

Abstract
In order to identify compounds with potential toxicity problems, particular attention is paid to structural alerts, which are high chemical reactivity molecular fragments or fragments that can be transformed via bioactivation by human enzymes into fragments with high chemical reactivity. The concept has been introduced in order to reduce the likelihood that future candidate substances as pharmaceuticals will have undesirable toxic effects. A significant proportion (∼78–86%) of drugs characterized by residual toxicity contain structural alerts; there is also evidence indicating the formation of active metabolites as a causal factor for the toxicity of 62–69% of these molecules. On the other hand, the pharmacological action of certain drugs depends on the formation of reactive metabolites. Detailed assessment of the potential for the formation of active metabolites is recommended to characterize a biologically active compound. Although many prescribed drugs frequently contain structural alerts and form reactive metabolites, the vast majority of these drugs are administered in low daily doses. Avoiding structural alerts has become almost a norm in new drug design. An in-depth review of the biochemical reactivity of these structural alerts for new drug candidates is critical from a safety point of view and is currently being monitored in the discovery of drugs. The chemical strategies applied to structural alerts in molecules to limit the toxicity are:
-
•
partial replacement or full replacement of the structural alert;
-
•
reduction of electronic density;
-
•
introduction of a structural element of metabolic interest (metabolic switching);
-
•
multiple approaches.
Therefore, chemical intervention strategies to eliminate bioactivation are often interactive processes; their success depends largely on a close working relationship between drug chemists, pharmacologists and researchers in metabolic science.
Keywords: Structural alerts, Active metabolites, Toxicity
1. Introduction
To identify compounds that can potentially create toxic effects, particular attention should be paid to structural alerts, i.e. information on potential toxicity of molecular fragments with high chemical reactivity. These fragments can be presented in the parent compound or its metabolites after biochemical transformation by human enzymes (bioactivation). The use of such structural alerts has been proposed in order to reduce the likelihood that future candidate drugs will have undesirable toxic effects.
The aim of this review is to present the data from the literature about structural alerts in order to help the medicinal chemists involved in early stages of drug discovery to identify the potential structural features that may cause adverse reactions, thus avoiding additional costs related to the development of the respective molecules. An additional objective is to present how molecular aspects may influence the bioactivation of drugs and to induce the occurrence of adverse effects. An in-depth review of the biochemical reactivity of these structural alerts, especially for new drug candidates and their metabolites, is essential from the point of view of therapeutic safety and drug discovery. However, there are molecules that contain structural alerts and will never generate toxic effects, as well as compounds that can be rejected as drugs due to manifested toxic effects without having a structural alert in their molecule [1]. In addition, there are cases where a drug can be used despite its toxic effects because the benefits are thought to outweigh the risks.
More and more questions are posed regarding when a molecule containing a structural alert may become a concern. It is assumed that removing compounds containing structural alerts from the bioactivity screening libraries and keeping short lists of candidates would reduce the risks in the new drug discovery process and failures in their further development. However, lately, there has been a growing concern that some structural alerts may be too strict, and their application would greatly diminish the chemical diversity that is so necessary to operate in drug discovery programs.
Besides the fact that a significant proportion (∼78–86%) of toxicity-associated drugs contain structural alerts, there is also evidence that the formation of active metabolites is the causal factor for the toxicity of 62–69% of these molecules. These active metabolites or toxophores can be considered as containing the structural alerts of concern [2]. On the other hand, the pharmacological action of certain drugs depends on the formation of active metabolites.
Elimination of active metabolites could be a useful starting point in mitigating the risks of idiosyncratic toxicity. Although not conclusively demonstrated, active metabolites play an important role in the appearance of idiosyncratic reactions. Idiosyncratic reactions cannot be anticipated based on the pharmacological properties of the drug, and they are unpredictable, dose-independent, and rarely tested on laboratory animals. A small amount can trigger an immune reaction. They can affect any organ, but especially the liver, skin and bone marrow. These reactions may occur when it is an exposure to new drugs that have not been completely tested for possible side effects. The mechanisms of most of these reactions are still unknown, most of them being immune mediated [3]. Active metabolites can be formed through the metabolism of a drug, with bioactivation, via oxidative, reductive or hydrolytic pathways, followed by glucuronic acid or glutathione conjugation, sulfonation, acetylation, amidation, etc. Mostly, active metabolites result from oxidation, usually mediated by cytochrome P450 enzymes, flavin monooxigenase (FMO), monoamine oxidase (MAO) and peroxidase (e.g., cyclooxygenase (COX) and myeloperoxidase (MPO) [4]. The active metabolites resulting from bioactivation are usually electron deficient or free radical molecules and can cause direct toxic effects. They can react with DNA, having mutagenic, carcinogenic, teratogenic effects or react with proteins, generating toxic effects on target organs or immune-idiosyncratic mediated reactions.
Identification of structural alerts, detailed assessment of the potential for the formation of active metabolites and the application of performance tests to early indication of in vivo toxicity problems have become necessary in the discovery of new drugs. Regulatory authorities ask for these tests to be performed for all new chemical entities before their use in human clinical trials [5]. Structural alerts should be combined with Quantitative Structure-Activity Relationships (QSAR) modeling and/or Chemical Biological Read-Across (CBRA) models to improve the accuracy of toxicity prediction [6].
The study of structural alerts has also been interesting since the acceptance of the Registration, Evaluation, Authorization, and Restriction of Chemicals (REACH) Regulation by the European Council and European Parliament on 18th December 2006, the date from which the concept has become a major one in chemical safety assessment and regulatory decision support. This law compels manufacturers to provide detailed information on chemicals that are manufactured, marketed, or imported [6,7]. In creative disruption with previous practice regarding chemical safety legislation, REACH allows the use of QSARs and read-across in the hazard evaluation of chemical compounds in or entering the European market. In this context, structural alerts become even more important, since they provide the sine qua non tool for computational toxicology in terms of hazard prediction and thus safe design of new chemicals.
Although many prescription drugs frequently contain structural alerts and form active metabolites, most of them are administered and are prescribed in low daily doses. A lower dose reduces the risk of toxic effects. Most drugs that were withdrawn from therapy or received a black-box warning, for example on hepatotoxicity, were prescribed in daily doses greater than 50 mg / day, because the doses are not known in the early stages of drug development. Any drug that is given at a daily dose of 10 mg or less is unlikely to be associated with idiosyncratic phenomena [8]. Clearly, total exposure to an active metabolite is an important aspect in this regard Fig. 1).
Fig. 1.

Structures of raloxifene and its metabolites.
To see how the dose affects the toxic effects, we can compare clozapine (daily dose 300 mg) with olanzapine (daily dose 10 mg). Although both form active metabolites, only clozapine has a risk of developing agranulocytosis. Therefore, a lower amount of active metabolites does not lead to toxic phenomena [9].
There are also medications that, although administered at doses higher than 50 mg / day, will never produce idiosyncratic reactions. Raloxifene, [6-hydroxy-2-(4-hydroxyphenyl)-benzothiophen-3-yl]-[4-[2-(1-piperidyl)ethoxy]phenyl]methanone, is a selective estrogen receptor modulator, useful in the treatment of osteoporosis, and bioactivated by cytochrome P4503A4, in hepatic microsomes. Its electrophilic active metabolite has a quinone structure; it is thus conjugated with glutathione (GSH). However, the main metabolic pathway remains the conjugation with glucuronic acid (the main enzyme involved being uridine diphosphate glucuronyl transferase-UGT), and the intestinal glucuronidation limits the amount of raloxifene which is bioactivated [10,11].
2. Methods
We used the keyword and cited reference searches in four databases (Scopus, PubMed, Web of Science, Google Scholar) and we found that there are several chemical strategies applied to determine structural alerts in drug molecules in order to attenuate or avoid their toxicity. These include:
-
(a)
replacement of the potential structural alert with substitutes that are generally resistant to metabolism;
-
(b)
reduction of electronic density;
-
(c)
introduction of a structural element of metabolic interest (metabolic switching); and
-
(d)
a multitude of various other approaches in combination.
The results of the review related to the strategies applied to structural alerts in drug molecules in order to attenuate or avoid their toxicity is presented and discussed below.
3. Discussion
3.1. Replacement of the potential structural alert with substitutes that are generally resistant to metabolism
The most important chemical strategy remains the full or partial direct replacement of the potential structural alert, or groups that can lead to creation of metabolites with structural alerts, with substitutes that are generally resistant to metabolism or functional groups that undergo biotransformation to non-reactive metabolites. A number of cases are given below:
3.1.1. Procainamide
Procainamide, 4-amino-N-(2-diethylaminoethyl)benzamide, an antiarrhythmic agent, can cause agranulocytosis, an idiosyncratic adverse reaction. Procainamide is metabolised in various ways, such as acetylation, or using the neutrophil myeloperoxidase (MPO) which activates the compound by forming a N-hydroxyaniline derivative by N-hydroxylation and a nitroso derivative by oxidation, which reacts with glutathione or covalently binds to proteins, being implicated in direct toxicity to bone marrow cells and may cause agranulocytosis [12,13] (Fig. 2).
Fig. 2.

Structures of procainamide and its metabolites.
The complete replacement of the toxophore aniline fragment resulted in the preparation of flecainide, (RS)-N-(2-piperidylmethyl)-2,5-bis(2,2,2-trifluoroethoxy)benzamide (Fig. 3).
Fig. 3.
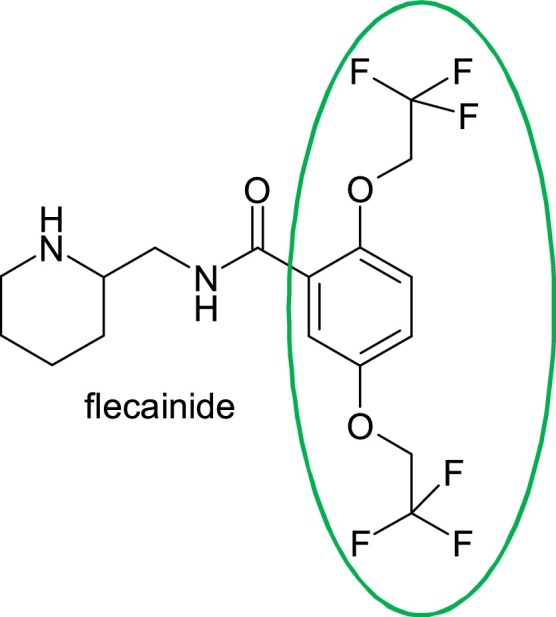
Structure of flecainide.
3.1.2. Carbutamide
Carbutamide, 4-amino-N-[(butylamino)carbonyl]benzenesulfonamide, is a sulfonylurea antidiabetic substance known to be associated with bone marrow toxicity. Moreover, because of medullary toxicity due to bioactivity to nitroso-derivative, it was withdrawn from therapeutics. Replacement of the aniline moiety, more precisely the amino group, with a methyl group, resulted in tolbutamide, 1-butyl-3-(p-tosylsulfonyl)urea [14] (Fig. 4).
Fig. 4.

Structures of carbutamide and tolbutamide.
3.1.3. Clozapine
Clozapine, 8-chloro-11-(4-methylpiperazin-1-yl)-5H-dibenzo[b,e][1,4]diazepine, is an antipsychotic (D2 / 5 / HT2 antagonist) bioactivated by neutrophil myeloperoxidase, with the formation of imine ion-reactive species, which are supposed to be responsible for idiosyncratic agranulocytosis with 1% incidence. The use of clozapine is limited not only to the production of agranulocytosis, but also to its hepatotoxicity and the high incidence of myocarditis. Also, in human neutrophils, clozapine is oxidized with nitrilium ion formation, having a delocalized positive charge. The resulting active metabolite reacts with glutathione to form its thioether adducts, which are irreversibly bounded to neutrophils [[15], [16], [17], [18], [19]] (Fig. 5).
Fig. 5.

Structures of clozapine and its metabolites.
Agranulocytosis is reduced by replacing one of the nitrogen atoms of the dibenzodiazepine nucleus (the structural alert) with oxygen or nitrogen giving the dibenzooxazepine nucleus in loxapine, (8-chloro-6-(4-methylpiperazin-1-yl)benzo[b]4]benzoxazepine), or dibenzothiazepine nucleus in quetiapine, (2-(2-(4-dibenz[b,f][1,4]thiazepin-11-yl-1-piperazinyl)ethoxy)ethanol, despite of the fact that they are administered at comparable doses of clozapine (Fig. 6).
Fig. 6.

Structures of loxapine and quetiapine.
3.1.4. Amodiaquine
Another example where the formation of the active metabolite can be eliminated by direct replacement of the toxic-causing group is represented by the study of the antimalarial agent amodiaquine, 4-[(7-chloroquinolin-4-yl)amino]-2-(diethylaminomethyl)phenol, which has been withdrawn from use due to cases of hepatotoxicity and agranulocytosis. Toxicity problems have been attributed to bioactivation, with the formation of a quinoneimine metabolite, which reacts preferentially with nucleophilic thiol group [20] (Fig. 7).
Fig. 7.

Structures of amodiaquine and its quinoneimine metabolite.
Tingle et al. [21] have been shown that replacement of the OH-phenolic group with a fluorine atom leads to a compound which does not undergo oxidation in the presence of peroxidase; in the case of amodiaquine containing the 4-aminophenolic moiety, the quinonimine is formed.
3.1.5. Nefazodone
Nefazodone, 2-[3-[4-(3-chlorophenyl)piperazin-1-yl]propyl]-5-ethyl-4-(2-phenoxyethyl)-1,2,4-triazol-3-one, is an antidepressant with a severe hepatotoxicity which has led to its withdrawal from the market. In the presence of cytochrome P4503A4, a hepatotoxic quinonimine is formed, and this gives an adduct with glutathione associated with the 3-chlorophenyl-piperazine moiety. The formation of the active metabolite and the fact that nefazodone inhibits the export of bile salts to the liver are responsible for the hepatotoxicity of the compound [22] (Fig. 8).
Fig. 8.

Structures of nefazodone and its metabolites.
The replacement of the metabolically labile chloro-phenyl and phenoxy groups with a substituted indole nucleus and a benzofuran nucleus resulted in vilazodone, 5-{4-[4-(5-cyano-3-indolyl)butyl]-1-piperazinyl}benzofuran-2-carboxamide (Fig. 9).
Fig. 9.

Structure of vilazodone.
3.1.6. Practolol
Practolol, (RS)-N-{4-[2-hydroxy-3-(isopropylaminopropoxy]phenyl}acetamide, a beta-1 blocker agent, caused severe skin and eye injuries (oculomucocutaneous syndrome), which led to its withdrawal from the market in 1975. The evidence indicates hydrolysis of the amide group and aniline oxidation, with the formation of N-hydroxyaniline derivative [23] (Fig. 10, Fig. 11).
Fig. 10.

Structures of practolol and atenolol.
Fig. 11.

Structures of practolol desacetylation and oxidation productions.
The applied chemical strategy was to replace the acetylaniline moiety with the inverse amide deriving to atenolol, (RS)-2-{4-[2-hydroxy-3-(propan-2-ylamino)propoxy]phenyl]acetamide, a selective beta-1 receptor antagonist which doesn’t have the aniline nitrogen atom.
3.1.7. Flunitrazepam
Flunitrazepam, 5-(2-fluorophenyl)-1-methyl-7-nitro-1H-benzo[e][1,4]diazepin-2(3 H)-one, is hypnotic, sedative and anxiolytic drug used to treat severe insomnia. CYP2C19 and CYP3A4 are involved in the metabolism of flunitrazepam to its major metabolites, desmethylflunitrazepam and 3-hydroxyflunitrazepam [24]. Bioactivation also occurs with the reduction of the nitro group and further N-acetylation of the resulting amino group [25]. The substitution of the nitro group with bromine, an electron attracting atom, resulted in bromazepam, 7-bromo-5-(pyridin-2-yl)-1H-benzo[e][1,4]diazepin-2(3 H)-one, with anxiolytic properties (Fig. 12).
Fig. 12.

Structures of flunitrazepam and bromazepam.
3.1.8. Prazosin
Prazosin, [4-(4-amino-6,7-dimethoxy-2-quinazolinyl)-1-piperazinyl](2-furyl)methanone, is a quinazoline derivative used as a peripheral vasodilator in the treatment of hypertension and congestive heart failure. This drug has been in use since 1976, with no evidence of safety issues despite the presence of 2-substituted furan ring in its structure. It is an example of a low dose drug (1 mg/ day) without idiosyncratic side effects despite the formation of an active metabolite with a γ-keto-propenal fragment by epoxidation of the furan nucleus followed by the opening of this heterocycle. Oxidation of the furan ring and ring opening is the major route of in vivo metabolism of this drug; this pathway does not necessarily lead to toxicity. The carboxylic acid derivative is the metabolite detected in the largest amount in urine and feces of rat. The rapid reduction or oxidation of the active metabolite is probably responsible for the absence of toxicity, despite the extensive metabolism of the furan ring [26] (Fig. 13).
Fig. 13.

Structures of prasozin metbolites.
By replacing the furanic ring with benzo[1,4]dioxane nucleus, gave doxazosin, (RS)-2-[4-(2,3-dihydro-1,4-benzodioxin-2-carbonyl)piperazin-1-yl]-4-amino-6,7-dimethoxyquinazoline (Fig. 14).
Fig. 14.

Structures of prasozin and dexazosin.
3.1.9. Halothane
Halothane, 2-bromo-2-chloro-1,1,1-trifluoroethane, is a volatile anesthetic used from 1959, and it has the highest incidence of hepatotoxicity, compared to isoflurane and desflurane (Fig. 15).
Fig. 15.

Structures of halothane, isoflurane and desflurane.
These compounds undergo oxidative metabolism which results in the formation of reactive acyl halides, but the degree to which these anesthetics are bioactivated is significantly higher in the case of halothane. Transformation in the highest proportion of the active trifluoroacyl chloride of halotane is attributed to the presence of the bromine atom in the structure, which is more easily released. In contrast, isoflurane and desflurane are less bioactively activated due to the change in electronic properties by replacing the bromine from halothane molecule with the difluoromethoxy group, which has a relatively low capacity of release compared to bromine and which generally reduces the affinity for metabolism. Replacing bromine with a difluoromethoxy group is an example of modulation of biochemical reactivity through changes in the electronic properties.
The activation mechanism of halothane is [27] (Fig. 16):
Fig. 16.

Structures of halothane, trifluoroacetyl chloride and trigluoroacetylated proteins.
3.1.10. Ibufenac
Ibufenac, 4-isobutylphenylacetic acid, has been withdrawn from therapy due to severe hepatotoxicity. The chosen chemical strategy to reduce biochemical reactivity was the substitution at 2-position of the acetic acid moiety with a methyl group with the occurrence of a chirality center, this being an example of modulation of biochemical reactivity by steric impediment. Ibuprofen, (RS)-2-(4-(2-methylpropyl)phenyl)propanoic acid, is one of the safest non-steroidal anti-inflammatory, analgesics, antipyretics and platelet antiaggregant drugs (Fig. 17).
Fig. 17.

Structures of ibufenac and ibuprofene.
Bioactivation of ibufenac takes place with the formation of acylglucuronoconjugates, trans-acylation of 1-O-acyl-glucuronoconjugate with proteins, and Schiff base formation and Amadori arrangement after opening of the 3-O-acylglucuronyl-conjugate heterocycle. Glucuronoconjugates of ibufenac are much more active metabolites than those of ibuprofen. In addition, the acyl-glucuronoconjugates of the 2-substituted propionic acid derivatives may exhibit a rate of spatial alignment and covalent protein binding slower than the corresponding acetic acid analogues due to steric impediment provided by the α-methyl substituent which probably slows the spatial rearrangement of the glucuronide conjugate [[28], [29], [30]] (Fig. 18).
Fig. 18.

Structures of products of ibufenac bioactivation.
3.2. Reducing electronic density
Antidepressants, a widely used drug group, are associated with a range of idiosyncratic reactions that affect liver, skin, haematological and central nervous system in particular. These reactions are mediated by active metabolites formed by the cytochrome P450 enzyme system, and the toxicity occurs directly or indirectly through an immune mechanism and due to their high electronic density. Individual susceptibility is determined by both, genetic and environmental factors, that lead to inappropriate detoxification of these toxic active metabolites. An example of overpassing toxicity issues by reducing electronic density is this of Imipramine. Imipramine, 3-(10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)-N,N-dimethylpropan-1-amine is metabolised by hydroxylation, demethylation (CYP 2D6) and N-oxidation (predominantly to imipramine compared to clomipramine). The epoxidation leads to a hepatotoxic epoxide metabolite. By replacing the hydrogen atom at position 2 of the benzazepine ring with an electron attracting chlorine atom, imipramine is converted into clomipramine, 3-(3-chloro-10,11-dihydro-5H-dibenzo[b,f]azepin-yl)-N,N-dimethylpropan-1-amine, a chemical with antidepressant action (Fig. 19).
Fig. 19.

Structures of imipramine and clomipramine.
3.3. The introduction of a structural element of metabolic interest (metabolic switching)
In addition to strategies that focus on replacing structural alerts or blocking metabolic sites excluding bioactivation, chemists can also introduce structural elements of metabolic interest to deviate metabolism.
3.3.1. Nifedipine & amlodipine
Nifedipine is a well-tolerated drug despite the fact that it contains a structural alert represented by the nitro group. The main metabolic pathways of nifedipine identified in humans include oxidation of the 1,4-dihydropyridine ring, hydrolysis of both methyl ester groups and / or hydroxylation of the methyl group, followed by lactonization. There is no evidence of formation of metabolites derived from the metabolic reduction of the nitro group in humans [31,32].
In the discovery of amlodipine, a calcium channel blocker with dihydropyridine structure, the strategy of modifying the neutral molecule nifedipine, was used making the molecule more alkaline. The alkalinity of amlodipine influences the plasma half-life (35 h). Pharmacokinetic parameters allow once-daily administration of amlodipine without requiring prolonged release technology. The large volume of distribution is achieved despite the moderate lipophilicity of amlodipine and can be compared to that of nifedipine which has a similar lipophilicity but is neutral. Changes in the structure of nifedipine do not cause a significant change in clearance [33,34] (Fig. 20).
Fig. 20.

Structures of nifedipine & amlodipine.
3.3.2. Tolcapone & entacapone
Tolcapone, 3,4-dihydroxy-5-nitrophenyl)(4-methylphenyl)methanone, and entacapone, (2E)-2-cyano-3-(3,4-dihydroxy-5-nitrophenyl)-N,N-diethylprop-2-enamide, are two catechol-O-methyltransferase inhibitors used for the treatment of Parkinson's disease, which also provide important information on the effectiveness of this strategy. The use of tolcapone has been associated with several problems, such as abnormal liver function tests and fatal hepatotoxicity. These issues have led to market withdrawal in some countries or the introduction of strong warning and monitoring requirements in the United States. These adverse reactions do not occur when using entacapone despite dosing similar to tolcapone. Both tolcapone and entacapone are metabolised. The biotransformation of tolcapone is accomplished by reducing the nitrobenzene moiety to form an aniline derivative which is then transformed with N-acetyl transferase into the corresponding anilide. Both metabolites of tolcapone suffer a slight oxidation of quinoneimine metabolites that react with glutathione [35,36]. On the contrary, entacapone, did not show a reduction in the nitrobenzene fragment, and the primary metabolic pathway involved N-deethylation of the amide substituent and isomerization of the active E isomer to the inactive Z isomer, followed by glucuronidation [37] (Fig. 21).
Fig. 21.

Structures of tolcapone and entacapone.
3.3.3. Alpidem & zolpidem
Alpidem, 2-[6-chloro-2-(4-chlorophenyl)imidazo[1,2-a]pyridin-3-yl]-N,N-dipropylacetamide, an anxiolytic agent, was withdrawn from the first year of use into therapy due to severe hepatotoxicity that led to deaths or required liver transplantation immediately. In contrast, N,N-dimethyl-2-[6-methyl-2 (4-methylphenyl)imidazo[1,2-a]pyridin-3-yl]acetamide, zolpidem, a structural analogue, does not possess hepatotoxicity (Fig. 22).
Fig. 22.
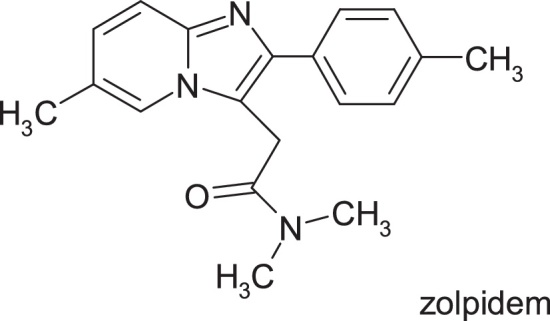
Structure of zolpidem.
Although the reason for this discrepancy remains unclear, it is interesting to note that the chlorid-imidazopyridine nucleus of alpidem is subjected to bioactivation in the presence of cytochrome P450, leading to the formation of a reactive epoxide that reacts with glutathione to produce conjugates (Fig. 23).
Fig. 23.

Structures of alpidem and its metabolites.
An essential structural difference between the two drugs is the replacement of the two chlorine atoms in the alpidem with two metabolically labile metabolic groups in zolpidem, that undergo oxidative metabolism to hydroxymethyl and carboxyl groups. Also, the daily dose of zolpidem is significantly lower than that of alpidem.
3.3.4. Ticlopidine & clopidogrel
An example that highlights the success of this strategy is clopidogrel, the structural model being provided by the ticlopidine molecule. Toxicity of ticlopidine, a platelet aggregation inhibitor, was linked to bioactivity with cytochrome P450 or myeloperoxidase (MPO) catalysing S-oxidation of thiophene ring to active S-oxides or other intermediates that bind covalently to hepatic proteins and neutrophils [38]. In the case of the structurally similar clopidogrel, introduction of the additional methyl ester fragment and move of C1 in the ring lead to a change in metabolism, so that the hydrolysis of the ester to the inactive acid derivative metabolite is the principal metabolic pathway of this drug [39]. Of great interest in this context is the observation that clopidogrel itself is inactive and requires biotransformation to produce inhibition of platelet aggregation in vivo. Pereillo et al. [40] have identified a thiophene ring cleavage product as active metabolite of clopidogrel. The formation of this active metabolite takes place by oxidation of the thiophenic nucleus to produce a 2-oxothiophene metabolite, and its hydrolytic cleavage generates the active component. Finally, it is important to note that the daily dose of clopidogrel is lower than that of ticlopidine; this feature may play a key role in the safety profile of clopidogrel that has significantly improved [40] (Fig. 24, Fig. 25).
Fig. 24.

Structures of ticlopidine and its metabolites.
Fig. 25.

Structures of clopidogrel and its metabolites.
3.4. Multiple approaches
3.4.1. Sudoxicam, piroxicam, and meloxicam
Sudoxicam, 4-hydroxy-2-methyl-N-(2-thiazolyl)-2H-1,2-benzothiazine-3-carboxamide, an enol-carboxamide-containing COX inhibitors for inflammation, has produced acute liver failure in the studies and was therefore withdrawn from Phase III clinical trials. The cause is the structural alert represented by the thiazole ring. The main metabolic pathway consists in opening this cycle (Fig. 26).
Fig. 26.

Structures of sudoxicam and 4-hydroxy-2-methyl-N-(2-thiazolyl)-2H-1,2-benzothiazine-3-carboxamide.
The chemical strategies applied to eliminate toxic effects consisted of the introduction of a methyl group, resulting in meloxicam or replacement of the thiazole nucleus with the pyridine nucleus, as in piroxicam (Fig. 27).
Fig. 27.

Structures of sudoxicam, piroxicam and meloxicam.
Meloxicam, 4-hydroxy-2-methyl-N-(5-methyl-2-thiazolyl)-2H-1,2-benzothiazine-3-carboxamide-1,1-dioxide, is used in therapy from 1996 for its anti-inflammatory, analgesic and antipyretic properties. Hepatic side effects are rare. Its main metabolic pathway is oxidation of the methyl group as a metabolic soft spot [41] (Fig. 28).
Fig. 28.

Structures of meloxicam and 4-hydroxy-2-methyl-N-(5-methyl-2-thiazolyl)-2H-1,2-benzothiazine-3-carboxamide-1,1-dioxide.
3.4.2. Histamine, burimamide, and metiamide
The structural model provided by the histamine molecule, 2-(1H-imidazol-4-yl)ethanamine, was the basis for the design of burimamide, 1-[4-(1H-imidazol-5-yl)butyl]-3-methylthiourea, an antagonist of histamine H2 receptors. The compound is largely inactive at physiological pH and for stabilization it was intended to modify the acidic exponent (pKa) by substituting a methylene group with a sulfur atom in tiaburimamide or by introducing a methyl group at the 4-position of the imidazole ring to favor the tautomeric form which binds better to the H2 receptor, in metiamide, (N-methyl-N '-(2-{[(4-methyl-1H-imidazol-5-yl)methyl]thio}ethyl)thiourea (Fig. 29).
Fig. 29.

Structures of histamine, burimamide, and metiamide.
The modifications brought increased the bioavailability of metiamide, being more active in the inhibition of histamine-stimulated gastric acid secretion, increasing the peptic ulcer healing rate. However, because of the toxic effects on the hematopoietic marrow, with the occurrence of agranulocytosis and nephrotoxicity, metiamide was discarded. The responsible for agranulocytosis is the thiourea fragment that is subjected to bioactivation. It was suggested to replace = S from the thiourea fragment with = O or = NH. The guanidine analogue of methiamide had less activity than metiamide. It has been found that by the addition of electron attracting groups such as -C≡N or -NO2, the protonation of the guanidine group at physiological pH is prevented, the bone marrow toxicity is eliminated, the acidity exponent of the neighboring nitrogen atoms is reduced and the activity of the drug in a physiological environment is preserved.
3.4.3. Cimetidine and ranitidine
Guanidine derivatives and their analogues were investigated, finally gave cimetidine, 2-cyano-1-methyl-3-(2-[(5-methyl-1H-imidazol-4-yl)methylthio]ethylguanidine [42].
The principles of bioisosterism were used for the design of the ranitidine molecule, N-(2-[(5-[(dimethylamino)methyl]furan-2 yl)methylthio]ethyl)-N'-methyl-2-nitroetenyl-1,1-diamine: the imidazole cycle of cimetidine has been isosterically replaced with a steric and electronic equivalent nucleus, as is the furan nucleus, and the thiourea fragment was replaced by 1,1-diamino-2-nitroethylene. It should be noticed that ranitidine, a possible hepatotoxic, does not suffer the typical metabolism of the 2,5-disubstituted furan ring. This is probably due to reduced lipophilicity, low dose and additional substitution. Its metabolism is by N-oxidation to the dimethylamino group (Fig. 30).
Fig. 30.

Structures of cimetidine and ranitidine.
4. Conclusions
Prevention of idiosyncratic reactions should be based either on the development of new compounds that are not transformed into toxic metabolites or on prediction of individual susceptibility before drug administration. Until we better understand the pathophysiological mechanisms associated with idiosyncratic toxicity phenomena, improving the pharmacokinetic properties and receptor affinity as a means of decreasing doses will remain one of the main objectives in discovering of new drugs. Therefore, chemical intervention strategies to eliminate bioactivity are often interactive processes the success of which depends largely on a close working relationship between drug chemistry specialists, pharmacologists, toxicologists and metabolic engineers.
References
- 1.Claesson A., Minidis A. Systematic approach to organizing structural alerts for reactive metabolite formation from potential drugs. Chem. Res. Toxicol. 2018 doi: 10.1021/acs.chemrestox.8b00046. [DOI] [PubMed] [Google Scholar]
- 2.Stepan A.F., Walker D.P., Bauman J., Price D.A., Baillie T.A., Kalgutkar A.S., Aleo M.D. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 2011;24:1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 3.Uetrecht J. Immune-mediated adverse drug reactions. Chem. Res. Toxicol. 2009;22:24–34. doi: 10.1021/tx800389u. [DOI] [PubMed] [Google Scholar]
- 4.Tafazoli S., O’Brien P.J. Peroxidases: a role in the metabolism and side effects of drugs. Drug Discov. Today. 2005;10:617–625. doi: 10.1016/S1359-6446(05)03394-5. [DOI] [PubMed] [Google Scholar]
- 5.EMEA . 2008. EMA/CHMP/ICH/126642/2008 - European Medicines Agency - ICH Guideline S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use. [Google Scholar]
- 6.Alves V., Muratov E., Capuzzi S., Politi R., Low Y., Braga R., Zakharov A.V., Sedykh A., Mokshyna E., Farag S., Andrade C., Kuz’min V., Fourches D., Tropsha A. Alarms about structural alerts. Green Chem. 2016;18(16):4348–4360. doi: 10.1039/C6GC01492E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corrigendum to Regulation (EC) No 1907/2006 Corrigendum to Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 Concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), Establishing a European Chemicals Agency, Amending Directive 1999/45/EC and Repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 As Well As Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC (OJ L 396, 30.12.2006) http://data.europa.eu/eli/reg/2006/1907/corrigendum/2007-05-29/oj.
- 8.Uetrecht J. Screening for the potential of a drug candidate to cause idiosyncratic drug reactions. Drug Discov. Today. 2003;8:832–837. doi: 10.1016/s1359-6446(03)02816-2. [DOI] [PubMed] [Google Scholar]
- 9.Gardner I., Leeder J.S., Chin T., Zahid N., Uetrecht J.P. A comparison of the covalent binding of clozapine and olanzapine to human neutrophils in vitro and in vivo. Mol. Pharmacol. 1998;53:999–1008. [PubMed] [Google Scholar]
- 10.Chen Q., Ngui J.S., Doss G.A., Wang R.W., Cai X., DiNinno F.P., Blizzard T.A., Hammond M.L., Stearns R.A., Evans D.C., Baillie T.A., Tang W. Cytochrome P450 3A4-mediated bioactivation of raloxifene: irreversible enzyme inhibition and thiol adduct formation. Chem. Res. Toxicol. 2002;15:907–914. doi: 10.1021/tx0200109. [DOI] [PubMed] [Google Scholar]
- 11.Kemp D.C., Fan P.W., Stevens J.C. Characterization of raloxifene glucuronidation in vitro: contribution of intestinal metabolism to presystemic clearance. Drug Metab. Dispos. 2002;30:694–700. doi: 10.1124/dmd.30.6.694. [DOI] [PubMed] [Google Scholar]
- 12.Siraki A.G., Deterding L.J., Bonini M.G., Jiang J., Ehrenshaft M., Tomer K.B., Mason R.P. Procainamide, but not N-acetylprocainamide, induces protein free radical formation on myeloperoxidase: a potential mechanism of agranulocytosis. Chem. Res. Toxicol. 2008;21:1143–1153. doi: 10.1021/tx700415b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uetrecht J.P. Reactivity and possible significance of hydroxylamine and nitroso metabolites of procainamide. J. Pharmacol. Exp. Ther. 1985;232:420–425. [PubMed] [Google Scholar]
- 14.Smith D.A., Van de Waterbeemd H., Walker D.K. American Chemical Society; 2001. Pharmacokinetics and Metabolism in Drug Design. Methods and Principles in Medicinal Chemistry. Volume 31. ISBN 3527313680. [Google Scholar]
- 15.Fischer V., Haar J.A., Greiner L., Lloyd R.V., Mason R.P. Possible role of free radical formation in clozapine (clozaril)-induced agranulocytosis. Mol. Pharmacol. 1991;40:846–853. [PubMed] [Google Scholar]
- 16.Kalgutkar A.S., Soglia J.R. Minimising the potential for metabolic activation in drug discovery. Expert Opin. Drug Metab. Toxicol. 2005;1:91–142. doi: 10.1517/17425255.1.1.91. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z.C., Uetrecht J.P. Clozapine is oxidized by activated human neutrophils to a reactive nitrenium ion that irreversibly binds to the cells. J. Pharmacol. Exp. Ther. 1995;275:1476–1483. [PubMed] [Google Scholar]
- 18.Maggs J.L., Williams D., Pirmohamed M., Park B.K. The metabolic formation of reactive intermediates from clozapine, a drug associated with agranulocytosis in man. J. Pharmacol. Exp. Ther. 1995;275:1463–1475. [PubMed] [Google Scholar]
- 19.Uetrecht J.P. New concepts in immunology relevant to idiosyncratic drug reactions: the "danger hypothesis" and innate immune system. Chem. Res. Toxicol. 1999;12:387–395. doi: 10.1021/tx980249i. [DOI] [PubMed] [Google Scholar]
- 20.Naisbitt D.J., Williams D.P., O’Neill P.M., Maggs J.L., Willock D.J., Pirmohamed M., Park B.K. Metabolism-dependent neutrophil cytotoxicity of amodiaquine: a comparison with pyronaridine and related antimalarial drugs. Chem. Res. Toxicol. 1998;11:1586–1595. doi: 10.1021/tx980148k. [DOI] [PubMed] [Google Scholar]
- 21.Tingle M.D., Jewell H., Maggs J.L., O’Neill P.M., Park B.K. The bioactivation of amodiaquine by human polymorphonuclear leucocytes in vitro: chemical mechanisms and the effects of fluorine substitution. Biochem. Pharmacol. 1995;50:1113–1119. doi: 10.1016/0006-2952(95)00236-s. [DOI] [PubMed] [Google Scholar]
- 22.Kalgutkar A.S., Vaz A.D., Lame M.E., Henne K.R., Soglia J., Zhao S.X., Abramov Y.A., Lombardo F., Collin C., Hendsch Z.S., Hop C.E. Bioactivation of the nontricyclic antidepressant nefazodone to a reactive quinone-imine species in human liver microsomes and recombinant cytochrome P450 3A4. Drug Metab. Dispos. 2005;33:243–253. doi: 10.1124/dmd.104.001735. [DOI] [PubMed] [Google Scholar]
- 23.Sim E., Stanley L., Gill E.W., Jones A. Metabolites of procainamide and practolol inhibit complement components C3 and C4. Biochem. J. 1988;251:323–326. doi: 10.1042/bj2510323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kilicarslan T., Haining R.L., Rettie A.E., Busto U., Tyndale R.F., Sellers E.M. Flunitrazepam metabolism by cytochrome P450S 2C19 and 3A4. Drug Metab. Dispos. 2001;29:460–465. [PubMed] [Google Scholar]
- 25.Zhou X. Handbook of Metabolic Pathways of Xenobiotics. 2014. Part 2. Compound articles. [Google Scholar]
- 26.Peterson L.A. Reactive metabolites in the biotransformation of molecules containing a furan ring. Chem. Res. Toxicol. 2013;26:6–25. doi: 10.1021/tx3003824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Njoku D., Laster M.J., Gong D.H., Eger E.I., 2nd, Reed G.F., Martin J.L. Biotransformation of halothane, enflurane, isoflurane, and desflurane to trifluoroacetylated liver proteins: association between protein acylation and hepatic injury. Anesth. Analg. 1997;84:173–178. doi: 10.1097/00000539-199701000-00031. [DOI] [PubMed] [Google Scholar]
- 28.Bolze S., Bromet N., Gay-Feutry C., Massiere F., Boulieu R., Hulot T. Development of an in vitro screening model for the biosynthesis of acyl glucuronide metabolites and the assessment of their reactivity toward human serum albumin. Drug Metab. Dispos. 2002;30:404–413. doi: 10.1124/dmd.30.4.404. [DOI] [PubMed] [Google Scholar]
- 29.Castillo M., Smith P.C. Disposition and reactivity of ibuprofen and ibufenac acyl glucuronides in vivo in the rhesus monkey and in vitro with human serum albumin. Drug Metab. Dispos. 1995;23:566–572. [PubMed] [Google Scholar]
- 30.Wang J., Davis M., Li F., Azam F., Scatina J., Talaat R. A novel approach for predicting acyl glucuronide reactivity via Schiff base formation: development of rapidly formed peptide adducts for LC/MS/MS measurements. Chem. Res. Toxicol. 2004;17:1206–1216. doi: 10.1021/tx049900+. [DOI] [PubMed] [Google Scholar]
- 31.Bocker R.H., Guengerich F.P. Oxidation of 4-aryl- and 4-alkyl-substituted 2,6-dimethyl-3,5-bis(alkoxycarbonyl)-1,4-dihydropyridines by human liver microsomes and immunochemical evidence for the involvement of a form of cytochrome P-450. J. Med. Chem. 1986;29:1596–1603. doi: 10.1021/jm00159a007. [DOI] [PubMed] [Google Scholar]
- 32.Guengerich F.P., Peterson L.A., Bocker R.H. Cytochrome P-450-catalyzed hydroxylation and carboxylic acid ester cleavage of Hantzsch pyridine esters. J. Biol. Chem. 1988;263:8176–8183. [PubMed] [Google Scholar]
- 33.Mason R.P., Rhodes D.G., Herbette L.G. Reevaluating equilibrium and kinetic binding parameters for lipophilic drugs based on a structural model for drug interaction with biological membranes. J. Med. Chem. 1991;34:869–877. doi: 10.1021/jm00107a001. [DOI] [PubMed] [Google Scholar]
- 34.Smith D.A., Jones B.C., Walker D.K. Design of drugs involving the concepts and theories of drug metabolism and pharmacokinetics. Med. Res. Rev. 1996;16:243–266. doi: 10.1002/(SICI)1098-1128(199605)16:3<243::AID-MED2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 35.Jorga K., Fotteler B., Heizmann P., Gasser R. Metabolism and excretion of tolcapone, a novel inhibitor of catechol-O-methyltransferase. Br. J. Clin. Pharmacol. 1999;48:513–520. doi: 10.1046/j.1365-2125.1999.00036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith K.S., Smith P.L., Heady T.N., Trugman J.M., Harman W.D., Macdonald T.L. In vitro metabolism of tolcapone to reactive intermediates: relevance to tolcapone liver toxicity. Chem. Res. Toxicol. 2003;16:123–128. doi: 10.1021/tx025569n. [DOI] [PubMed] [Google Scholar]
- 37.Wikberg T., Vuorela A., Ottoila P., Taskinen J. Identification of major metabolites of the catechol-O-methyltransferase inhibitor entacapone in rats and humans. Drug Metab. Dispos. 1993;21:81–92. [PubMed] [Google Scholar]
- 38.Liu Z.C., Uetrecht J.P. Metabolism of ticlopidine by activated neutrophils: implications for ticlopidine-induced agranulocytosis. Drug Metab. Dispos. 2000;28:726–730. [PubMed] [Google Scholar]
- 39.Reist M., Roy-de Vos M., Montseny J.P., Mayer J.M., Carrupt P.A., Berger Y., Testa B. Very slow chiral inversion of clopidogrel in rats: a pharmacokinetic and mechanistic investigation. Drug Metab. Dispos. 2000;28:1405–1410. [PubMed] [Google Scholar]
- 40.Pereillo J.M., Maftouh M., Andrieu A., Uzabiaga M.F., Fedeli O., Savi P., Pascal M., Herbert J.M., Maffrand J.P., Picard C. Structure and stereochemistry of the active metabolite of clopidogrel. Drug Metab. Dispos. 2002;30:1288–1295. doi: 10.1124/dmd.30.11.1288. [DOI] [PubMed] [Google Scholar]
- 41.Kalgutkar A.S., Gardner I., Obach R.S., Shaffer C.L., Callegari E., Henne K.R., Mutlib A.E., Dalvie D.K., Lee J.S., Nakai Y., O’Donnell J.P., Boer J., Harriman S.P. A comprehensive listing of bioactivation pathways of organic functional groups. Curr. Drug Metab. 2005;6:161–225. doi: 10.2174/1389200054021799. [DOI] [PubMed] [Google Scholar]
- 42.Durant G.J., Emmett J.C., Ganellin C.R., Miles P.D., Parsons M.E., Prain H.D., White G.R. Cyanoguanidine-thiourea equivalence in the development of the histamine H2-receptor antagonist, cimetidine. J. Med. Chem. 1977;20:901–906. doi: 10.1021/jm00217a007. [DOI] [PubMed] [Google Scholar]