR2 expression, as well treprostinil did not have any effect on BMPR2 expression. Moreover, contrary to the MCT model, treprostinil did not alter phospho‐SMAD 1/5/9 expression in the lungs. These findings highlight important signalling differences between the two widely used animal models of PAH. Further, we also explored the effects of treprostinil on cleaved caspase‐3 and VEGFR‐2 expression in the lungs of SUHx rats. Increased cleaved caspase‐3 expression has been shown in the lungs of SUHx rats up to 3 weeks post‐SU (Taraseviciene‐Stewart et al., 2001; Jiang et al., 2016). Interestingly, in the present study, we observed that the caspase activation in the lungs by SUHx persists for a longer period, and it was evident at least until 7 weeks post‐SU. However, treprostinil had no effect on the cleaved caspase‐3 expression or activity in the SUHx lungs. Consistent with earlier reports (Taraseviciene‐Stewart et al., 2001), we observed decrease in VEGFR‐2 expression in the lungs of SUHx rats; however, this decrease in VEGFR‐2 expression was also not altered by treprostinil treatment. Together, treprostinil does not affect BMPR2, cleaved caspase‐3 or VEGFR‐2 in the SUHx model.
Overall, our data suggest that treprostinil treatment improves pulmonary haemodynamic and cardiac function and structure in the SUHx model of severe PAH. These effects appear to be due primarily to pulmonary vasodilatation induced by treprostinil and not due to improvement in vascular remodelling. Moreover, the protective effects of treprostinil in the SUHx model are also independent of BMPR2, caspase and VEGFR‐2 signalling.
Author contributions
K.R.C., T.H.P., S.H.J.M. and D.J.S. participated in the research design; K.R.C., Y.D., C.M.S. and M.T. conducted the experiments; K.R.C., Y.D. and D.J.S. performed the data analysis; and K.R.C. and D.J.S. wrote the manuscript. All authors reviewed and revised the final version of manuscript and approved manuscript submission.
Conflict of interest
This study was partially funded by United Therapeutics Corp. and Northern Therapeutics Inc. United Therapeutics Corp. owns the patent rights for treprostinil sodium (Remodulin®) for treatment of PAH. T.H.P. is an employee of United Therapeutics Corp. D.J.S. is a consultant, and S.H.J.M. is the chief operating officer of Northern Therapeutics Inc. The other authors report no conflicts.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
References
- Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD et al (2010). Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 121: 2747–2754. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide To PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelgaard S, Holmboe S, Ringgaard S, Hillgaard TK, Andersen S, Hansen MS et al (2017). Effects of chronic treprostinil treatment on experimental right heart hypertrophy and failure. Cardiol Young 27: 90–100. [DOI] [PubMed] [Google Scholar]
- Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM et al (1992). An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med 327: 70–75. [DOI] [PubMed] [Google Scholar]
- Clapp LH, Finney P, Turcato S, Tran S, Rubin LJ, Tinker A (2002). Differential effects of stable prostacyclin analogs on smooth muscle proliferation and cyclic AMP generation in human pulmonary artery. Am J Respir Cell Mol Biol 26: 194–201. [DOI] [PubMed] [Google Scholar]
- Farber HW, Gin‐Sing W (2016). Practical considerations for therapies targeting the prostacyclin pathway. Eur Respir Rev 25: 418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Arroyo J, Sakagami M, Syed AA, Farkas L, Van Tassell B, Kraskauskas D et al (2015). Iloprost reverses established fibrosis in experimental right ventricular failure. Eur Respir J 45: 449–462. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS guide to pharmacology in 2018: updates and expansion to encompass the new guide to immunopharmacology. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmboe S, Andersen A, Jensen RV, Kimose HH, Ilkjaer LB, Shen L et al (2017a). Prostacyclins have no direct inotropic effect on isolated atrial strips from the normal and pressure‐overloaded human right heart. Pulm Circulation 7: 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmboe S, Andersen A, Johnsen J, Nielsen JM, Norregaard R, Botker HE et al (2017b). Inotropic effects of prostacyclins on the right ventricle are abolished in isolated rat hearts with right‐ventricular hypertrophy and failure. J Cardiovasc Pharmacol 69: 1–12. [DOI] [PubMed] [Google Scholar]
- Holmboe S, Andersen A, Vildbrad MD, Nielsen JM, Ringgaard S, Nielsen‐Kudsk JE (2013). Iloprost improves ventricular function in the hypertrophic and functionally impaired right heart by direct stimulation. Pulm Circulation 3: 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Deng Y, Suen C, Taha M, Chaudhary KR, Courtman DW et al (2016). Marked strain‐specific differences in the SU5416 rat model of severe pulmonary arterial hypertension. Am J Respir Cell Mol Biol 54: 461–468. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Thudium E, Laliberte K, Zaccardelli D, Nelsen A (2016). A comprehensive review of treprostinil pharmacokinetics via four routes of administration. Clin Pharmacokinet 55: 1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau EMT, Giannoulatou E, Celermajer DS, Humbert M (2017). Epidemiology and treatment of pulmonary arterial hypertension. Nat Rev Cardiol 14: 603–614. [DOI] [PubMed] [Google Scholar]
- LeVarge BL (2015). Prostanoid therapies in the management of pulmonary arterial hypertension. Ther Clin Risk Manag 11: 535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSwain CS, Benza R, Shapiro S, Hill N, Schilz R, Elliott CG et al (2008). Dose proportionality of treprostinil sodium administered by continuous subcutaneous and intravenous infusion. J Clin Pharmacol 48: 19–25. [DOI] [PubMed] [Google Scholar]
- Nikam VS, Schermuly RT, Dumitrascu R, Weissmann N, Kwapiszewska G, Morrell N et al (2010). Treprostinil inhibits the recruitment of bone marrow‐derived circulating fibrocytes in chronic hypoxic pulmonary hypertension. Eur Respir J 36: 1302–1314. [DOI] [PubMed] [Google Scholar]
- Ogura S, Shimosawa T, Mu S, Sonobe T, Kawakami‐Mori F, Wang H et al (2013). Oxidative stress augments pulmonary hypertension in chronically hypoxic mice overexpressing the oxidized LDL receptor. Am J Physiol Heart Circ Physiol 305: H155–H162. [DOI] [PubMed] [Google Scholar]
- Oka M, Homma N, Taraseviciene‐Stewart L, Morris KG, Kraskauskas D, Burns N et al (2007). Rho kinase‐mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100: 923–929. [DOI] [PubMed] [Google Scholar]
- Pogoriler JE, Rich S, Archer SL, Husain AN (2012). Persistence of complex vascular lesions despite prolonged prostacyclin therapy of pulmonary arterial hypertension. Histopathology 61: 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg‐Maitland M, Archer SL (2010). Long‐term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest 138: 1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan J, Bloch K, Archer SL (2011). Rodent models of pulmonary hypertension: harmonisation with the world health organisation's categorisation of human PH. Int J Clin Pract Suppl 172: 15–34. [DOI] [PubMed] [Google Scholar]
- Schermuly RT, Yilmaz H, Ghofrani HA, Woyda K, Pullamsetti S, Schulz A et al (2005). Inhaled iloprost reverses vascular remodeling in chronic experimental pulmonary hypertension. Am J Respir Crit Care Med 172: 358–363. [DOI] [PubMed] [Google Scholar]
- Simonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC et al (2002). Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double‐blind, randomized, placebo‐controlled trial. Am J Respir Crit Care Med 165: 800–804. [DOI] [PubMed] [Google Scholar]
- Sitbon O, Vonk Noordegraaf A (2017). Epoprostenol and pulmonary arterial hypertension: 20 years of clinical experience. Eur Respir Rev 26: 160055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraseviciene‐Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J et al (2001). Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death‐dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 15: 427–438. [DOI] [PubMed] [Google Scholar]
- Tonelli AR, Conci D, Tamarappoo BK, Newman J, Dweik RA (2014). Prognostic value of echocardiographic changes in patients with pulmonary arterial hypertension receiving parenteral prostacyclin therapy. J Am Soc Echocardiogr 27: 733–741.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L et al (1999). Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 159: 1925–1932. [DOI] [PubMed] [Google Scholar]
- Urboniene D, Haber I, Fang YH, Thenappan T, Archer SL (2010). Validation of high‐resolution echocardiography and magnetic resonance imaging vs. high‐fidelity catheterization in experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 299: L401–L412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Albada ME, van Veghel R, Cromme‐Dijkhuis AH, Schoemaker RG, Berger RM (2006). Treprostinil in advanced experimental pulmonary hypertension: beneficial outcome without reversed pulmonary vascular remodeling. J Cardiovasc Pharmacol 48: 249–254. [DOI] [PubMed] [Google Scholar]
- Whittle BJ, Silverstein AM, Mottola DM, Clapp LH (2012). Binding and activity of the prostacyclin receptor (IP) agonists, treprostinil and iloprost, at human prostanoid receptors: treprostinil is a potent DP1 and EP2 agonist. Biochem Pharmacol 84: 68–75. [DOI] [PubMed] [Google Scholar]
- Yang J, Li X, Al‐Lamki RS, Southwood M, Zhao J, Lever AM et al (2010). Smad‐dependent and Smad‐independent induction of id1 by prostacyclin analogues inhibits proliferation of pulmonary artery smooth muscle cells in vitro and in vivo . Circ Res 107: 252–262. [DOI] [PubMed] [Google Scholar]
- Yigitaslan S, Sirmagul B (2012). Relation of bosentan, iloprost, and sildenafil with growth factor levels in monocrotaline‐induced pulmonary hypertension. Clin Exp Hypertens 34: 222–229. [DOI] [PubMed] [Google Scholar]
- Zhou L, Chen Z, Vanderslice P, So SP, Ruan KH, Willerson JT et al (2013). Endothelial‐like progenitor cells engineered to produce prostacyclin rescue monocrotaline‐induced pulmonary arterial hypertension and provide right ventricle benefits. Circulation 128: 982–994. [DOI] [PubMed] [Google Scholar]
Supporting information
Figure S1 Effect of low‐dose treprostinil (Trep‐100) on SUHx induced PAH. A. Baseline RVSP (week‐4); B. RVSP at end study (week‐7); C. RV hypertrophy (Fulton Index, RV/LV + S); and E. Change in RVSP from baseline (4‐week) to end study (7‐week).
Figure S2 Echocardiographic parameters to estimate pulmonary haemodynamics in SUHx rats treated with Trep‐100. Bar graphs showing A. PAAT B. PET and C. PAAT/PET. D. Bar graph demonstrating RVSP/CO as a surrogate marker for pulmonary vascular resistance. E. Cardiac output (CO), F. stroke volume (SV), G. heart rate, H. RVIDd/LVIDd.
Acknowledgements
The authors would like to thank Anli Yang for the technical support for the animal experiments and lung histology and Sophie Wen for technical support during the animal experiments. We would also like to thank Tendam Labs for the help in plasma treprostinil analysis. This work was supported by a grant from the Canadian Institute of Health Research. Additional funding was provided by an unrestricted grant from Northern Therapeutics Inc. and United Therapeutics Corp. K.R.C. is a recipient of Research Fellowship Award from Heart and Stroke Foundation of Canada and Canadian Vascular Network.
Chaudhary, K. R. , Deng, Y. , Suen, C. M. , Taha, M. , Petersen, T. H. , Mei, S. H. J. , and Stewart, D. J. (2018) Efficacy of treprostinil in the SU5416‐hypoxia model of severe pulmonary arterial hypertension: haemodynamic benefits are not associated with improvements in arterial remodelling. British Journal of Pharmacology, 175: 3976–3989. 10.1111/bph.14472.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of low‐dose treprostinil (Trep‐100) on SUHx induced PAH. A. Baseline RVSP (week‐4); B. RVSP at end study (week‐7); C. RV hypertrophy (Fulton Index, RV/LV + S); and E. Change in RVSP from baseline (4‐week) to end study (7‐week).
Figure S2 Echocardiographic parameters to estimate pulmonary haemodynamics in SUHx rats treated with Trep‐100. Bar graphs showing A. PAAT B. PET and C. PAAT/PET. D. Bar graph demonstrating RVSP/CO as a surrogate marker for pulmonary vascular resistance. E. Cardiac output (CO), F. stroke volume (SV), G. heart rate, H. RVIDd/LVIDd.