

Abstract
Background and Purpose
Haem oxygenase‐1 (HO‐1) is induced by thiazolidinediones including rosiglitazone and exerts anti‐inflammatory effects in various models. However, the molecular mechanisms underlying rosiglitazone‐induced HO‐1 expression remain largely unknown in human pulmonary alveolar epithelial cells (HPAEpiCs).
Experimental Approach
HO‐1 expression was determined by real time‐PCR, Western blotting and promoter reporter analyses. Signalling pathways were investigated using pharmacological inhibitors or specific siRNAs. Interactions between nuclear factor erythroid‐2‐related factor (Nrf2) and antioxidant response elements (ARE) binding site of the HO‐1 promoter were investigated with chromatin immunoprecipitation assays.
Key Results
Up‐regulation of HO‐1 in HPAEpiCs or in mice by rosiglitazone blunted ICAM‐1 expression and monocyte adhesion to HPAEpiCs challenged with LPS. Rosiglitazone‐induced HO‐1 expression was significantly attenuated by NADPH oxidase (NOX) inhibitors (apocynin and diphenyleneiodonium) or ROS scavenger (N‐acetyl cysteine). The involvement of NOX activity and ROS generation in rosiglitazone‐induced HO‐1 expression was confirmed by transfection with p47phox or NOX2 siRNA. Moreover, pretreatment with the inhibitors of c‐Src (c‐Srci II), proline‐rich tyrosine kinase 2 (Pyk2) (PF431396), Akt (Akti VIII) or PPARγ (GW9662) and transfection with siRNA of c‐Src, Pyk2, Akt or PPARγ abolished the rosiglitazone‐induced HO‐1 expression in HPAEpiCs. Subsequently, Nrf2 was activated by phosphorylation of c‐Src, Pyk2 and Akt, which turned on transcription of HO‐1 gene by binding to AREs binding site and enhancing ARE promoter activity.
Conclusions and Implications
Rosiglitazone induces HO‐1 expression via either NOX/ROS/c‐Src/Pyk2/Akt‐dependent Nrf2 activation or PPARγ in HPAEpiCs and suppresses LPS‐mediated inflammatory responses, suggesting that PPARγ agonists may be useful for protection against pulmonary inflammation.
Abbreviations
- AREs
antioxidant response elements
- ChIP
chromatin immunoprecipitation
- DHE
dihydroethidium
- H2DCFDA
2′,7′‐dichlorodihydrofluorescein diacetate
- HO‐1
haem oxygenase‐1
- HPAEpiCs
human pulmonary alveolar epithelial cells
- ICAM‐1
intercellular adhesion molecule
- IF
immunofluorescence
- IHC
immunohistochemistry
- Keap1
Kelch like ECH associated protein 1
- Nrf2
nuclear factor erythroid‐2‐related factor
- Pyk2
proline‐rich tyrosine kinase 2
- TZDs
thiazolidinediones
Introduction
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5019 triggers acute lung inflammation during bacterial infections, through up‐regulation of adhesion molecules, such as intercellular adhesion molecule (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6757) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6758, leading to the recruitment of polymorphonuclear cells to inflammatory tissues and airway fluid (Liou et al., 2016; Woodfin et al., 2016). The up‐regulation of adhesion molecules is involved in pathological changes in the lung. Thus, either ICAM‐1 or VCAM‐1 could serve as an inflammatory marker and have been used to evaluate the anti‐inflammatory effects of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1441 in our studies. We also confirmed that LPS‐induced ICAM‐1 or VCAM‐1 expression and monocytes (THP‐1) adhesion might be used to evaluate the progression of lung inflammation (Cho et al., 2016). Therefore, pursuing an effective medicine to target these critical components is important in the management of pulmonary inflammatory diseases.
Thiazolidinediones (TZD) have been shown to activate http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=595 and have potential value in treatment of animal models of inflammatory diseases. Their anti‐inflammatory effects result from up‐regulation of HO‐1 which suppresses pulmonary artery cell proliferation and vascular remodelling (Kronke et al., 2007; Li et al., 2010) and decreases LPS‐induced lung inflammatory responses (Liu et al., 2005; Wang et al., 2014). We recently also confirmed that suppression of LPS‐mediated inflammation (VCAM‐1 expression and cell adhesion) by HO‐1, induced by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1056, was mediated through PKCα/AMPKα/p38 MAPKα/SIRT1‐dependent deacetylation of Ac‐PGC1α and fragmentation of NCoR/PPARγ activation in human pulmonary alveolar epithelial cells (HPAEpiCs) (Cho et al., 2018). These studies demonstrate that PPARγ agonists could exert anti‐inflammatory effects through a PPARγ‐dependent HO‐1 up‐regulation.
HO‐1 induced by various stimuli and oxidative stresses protects against inflammatory responses (Rushworth et al., 2005; Huang et al., 2014). The ROS‐dependent activation of signalling kinases (Polvani et al., 2012) by PPAR agonists could regulate the expression of various genes (Gardner et al., 2005). PPARγ ligands could also inhibit airway inflammation and hyperresponsiveness via a PPARγ‐independent mechanism in mouse models challenged with allergen (Donovan et al., 2012). In addition, the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=993ROS system could induce HO‐1 expression through different signalling components. Indeed, c‐http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2206 is an upstream component of proline‐rich tyrosine kinase 2 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2181) phosphorylation stimulated by TZDs (Peng et al., 2003; Dewar et al., 2007), which is regulated by ROS (Ke et al., 2014), leading to HO‐1 expression in various cell types (Han et al., 2009; Chi et al., 2015; Yang et al., 2015). Activated http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=285 triggers nuclear factor erythroid‐2‐related factor (Nrf2)/HO‐1 expression which could protect against cellular injury and promote cell survival (Deng et al., 2013; Xu et al., 2015b). However, whether rosiglitazone‐induced HO‐1 expression mediated through NOX/ROS, c‐Src, Pyk2, Akt and Nrf2 was still unknown in HPAEpiCs.
Nrf2 is involved in the HO‐1 gene transcription through binding to antioxidant response elements (AREs) in the promoter region in response to ROS (Alam and Cook, 2007; Loboda et al., 2016). On exposure to ROS, Nrf2 dissociates from kelch like ECH associated protein 1 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2757) and translocates into the nucleus, wherein Nrf2 oligomerizes with the protein Maf binding to the ARE sites of target genes and leading to gene transcription (Itoh et al., 1999; Baird et al., 2013; Foresti et al., 2013). However, the detailed mechanisms of intracellular signalling pathways involved in the rosiglitazone‐induced HO‐1 expression in HPAEpiCs were not completely defined. Here, we have shown that the expression of HO‐1 induced by rosiglitazone is mediated through not only PPARγ‐dependent but also through a separate NOX/ROS/c‐Src/Pyk2/Akt‐dependent Nrf2 activation pathway and protects against the LPS‐mediated inflammatory responses.
Methods
Animal care and experimental procedures
All animal care and experimental procedures complied with the guidelines of the Animal Care Committee of Chang Gung University (Approval Document No. CGU 16‐046) and NIH Guides for the Care and Use of Laboratory Animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Male ICR mice aged 6–8 weeks were purchased from the National Laboratory Animal Centre (Taipei, Taiwan). Mice were assigned randomly into three groups: sham [0.1 mL of DMSO‐PBS (1:100) with 0.1% (W/V) BSA treated mice], LPS (LPS‐treated mice) and Rosi + LPS (rosiglitazone plus LPS mice); five mice in each group/cage and kept in standard individually ventilated cages in an animal facility under standardized conditions (12 h light/dark cycle, 21–24°C, humidity of 50–60%) with food and water ad libitum. Mice treated with rosiglitazone (i.p.; 0.1 mg·kg−1) and 1 h later were anaesthetized with pentothal (i.p.; 50 mg·kg−1). Mice were placed individually on a board in a near vertical position and the tongues withdrawn with lined forceps. LPS (3 mg·kg−1; 45 μL in 30g mouse) was placed posteriorly in the throat and aspirated into lungs for 16 h to provide a model of lung inflammation (Hsu et al., 2014). At the end of the experimental period, mice were killed by an overdose of pentothal (i.p.; 100 mg·kg−1) for collection of lung tissues. These tissues were extracted for protein (right superior lobe + post caval lobe) and mRNA (right middle lobe + right inferior lobe) for ICAM‐1, HO‐1 or β‐actin. Data collection and evaluation of all in vivo and in vitro experiments were performed without knowledge of the treatment of the group.
Immunohistochemical (IHC) staining
To examine the cellular expression and location of the ICAM‐1 or HO‐1 protein, immunohistochemical (IHC) staining was performed on the sections of the lung tissues (left lung), which were deparaffinized, rehydrated and washed with TTBS (composition; 50 mM Tris‐HCl, 150 mM NaCl, 0.05% (w/v) Tween 20, pH7.4). Non‐specific binding was blocked by preincubation with PBS containing 5 mg·mL−1 of BSA for 1 h at room temperature. The sections were incubated with an anti‐ICAM‐1 or anti‐HO‐1 antibody (1:100 dilution) at 4°C for 16 h and then with anti‐mouse or anti‐rabbit HRP antibody at room temperature for 1 h. Bound antibodies were detected by incubation in 0.5 mg·mL−1 of 3,3‐diaminobenzidine/0.01% (v/v) hydrogen peroxide (H2O2) in 0.1 M Tris–HCl buffer, as chromogen (Vector Lab, Burlingame, CA, USA).
Cell culture
HPAEpiCs were purchased from the ScienCell Research Laboratories (San Diego, CA, USA). Cells were cultured in DMEM/F12 medium containing 10% (v/v) FBS and antibiotics (100 U·mL−1 penicillin G, 100 μg·mL−1 gentamicin and 250 ng·mL−1 fungizone) at 37°C in a humidified 5% CO2. Experiments were performed with cells from passages 4 to 7. The growth medium was changed after 24 h and then every 3 days. The viability of HPAEpiCs after treatment with 1% (v/v) DMSO or the pharmacological inhibitors alone was determined by a cell counting Kit‐8 assay, which showed no significant differences (data not shown).
Protein preparation and Western blot analysis
Cells were incubated with or without different concentrations of rosiglitazone at 37°C for the indicated time periods. When inhibitors used, they were added 1 h prior to the application of rosiglitazone. The collected cells were lysed with a lysis buffer. The mixed samples (15 μL) were subjected to 10% SDS‐PAGE and transferred to nitrocellulose membrane. Membranes were washed with TTBS four times for 5 min each, incubated with anti‐rabbit or anti‐mouse HRP antibody (1:2000 dilution) for 1 h. The immunoreactive bands were detected by ECL reagents and captured by a UVP BioSpectrum 500 Imaging System (Upland, CA, USA). The values of Western blot were quantified by UN‐SCAN‐IT gel version 6.1 (Utah, USA) and normalized to GAPDH or total protein for five individual experiments. Changes after LPS are expressed as fold change, relative to the mean basal value.
Real‐time PCR analysis
Total RNA was extracted from HPAEpiC using TRIzol reagent. The mRNA was reverse‐transcribed into cDNA and analysed by real‐time PCR. Real‐time PCR was performed with a StepOnePlus™ real‐time PCR system (ThermoScientific‐Applied Biosystems, Waltham, MA, USA) and Kapa Probe Fast qPCR Kit Master Mix (2X) Universal (KK4705; KAPA Biosystems, Wilmington, MA, USA). The expression of HO‐1 and ICAM‐1 was quantified by normalization to the GAPDH expression. Relative gene expression was determined by the ΔΔCt method, where Ct meant threshold cycle. All experiments were performed five times (n = 5).
Adhesion assay
HPAEpiCs were plated on six‐well culture plates with coverslips and pretreated with rosiglitazone for 1 h, and then incubated with or without ZnPP IX before incubation with LPS for 16 h at 37°C in a humidified 5% CO2 atmosphere. THP‐1 cells (human monocytic cell line, BCRC Cat# 60430, RRID:CVCL_0006) were purchased from Bioresource Collection and Research Center (Hsinchu, Taiwan) and maintained in suspension in DMEM/F‐12 containing 10% (v/v) FBS medium. Before labelling, THP‐1 cells were washed and resuspended in warm PBS. BCECF‐AM (10 μM) was added to THP‐1 in warm PBS for 1 h at 37°C. After labelling, cells were washed thrice and resuspended in warm PBS and kept in the dark at room temperature and then the labelled THP‐1 cells were added to HPAEpiCs. The cultures were incubated in a CO2 incubator for 1 h. Non‐adherent cells were removed from the plate by gentle washing with warm PBS. The numbers of adherent THP‐1 cells were determined by counting four fields per 200X high‐power field well using a fluorescence microscope (Axiovert 200M, Zeiss, Jena, Germany). Experiments were performed in triplicate and repeated five times (n = 5).
Transient transfection with siRNAs in HPAEpiCs
Human siRNAs of scrambled, SMARTpool RNA duplexes corresponding to p47phox (SASI_Hs02_00302212), NOX2 (SASI_Hs01_00086110), c‐Src (SASI_Hs01_00112905), Pyk2 (SASI_Hs01_00032249), Akt (SASI_Hs01_00105954), Nrf2 (SASI_Hs02_00302212), Keap1 (SASI_Hs01_00080908) and Scrambled control siRNA were from Sigma‐Aldrich (St. Louis, MO, USA). Briefly, siRNA (100 nM) was formulated with Lipofectamine 2000 transfection reagent according to the manufacturer's instruction.
NADPH oxidase activity assay
After exposure to 30 μM rosiglitazone for the indicated time intervals, cells were gently scraped and centrifuged at 400× g for 10 min at 4°C. The cell pellet was resuspended with 35 μL of ice‐cold PBS, and the cell suspension was kept on ice. To a final 200 μL volume of pre‐warmed (37°C) PBS containing either NADPH (1 μM) or lucigenin (20 μM), 5 μL of cell suspension (2 × 104 cells) was added to initiate the reaction followed by immediate measurement of chemiluminescence in a luminometer (SynergyH1 Hybird Reader, BioTek, Winooski, VT, USA).
Measurement of intracellular ROS accumulation
Cells were cultured in DMEM/F‐12 for 24 h and then treated with rosiglitazone. When inhibitors were used, they were added 1 h prior to the application of rosiglitazone. After washing twice with warm PBS, the cells were stained with 2′,7′‐dichlorodihydrofluorescein diacetate (H2DCFDA) (10 μM) or DHE (5 μM) for 30 or 10 min. After staining, the cells were recovered with DMEM/F‐12 containing 10% (v/v) FBS for 30 min in H2DCFDA group. For ELISA assay, the fluorescence for DCF and DHE staining was detected at 495/529 and 518/605 nm using a fluorescence microplate reader (SynergyH1 Hybird Reader, BioTek) and FACSCalibur equipped with CellQuest software (BD Biosciences, San Jose, CA, USA). For immunofluorescence staining, the stained cells were washed three times with cold‐PBS and then the fluorescence for DCF and DHE staining was detected at 495/529 and 518/605 nm, respectively, using a fluorescence microscope (Axiovert 200M, Zeiss).
Transfection and promoter luciferase assay
ARE‐luc reporter construct plasmids were transiently transfected at a concentration of 0.8 μg·mL−l, and the control pGal encoding for β‐galactosidase presented at 0.2 μg·mL−1 to normalize the transfection efficiency. ARE‐luc luciferase activities were determined by using a luciferase assay system (Abcam, Cambridge, UK) according to the manufacturer's instructions. Detected firefly luciferase activities were standardized with β‐galactosidase activity.
Isolation of subcellular fractions
The cytosolic and nuclear fractions were isolated, as described previously (Cho et al., 2016). The cells were harvested, sonicated for 10 s at output 1.5 with a sonicator (Misonix, Farmingdale, NY, USA) and centrifuged at 5000× g at 4°C for 15 min. The pellet was collected as the nuclear fraction and the supernatant collected as the cytosolic fraction.
Chromatin immunoprecipitation (ChIP) assay
To detect the association of transcription factors with the human HO‐1 promoter, the ChIP analysis was performed as previously described (Cho et al., 2016). Briefly, HPAEpiCs were cross‐linked with 1% (v/v) formaldehyde at 37°C for 30 min and stop this reaction with 0.125 M glycine, then washed three times with ice‐cold PBS containing 1 mM PMSF, 1% (v/v) aprotinin and 1% (v/v) leupetin. Soluble chromatin was prepared using a ChIP assay kit (Upstate) according to the instructions of the manufacturer and immunoprecipitated without (control) or with an anti‐Nrf2 antibody and normal goat IgG. Following washing and elution, immunoprecipitates were heated overnight at 65°C to reverse cross‐linking of DNA and protein. To avoid the possibility of amplification artefacts, PCR products for all SYBR Green primer pairs were verified to produce single products by agarose electrophoresis and high resolution melt curve. The relative mRNA levels were calculated using the comparative Ct method (ΔΔCt). The DNA was extracted and resuspended in H2O and subjected to PCR amplification with the ARE primers:
ARE (327 bp)
F: 5′‐GCTGC CCAAA CCACT TCTGT‐3′
R: 5′‐GCCCT TTCAC CTCCC ACCTA‐3′
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). Statistical analysis was performed only when n = 5 independent samples were acquired. All data are expressed as means ± SEM, from five individual experiments (n ≥ 5). Statistical analysis was performed by using GraphPad Prizm Program 6.0 software (GraphPad, San Diego, CA). We used one‐way ANOVA followed by Dunnett's post hoc test when comparing more than two groups of data and one‐way ANOVA with a non‐parametric Kruskal–Wallis test, followed by Dunnett's post hoc test when comparing multiple independent groups. Post tests were run only if F achieved P < 0.05 and there was no significant variance inhomogeneity. P < 0.05 was considered to show statistical significance. Error bars are omitted when they fell within the dimensions of the symbols.
Materials
Apocynin (APO), diphenyleneiodonium chloride (DPI) was from Biomol (Plymouth Meeting, PA, USA). Anti‐GAPDH (mouse monoclonal antibody, Cat# MCA‐1D4, http://antibodyregistry.org/search.php?q=AB_2107599) antibody was from EnCor Biotechnology (Gainesville, FL, USA). Anti‐β‐actin (C4) (mouse monoclonal antibody, Cat# sc‐47778 HRP, RRID:AB_2714189), anti‐c‐Src (SRC 2) (rabbit polyclonal antibody, Cat# sc‐18, RRID:AB_631324), anti‐Akt1/2/3 (H‐136) (rabbit monoclonal antibody, Cat# sc‐8312, RRID:AB_671714), anti‐Nrf2 (C‐20) (rabbit polyclonal antibody, Cat# sc‐722, RRID:AB_2108502), anti‐Keap1 (H‐190) (rabbit polyclonal antibody, Cat# sc‐33569, RRID:AB_2280949), anti‐Lamin A (H‐102) (rabbit polyclonal antibody, Cat# sc‐20680, RRID:AB_648148), anti‐ICAM‐1 (H‐108) (rabbit polyclonal antibody, Cat# sc‐7891, RRID:AB_647486) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti‐HO‐1 pAb (rabbit polyclonal antibody, Cat# ADI‐SPA‐895, RRID:AB 10618757) was from Enzo Life Sciences (Farmingdale, NY, USA). Anti‐NOX2/gp91phox [EPR6991] (rabbit monoclonal antibody, Cat# ab129068, RRID:AB_11144496), anti‐Pyk2 [E354] (rabbit monoclonal antibody, Cat# ab32448, RRID:AB_777568), anti‐ICAM‐1 [EPR16608] (rabbit monoclonal antibody, Cat# ab179707) (animal experimentation used) and Anti‐Nrf2 (phospho S40) [EP1809Y] (rabbit monoclonal antibody, Cat# ab76026, RRID:AB_1524049) were from Abcam. Anti‐p47phox (Phospho‐Ser370) (rabbit polyclonal antibody, Cat# A1171, RRID:AB_10696129) was from Assay Biotech (Sunnyvale, CA, USA). Anti‐p47phox (D21F6) (rabbit monoclonal antibody, Cat# 4301S, RRID:AB_2150286), anti‐phospho‐c‐Src family (Tyr416) (rabbit polyclonal antibody, Cat# 2101, RRID:AB_331697), anti‐phospho‐Pyk2 (Tyr402) (rabbit polyclonal antibody, Cat# 3291, RRID:AB_2300530) and anti‐phospho‐Akt (Ser473) (rabbit polyclonal antibody, Cat# 9271, RRID:AB_329825) were from Cell Signaling Technology (Danvers, MA, USA). Rosiglitazone, GW590735, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2686, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2711, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2694, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2693, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3442 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8903 were from Cayman Chemical (Ann Arbor, MI, USA). PF431396, c‐Srci II and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5921 VIII were from Merck (Merck Millipore, Billerica, MA, USA). BCECF‐AM, dihydroethidium (DHE) and CM‐H2DCFDA were from Molecular Probes (Eugene, OR, USA). N‐acetyl‐L‐cysteine (NAC), LPS (L2630) and other chemicals were from Sigma‐Aldrich. SDS‐PAGE supplies were from MDBio Inc (Taipei, Taiwan).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c)
Results
Up‐regulation of HO‐1 inhibits inflammatory responses in mice challenged with LPS
HO‐1 has been shown to protect against the inflammatory responses induced by various insults including LPS (Liu et al., 2005; Xu et al., 2015a). Therefore, we confirmed these results in an in vivo study. We observed that LPS markedly induced ICAM‐1 expression, which was attenuated by rosiglitazone through up‐regulation of HO‐1, determined by IHC staining (Figure 1A). We also found that LPS significantly enhanced ICAM‐1 mRNA and protein expression, which were attenuated by rosiglitazone via up‐regulation of HO‐1 (Figure 1B,C). These results suggested that up‐regulation of HO‐1 by rosiglitazone protects lung tissues against the expression of inflammatory proteins such as ICAM‐1, following exposure to LPS.
Figure 1.

Rosiglitazone alleviates LPS‐induced pulmonary inflammatory responses in vivo. (A) Mice were pre‐treated with rosiglitazone (i.p., 0.1 mg·kg−1) or vehicle and 1 h later treated with or without LPS (intra‐tracheally; 3 mg·kg−1) for 16 h. H&E and immunohistochemical staining for ICAM‐1 and HO‐1 in serial sections of the lung tissues from Sham [0.1 mL of DMSO‐PBS (1:100) with 0.1% (W/V) BSA treated mice], LPS (LPS‐treated mice) and Rosi + LPS (rosiglitazone plus LPS mice). The arrows indicate the ICAM‐1 and HO‐1 expression on pulmonary alveolar cells. All images are representative of five mice per group. (B, C) Lung tissues were homogenized to extract protein and mRNA. The protein and mRNA levels of ICAM‐1 and HO‐1 were determined by Western blot and real‐time PCR. Data are expressed as mean ± SEM, from five independent experiments (n = 5). # P < 0.05, significantly different as indicated.
PPARs stimulate expression of HO‐1 protein
Activation of PPARs by agonists could inhibit the proliferation of pulmonary artery cells and reduce pulmonary vascular remodelling by HO‐1 up‐regulation (Kronke et al., 2007; Li et al., 2010). To investigate whether http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=593 http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=594 or PPARγ agonists could induce HO‐1 expression in HPAEpiCs, the cells were treated with the agonist of PPARα (GW590735), PPARδ (GW0742) or PPARγ (rosiglitazone) for the indicated time intervals. These agonists time‐dependently induced HO‐1 protein expression (Figure 2A). Other PPARγ agonists (ciglitazone, pioglitazone and troglitazone) over a range of concentrations (0.1 ‐ 30 μM) were used to induce HO‐1 expression. At the highest concentration (30 μM), these three agonists, but not rosiglitazone, induced cell death of HPAEpiCs. We therefore chose lower concentrations of ciglitazone, pioglitazone and troglitazone to induce the expression of HO‐1 (Figure 2B). As the level of HO‐1 expression by rosiglitazone (3.6 ± 0.1 fold; Figure 3A) was greater than those after ciglitazone (2.7 ± 0.2 fold), pioglitazone (2.8 ± 0.1 fold) and troglitazone (3.0 ± 0.3 fold) over 16 h (all at 10 μM). Therefore, we chose rosiglitazone as an inducer for the following experiments in this study.
Figure 2.
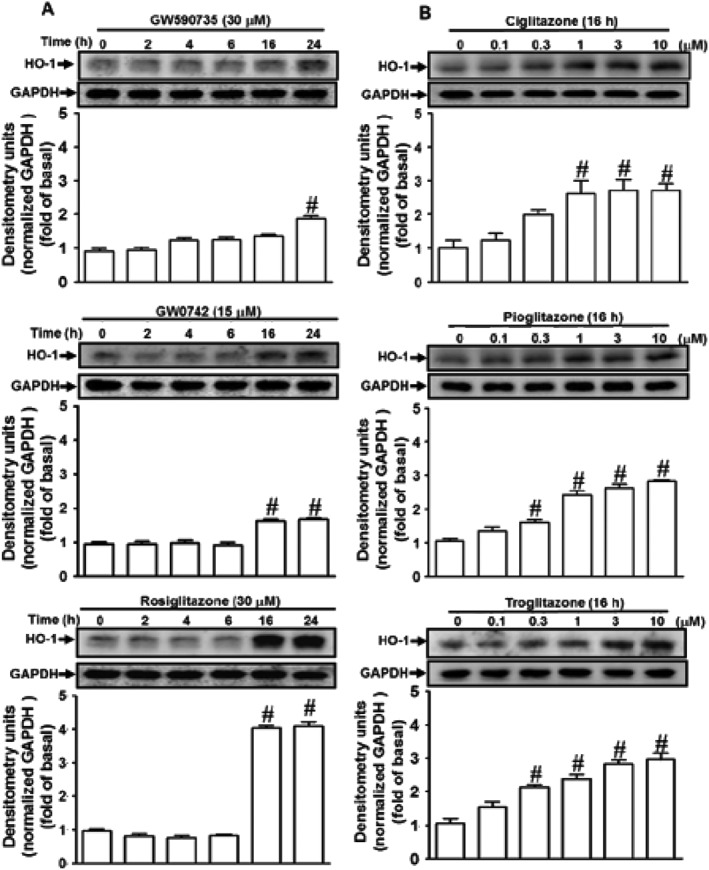
PPAR agonists induce HO‐1 expression in HPAEpiCs. (A) The effects of PPARα, PPARδ and PPARγ agonists on HO‐1 expression, the cells were treated with the agonist of PPARα (GW590735), PPARδ (GW0742) or PPARγ (rosiglitazone) for the indicated time intervals. (B) The effects of PPARγ agonists on HO‐1 expression, the cells were treated with various concentrations of ciglitazone, pioglitazone or troglitazone for 16 h. The levels of HO‐1 and GAPDH protein were determined by Western blot. Data are expressed as mean ± SEM, from five independent experiments (n = 5). # P < 0.05, significantly different from vehicle (0 h) alone.
Figure 3.
Overexpression of HO‐1 inhibits LPS‐mediated monocyte adhesion in vitro. (A) Cells were incubated with different concentrations of rosiglitazone (30, 10, 3 or 1 μM) for various time intervals. The levels of HO‐1 and GAPDH expression were determined by Western blot. (B) Total RNA was isolated from HPAEpiCs treated with rosiglitazone (30 μM) for the indicated time intervals. The levels of HO‐1 and GAPDH mRNA were determined by real‐time PCR. (C–E) Cells were pretreated rosiglitazone for 1 h, then incubated with ZnPPIX for 1 h and finally stimulated with LPS for 16 h (protein and cell adhesion) or 4 h (mRNA). The levels of ICAM‐1, HO‐1 and GAPDH protein and mRNA were determined by Western blot and real‐time PCR respectively. (E) The adhesion of THP‐1 cells was measured. Data are expressed as mean ± SEM, from five independent experiments (n = 5). # P < 0.05, significantly different from vehicle alone (A, B); or significantly different as indicated (C–E).
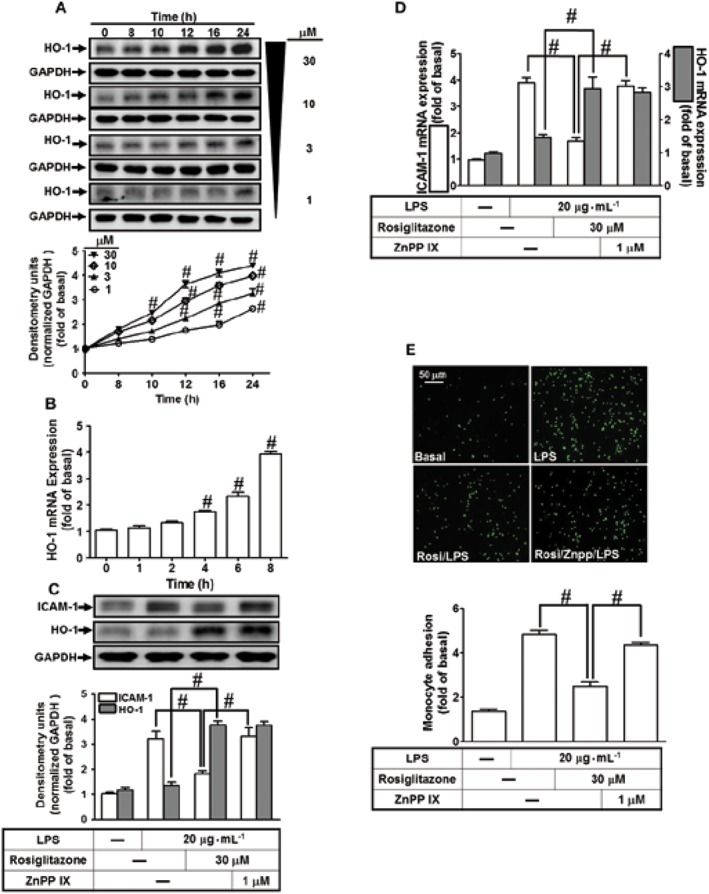
Rosiglitazone induces HO‐1 expression
The mechanisms underlying rosiglitazone‐induced HO‐1 gene expression were dissected in HAPEpiCs. We found that rosiglitazone time‐ and concentration‐dependently induced HO‐1 protein expression (Figure 3A). There was a significant increase within 10 h and reaching a maximal response within 24 h. Moreover, the levels of HO‐1 mRNA were also enhanced by rosiglitazone (Figure 3B). We also evaluated the effect of HO‐1 up‐regulation induced by rosiglitazone on LPS‐induced ICAM‐1 expression. HPAEpiCs were pretreated with 30 μM rosiglitazone for 1 h and then continuously incubated with or without 1 μM ZnPPIX (a HO‐1 activity inhibitor) for 1 h, and finally treated with 20 μg·mL−1 LPS for 16 h. As shown in Figure 3C,D the levels of HO‐1 protein and mRNA were significantly increased in HPAEpiCs challenged with rosiglitazone, which accompanied with down‐regulation of ICAM‐1 expression induced by LPS and was reversed by treatment with ZnPPIX. In addition, treatment with ZnPPIX also blunted the reduction, induced by rosiglitazone, of the adhesion of THP‐1 cells to HPAEpiCs challenged with LPS (Figure 3E), suggesting that HO‐1 induction by rosiglitazone protects against monocyte adhesion to HPAEpiCs, challenged with LPS.
NOX‐derived ROS generation is required for rosiglitazone‐induced HO‐1 expression
HO‐1 is induced by intracellular oxidative stress or imbalance between intracellular redox conditions (Loboda et al., 2016). Activation of NOX is one of the sources of ROS which could act as second messengers leading to HO‐1 expression (Seo et al., 2011). To determine whether NOX activation and ROS generation were involved in the HO‐1 expression, HPAEpiCs were pretreated with a scavenger of ROS (NAC), or inhibitors of p47phox (APO) or NOX (DPI). As shown in Figure 4A,B, these treatments concentration‐dependently attenuated the induction of HO‐1 protein (Figure 4A) and mRNA (Figure 4B) by rosiglitazone. In NOX enzymes, NOX2 has been shown to distinctly recruit p22phox (presence) and p47phox (regulatory subunits) to produce superoxide (O2 •−) in pulmonary alveolar epithelial cells (Tickner et al., 2011). To confirm the roles of p47phox and NOX2 in rosiglitazone‐induced responses, the levels of p47phox and NOX2 protein were knocked down by their respective siRNAs. As shown in Figure 4C, transfection with either p47phox or NOX2 siRNA knocked down the of level p47phox or NOX2 protein and attenuated the rosiglitazone‐induced HO‐1 expression in HPAEpiCs.
Figure 4.
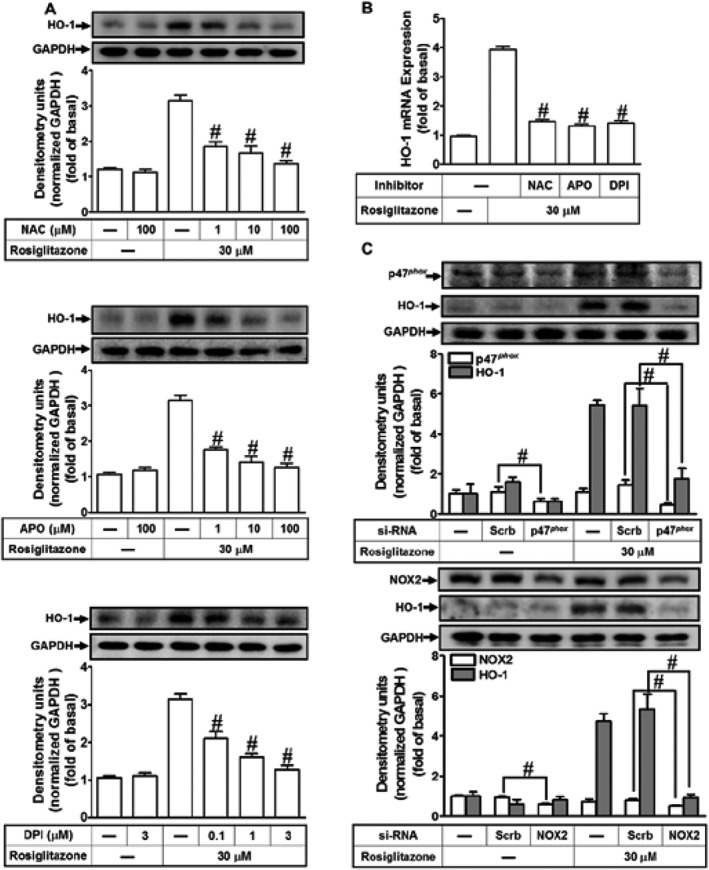
Activated NADPH oxidase and ROS generation are involved in rosiglitazone‐increased HO‐1 expression. (A) The cells were pretreated with various concentrations of NAC, APO or DPI for 1 h and then incubated with vehicle or rosiglitazone (30 μM) for 16 h. The levels of HO‐1 and GAPDH protein were determined by Western blot. (B) The cells were pretreated with NAC (100 μM), APO (100 μM) and DPI (3 μM) for 1 h and then incubated with vehicle or rosiglitazone (30 μM) for 8 h. The levels of HO‐1 and GAPDH mRNA were determined by real‐time PCR. (C) HPAEpiCs were transfected with p47phox or NOX2 siRNA and then incubated with rosiglitazone for 16 h. The levels of HO‐1, p47phox, NOX2 or GAPDH were determined by Western blot. Data are expressed as mean ± SEM, from five independent experiments (n = 5), # P < 0.05, significantly different from rosiglitazone alone (A, B); or significantly different as indicated (C).
Phosphorylation of p47phox is a key event in NOX activation and ROS generation (Lam et al., 2010). We found that, in HPAEpiCs, rosiglitazone‐stimulated p47phox phosphorylation, which was significantly inhibited by APO (Figure 5A). To further investigate whether rosiglitazone stimulates NOX activity leading to ROS generation, as shown in Figure 5B, rosiglitazone‐stimulated NOX activity was blocked by pretreatment with APO (100 μM) or DPI (3 μM). Furthermore, to directly detect ROS generation in rosiglitazone‐treated HPAEpiCs, the levels of intracellular H2O2 and O2 •− were determined by H2DCFDA and DHE respectively. Pretreatment with either APO or DPI inhibited the ROS generation induced by rosiglitazone (Figure 5C). These results were further supported by the data of DCF and DHE fluorescence images obtained by fluorescence microscopy (Figure 5D). These results suggested that rosiglitazone‐induced HO‐1 expression is mediated through NOX2‐dependent ROS generation in HPAEpiCs.
Figure 5.
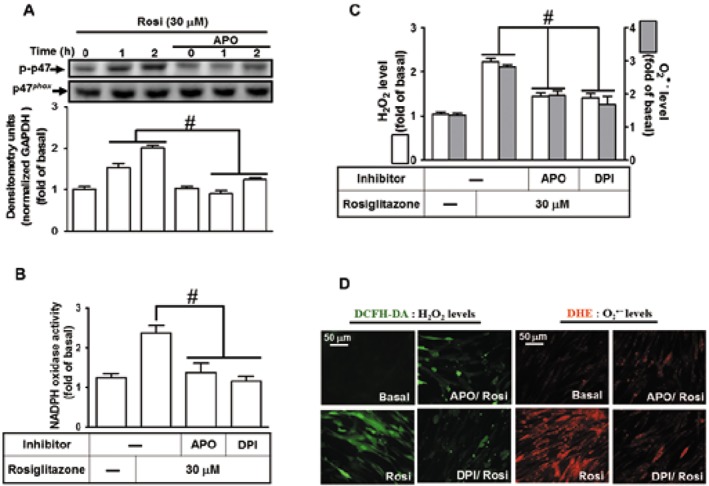
Activation of p47phox/NOX contributes to rosiglitazone‐induced HO‐1 expression. (A) HPAEpiCs were pretreated with APO and then incubated with vehicle or rosiglitazone (30 μM) for the indicated time intervals. Western blot was performed by using an anti‐phospho‐p47phox or anti‐p47phox antibody. (B–D) Rosiglitazone‐induced NADPH oxidase activity and ROS generation (ELISA and immunostaining) were reduced by pretreatment with APO or DPI in HPAEpiCs. Data are expressed as mean ± SEM, from five independent experiments (n = 5). # P < 0.05, significantly different from rosiglitazone alone.
Rosiglitazone‐induced HO‐1 expression is mediated via a NOX/ROS‐dependent c‐Src/Pyk2/Akt pathway
NOX/ROS production activates downstream components such as c‐Src and Pyk2 to modulate cellular functions (Peng et al., 2003; Ke et al., 2014). In addition, rosiglitazone also attenuated endothelial progenitor cell dysfunction and increased NO production via phosphorylation of Akt, which is one of downstream signalling components of the c‐Src/Pyk2 pathway (Liang et al., 2009; Deng et al., 2013). Therefore, we investigated whether the rosiglitazone‐induced HO‐1 expression is mediated through activation of ROS‐dependent c‐Src, Pyk2 and Akt signalling components in HPAEpiCs. The inhibitors of c‐Src (c‐Srci II), Pyk2 (PF431396) and Akt (Akti VIII) were used for these purposes. Pretreatment with c‐Srci II, PF431396 or Akti VIII significantly attenuated the rosiglitazone‐induced HO‐1 protein and mRNA expression in HPAEpiCs (Figure 6A,B). To confirm the involvement of c‐Src, Pyk2 and Akt in rosiglitazone‐mediated responses, as shown in Figure 6C, transfection with c‐Src, Pyk2 or Akt siRNA down‐regulated the level of c‐Src, Pyk2 or Akt protein and then attenuated the rosiglitazone‐induced HO‐1 protein expression.
Figure 6.
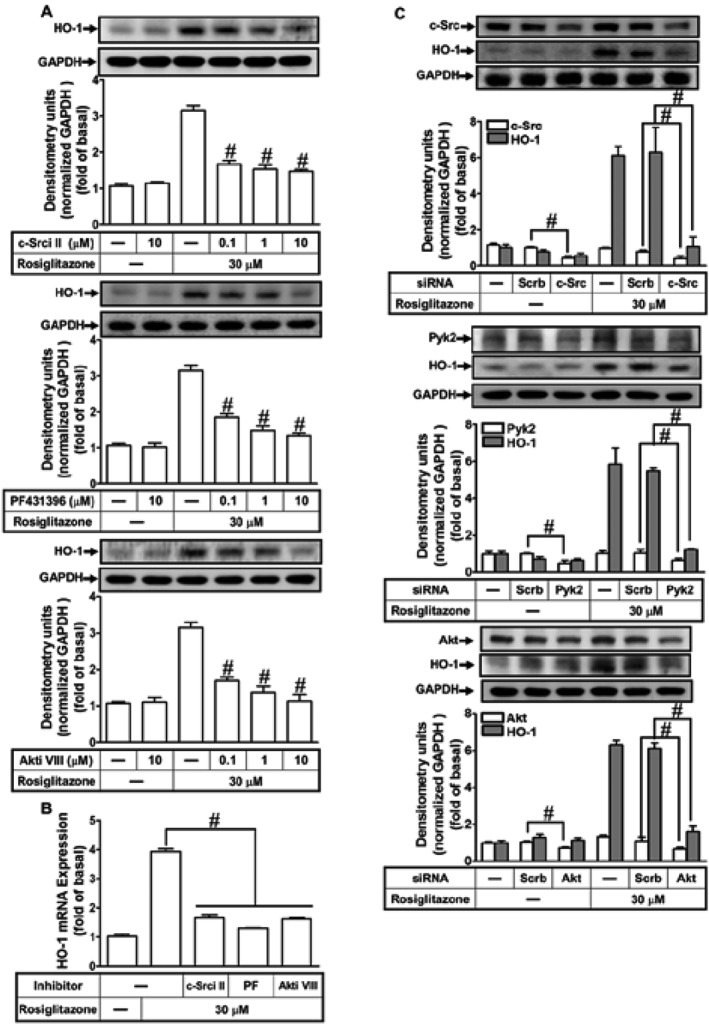
Activated c‐Src, Pyk2 and Akt play crucial roles in rosiglitazone‐induced HO‐1 expression. (A) The cells were incubated with various concentrations of c‐Srci II, PF431396 or Akti VIII for 1 h and then stimulated by vehicle or rosiglitazone (30 μM) for 16 h. The levels of HO‐1 and GAPDH protein were determined by Western blot. (B) The cells were pretreated with c‐Srci II (10 μM), PF431396 (10 μM) or Akti VIII (10 μM) for 1 h and then incubated with vehicle or rosiglitazone (30 μM) for 8 h. The levels of HO‐1 and GAPDH mRNA were determined by real‐time PCR. (C) HPAEpiCs were transfected with c‐Src, Pyk2 or Akt siRNA and then incubated with rosiglitazone for 16 h. The levels of HO‐1, c‐Src, Pyk2 and GAPDH protein were determined by Western blot. Data are expressed as mean ± SEM, from five independent experiments (n = 5). # P < 0.05, significantly different from rosiglitazone alone (A, B); or significantly different as indicated (C).
We further determined whether phosphorylation of c‐Src, Pyk2 and Akt participated in rosiglitazone‐induced HO‐1 expression in HPAEpiCs, by Western blot, using an antibody specific for the phosphorylated form of c‐Src, Pyk2 or Akt. Rosiglitazone‐stimulated the phosphorylation of c‐Src which was attenuated by the siRNA for p47phox or NOX2 and APO, DPI or c‐Srci II (Figure 7A,B). Similarly, pretreatment with c‐Srci II inhibited rosiglitazone‐stimulated phosphorylation of Pyk2, but not p47phox (Figure 7B), suggesting that rosiglitazone‐stimulated c‐Src‐dependent Pyk2 phosphorylation is mediated through NOX/ROS. Moreover, pretreatment with PF431396 attenuated rosiglitazone‐stimulated Pyk2 phosphorylation, but not c‐Src (Figure 7C), implying that Pyk2 was a downstream component of NOX/ROS/c‐Src cascade. Further, rosiglitazone time‐dependently stimulated Akt phosphorylation which was attenuated by pretreatment with either PF431396 or Akti VIII (Figure 7C,D). Akti VIII failed to inhibit the Pyk2 phosphorylation. Collectively, these data suggested that rosiglitazone‐induced HO‐1 expression is mediated through a NOX/ROS/c‐Src/Pyk2/Akt cascade in HPAEpiCs.
Figure 7.

Rosiglitazone‐induced HO‐1 expression via NOX/ROS‐dependent c‐Src/Pyk2/Akt pathway. HPAEpiCs were (A) transfected with p47phox or NOX2 siRNA, and pretreated with APO or DPI, (B) c‐Srci II, (C) PF431396 or (D) Akti VIII for 1 h, and then incubated with vehicle or rosiglitazone (30 μM) for the indicated time intervals. Western blot was performed by using an anti‐phospho‐c‐Src, anti‐c‐Src, anti‐phospho‐Pyk2, anti‐Pyk2, anti‐phospho‐Akt or anti‐Akt antibody. Data are expressed as mean ± SEM, from five independent experiments (n = 5). # P < 0.05, significantly different from rosiglitazone alone.
Up‐regulation of ARE promoter activity is mediated through NOX/ROS/c‐Src/Pyk2/Akt/Nrf2 cascade
Nrf2, an oxidative stress sensitive transcription factor, regulates the expression of antioxidant genes including HO‐1 (Alam and Cook, 2007; Loboda et al., 2016). As addressed, Keap1 accelerates the degradation of Nrf2 via facilitating the binding of E3 ligase, and the modification of cysteine residues of Keap1 releases functional Nrf2 (Baird et al., 2013). To assess the involvement of Keap1/Nrf2 in our model, HPAEpiCs were transfected with Nrf2 or Keap1 siRNA. As shown in Figure 8A (left panel), knockdown of Nrf2 protein expression by Nrf2 siRNA attenuated HO‐1 expression induced by rosiglitazone. In contrast, transfection with Keap1 siRNA down‐regulated the levels of Keap1 protein and enhanced the rosiglitazone‐induced HO‐1 expression (Figure 8A, right panel). Accumulated nuclear Nrf2 further binds to ARE on HO‐1 promoter and initiates HO‐1 gene expression. We found that rosiglitazone enhanced the recruitment of Nrf2 to the proximal ARE binding site on the HO‐1 promoter (Figure 8C). The subcellular fractions and real‐time PCR analyses further confirmed that pretreatment with APO, DPI, Srci II, PF431396 or Akti VIII significantly attenuated the rosiglitazone‐stimulated Nrf2 phosphorylation and translocation into the nucleus and its association with the ARE binding site on the HO‐1 promoter (Figure 8B,D). Further, to investigate whether rosiglitazone stimulated transcriptional activation of ARE, HPAEpiCs were co‐transfected with an ARE luciferase reporter construct (ARE‐Luc) and an internal control (pGal‐Luc) and then stimulated with rosiglitazone. Rosiglitazone significantly stimulated ARE‐Luc reporter activity which was attenuated by pretreatment with the respective inhibitors of NOX, c‐Src, Pyk2 and Akt (Figure 8D). These results suggested that rosiglitazone‐induced HO‐1 expression is mediated through a NOX/ROS/c‐Src/Pyk2/Akt pathway which then activates the Nrf2‐ARE axis in HPAEpiCs.
Figure 8.
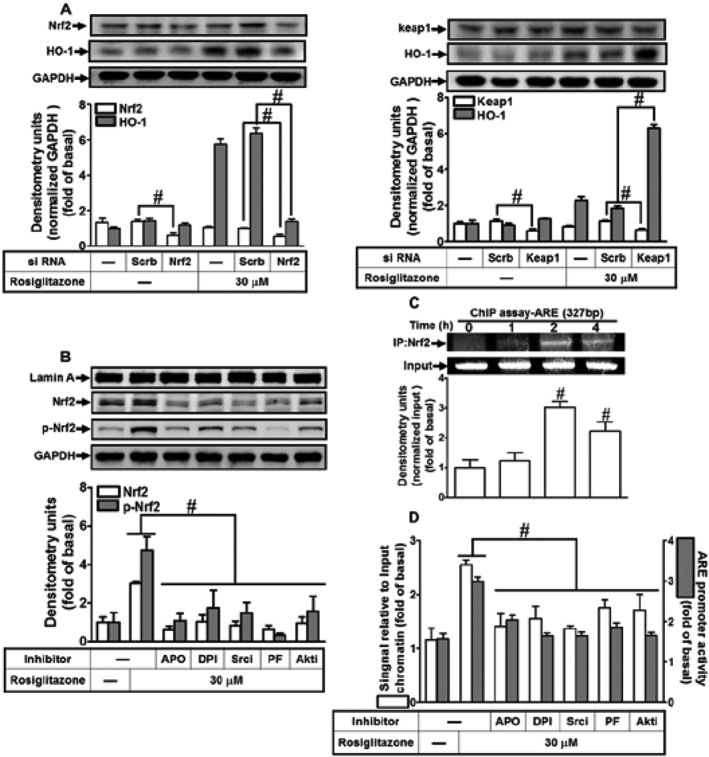
The involvement of NOX/ROS/c‐Src/Pyk2/Akt/Nrf2 cascade in the rosiglitazone‐stimulated ARE promoter activity. (A) HPAEpiCs were transfected with Nrf2 or Keap1 siRNA and then incubated with rosiglitazone for 16 h. The levels of Nrf2, Keap1, HO‐1 and GADPH proteins were determined by Western blot. (B) Cells were pretreated with APO, DPI, c‐Srci II, PF431396 or Akti VIII for 1 h and then stimulated with rosiglitazone for 2 h. The cytoplasmic and nuclear fractions were extracted and analysed by Western blot by using an anti‐Nrf2, anti‐phospho‐Nrf2, anti‐GAPDH or anti‐Lamin A antibody. (C, D) Cells were treated with 30 μM of rosiglitazone for the indicated time points or pretreated with APO, DPI, c‐Srci II, PF431396 and Akti VIII for 1 h and then stimulated by rosiglitazone for 2 h. The levels of Nrf2 binding to ARE region of the HO‐1 promoter were detected by a ChIP assay (C) or SYBR green real‐time PCR (D, open bars). ARE‐luc plasmids transfected HPAEpiCs were pretreated with APO, DPI, c‐Srci II, PF431396 or Akti VIII for 1 h and then incubated with vehicle or rosiglitazone (30 μM) for 2 h. ARE promoter luciferase activity was determined. Data are expressed as mean ± SEM, from five independent experiments (n = 5). # P < 0.05, significantly different as indicated (A); significantly different from rosiglitazone alone (B, D); or significantly different from vehicle (0 h) alone (C).
Involvement of PPARγ in rosiglitazone‐induced HO‐1 expression
PPARγ is known to contribute to several cellular functions in different cell types. However, PPARγ ligands may act through PPARγ‐dependent and ‐independent manners to protect against inflammatory responses (Chawla et al., 2001). As shown in Figure 9A, pretreatment of HPAEpiCs with the PPARγ antagonist, GW9662, concentration‐dependently inhibited the rosiglitazone‐induced HO‐1 protein and mRNA expression. These results were further confirmed by transfection with PPARγ siRNA, which markedly knocked down the level of PPARγ protein of either control or rosiglitazone treatment (control: 44 ± 7%; rosiglitazone: 51 ± 6%, si‐PPARγ vs. Scrb) and also reduced the rosiglitazone‐induced HO‐1 expression by 33 ± 4% in HPAEpiCs (Figure 9B). Further, we observed that rosiglitazone time‐dependently enhanced PPARγ promoter activity, which was inhibited by pretreatment with PPARγ antagonist GW9662 (Figure 9C). These results suggested the participation of PPARγ in the rosiglitazone‐induced HO‐1 expression in HPAEpiCs. Phosphorylation on Ser112 residue of PPARγ has been shown to correlate to its binding affinity with PPARγ ligands and post‐translational events (Burns and Vanden Heuvel, 2007; Rai et al., 2014). Data in Figure 9D showed that both PPARγ antagonist (GW9662) and PPARγ siRNA markedly attenuated phosphorylation of PPARγ stimulated by rosiglitazone. Thus, whether rosiglitazone stimulated PPARγ phosphorylation via activation of NOX/ROS/c‐Src/Pyk2/Akt/Nrf2 cascade was determined in HPAEpiCs. We found that transfection with p47phox, NOX2, c‐Src, Pyk2, Akt or Nrf2 siRNA had no significant effect on PPARγ phosphorylation stimulated by rosiglitazone (Figure 9E). Further, we also differentiated whether rosiglitazone‐induced HO‐1 expression was mediated through a PPARγ‐dependent c‐Src/Pyk2/Akt \cascade. We found that pretreatment with GW9662 or transfection with PPARγ siRNA failed to reduce the phosphorylation of c‐Src, Pyk2 or Akt (Supporting Information Figure S1). These results suggested that rosiglitazone‐induced HO‐1 expression is mediated through either PPARγ‐dependent or ‐independent manner in HPAEpiCs.
Figure 9.
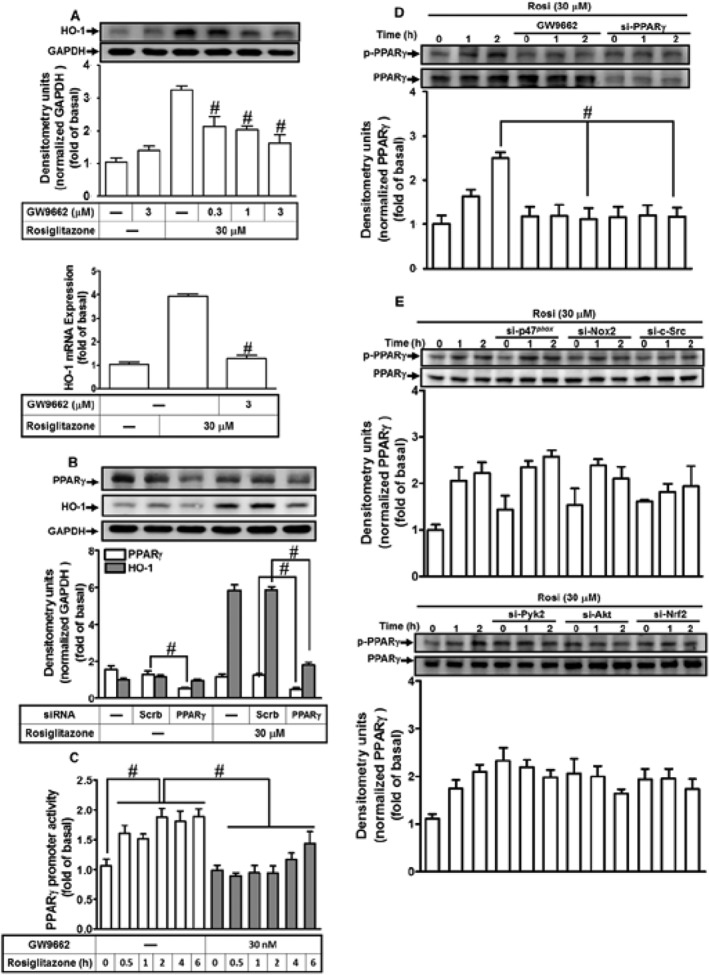
Rosiglitazone induces HO‐1 expression through a PPARγ‐dependent manner. (A) HPAEpiCs were pretreated with various concentrations of GW9662 for 1 h and then incubated with vehicle or rosiglitazone (30 μM) for 16 h. The levels of HO‐1 and GADPH proteins were determined by Western blot. (B) Cells were transfected with PPARγ siRNA and then incubated with rosiglitazone for 16 h. The levels of PPARγ, HO‐1 and GAPDH were detected by Western blot. (C) PPARγ‐RE reporter construct‐transfected cells were pretreated with GW9662 (30 nM) for 1 h and then incubated rosiglitazone (30 μM) for 2 h. PPARγ RE promoter luciferase activity was determined in the cell lysates. (D, E) HPAEpiCs were pretreated with GW9662 and transfected with PPARγ, p47phox, NOX2, c‐Src, Pyk2, Akt or Nrf2 siRNA, and then incubated with vehicle or rosiglitazone (30 μM) for the indicated time intervals. Western blot was used to detect the phosphorylation of PPARγ and the levels of total PPARγ. Data are expressed as mean ± SEM, from five independent experiments (n = 5). # P < 0.05, significantly different from rosiglitazone alone (A, D); or significantly different as indicated (B–D).
Discussion
Accumulating evidence indicates that rosiglitazone exerts anti‐inflammatory effects through up‐regulation of HO‐1 in the pulmonary system (Liu et al., 2005; Kronke et al., 2007; Li et al., 2010; Xu et al., 2015b), suggesting a possible therapeutic application for treatment of inflammatory diseases (Belvisi and Mitchell, 2009). In our previous study, we found that rosiglitazone suppressed LPS‐induced nuclear translocation of phosphorylated NF‐κB (p65) and expression of adhesion molecules through a PPARγ‐dependent, up‐regulation of HO‐1 (Cho et al., 2018). Here, we confirmed that rosiglitazone inhibited LPS‐induced lung inflammation by inducing HO‐1 independently of PPARγ. Further, application of pharmacological inhibitors and transfection with siRNAs inhibited rosiglitazone‐induced HO‐1 expression in HPAEpiCs. Our results demonstrated that HO‐1 expression induced by rosiglitazone was mediated through activation of either a NOX/ROS/c‐Src/Pyk2/Akt/Nrf2 pathway or a PPARγ cascade to suppress the inflammatory responses triggered by LPS (Figure 10).
Figure 10.
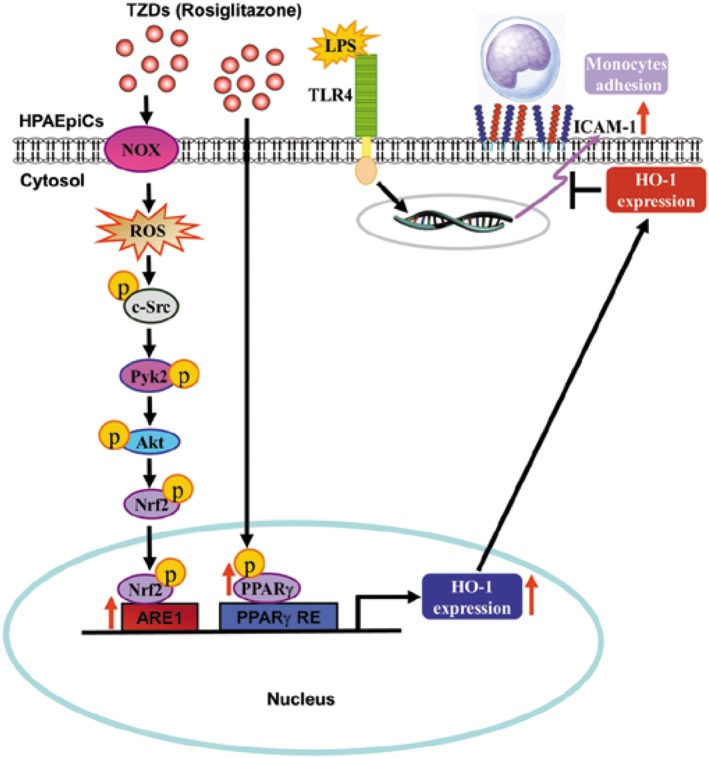
A schematic pathway for rosiglitazone induced HO‐1 expression in HPAEpiCs. Rosiglitazone attenuated LPS‐induced ICAM‐1 expression and lung monocyte/leukocyte accumulation by up‐regulating HO‐1 expression via two manners: PPARγ dependence and PPARγ independence. Activated PPARγ directly bound to PPARγ responsive element of HO‐1 promoter, which increased the expression of HO‐1 gene. In the PPARγ independent pathway, rosiglitazone enhanced NADPH oxidase activity, which resulted in the accumulation of intracellular ROS. Imbalance of oxidative stress promoted the phosphorylation of c‐Src/Pyk2/Akt and the activation of Nrf2. After nuclear translocation, Nrf2 binds to the ARE region of HO‐1 promoter and increases the expression of the HO‐1 gene.
PPAR subtypes are expressed in many tissues (Kota et al., 2005), and regulate several cellular functions (Michalik et al., 2006). PPAR agonists such as WY‐14643 (PPARα), GW501516 (PPARβ/δ) and rosiglitazone (PPARγ) induce HO‐1 expression to inhibit inflammatory responses (Kronke et al., 2007; Sodhi et al., 2014). Consistent with these findings, our results showed that agonitsts of these three subtypes of PPAR enhanced HO‐1 expression in HPAEpiCs. Particularly, rosiglitazone possesses potential effects against acute lung injury and inflammation via HO‐1 expression (Lin et al., 2014; Lv et al., 2016). Our results also confirmed that treatment of mice or HPAEpiCs with rosiglitazone significantly attenuated LPS‐induced ICAM‐1 expression and leukocyte adhesion, via up‐regulation of HO‐1.
ROS act as messengers in normal physiological functions or as inflammatory mediators, dependent on their concentration (Kamata and Hirata, 1999). NOX/ROS‐dependent HO‐1 expression is induced by different stimuli (Srisook et al., 2006; Jamal Uddin et al., 2016). Recently, an endogenous PPARγ ligand, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1877, induced ROS generation which promoted Nrf2‐dependent expression of HO‐1 (Koyani et al., 2016). Therefore, NOX‐dependent ROS generation may be involved in HO‐1 up‐regulation induced by rosiglitazone in HPAEpiCs. This was confirmed by the results that rosiglitazone‐induced HO‐1 expression was inhibited by NAC, APO and DPI. Moreover, NOX inhibitors prevent p47phox phosphorylation or translocation to the membrane and thus inhibit NOX activation and ROS generation (Barbieri et al., 2004). In line with these reports, our results demonstrated that pretreatment with APO or DPI attenuated rosiglitazone‐induced NOX/ROS generation associated with HO‐1 expression, suggesting that NOX/ROS play an important role in these responses.
NOX/ROS‐dependent HO‐1 expression has been shown to be mediated through the activation of c‐Src and Pyk2 (Han et al., 2009; Chi et al., 2015) and PI3K/Akt (Deng et al., 2013; Xu et al., 2015b), which protects against oxidative injury. Our results confirmed that c‐Src, Pyk2 and Akt are involved in HO‐1 expression in HPAEpiCs, which may result from the phosphorylation of c‐Src, Pyk2 and Akt stimulated by rosiglitazone. We also demonstrated that c‐Src, Pyk2 and Akt are downstream targets of NOX/ROS, as rosiglitazone‐stimulated phosphorylation of c‐Src was attenuated by NOX inhibitors or siRNA. The relationship among c‐Src, Pyk2 and Akt in rosiglitazone‐mediated responses was further differentiated by using the corresponding inhibitors. Pretreatment with c‐Srci II inhibited the rosiglitazone‐stimulated phosphorylation of c‐Src and Pyk2, whereas PF43139 only attenuated phosphorylation of Pyk2, suggesting that Pyk2 is downstream of c‐Src in rosiglitazone‐mediated responses. We also observed that rosiglitazone‐stimulated Akt phosphorylation was attenuated by PF431396 or Akti VIII, implying that Akt is downstream of c‐Src/Pyk2. It is worth noting that phosphorylation of c‐Src, Pyk2 and Akt was not changed by GW9662 or PPARγ siRNA. These results confirmed that rosiglitazone‐induced HO‐1 expression through a NOX/ROS‐mediated c‐Src/Pyk2/Akt phosphorylation is independent on PPARγ in HPAEpiCs.
Nrf2 controls the basal and inducible expression of an array of ARE‐dependent genes to regulate the pathophysiological outcomes of oxidant exposure (Espinosa‐Diez et al., 2015). In our study, rosiglitazone mediated Nrf2 phosphorylation by promoting the degradation of Keap1 and nuclear accumulation of Nrf2 in HPAEpiCs. Phosphorylated Nrf2 translocation from the cytosol into the nucleus and binding to ARE were essential for HO‐1 induction (Yang et al., 2015). Indeed, ChIP assay suggested that rosiglitazone increases Nrf2 interactions with ARE binding sites on the HO‐1 promoter. Silencing Nrf2 expression by its siRNA attenuated HO‐1 expression, but down‐regulated Keap1 expression increased HO‐1 protein levels in rosiglitazone‐stimulated HPAEpiCs. These results are consistent with the reports that expression of HO‐1, in response to up‐regulated Nrf2 in post‐transcriptional sites, protects cells from apoptosis in an Nrf2‐denpendent manner (Foresti et al., 2013). Moreover, the Akt/Nrf2 signalling pathway protects against toxicity (de Oliveira et al., 2015). Here, we reported that blockade of the NOX/ROS/c‐Src/Pyk2/Akt cascade by their respective inhibitors reduced rosiglitazone‐stimulated phosphorylation/nuclear translocation of Nrf2, HO‐1 promoter binding and ARE promoter activity in HPAEpiCs. These results suggested that Nrf2 is an acceptor of these protein kinases in rosiglitazone‐mediated responses.
On the other hand, PPARγ activation by rosiglitazone significantly increases HO‐1 expression and attenuates airway inflammation and remodelling (Xu et al., 2015a). In this study, blockade of PPARγ by its antagonist (GW9662) or siRNA, diminished rosiglitazone‐mediated PPARγ phosphorylation and promoter activity in HPAEpiCs, suggesting the dependence of PPARγ on HO‐1 expression. These results were consistent with the report indicating that PPARγ agonists block cigarette smoke extract‐induced inflammatory responses through multiple PPARγ‐mediated mechanisms (Lakshmi et al., 2014). However, phosphorylation of PPARγ was independent on Nox/ROS/c‐Src/Pyk2/Akt/Nrf2 cascade in rosiglitazone‐stimulated HPAEpiCs. This was confirmed by transfection with respective siRNAs which had no significant effect on the rosiglitazone‐stimulated phosphorylation of PPARγ. We also noted that both PPARγ‐dependent and PPARγ‐independent pathways are regulated by rosiglitazone. Our previous report indicated that rosiglitazone activated PPARγ binding to PPRE on HO‐1 promoter to induce HO‐1 transcription (Cho et al., 2018). Moreover, in the present study, we focused on the Nrf2 activity in HO‐1 induction. The studies of promoter activity indicated that PPARγ‐PPRE (−1740 to −1881 bp) and Nrf2‐ARE (−4019 to −4082 bp) response elements exhibited a similar trend in the transcriptional regulation (Kronke et al., 2007; Wang et al., 2013). Our previous report also indicated that arachidonic acid increases HO‐1 expression through the association of Nrf2 and PPARγ with ARE in rat astrocytes (Lin et al., 2018). Rosiglitazone shared the similar mechanism to induce the cooperation of PPARγ and Nrf2 on HO‐1 expression.
In summary, these findings suggest that rosiglitazone‐induced HO‐1 expression might play a protective role in lung inflammatory diseases, mediated via either NOX/ROS‐dependent c‐Src/Pyk2/Akt activation of Nrf2 which binds to ARE regions or PPARγ. Although several detailed mechanisms should be elucidated in the future, these findings expand the application of PPARγ agonists as potential interventions for the prevention or treatment of pulmonary inflammatory diseases. Treatment with rosiglitazone at 2–4 weeks was associated with a modest improvement in pre‐bronchodilator FEV1 (forced expiratory volume in 1 s), peak expiratory flow and FEF25–75 (forced expiratory flow between 25 and 75% of the forced vital capacity) of asthmatic reaction (Spears et al., 2009; Richards et al., 2010). Compared with rosiglitazone, administration of pioglitazone in mild asthma failed to improve any markers of airway inflammation, although it produced significant weight gains in asthmatic who were obese (Dixon et al., 2015; Anderson et al., 2016). Rosiglitazone has been reported to be associated with an increased risk of congestive heart failure, acute myocardial infarction and mortality in the patients during treatment for Type 2 diabetes. However, they are still of research value because of their anti‐inflammatory activity in lung diseases (Home et al., 2007; Azimova et al., 2014).
Author contributions
R.L.C., C.C.Y., H.C.T., L.D.H., C.C.L. and C.M.Y. conceived and designed the experiments. R.L.C., H.C.T., C.C.Y., L.D.H. and C.C.L. performed the experiments. R.L.C., C.C.Y., H.C.T., L.D.H., C.C.L. and C.M.Y. analysed the data and drafted relevant text. R.L.C., C.C.L. and C.M.Y. wrote the manuscript. All authors have read and approved the final version of this manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Rosilitazone‐induced c‐Src/Pyk2/Akt pathway through the PPARγ‐independent manner. HPAEpiCs were pretreated with GW9662 or transfected with PPARγ siRNA, and then inculated with vehicle or rosiglitazone (30 μM) for the indicated time intervals. Western blot was performed by using an anti‐phospho‐c‐Src, anti‐c‐Src, anti‐phospho‐Pyk2, anti‐Pyk2, anti‐phospho‐Akt or anti‐Akt antibody, Data are expressed as mean ± SEM, from five independent experiments (n = 5). # P < 0.05, as compared with the cells exposed to rosiglitazone alone.
Acknowledgements
We thank Ms. Yu‐Wen Chen for her technical assistance. This work was supported by the Ministry of Education, Taiwan, grant numbers: EMRPD1H032 and EMRPD1H055; the Ministry of Science and Technology, Taiwan, grant numbers: MOST104‐2320‐B‐182A‐003‐MY3, MOST105‐2320‐B‐182‐005‐MY3, MOST107‐2320‐B‐182A‐011 and MOST107‐2320‐B‐182‐020‐MY2; Chang Gung Medical Research Foundation, Taiwan, grant numbers: CMRPD1F0022, 0CMRPD1F0023, CMRPD1F0551, CMRPD1F0552, CMRPG3E2232, CMRPG3F1532, CMRPG3F1533, CMRPG3H0061 and CMRPG5F0202.
Cho, R.‐L. , Yang, C.‐C. , Tseng, H.‐C. , Hsiao, L.‐D. , Lin, C.‐C. , and Yang, C.‐M. (2018) Haem oxygenase‐1 up‐regulation by rosiglitazone via ROS‐dependent Nrf2‐antioxidant response elements axis or PPARγ attenuates LPS‐mediated lung inflammation. British Journal of Pharmacology, 175: 3928–3946. 10.1111/bph.14465.
Contributor Information
Chih‐Chung Lin, Email: chihchung@adm.cgmh.org.tw.
Chuen‐Mao Yang, Email: chuenmao@mail.cgu.edu.tw.
References
- Alam J, Cook JL (2007). How many transcription factors does it take to turn on the heme oxygenase‐1 gene? Am J Respir Cell Mol Biol 36: 166–174. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–-S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JR, Mortimer K, Pang L, Smith KM, Bailey H, Hodgson DB et al (2016). Evaluation of the PPARγ agonist pioglitazone in mild asthma: a double‐blind randomized controlled trial. PLoS One 11: e0160257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimova K, San Juan Z, Mukherjee D (2014). Cardiovascular safety profile of currently available diabetic drugs. Ochsner J 14: 616–632. [PMC free article] [PubMed] [Google Scholar]
- Baird L, Lleres D, Swift S, Dinkova‐Kostova AT (2013). Regulatory flexibility in the Nrf2‐mediated stress response is conferred by conformational cycling of the Keap1‐Nrf2 protein complex. Proc Natl Acad Sci U S A 110: 15259–15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri SS, Cavalca V, Eligini S, Brambilla M, Caiani A, Tremoli E et al (2004). Apocynin prevents cyclooxygenase 2 expression in human monocytes through NADPH oxidase and glutathione redox‐dependent mechanisms. Free Radic Biol Med 37: 156–165. [DOI] [PubMed] [Google Scholar]
- Belvisi MG, Mitchell JA (2009). Targeting PPAR receptors in the airway for the treatment of inflammatory lung disease. Br J Pharmacol 158: 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KA, Vanden Heuvel JP (2007). Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta 1771: 952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM (2001). PPAR‐γ dependent and independent effects on macrophage‐gene expression in lipid metabolism and inflammation. Nat Med 7: 48–52. [DOI] [PubMed] [Google Scholar]
- Chi PL, Lin CC, Chen YW, Hsiao LD, Yang CM (2015). CO induces Nrf2‐dependent heme oxygenase‐1 transcription by cooperating with Sp1 and c‐Jun in rat brain astrocytes. Mol Neurobiol 52: 277–292. [DOI] [PubMed] [Google Scholar]
- Cho RL, Lin WN, Wang CY, Yang CC, Hsiao LD, Lin CC et al (2018). Heme oxygenase‐1 induction by rosiglitazone via PKCα/AMPKα/p38 MAPKα/SIRT1/PPARγ pathway suppresses lipopolysaccharide‐mediated pulmonary inflammation. Biochem Pharmacol 148: 222–237. [DOI] [PubMed] [Google Scholar]
- Cho RL, Yang CC, Lee IT, Lin CC, Chi PL, Hsiao LD et al (2016). Lipopolysaccharide induces ICAM‐1 expression via a c‐Src/NADPH oxidase/ROS‐dependent NF‐κB pathway in human pulmonary alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 310: L639–L657. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Brit J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira MR, Ferreira GC, Schuck PF, Dal Bosco SM (2015). Role for the PI3K/Akt/Nrf2 signaling pathway in the protective effects of carnosic acid against methylglyoxal‐induced neurotoxicity in SH‐SY5Y neuroblastoma cells. Chem Biol Interact 242: 396–406. [DOI] [PubMed] [Google Scholar]
- Deng X, Rui W, Zhang F, Ding W (2013). PM2.5 induces Nrf2‐mediated defense mechanisms against oxidative stress by activating PIK3/AKT signaling pathway in human lung alveolar epithelial A549 cells. Cell Biol Toxicol 29: 143–157. [DOI] [PubMed] [Google Scholar]
- Dewar BJ, Gardner OS, Chen CS, Earp HS, Samet JM, Graves LM (2007). Capacitative calcium entry contributes to the differential transactivation of the epidermal growth factor receptor in response to thiazolidinediones. Mol Pharmacol 72: 1146–1156. [DOI] [PubMed] [Google Scholar]
- Dixon AE, Subramanian M, DeSarno M, Black K, Lane L, Holguin F (2015). A pilot randomized controlled trial of pioglitazone for the treatment of poorly controlled asthma in obesity. Respir Res 16: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan C, Tan X, Bourke JE (2012). PPARγ ligands regulate noncontractile and contractile functions of airway smooth muscle: implications for asthma therapy. PPAR Res 2012: 809164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa‐Diez C, Miguel V, Mennerich D, Kietzmann T, Sanchez‐Perez P, Cadenas S et al (2015). Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol 6: 183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foresti R, Bains SK, Pitchumony TS, de Castro Bras LE, Drago F, Dubois‐Rande JL et al (2013). Small molecule activators of the Nrf2‐HO‐1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol Res 76: 132–148. [DOI] [PubMed] [Google Scholar]
- Gardner OS, Dewar BJ, Graves LM (2005). Activation of mitogen‐activated protein kinases by peroxisome proliferator‐activated receptor ligands: an example of nongenomic signaling. Mol Pharmacol 68: 933–941. [DOI] [PubMed] [Google Scholar]
- Han Z, Varadharaj S, Giedt RJ, Zweier JL, Szeto HH, Alevriadou BR (2009). Mitochondria‐derived reactive oxygen species mediate heme oxygenase‐1 expression in sheared endothelial cells. J Pharmacol Exp Ther 329: 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Home PD, Pocock SJ, Beck‐Nielsen H, Gomis R, Hanefeld M, Jones NP et al (2007). Rosiglitazone evaluated for cardiovascular outcomes‐an interim analysis. N Engl J Med 357: 28–38. [DOI] [PubMed] [Google Scholar]
- Hsu CK, Lee IT, Lin CC, Hsiao LD, Yang CM (2014). Nox2/ROS‐dependent human antigen R translocation contributes to TNF‐α‐induced SOCS‐3 expression in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 306: L521–L533. [DOI] [PubMed] [Google Scholar]
- Huang J, Shen XD, Yue S, Zhu J, Gao F, Zhai Y et al (2014). Adoptive transfer of heme oxygenase‐1 (HO‐1)‐modified macrophages rescues the nuclear factor erythroid 2‐related factor (Nrf2) antiinflammatory phenotype in liver ischemia/reperfusion injury. Mol Med 20: 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD et al (1999). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino‐terminal Neh2 domain. Genes Dev 13: 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal Uddin M, Joe Y, Kim SK, Oh Jeong S, Ryter SW, Pae HO et al (2016). IRG1 induced by heme oxygenase‐1/carbon monoxide inhibits LPS‐mediated sepsis and pro‐inflammatory cytokine production. Cell Mol Immunol 13: 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata H, Hirata H (1999). Redox regulation of cellular signalling. Cell Signal 11: 1–14. [DOI] [PubMed] [Google Scholar]
- Ke K, Sul OJ, Choi EK, Safdar AM, Kim ES, Choi HS (2014). Reactive oxygen species induce the association of SHP‐1 with c‐Src and the oxidation of both to enhance osteoclast survival. Am J Physiol Endocrinol Metab 307: E61–E70. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota BP, Huang TH, Roufogalis BD (2005). An overview on biological mechanisms of PPARs. Pharmacol Res 51: 85–94. [DOI] [PubMed] [Google Scholar]
- Koyani CN, Kitz K, Rossmann C, Bernhart E, Huber E, Trummer C et al (2016). Activation of the MAPK/Akt/Nrf2‐Egr1/HO‐1‐GCLc axis protects MG‐63 osteosarcoma cells against 15d‐PGJ2‐mediated cell death. Biochem Pharmacol 104: 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke G, Kadl A, Ikonomu E, Bluml S, Furnkranz A, Sarembock IJ et al (2007). Expression of heme oxygenase‐1 in human vascular cells is regulated by peroxisome proliferator‐activated receptors. Arterioscler Thromb Vasc Biol 27: 1276–1282. [DOI] [PubMed] [Google Scholar]
- Lakshmi SP, Reddy AT, Zhang Y, Sciurba FC, Mallampalli RK, Duncan SR et al (2014). Down‐regulated peroxisome proliferator‐activated receptor γ (PPARγ) in lung epithelial cells promotes a PPARγ agonist‐reversible proinflammatory phenotype in chronic obstructive pulmonary disease (COPD). J Biol Chem 289: 6383–6393. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lam GY, Huang J, Brumell JH (2010). The many roles of NOX2 NADPH oxidase‐derived ROS in immunity. Semin Immunopathol 32: 415–430. [DOI] [PubMed] [Google Scholar]
- Li M, Li Z, Sun X, Yang L, Fang P, Liu Y et al (2010). Heme oxygenase‐1/p21WAF1 mediates peroxisome proliferator‐activated receptor‐γ signaling inhibition of proliferation of rat pulmonary artery smooth muscle cells. FEBS J 277: 1543–1550. [DOI] [PubMed] [Google Scholar]
- Liang C, Ren Y, Tan H, He Z, Jiang Q, Wu J et al (2009). Rosiglitazone via upregulation of Akt/eNOS pathways attenuates dysfunction of endothelial progenitor cells, induced by advanced glycation end products. Br J Pharmacol 158: 1865–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, Yang CC, Chen YW, Hsiao LD, Yang CM (2018). Arachidonic acid induces ARE/Nrf2‐dependent heme oxygenase‐1 transcription in rat brain astrocytes. Mol Neurobiol 55: 3328–3343. [DOI] [PubMed] [Google Scholar]
- Lin CF, Young KC, Bai CH, Yu BC, Ma CT, Chien YC et al (2014). Rosiglitazone regulates anti‐inflammation and growth inhibition via PTEN. Biomed Res Int 2014: 787924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou CJ, Lai YR, Chen YL, Chang YH, Li ZY, Huang WC (2016). Matrine attenuates COX‐2 and ICAM‐1 expressions in human lung epithelial cells and prevents acute lung injury in LPS‐induced mice. Mediators Inflamm 2016: 3630485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Zeng BX, Zhang SH, Yao SL (2005). Rosiglitazone, an agonist of peroxisome proliferator‐activated receptor γ, reduces pulmonary inflammatory response in a rat model of endotoxemia. Inflamm Res 54: 464–470. [DOI] [PubMed] [Google Scholar]
- Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J (2016). Role of Nrf2/HO‐1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci 73: 3221–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H, Yu Z, Zheng Y, Wang L, Qin X, Cheng G et al (2016). Isovitexin exerts anti‐inflammatory and anti‐oxidant activities on lipopolysaccharide‐induced acute lung injury by inhibiting MAPK and NF‐κB and activating HO‐1/Nrf2 pathways. Int J Biol Sci 12: 72–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ et al (2006). International union of pharmacology. LXI. peroxisome proliferator‐activated receptors. Pharmacol Rev 58: 726–741. [DOI] [PubMed] [Google Scholar]
- Peng G, Huang J, Boyd M, Kleinberg ME (2003). Properties of phagocyte NADPH oxidase p47phox mutants with unmasked SH3 (Src homology 3) domains: full reconstitution of oxidase activity in a semi‐recombinant cell‐free system lacking arachidonic acid. Biochem J 373: 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polvani S, Tarocchi M, Galli A (2012). PPARγ and oxidative stress: Con(β) catenating NRF2 and FOXO. PPAR Res 2012: 641087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai A, Tripathi S, Kushwaha R, Singh P, Srivastava P, Sanyal S et al (2014). CDK5‐induced p‐PPARγ(Ser 112) downregulates GFAP via PPREs in developing rat brain: effect of metal mixture and troglitazone in astrocytes. Cell Death Dis 5: e1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DB, Bareille P, Lindo EL, Quinn D, Farrow SN (2010). Treatment with a peroxisomal proliferator activated receptor γ agonist has a modest effect in the allergen challenge model in asthma: a randomised controlled trial. Respir Med 104: 668–674. [DOI] [PubMed] [Google Scholar]
- Rushworth SA, Chen XL, Mackman N, Ogborne RM, O'Connell MA (2005). Lipopolysaccharide‐induced heme oxygenase‐1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J Immunol 175: 4408–4415. [DOI] [PubMed] [Google Scholar]
- Seo WY, Goh AR, Ju SM, Song HY, Kwon DJ, Jun JG et al (2011). Celastrol induces expression of heme oxygenase‐1 through ROS/Nrf2/ARE signaling in the HaCaT cells. Biochem Biophys Res Commun 407: 535–540. [DOI] [PubMed] [Google Scholar]
- Sodhi K, Puri N, Kim DH, Hinds TD, Stechschulte LA, Favero G et al (2014). PPAR δbinding to heme oxygenase 1 promoter prevents angiotensin II‐induced adipocyte dysfunction in Goldblatt hypertensive rats. Int J Obes (Lond) 38: 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Spears M, Donnelly I, Jolly L, Brannigan M, Ito K, McSharry C et al (2009). Bronchodilatory effect of the PPARγ agonist rosiglitazone in smokers with asthma. Clin Pharmacol Ther 86: 49–53. [DOI] [PubMed] [Google Scholar]
- Srisook K, Han SS, Choi HS, Li MH, Ueda H, Kim C et al (2006). CO from enhanced HO activity or from CORM‐2 inhibits both O2‐ and NO production and downregulates HO‐1 expression in LPS‐stimulated macrophages. Biochem Pharmacol 71: 307–318. [DOI] [PubMed] [Google Scholar]
- Tickner J, Fan LM, Du J, Meijles D, Li JM (2011). Nox2‐derived ROS in PPARγ signaling and cell‐cycle progression of lung alveolar epithelial cells. Free Radic Biol Med 51: 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Liu L, Zhang Y, Han D, Liu J, Xu J et al (2014). Activation of PPARγ attenuates LPS‐induced acute lung injury by inhibition of HMGB1‐RAGE levels. Eur J Pharmacol 726: 27–32. [DOI] [PubMed] [Google Scholar]
- Wang S, Hannafon BN, Zhou J, Ding WQ (2013). Clofibrate induces heme oxygenase 1 expression through a PPARα‐independent mechanism in human cancer cells. Cell Physiol Biochem 32: 1255–1264. [DOI] [PubMed] [Google Scholar]
- Woodfin A, Beyrau M, Voisin MB, Ma B, Whiteford JR, Hordijk PL et al (2016). ICAM‐1‐expressing neutrophils exhibit enhanced effector functions in murine models of endotoxemia. Blood 127: 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhu YT, Wang GZ, Han D, Wu YY, Zhang DX et al (2015a). The PPARγ agonist, rosiglitazone, attenuates airway inflammation and remodeling via heme oxygenase‐1 in murine model of asthma. Acta Pharmacol Sin 36: 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Li H, Hou X, Li D, He S, Wan C et al (2015b). Punicalagin induces Nrf2/HO‐1 expression via upregulation of PI3K/AKT pathway and inhibits LPS‐induced oxidative stress in RAW264.7 macrophages. Mediators Inflamm 2015: 380218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CM, Lin CC, Lee IT, Hsu CK, Tai YC, Hsieh HL et al (2015). c‐Src‐dependent transactivation of EGFR mediates CORM‐2‐induced HO‐1 expression in human tracheal smooth muscle cells. J Cell Physiol 230: 2351–2361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Rosilitazone‐induced c‐Src/Pyk2/Akt pathway through the PPARγ‐independent manner. HPAEpiCs were pretreated with GW9662 or transfected with PPARγ siRNA, and then inculated with vehicle or rosiglitazone (30 μM) for the indicated time intervals. Western blot was performed by using an anti‐phospho‐c‐Src, anti‐c‐Src, anti‐phospho‐Pyk2, anti‐Pyk2, anti‐phospho‐Akt or anti‐Akt antibody, Data are expressed as mean ± SEM, from five independent experiments (n = 5). # P < 0.05, as compared with the cells exposed to rosiglitazone alone.