

 Antibiotic resistance caused by β-lactamase production continues to present a growing challenge to the efficacy of β-lactams and their role as the most important class of clinically used antibiotics.
Antibiotic resistance caused by β-lactamase production continues to present a growing challenge to the efficacy of β-lactams and their role as the most important class of clinically used antibiotics.
Abstract
Antibiotic resistance caused by β-lactamase production continues to present a growing challenge to the efficacy of β-lactams and their role as the most important class of clinically used antibiotics. In response to this threat however, only a handful of β-lactamase inhibitors have been introduced to the market over the past thirty years. The first-generation β-lactamase inhibitors (clavulanic acid, sulbactam and tazobactam) are all β-lactam derivatives and work primarily by inactivating class A and some class C serine β-lactamases. The newer generations of β-lactamase inhibitors including avibactam and vaborbactam are based on non-β-lactam structures and their spectrum of inhibition is extended to KPC as an important class A carbapenemase. Despite these advances several class D and virtually all important class B β-lactamases are resistant to existing inhibitors. The present review provides an overview of recent FDA-approved β-lactam/β-lactamase inhibitor combinations as well as an update on research efforts aimed at the discovery and development of novel β-lactamase inhibitors.
1. Introduction
There is an urgent need to develop new therapeutic options to combat the increasing number of pathogens that have become resistant to β-lactam antibiotics by gaining the ability to express β-lactamase enzymes. The β-lactamases are classified by both structural approaches (Ambler)1 and functional approaches (Bush–Jacoby–Medeiros).2 Throughout this review, the Ambler classification will be used to describe the β-lactamases. Class A is represented by the classic β-lactamases such as the TEM and SHV families which inactivate penicillins and narrow-spectrum cephalosporins. Some member of the TEM and SHV families, along with the CTX-M class, are also able to inactivate extended-spectrum β-lactams and are therefore referred to as extended-spectrum β-lactamases (ESBLs). There are also carbapenemases among class A enzymes which include KPC, IMI and SME.3,4 Unlike members of class A/C/D families which hydrolyze β-lactams by action of a serine nucleophile, class B β-lactamases are metalloenzymes that contain zinc ion in their active site. In these so-called metallo-β-lactamases a water molecule, activated via coordination to zinc, serves as a nucleophile and hydrolyzes the β-lactam ring rendering the antibiotic inactive (Fig. 1). With the exception of monobactams, class B metallo-β-lactamases (MBLs) are able to hydrolyze all classes of β-lactams. The rapidly emerging NDM along with VIM and IMP are among the most clinically important MBLs which possess carbapenemase activity.5–8 Class C is represented by CMY, ACT and DHA which are also known as AmpC enzymes. Gram-negative bacteria producing this class of enzymes are often resistant to penicillins and some cephalosporins. Class D contains OXA family the members of which are able to metabolize penicillins, cephalosporins, and carbapenems. In this regard, the emergence of OXA-producing Pseudomonas aeruginosa and Acinetobacter baumannii is of particular concern.9
Fig. 1. A. β-lactam inactivation mediated by serine β-lactamases (ambler class A, C and D) is facilitated by the attack of a nucleophilic serine. B. MBL (class B)-mediated inactivation of β-lactams involves a nucleophilic attack by an activated water molecule coordinated to zinc ions.

For the purpose of clarity, Fig. 2 provides an overview of the various antibiotics 1–13 that have been tested in combination with the β-lactamase inhibitors covered in this review. The first generation of β-lactamase inhibitors including clavulanic acid 14, sulbactam 15, and tazobactam 16 (Fig. 3) were granted FDA-approval between 1984 and 1993. They were formulated with penicillins and include amoxicillin 1-clavulanic acid, ticarcillin 2-clavulanic acid, ampicillin 3-sulbactam and piperacillin 4-tazobactam combinations.10 However, the spectrum of activity of these inhibitor/β-lactam combinations covers primarily the β-lactamases of class A (with the exception of KPC). In addition, the emergence of inhibitor-resistant TEM variants with lowered susceptibility to clavulanic acid, sulbactam, and tazobactam has been documented.11
Fig. 2. β-lactam antibiotics evaluated in combination with β-lactamase inhibitors.

Fig. 3. First generation of β-lactamase inhibitors; clavulanic acid 14, sulbactam 15 and tazobactam 16.

In response to the increasing risk of drug-resistant bacterial infections, new generations of β-lactamase inhibitors including avibactam and vaborbactam have been added to our arsenal in recent years. Despite these advances, carbapenem-resistant Enterobacteriaceae12–15 and difficult to treat microorganisms such as P. aeruginosa and A. baumannii produce a variety of β-lactamases and exhibit other resistance mechanisms that continue to challenge existing antibiotic treatments.9,16,17 The aim of his review is to provide an update on the β-lactam/β-lactamase inhibitor (BL/BLI) combinations being marketed or under clinical development. It is then followed by an overview of the research articles and patents published on the topic of small-molecule inhibitors of β-lactamases with particular attention paid to progress made in the past five years.
2. Recent FDA-approved BL/BLI combinations
2.1. Vabomere® (meropenem + vaborbactam)
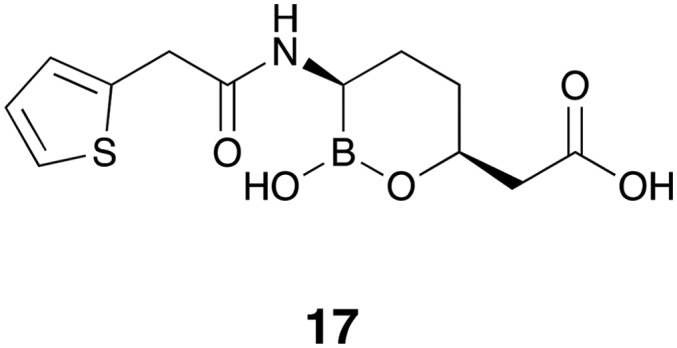
Vaborbactam 17 (formerly known as RPX7009, Fig. 4) is the first FDA-approved β-lactamase inhibitor containing a cyclic boronate pharmacophore.18–21 The design of vaborbactam is the result of medicinal chemistry efforts to develop a cyclic boronate analog with selectivity towards bacterial β-lactamases over mammalian serine hydrolases. X-ray crystallography studies confirmed that vaborbactam forms a covalent adduct with the catalytic serine residue of CTX-M-15 and AmpC. In addition, vaborbactam inhibited various Class A/C β-lactamases with sub-μM IC50 values.22 The combination of vaborbactam and meropenem 5 was tested against more than 300 Enterobacteriaceae clinical isolates, the majority of which carried KPC genes. A fixed vaborbactam concentration of 8 μg mL–1 potentiated the activity of meropenem 5 by at least 64-fold leading to MIC50 and MIC90 values of ≤0.06 and 1 μg mL–1 respectively.23 A follow-up study on a larger number of non-fastidious Gram-negative bacteria collected worldwide confirmed the potent activity of meropenem-vaborbactam against KPC-producing Enterobacteriaceae (MIC50 and MIC90 values of 0.12 and 0.5 μg mL–1 respectively), however vaborbactam did not reduce the MIC of meropenem 5 against bacterial strains expressing MBLs (Ambler class B) or OXA-48 (Ambler class D).24 In a complimentary study, Lomovskaya and co-workers used a panel of engineered E. coli strains producing β-lactamases of all four Ambler classes to assess ability of vaborbactam to potentiate a number of antibiotics.25 Since most of the strains producing β-lactamases of Ambler class A and C are already susceptible to meropenem 5, adding ceftazidime 6 and aztreonam 7 to their panel allowed them to fully characterize the inhibition spectrum of combinations with vaborbactam. Their findings reveal a broad spectrum synergistic effect against Escherichia coli strains producing β-lactamases of Ambler class A (KPC, SME, NMC, SHV, TEM, CTX) and class C (DHA, MIR, FOX, AmpC-ECL, CMY) when 4 μg mL–1 vaborbactam is added to meropenem 5, ceftazidime 6, or aztreonam 7. In line with studies employing clinical isolates, vaborbactam did not decrease the MIC of β-lactams against engineered E. coli strains producing class B (NDM-1, VIM-1) or class D (OXA) enzymes.25 In addition to strong in vitro activity, meropenem-vaborbactam exhibited promising results in clinical trials which indicated the safety, tolerability, and efficacy of the combination.26,27 In a randomized clinical trial meropenem-vaborbactam along with its comparator drug combination (piperacillin-tazobactam) were evaluated for the treatment of complicated urinary tract infection. Meropenem-vaborbactam was well tolerated by patients and proved to be non-inferior to the comparator therapy.27 Vaborbactam in combination with meropenem (Vabomere®) was approved by FDA in 2017 for treating complicated urinary tract infections and is marketed by Melinta therapeutics as an injectable solution with each vial containing 1 g of meropenem and 1 g of vaborbactam.28,29 At present other vaborbactam-antibiotic combinations are under clinical evaluation.
Fig. 4. Vaborbactam 17.
2.2. Avycaz® (ceftazidime + avibactam)
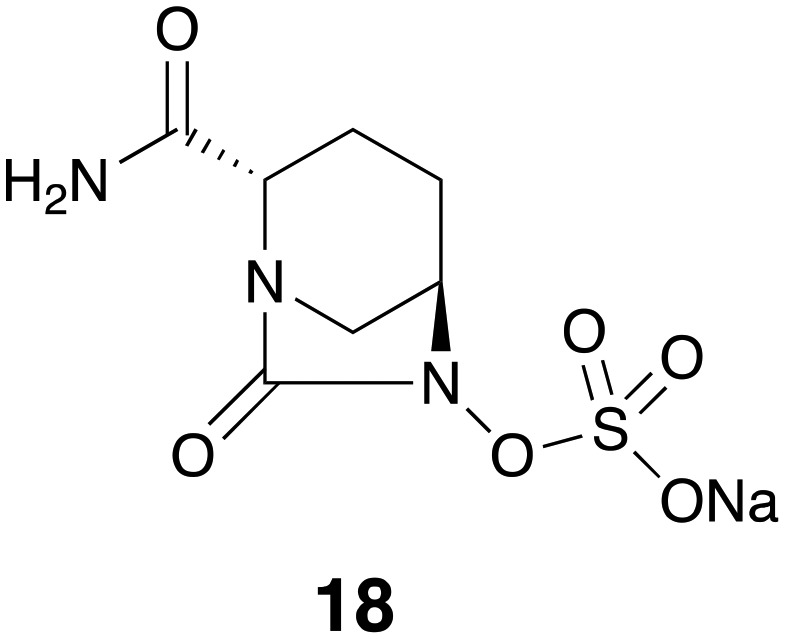
The avibactam/ceftazidime combination marketed as Avycaz was granted FDA-approval in 2015 for the treatment of complicated intra-abdominal infection (cIAI) and complicated urinary tract infection (cUTI). Structurally, avibactam 18 (formerly NXL104, Fig. 5) is a first-in-class SBL inhibitor with a cyclic urea replacing the β-lactam pharmacophore present in the older generation of β-lactamase inhibitors.30 Using a variety of biophysical techniques including UV spectroscopy, MS, and NMR, Ehmann and co-workers31 found that avibactam employs a mechanism based on covalent inhibition of TEM-1 with slow regeneration of the inhibitor. This covalent acylation with reversible deacylation through recyclization is unique to avibactam among β-lactamase inhibitors. When avibactam was tested against a larger panel of β-lactamases including TEM-1, CTX-M-15, KPC-2 (class A), Enterobacter cloacae P99 AmpC, P. aeruginosa PAO1 AmpC (class C), OXA-10 and OXA-48 (class D), it was confirmed that acylation of enzymes followed by slow release of inhibitor through cyclization could be considered as a general mechanism of inhibition by avibactam.32 In the case of KPC-2 inhibition however, it was found that recyclization competes with desulfation of avibactam followed by further degradation steps.32 Studies of avibactam in complex with class A and class C β-lactamases using X-ray crystallography suggest the stability of carbamate bond upon avibactam addition and the substrate-like conformation of the enzyme bound avibactam as the explanations for the favorability of recyclization over hydrolytic cleavage.33–35 There are multiple reports on the in vitro activity of avibactam combined with cephalosporins, carbapenems, and monobactams against both Gram-negative and Gram-positive bacterial pathogens. When tested against 126 P. aeruginosa clinical isolates, avibactam at 4 μg mL–1 reduced the MIC90 of ceftazidime 6 from 64 to 8 μg mL–1, superior to the effect of clavulanic acid 14 and tazobactam which led to no change and two-fold reduction of MIC90 respectively. Avibactam also potentiated imipenem 8 with an MIC90 reduction of 16 to 2 μg mL–1.36 The combination of avibactam with ceftaroline 9 inhibited Enterobacteriaceae strains containing multiple β-lactamases of class A and C. In addition, avibactam did not appear to adversely affect the activity of ceftaroline 9 against methicillin-resistant Staphylococcus aureus (MRSA) strains. The avibactam/ceftaroline 9 combination however showed little activity against Acinetobacter spp. and P. aeruginosa strains containing OXA (class D) enzymes or MBL-producing strains.37 Another study found the same trend of limited potency of ceftazidime-avibactam combination against A. baumannii strains producing PER-1, OXA-51 and OXA-58, while promising activity was observed against Klebsiella pneumoniae strains producing CTX-M-15 or OXA-48 and E. coli strains producing CTX-M-15.38 Susceptibility screening of 701 Enterobacteriaceae isolates with positive ESBL-phenotype collected from U.S. hospitals showed potent activity of ceftazidime-avibactam as well as tigecycline.39 Another published screening of 8640 Enterobacteriaceae collected from U.S. medical centers found the similar results with ceftazidime-avibactam, although the combination showed limited activity against Acinetobacter spp. isolates and MBL-producers.40 Avibactam restored the activity of ceftazidime 6 against isolates producing KPC, CTX-M-15-like, CTX-M-14-like and SHV ESBLs and CMY-2-like enzymes (MIC90 ≤ 2 μg mL–1 in all the cases).39 Wang and co-workers performed a series of in vitro assays with avibactam combined with ceftazidime 6 or aztreonam 7 revealing similar trends.41 The same study also found that avibactam resensitized Enterobacteriaceae isolates producing Ambler class A and C to ceftazidime 6 and aztreonam 7.41 Combining avibactam with aztreonam 7 appears to be an appealing strategy to extend the activity to MBL-producers, since aztreonam 7 is a poor substrate for MBLs.3,42,43 Wang and co-workers found that unlike ceftazidime-avibactam, aztreonam-avibactam did retain potency against the isolates co-producing IMP or NDM.41 Based on these findings and further in vitro susceptibility screenings44–46 it can be concluded that avibactam greatly potentiates ceftazidime 6 against bacterial pathogens producing class A, C, and some class D β-lactamases and outperforms older generation β-lactamase inhibitors such as clavulanic acid 14 and tazobactam. The ceftazidime-avibactam combination does however exhibit a higher range of MICs against P. aeruginosa strains and poor activity against Acinetobacter spp. and MBL-producer strains.37,38,40,47 Overproduction of efflux pumps and reduced outer membrane permeability has been suggested to be responsible for ceftazidime-avibactam resistance in P. aeruginosa isolates.48 Ceftazidime-avibactam has also been evaluated in a number of clinical trials for the treatment of complicated urinary tract infections (cUTI) and complicated intra-abdominal infections (cIAI). The published data indicate that overall the combination is well-tolerated by patients and noninferiority to its comparator drugs such as imipenem-cilastatin, meropenem 5 and doripenem was demonstrated.49–52 Avycaz® is manufactured and marketed by Allergan as a powder for injection containing a 4 : 1 ratio of ceftazidime 6 to avibactam based on dry weight.53 Clinical trials are ongoing to evaluate the efficacy of avibactam in combination with other β-lactam partners including ceftaroline 9 and aztreonam 7 for a number of other indications (; ClinicalTrials.gov identifiers: NCT01624246, NCT01281462, NCT01689207 and NCT03329092).
Fig. 5. Avibactam 18.
2.3. Zerbaxa® (ceftolozane + tazobactam)
Zerbaxa® received FDA approval in 2014 for the treatment of cIAI and cUTI. The drug consists of the novel fifth-generation cephalosporin antibiotic ceftolozane 10 (Fig. 2) and the established β-lactamase inhibitor tazobactam. Considering that this BL/BLI combination has been the focus of a number of detailed reviews,54–60 here only the structural features of ceftolozane 10 as well as an overview of the antibacterial spectrum of its combination with tazobactam, including key outcomes of clinical trials, is covered. Ceftolozane 10 was evolved as the result of a medicinal chemistry efforts aimed at developing a cephalosporin with improved potency against AmpC-producing P. aeruginosa strains.61–63 This was achieved by a series of structural modifications of the substituents at C3 and C7 position of the cephalosporin core. On C-7 position, in addition to the thiadiazole ring and oxyimino moiety, which are believed to be responsible for the extended spectrum of anti-Gram-negative activity and resistance to some β-lactamases,64 ceftolozane 10 also contains a dimethylacetic acid moiety which increases affinity to some PBPs, especially PBP3. After evaluating a number of protomolecules, it was eventually established that placement of a pyrazolium ring containing a basic side chain improves permeability, stability against Pseudomonal AmpC, and minimizes off target effects associated with the positively charged moiety.61,62 To determine to what extent the activity of ceftolozane 10, then known as FR264205, was affected by major resistance mechanisms of P. aeruginosa, it was assayed against variants producing AmpC, overexpressing efflux pumps, and lacking OprD. These studies revealed that ceftolozane 10 showed superior performance to ceftazidime 6 against all the resistant mutants and its activity was not affected by efflux pump overexpression and OprD loss.63 The inhibitory activity of tazobactam on the other hand, is highest against class A β-lactamases such as TEM, SHV, CTX-M enzymes.3 In doing so this inhibitor extends the activity spectrum of ceftolozane 10 against ESBL-producing Gram-negative bacteria. Indeed, when tazobactam was combined with ceftolozane 10, it strongly enhanced the activity of ceftolozane 10 against ESBL-producer and AmpC-hyperproducing Gram-negative bacteria in a concentration-dependent manner. Notably, strains producing KPC were not susceptible to the combination.65 Farrel and co-workers reported the screening results of 7071 Enterobacteriaceae strains isolated from U.S. hospitals. Overall, ceftolozane-tazobactam (TOL-TAZ) showed potent activity with an MIC90 of 1 μg mL–1 making it equipotent to cefepime and tigecycline. Also noteworthy was the performance of the ceftolozane-tazobactam combination against E. coli isolates with an ESBL phenotype (MIC90 = 4 μg mL–1) as well as 1971 tested P. aeruginosa isolates (MIC90 = 2 μg mL–1) showing it to be superior to combinations of ceftazidime 6 or piperacillin 4 with tazobactam (MIC90 = 32 and >64 μg mL–1 respectively).20 These findings were in agreement with the screening results against 2435 P. aeruginosa strains isolated from patients in Canadian hospitals.66 The MIC90 of 1 μg mL–1 for TOL-TAZ was found to be superior to those of colistin (MIC90 = 2 μg mL–1) and meropenem (MIC90 = 8 μg mL–1) among the panel of tested antibiotics.66 Tazobactam also potentiates the activity of ceftolozane 10 against anaerobes. Using a collection of 605 Gram-negative and Gram-positive anaerobic isolates, Snydman and co-workers observed high activity for TOL-TAZ against Bacteroides spp. specially Bacteroides fragilis (MIC90 = 4 μg mL–1) and excellent activity against Gram-negative anaerobes Prevotella spp. and Fusobacterium spp. (MIC90 ≤ 0.125 μg mL–1).67 The same study also revealed that ceftolozane-tazobactam has very little activity against Clostridium spp. Based on the results described above, TOL-TAZ can be viewed as a new carbapenem-sparing therapeutic option when facing clinically important pathogens such as ESBL-producing Enterobacteriaceae and P. aeruginosa including AmpC-hyperproducers. However, the antibiotic activity of the combination is expected to be compromised by pathogens expressing highly active carbapenemases and/or MBLs. In this regard a recent the phase III clinical trial named ASPECT-cIAI evaluated TOL-TAZ plus metronidazole in patients with complicated intra-abdominal infections (cIAI).68 The combination showed efficacy against infections with Enterobacteriacea producing CTX-M-type ESBLs and proved to be non-inferior to meropenem 5 as the comparator drug. For the treatment of cUTI including pyelonephritis, another phase III clinical trial known as ASPECT-cUTI was conducted to compare the efficacy of TOL-TAZ with that of levofloxacin. Overall, TOL-TAZ proved to be non-inferior to levofloxacin and adverse events were moderate.69 Zerbaxa® is manufactured by Merck as powder for injection comprised of a 2 : 1 (by weight) mixture of ceftolozane 10 and tazobactam.70
3. SBL inhibitors: recent and ongoing developments
Summarized in Table 1 are the drug candidates currently being evaluated in clinical trials spanning the past 10 years. These SBLIs can be structurally classified into β-lactams and non-β-lactams. BLIs with β-lactam structure are represented by the classic inhibitors such as clavulanic acid 14, sulbactam 15, and tazobactam 16. Recently, a structurally similar analog of tazobactam known as AAI101 successfully completed a phase II clinical trial (EudraCT Number in EU clinical trials register: 2016-005161-31). Efforts to discover BLIs among novel scaffolds have also resulted in two important new classes of SBLIs include the diazabicyclooctanes (represented by avibactam) and cyclic boronates (represented by vaborbactam). The following section covers these new SBLIs classes and their current state of clinical development.
Table 1. BLIs currently in the clinical development stage.
| Name/code | Chemical class | Clinical development phase |
| Nacubactam | Diazabicyclooctane | Phase I in combination with meropenem |
| Zidebactam | Diazabicyclooctane | Phase I in combination with cefepime |
| ETX2514 | Diazabicyclooctane | Phase II in combination with sulbactam |
| Relebactam | Diazabicyclooctane | Phase III in combination with imipenem/cilastatin |
| Avibactam | Diazabicyclooctane | Approved in combination with ceftazidime |
| Phase II in combination with ceftaroline fosamil | ||
| Phase I in combination with aztreonam | ||
| Vaborbactam | Cyclic boronate | Approved in combination with meropenem |
| Phase I in combination with biapenem | ||
| AAI101 | Penam sulfone | Phase II in combination with cefepime |
3.1. β-lactams
As far as can be gleaned from published reports, AAI101 (19, Fig. 6) is being evaluated in clinical trials as a combination with the fourth-generation cephalosporin cefepime (EudraCT Number in EU clinical trials register: 2016-005161-31). The results of MIC screening using cefepime 11 and various concentrations of AAI101 showed a concentration-dependent synergistic effect against K. pneumoniae and E. coli strains with carbapenem-resistance phenotypes.71 Another study found high activity for the combination particularly against ESBL-producing Enterobacteriaceae (MIC50/90 = 0.125/0.5 μg mL–1).72
Fig. 6. SBL inhibitor penam sulfones AAI101 (19) and LN-1-255 (20).

LN-1-255 (20, Fig. 6) is a penicillin sulfone inhibitor which has been reported to inhibit multiple class of SBLs.73 Pattanaik and co-workers reported strong inhibition of SHV-1 and SHV-2 (class A) by LN-1-255 and potentiation of ceftazidime 6 against strains producing TEM, SHV, CTX-M and Sme-1 enzymes.74 Crystallographic data obtained for SHV-1 suggests that LN-1-255 acylates the enzyme followed by rearrangement to a bicyclic indolizine adduct.74 Also interesting was the potent activity of this inhibitor against multiple enzymes of OXA family and its ability to reduce the MIC of carbapenems against OXA-producing E. coli, K. pneumoniae and A. baumannii strains.75–77
3.2. Diazabicyclooctanes
Relebactam
As recently summarized by Zhanel and co-workers,78 the diazabicyclooctane (DBO) analogue relebactam 21 (Fig. 7) has a spectrum of β-lactamase inhibition similar to that of the preeminent DBO-based SBL inhibitor avibactam. Relebactam is active against β-lactamases of Ambler class A including KPC and class C. Again as observed with avibactam, metallo-β-lactamases of class B and OXA-type enzymes of class D are not affected by relebactam.78 This inhibition spectrum is well reflected in the results of susceptibility screenings using a combination of relebactam and imipenem 8. Used at 4 μg mL–1, relebactam potentiated imipenem 8 against Gram-negative clinical isolates.79 While MIC50/90 against E. coli strains were retained at 0.25/0.25 μg mL–1 upon addition of relebactam, the combination was effectively synergistic against K. pneumoniae, Enterobacter spp., and P. aeruginosa isolates with MIC90/50 reduced to 0.25/0.25 μg mL–1, 0.25/0.5 μg mL–1, and 0.5/2 μg mL–1 respectively. Relebactam also successfully reduced the MIC90/50 of KPC-producing K. pneumoniae and imipenem-resistant P. aeruginosa isolates from 16/>16 μg mL–1 and 8/>16 μg mL–1 to 0.25/1 μg mL–1 and 1/2 μg mL–1 respectively. However, the combination was not active against A. baumannii strains producing OXA-23.79 Further screenings of Gram-negative pathogens collected in U.S. and European hospitals confirmed that A. baumannii, along with other organisms that produce MBLs or OXA-type enzymes are likely to present a challenge in the use of imipenem-relebactam.80,81 The in vitro performance of imipenem-relebactam was also evaluated against anaerobic Gram-negatives of Bacteroides group. Among the tested panel of antibiotics, imipenem 8 was found to be most potent with an MIC90 of ≤1 μg mL–1 against all the Bacteroides species. However, addition of relebactam did not lead to a further improvement in the activity of imipenem 8.82 Similarly, the combination showed excellent activity against Gram-positive anaerobes although overall it did not outperform imipenem 8 alone.83 Phase II studies were conducted in which imipenem-cilastatin plus relebactam or placebo were administered to patients with cIAI84 and cUTI.85 Both studies proved non-inferiority of relebactam combination with similar adverse effects profile to the placebo group. Phase III clinical evaluation of relebactam in combination with imipenem-cilastatin is currently ongoing (; ClinicalTrials.gov identifier: NCT03293485).
Fig. 7. Diazabicyclooctanes in clinical development: relebactam 21, zidebactam 22, nacubactam 23, ETX2514 24.

Zidebactam
The acyl hydrazide DBO analogue of the DBO family, zidebactam 22 (Fig. 7) belongs to the newest generation of DBO-based SBLIs with potent PBP inhibitory activity. Although not an inhibitor of class D β-lactamases,86 zidebactam selectively inhibited P. aeruginosa PAO1 PBP2 enzyme. A combination of zidebactam and cefepime 11 effectively inhibited growth of the P. aeruginosa PAO1 strain and its knock-outs with defective porins.87 Also interesting was the increased activity of the combination of zidebactam with selected β-lactams against VIM-1/VIM-2-producing P. aeruginosa clones. The most potent activities were observed when the monobactam agent aztreonam 7 was used as β-lactam partner.87 Likewise, an enzymatic study focusing on A. baumannii showed strong and selective inhibition of A. baumannii PBP2 by zidebactam, while no inhibition was observed against OXA-23. Interestingly, in antibacterial assays, 8 μg mL–1 of zidebactam was found to reduce the MIC of cefepime 11 and sulbactam 15 against OXA-23 producing A. baumannii to 16 μg mL–1 (4-fold reduction) and 2 μg mL–1 (8-fold reduction) respectively. The enhancing effect in this case could be attributed to the contribution of zidebactam to PBP (and not β-lactamase) inhibition.88 Zidebactam in combination with cefepime 11 showed excellent in vitro inhibition when evaluated against 7876 Gram-negative clinical isolates collected worldwide.89 Overall, the 1 : 1 combination effectively inhibited Enterobacteriaceae isolates with an MIC90 of 0.12 μg mL–1 compared with 16 μg mL–1 when cefepime 11 was tested alone. The combination also largely enhanced the potency of cefepime 11 by at least 16-fold against clinically important sub-classes including carbapenem-resistant Enterobacteriaceae, ESBL phenotype E. coli, and ESBL phenotype Klebsiella spp. Zidebactam reduced the MIC90 of cefepime 11 from 32 to 4 μg mL–1 against P. aeruginosa and from >64 to 32 μg mL–1 against Acinetobacter spp.89 Another study demonstrated the strong antibacterial activity of a 1 : 1 mixture of cefepime-zidebactam against a number of Enterobacteriaceae expressing various β-lactamases including: CTX-M-15 (MIC90 = 1 μg mL–1), SHV (MIC90 = 0.25 μg mL–1), ESBLs (MIC90 = 1 μg mL–1), plasmid AmpC (MIC90 ≤0.06 μg mL–1), derepressed AmpC (MIC90 = 0.5 μg mL–1), KPC (MIC90 = 1 μg mL–1) and MBLs (MIC90 = 8 μg mL–1). The inhibitory activity of the same combination had only moderate activity against P. aeruginosa and A. baumannii isolates.90 Currently, two phase I clinical trials evaluating the safety, tolerability, and pharmacokitenics of zidebactam have been completed with a third study currently recruiting patients (; ClinicalTrials.gov identifiers: NCT02674347, NCT02707107 and NCT02942810).
Nacubactam
Also known as OP0595, nacubactam 23 (Fig. 7) is an aminoethoxy-substituted analogue of avibactam which inhibits class A/C β-lactamase and PBP2. Nitrocefin-based enzyme assays showed inhibition of TEM, CTX-M, KPC-2 (class A), AmpC and CMY-2 (class C) by nacubactam with sub-μM IC50 values. This inhibitor showed relatively weak activity against OXA enzymes and none against IMP-1. Similar to zidebactam, nacubactam selectively inhibited PBP2 (IC50 = 0.12 μg mL–1) and upon incubation with E. coli, it induced the formation of spherical cells which is an expected result of PBP2 inhibition.91 Interestingly, nacubactam has been reported to possess antibacterial activity when tested alone.92–94 A recent study found that when administered at ≤4 μg mL–1, nacubactam inhibited most of the E. coli, Enterobacter spp., Citrobacter spp., and Klebsiella spp. strains tested although it had a poor performance against Serratia spp., P. aeruginosa and A. baumannii.94 Against those strains with an MIC > 4 μg mL–1, nacubactam strongly enhanced the activity of aztreonam 7, cefepime 11, biapenem 12 and piperacillin 4 in a concentration-dependent manner. In addition, the activity of nacubactam combined with the above-mentioned antibiotics against Enterobacteriaceae producing carbapenemases (KPC, OXA-48 and MBLs) was significant and superior to that of ceftazidime-avibactam. However, nacubactam did not potentiate the same antibiotics when tested against A. baumannii strains and MBL-producing P. aeruginosa.94 Since in vitro studies of β-lactamase inhibition by nacubactam is complicated due to its inherent antibacterial activity, Livermore and co-workers95 prepared nacubactam-resistant Enterobacteriaceae mutants with elevated MIC values of 8 to >32 μg mL–1. When nacubactam was tested against these mutants producing ESBLs, KPC, and OXA enzymes, use of 2 μg mL–1 of nacubactam, greatly enhanced the activity of piperacillin 4, cefepime 11, and aztreonam 7 leading to mean MIC values of <1 μg mL–1 for these three β-lactamase families. A similar reduction of mean MIC (From 8.43 μg mL–1 to <1 μg mL–1) was observed when nacubactam was combined with meropenem 5 and assayed against KPC-producing mutants. Also interesting was the finding that nacubactam at 1 μg mL–1 reduced the mean MIC of aztreonam 7 against MBL-producing mutants from 4.68 to 0.072 μg mL–1. Taken together the study suggests that the synergy observed by nacubactam is not limited to its PBP2 inhibition but also its inhibition of class A/C β-lactamase. In addition, nacubactam in combination with aztreonam 7 might provide a viable therapeutic option against MBL-producing Gram-negative pathogens.95 To date, two clinical trials evaluating safety, pharmacokinetics, and intrapulmonary lung penetration of nacubactam have been completed (; ClinicalTrials.gov identifiers: NCT02134834 and NCT03182504).
ETX2514
Another recently described DBO-based β-lactamase inhibitor known as ETX2514 (24, Fig. 7) has demonstrated a very broad spectrum of activity including inhibition of class A/C/D β-lactamases and PBP2.96,97 In preparing ETX2514 Durand-Réville and co-workers96 modified avibactam with the aim of introducing activity against a broader panel of OXA enzymes known to complicate the treatment of resistant A. baumannii isolates. Introduction of an endocyclic double bond was implemented to increase chemical reactivity of the ring and addition a of methyl group at C-3 (Fig. 7) led to ETX2514 which displayed a potent inhibitory activity against OXA-24 (IC50 = 0.19 μg mL–1) along with enhanced biochemical and antibacterial activity. This finding was supported by X-ray crystallography data and molecular modeling of ETX2514 and avibactam which revealed the mode of binding to OXA-24.96 Another interesting finding was the inhibitory activity of ETX2514 against PBPs with preference to PBP2 of E. coli and A. baumannii. Use of 4 μg mL–1 of this inhibitor, decreased the MIC90 of imipenem 8 by 8-fold to 2 μg mL–1, while its combination with sulbactam 15 most effectively inhibited growth of A. baumannii reducing the MIC90 of sulbactam 15 from 64 μg mL–1 to 4 μg mL–1. The intrinsic activity of sulbactam 15 against PBP3 plus the dual BL/PBP inhibition by ETX2514 may explain the excellent activity of their combination against A. baumannii a challenging nosocomial pathogen that is often multi-drug resistant.96 A follow-up study showed that similar to avibactam, ETX2514 acylates β-lactamases of class A, C and D.98 Mass-spectrometry analysis of the resulting enzyme-inhibitor complexes suggested that ETX2514 can recyclize and is released in intact form when incubated with AmpC, CTX-M-15, P99, SHV-5 and TEM-1. On the other hand, interaction with KPC-2, OXA-10, OXA-23, OXA-24 and OXA-48 was accompanied by desulfation and irreversible degradation of the inhibitor. A combination of ETX2514 with imipenem 8 and piperacillin 4 was highly active against isogenic P. aeruginosa producing class A, C and D β-lactamases. Compared to avibactam, ETX2514 displayed superior and broader spectrum of activity specially against OXA family of enzymes.98 Additionally, Iyer and co-workers demonstrated that ETX2514 uses the outer membrane porin OmpAAb to permeate the A. baumannii membrane and synergize with sulbactam 15.99 ETX2514 in combination with sulbactam 15 is currently undergoing a phase II clinical trial for the patients with cUTI including acute pyelonephritis (; ClinicalTrials.gov identifier: NCT03445195).
Review of the recent patent literature reveals a number of other functionalized DBO analogs with SBL inhibitory activity (Fig. 8). Chang and co-workers reported isoxazoline analogs 25 among which compound 26 was particularly active, reducing the MIC of meropenem 5 against K. pneumoniae strains producing class A/C/D enzymes by up to 1024-fold.100 Gu and co-workers reported another group of oxadiazole-substituted analogs 27 as SBL inhibitors.101 Hydroxamate and hydrazide analogs 28-30 were reported by Maiti and co-workers, as exhibiting potent inhibition of class A and C enzymes with <19 nM IC50 values. Of note, compound 30 not only demonstrated high intrinsic antibacterial activity but also when combined with meropenem 5 inhibited E. coli and K. pneumoniae strains expressing several β-lactamases of class A, B and C.102 Also noteworthy is the report by Comita-prevoir and co-workers of a large library of DBO analogs closely related to ETX2514 wherein the sulfate moiety is replaced by functionalized glycolates.103 Several compounds with the general structure of 31 demonstrated potent inhibition of TEM-1, AmpC, and OXA-48. These analogues also synergized with cefpodoxime 13 against Citrobacter freundii, E. coli, and K. pneumoniae strains producing multiple β-lactamases of class A, C and D, and showed in vivo efficacy in mouse models of infection.103
Fig. 8. DBO analogs as β-lactamase inhibitors reported in the recent patents.

3.3. Boronates
Boronate-based β-lactamase inhibitors have long been of interest given to their resemblance to the tetrahedral intermediate formed upon β-lactam ring attacked by nucleophilic serine of β-lactamases.104 For this reason these BLIs are sometimes referred to as boronic acid transition-state inhibitors (BATSIs).105Fig. 9 shows the chemical structures of a number of such boronates that have been investigated for SBL inhibition including acyclic boronic acids (represented by 32–34)106–108 or cyclic boronate analogs (represented by 35, 37 and 38).109–111 Of particular note are recent studies aiming at developing cyclic boronates as pan-β-lactamase inhibitors, the rationale being that both MBL- and SBL-mediated hydrolysis of β-lactams involve a tetrahedral transition state that precedes ring opening. Therefore, structures mimicking the transition state have the potential to exert cross-class β-lactamase inhibition (Fig. 10). Validation of this idea is found in the structural diversity of boronates contained in a number of patent applications claiming both SBL and MBL inhibition. Of note are the acyclic boronic acids represented by 36 which show inhibition of some SBLs and VIM-2 enzyme of class B112 as well as the cyclic boronates 37 (ref. 113 and 114) and 38 (ref. 115–118) (Fig. 9) which display sub-μM IC50 values for both SBLs and MBLs. By screening a series of cyclic boronates, Brem and co-workers identified a series of SBL-inhibitor analogs with potent activity against MBLs, specifically VIM-2 and NDM-1.110 Interestingly, 38b was found to exert potent inhibition of PBP-5. X-ray crystallography studies with 38b on BcII, VIM-2, OXA-10, and PBP-5 confirmed that the cyclic boronate structure interacts with the crucial β-lactamase residues (and coordinates with Zn2+ of MBLs) in the way that mimics the high energy transition state intermediates formed in each case. In addition, 38b largely enhanced the activity of meropenem 5 towards Enterobacteriaceae expressing multiple β-lactamases.110 A follow-up study confirmed nM range IC50s for the activity of cyclic boronate analogs against TEM-1, CTX-M-15, and AmpC. Compound 38b exhibited a synergistic relationship with carbapenems against Enterobacteriaceae producing multiple β-lactamases including KPC-2, OXA-181 (meropenem only), VIM-1, and VIM-4. However, carbapenemase-producing P. aeruginosa and A. baumannii strains remained resistant to all combinations.111
Fig. 9. Representative boronic acids as β-lactamase inhibitors.

Fig. 10. A. Tetrahedral intermediate formed by nucleophilic attack of SBLs (Nu: = serine-OH) and MBLs (Nu: is zinc-coordinated OH–) on β-lactam ring. B. Cyclic boronates mimicking the tetrahedral transition state of β-lactam hydrolysis.110,111 .

4. Recent advances in the development of MBL inhibitors
Based on their catalytic activities, β-lactamases are classified as serine β-lactamases (SBLs, Ambler class A, C and D) and metallo-β-lactamases (MBLs, Ambler class B). The latter contains zinc ion(s) in the active site which is stabilized by histidine, cysteine and aspartate residues and is also bound to an active water molecule responsible for hydrolyzing β-lactams. MBLs in turn are divided into subclasses B1, B2, and B3. While enzymes of class B1 and B3 contain two zinc ions, B2 functions with only one.5,119 The most clinically relevant MBLs include NDM, VIM, and IMP enzymes of class B1 which inactivate a broad range of β-lactams but have a low affinity for monobactams.120 Due to their carbapenemase activity and rapid dissemination, MBLs pose a serious challenge to the antibiotic treatment of infections caused by Gram-negative bacteria. The design and development of broad-spectrum MBL inhibitors is challenged by the high active site heterogenicity of the different enzymes of this family.3,8,121,122 As a result, to date, there are no effective MBL inhibitors currently in clinical use.
Compounds classes with the potential to inhibit MBLs have been the subject of several detailed reviews.5,120–125 In order to avoid redundancy and to build upon previous reviews, we here focus on new developments in the MBL inhibitors field over the past five years.
Traditionally, sulfur-containing compounds have been one of the most studied classes of small molecules in the search for MBL-inhibitors. Compounds containing a variety of free thiols, thioethers, thioesters, thioketones, and thioureas have been recently reported to possess inhibitory activity against different class of MBLs.121 Also of note are thiol-containing drugs that while approved for other indications have shown some capacity to inhibit MBLs. In this regard Klingler and co-workers found that thiorphan (39, Fig. 11), the active metabolite of the antidiarrheal racecadotril, inhibits NDM-1, IMP-7, and VIM-1 with low-μM IC50 values and also markedly enhances the activity of imipenem 8 against MBL-producing strains.126 In addition, captopril 40 an FDA-approved drug used for the treatment of hypertension, has also received some attention for its ability to inhibit NDM-1 (IC50 = 7.9 μM).127 Building upon these findings efforts have been made to replace the prolyl residue of captopril with various other functional groups,127–130 as well as modification of the thiolated acyl residue, and/or ring size.129,131 Brem and co-workers also found the MBL inhibition of d-captopril to be superior to that of its other stereoisomers when evaluated against BCII, IMP-1, VIM-2, NDM-1, and SPM-1.132 These findings were further supported and could be rationalized by X-ray crystallography studies reported in the same paper.132
Fig. 11. Thiol-containing MBL inhibitors, thiorphan 39, captopril 40 and substituted mercaptoacetamides 41.

It has long been known that mercaptoacetic acid and its related structural analogs are among the potent MBL-inhibitors.121 Recent reports have described the development of aminoacid thioesters of mercaptoacetic acid as inhibitors of L1, an MBL of the B3 class.133,134 Substituted amide derivatives of mercaptoacetic acid (mercaptoacetamides, 41) are also prominent in a number of recent studies: Arjomandi and co-workers reported a series of amino acid conjugates of mercaptoacetamide and some longer chain homologs (mercaptpropionamide and mercaptobutyramide) which display IMP-1 inibition.135 Other studies employed mercaptoacetamide thioethers containing acetate136 and azolyl ring137–140 substituents. The diverse library of thiol-containing MBL-inhibitors also include thiomethylbenzoic acids,141 bisthiazolidines,142,143 rhodanines and its related thioenolates,144–147 cysteine-containing oligopeptides,148,149 phosphonate and tetrazole bioisosters of mercaptoacids150 and thiones.151–154 Finally, it should be added that although thiols are among the most potent and broad-spectrum inhibitors of MBLs, their tendency to rapidly oxidize to disulfides poses a serious challenge to further clinical developments. This is important since studies suggest that upon disulfide formation zinc-binding affinity is greatly reduced leading to a loss of MBL-inhibition and in vitro synergistic activity.155,156 Creative chemical modifications to enhance the biological stability of thiol-based inhibitors may be the key to develop such compounds as clinically viable drug candidates.
Picolinic acid derivatives are another well-known class of zinc chelators and act via the same metal-sequestration mechanism as EDTA to inhibit MBLs.157 In fact pyridine-2,6-dicarboxylic acid also known as dipicolinic acid or DPA (42, Fig. 12) is a commonly used reagent for the phenotypic detection of MBL-producing pathogens.158–161 By evaluating a series of DPA analogues– represented by compound 43 – Chen and co-workers identified compounds with enhanced NDM-1 inhibition that retained MBL-selectivity over other zinc-dependent metalloenzymes.162 Compound 43 inhibited NDM-1, VIM-2, and IMP-1 with IC50 values of 0.080, 0.21 and 0.24 μM respectively and demonstrated synergistic relationship with imipenem 8 when tested in vitro against NDM-1 producing E. coli and K. pneumoniae isolates. Also of note are the results of various experiments including NMR and equilibrium dialysis suggesting that compound 43 engages in a ternary complex with zinc and NDM-1 unlike its parent compound DPA and EDTA.162 In a complimentary study, Hinchliffe and co-workers investigated the potential of phosphonate analogs of 2-picolinic acid to inhibit MBLs of B1 and B3 sub-class. They found potent and broad-spectrum inhibition of NDM-1, VIM-2, IMP-1, and L1 by compounds 44-46. Compound 44 reduced the MIC of meropenem 5 down to 8 to <0.125 μg mL–1 against both recombinant and clinically isolated Gram-negative strains producing the earlier mentioned MBLs.163
Fig. 12. Pyridine derivatives as MBL-inhibitors.

Recently, Antabio Inc. reported the discovery of the sulfonamide small-molecule ANT431 (47) which was also evolved from 2-picolinic acid.164 After demonstrating strong inhibition of NDM-1 (Ki = 0.29 μM) and VIM-2 (Ki = 0.19 μM) and the potentiation of meropenem 5 against the BL21 E. coli producing the mentioned enzymes, ANT431 was tested against 94 MBL-producing clinical isolates of Enterobacteriaceae family. When used at 30 μg mL–1 this compound resensitized 72% of the isolates to meropenem 5. ANT431 also demonstrated in vivo efficacy in a mouse model of infection with NDM-1 producing E. coli and is currently being considered as a suitable starting point for further lead optimization.164
There are multiple reports on the MBL inhibitory activity of dicarboxylic acids.121 Guided by an X-ray crystallography study of compound 48 (Fig. 13) in complex with IMP-1, Hiraiwa and co-workers designed and synthesized di-substituted phthalic acids among which the bis(4-hydroxypiperidine) derivative 49 showed strongest inhibition towards IMP-1 (IC50 = 0.270 μM) and reduced the MIC of biapenem 12 against IMP-1 producing P. aeruginosa strains by at least 128-fold to ≤0.5 μg mL–1.165 Also recently described as MBL inhibitors are dicarboxylate substituted, five-membered heterocycles with 2,5-pyrrolidinedicarboxylic acid 50 identified as a potent competitive inhibitor of CcrA (Ki = 0.73 μM) and L1 (Ki = 0.69 μM).166 Notably, compound 50 reduced the MIC of cefazolin against CcrA and L1 producing E. coli strains to <1 μg mL–1 concentrations.166 Tetrazolylpropionic acids such as compound 51 have also been explored as bioisosters of dicarboxylates and reported to possess potent MBL activity with sub-μM IC50 values against NDM-1, IMP-1, and VIM-1.167
Fig. 13. Dicarboxylic acid analogs as MBL-inhibitors.

As described above, Zn2+ plays a vital role in the catalytic activity of MBLs and a variety of chelating agents have been shown to inhibit this class of enzymes and resensitize MBL-producing pathogens to β-lactam antibiotics. The MBL-inhibitory activity of aspergillomarasmine A (AMA) – a fungal metabolite with strong zinc chelating ability – was recently reported by King and co-workers.168 After screening a collection of fungal extracts using a phenotypic assay for synergy with meropenem 5, they isolated and characterized the active component, AMA (52, Fig. 14) and identified it as an inhibitor of NDM-1 (IC50 = 4.0 μM) and VIM-2 (IC50 = 9.6 μM). AMA greatly reduced the MIC of meropenem 5 to ≤2 μg mL–1 against Gram-negative strains producing NDM and VIM enzymes and demonstrated promising in vivo results in a mouse model of infection with NDM-1 producing K. pneumoniae.168 Soon after this report, multiple chemical169–172 and chemoenzymatic173 methodologies were developed to synthesize AMA and its closely related analogs. It was found that the diastereomers of AMA possessed similar activities against NDM-1 and VIM-2.170 The work by Bergstrom and co-workers174 shed light upon the action mechanism of AMA as it was shown by isothermal titration calorimetry that AMA strongly binds to Zn2+ (Kd = 200 nM). In addition, membrane dialysis and NMR experiments demonstrated that AMA inhibits NDM-1, VIM-2, and IMP-7 by stripping zinc from these enzymes.174
Fig. 14. Zinc chelators 52–57 and other unique compounds with MBL inhibitory activity.

The semicarbazide moiety is a well-known metal chelator and has been employed in the search for MBL inhibitors.175 As an example, compound 53 found in the recent patent literature exhibits strong inhibition of NDM-1 (IC50 = 35 nM).175 Other well established metal-chelators such as 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA, 54) and 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA, 55) and their analogs have also been described as MBL inhibitors with the ability to potentiate carbapenems against Gram-negative strains producing NDM, IMP or VIM enzymes.176,177 Similarly, the well-known zinc binder N,N,N′,N′-tetrakis-(2-pyridylmethyl)ethylenediamine (TPEN, 56) also been shown to synergize with β-lactam antibiotics to kill strains expressing various MBLs.178 While such chelating agents have also been described as nonhemolytic and nontoxic to mammalian cells in vitro, their potential to be advanced to clinical application should be viewed with caution due to their presumed lack of target specificity. In an attempt to address this problem, Yarlagadda and co-workers covalently linked the zinc binding motif dipicolylamine to vancomycin in an attempt to produce bacterial cell-specific hybrid. Given that vancomycin's inability to effectively kill Gram-negative pathogens is generally ascribed to its inability to penetrate the Gram-negative outer membrane, it is somewhat surprising that the vancomycin derivative 57 showed activity against strains expressing NDM-1 and restored the activity of meropenem 5 in both in vitro and in vivo experiments.179
Another class of MBL inhibitors based on the 3,7-substituted-indole-2-carboxylic acid scaffold was recently reported in the patent literature by Berm et al. who screened a large library of analogs for activity against VIM-2, IMP-1, and NDM-1.180 Several examples were found to possess sub-μM activities among which compounds 58 and 59 were found to be most potent against NDM-1 (IC50 values of 0.35 and 0.5 nM respectively). Researchers at Merck have also assessed numerous sulfonamides for MBL inhibitory activity and in a series of patents describe compounds such as 2-tetrazolylbenzenesulfonamides 60 (ref. 181 and 182) as potent inhibitors of IMP-1, VIM-1, and NDM-1. Using a related approach Fast and co-workers also found indoline-7-sulfonamides such as compound 61 to possess single-digit μM IC50 values against NDM-1.183
In addition to MBL inhibitors discovered by dedicated screening approaches, a range of other compounds have also been reported to possess anti-MBL activity including: the β-lactam antibiotic cefaclor,184 3-formylchromone,185 ebselen,186 as well as various hydrazones,187 phosphonic acids,188 oxoisoindolines,189 diphenylpyrroles,190 and bismuth complexes.191
5. Conclusions
In summary, the new generation of SBL inhibitors including avibactam and vaborbactam were significant breakthroughs in that they were developed from non-β-lactam structural backbones. While the activity spectrum of classic β-lactamase inhibitors was limited to non-carbapenemase enzymes of class A and some class C SBLs, avibactam and vaborbactam proved to be potent inhibitors of KPC carbapenemase as well as other class A/C enzymes. Building up the success of this compound class the advanced generation of DBO analogs has provided progress towards achieving broad spectrum SBL inhibitors with activity extending to the clinically important class D OXA enzymes and PBPs. In addition, the advent of cyclic boronate analogues could lead to the first pan-β-lactamase inhibitors due to their structural resemblance to the common transition state formed upon both SBL- and MBL-mediated hydrolysis of β-lactams. For the various other compound classes recently described as MBL inhibitors, challenges including stability in physiological conditions (i.e. for thiol-based inhibitors) and site-specificity (as for metal chelators) must first be addressed before their clinical relevance can be properly assessed.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
Financial support was provided by The Netherlands Organization for Scientific Research (NWO) and The European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 725523).
References
- Ambler R. P. Philos. Trans. R. Soc., B. 1980;289:321–331. doi: 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- Bush K., Jacoby G. A., Medeiros A. A. Antimicrob. Agents Chemother. 1995;39:1211–1233. doi: 10.1128/aac.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drawz S. M., Bonomo R. A. Clin. Microbiol. Rev. 2010;23:160–201. doi: 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K. Antimicrob. Agents Chemother. 2018 doi: 10.1128/AAC.01076-18. [DOI] [Google Scholar]
- Wang J.-F., Chou K.-C. Curr. Top. Med. Chem. 2013;13:1242–1253. doi: 10.2174/15680266113139990011. [DOI] [PubMed] [Google Scholar]
- Shakil S., Azhar E. I., Tabrez S., Kamal M. A., Jabir N. R., Abuzenadah A. M., Damanhouri G. A., Alam Q. J. Chemother. 2011;23:263–265. doi: 10.1179/joc.2011.23.5.263. [DOI] [PubMed] [Google Scholar]
- Johnson A. P., Woodford N. J. Med. Microbiol. 2013;62:499–513. doi: 10.1099/jmm.0.052555-0. [DOI] [PubMed] [Google Scholar]
- Wang Z., Fast W., Valentine A. M., Benkovic S. J. Curr. Opin. Chem. Biol. 1999;3:614–622. doi: 10.1016/s1367-5931(99)00017-4. [DOI] [PubMed] [Google Scholar]
- Potron A., Poirel L., Nordmann P. Int. J. Antimicrob. Agents. 2015;45:568–585. doi: 10.1016/j.ijantimicag.2015.03.001. [DOI] [PubMed] [Google Scholar]
- Marques D. de A. Viana, Machado S. E. F., Ebinuma V. C. S., Duarte C. de A. L., Converti A., Porto A. L. F. Antibiotics. 2018;7:61. doi: 10.3390/antibiotics7030061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaibi E. B., Sirot D., Paul G., Labia R. J. Antimicrob. Chemother. 1999;43:447–458. doi: 10.1093/jac/43.4.447. [DOI] [PubMed] [Google Scholar]
- van Duin D., Doi Y. Virulence. 2017;8:460–469. doi: 10.1080/21505594.2016.1222343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomo R. A., Burd E. M., Conly J., Limbago B. M., Poirel L., Segre J. A., Westblade L. F. Clin. Infect. Dis. 2018;66:1290–1297. doi: 10.1093/cid/cix893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins R. R., Deresinski S. Expert Rev. Anti-infect. Ther. 2017;15:893–895. doi: 10.1080/14787210.2017.1380519. [DOI] [PubMed] [Google Scholar]
- Bush K. ACS Infect. Dis. 2018;4:84–87. doi: 10.1021/acsinfecdis.7b00243. [DOI] [PubMed] [Google Scholar]
- Clark N. M., Zhanel G. G., Lynch 3rd J. P. Curr. Opin. Crit. Care. 2016;22:491–499. doi: 10.1097/MCC.0000000000000337. [DOI] [PubMed] [Google Scholar]
- Tillotson G. S., Zinner S. H. Expert Rev. Anti-infect. Ther. 2017;15:663–676. doi: 10.1080/14787210.2017.1337508. [DOI] [PubMed] [Google Scholar]
- Lee Y. R., Baker N. T. Eur. J. Clin. Microbiol. Infect. Dis. 2018;37:1411–1419. doi: 10.1007/s10096-018-3260-4. [DOI] [PubMed] [Google Scholar]
- Jorgensen S. C. J., Rybak M. J. Pharmacotherapy. 2018;38:444–461. doi: 10.1002/phar.2092. [DOI] [PubMed] [Google Scholar]
- Farrell D. J., Flamm R. K., Sader H. S., Jones R. N. Antimicrob. Agents Chemother. 2013;57:6305–6310. doi: 10.1128/AAC.01802-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J. C., Zmarlicka M. T., Shaeer K. M., Pardo J. Ann. Pharmacother. 2018;52:769–779. doi: 10.1177/1060028018763288. [DOI] [PubMed] [Google Scholar]
- Hecker S. J., Reddy K. R., Totrov M., Hirst G. C., Lomovskaya O., Griffith D. C., King P., Tsivkovski R., Sun D., Sabet M., Tarazi Z., Clifton M. C., Atkins K., Raymond A., Potts K. T., Abendroth J., Boyer S. H., Loutit J. S., Morgan E. E., Durso S., Dudley M. N. J. Med. Chem. 2015;58:3682–3692. doi: 10.1021/acs.jmedchem.5b00127. [DOI] [PubMed] [Google Scholar]
- Castanheira M., Rhomberg P. R., Flamm R. K., Jones R. N. Antimicrob. Agents Chemother. 2016;60:5454–5458. doi: 10.1128/AAC.00711-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanheira M., Huband M. D., Mendes R. E., Flamm R. K. Antimicrob. Agents Chemother. 2017;61:e00567. doi: 10.1128/AAC.00567-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomovskaya O., Sun D., Rubio-Aparicio D., Nelson K., Tsivkovski R., Griffith D. C., Dudley M. N. Antimicrob. Agents Chemother. 2017;61:e01443. doi: 10.1128/AAC.01443-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith D. C., Loutit J. S., Morgan E. E., Durso S., Dudley M. N. Antimicrob. Agents Chemother. 2016;60:6326–6332. doi: 10.1128/AAC.00568-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye K. S., Bhowmick T., Metallidis S., Bleasdale S. C., Sagan O. S., Stus V., Vazquez J., Zaitsev V., Bidair M., Chorvat E., Dragoescu P. O., Fedosiuk E., Horcajada J. P., Murta C., Sarychev Y., Stoev V., Morgan E., Fusaro K., Griffith D., Lomovskaya O., Alexander E. L., Loutit J., Dudley M. N., Giamarellos-Bourboulis E. J. JAMA, J. Am. Med. Assoc. 2018;319:788–799. doi: 10.1001/jama.2018.0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabomere webpage, www.vabomere.com.
- Vabomere product sheet, http://www.vabomere.com/media/pdf/P-VAB-US-00050_R01_MVAB_HCP_OrderSheet_Web.pdf.
- Ball M., Boyd A., Ensor G. J., Evans M., Golden M., Linke S. R., Milne D., Murphy R., Telford A., Kalyan Y., Lawton G. R., Racha S., Ronsheim M., Zhou S. H. Org. Process Res. Dev. 2016;20:1799–1805. [Google Scholar]
- Ehmann D. E., Jahic H., Ross P. L., Gu R.-F., Hu J., Kern G., Walkup G. K., Fisher S. L. Proc. Natl. Acad. Sci. U. S. A. 2012;109:11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmann D. E., Jahić H., Ross P. L., Gu R. F., Hu J., Durand-Réville T. F., Lahiri S., Thresher J., Livchak S., Gao N., Palmer T., Walkup G. K., Fisher S. L. J. Biol. Chem. 2013;288:27960–27971. doi: 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S. D., Johnstone M. R., Ross P. L., McLaughlin R. E., Olivier N. B., Alm R. A. Antimicrob. Agents Chemother. 2014;58:5704–5713. doi: 10.1128/AAC.03057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S. D., Mangani S., Durand-Reville T., Benvenuti M., De Luca F., Sanyal G., Docquier J. D. Antimicrob. Agents Chemother. 2013;57:2496–2505. doi: 10.1128/AAC.02247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi C., Di Pisa F., De Luca F., Benvenuti M., Docquier J. D., Mangani S. ChemMedChem. 2018;13:1437–1446. doi: 10.1002/cmdc.201800213. [DOI] [PubMed] [Google Scholar]
- Levasseur P., Girard A.-M., Claudon M., Goossens H., Black M. T., Coleman K., Miossec C. Antimicrob. Agents Chemother. 2012;56:1606–1608. doi: 10.1128/AAC.06064-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanheira M., Sader H. S., Farrell D. J., Mendes R. E., Jones R. N. Antimicrob. Agents Chemother. 2012;56:4779–4785. doi: 10.1128/AAC.00817-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktaş Z., Kayacan C., Oncul O. Int. J. Antimicrob. Agents. 2012;39:86–89. doi: 10.1016/j.ijantimicag.2011.09.012. [DOI] [PubMed] [Google Scholar]
- Castanheira M., Farrell S. E., Krause K. M., Jones R. N., Sader H. S. Antimicrob. Agents Chemother. 2014;58:833–838. doi: 10.1128/AAC.01896-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sader H. S., Castanheira M., Flamm R. K., Farrell D. J., Jones R. N. Antimicrob. Agents Chemother. 2014;58:1684–1692. doi: 10.1128/AAC.02429-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhang F., Zhao C., Wang Z., Nichols W. W., Testa R., Li H., Chen H., He W., Wang Q., Wang H. Antimicrob. Agents Chemother. 2014;58:1774–1778. doi: 10.1128/AAC.02123-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall S., Hujer A. M., Rojas L. J., Papp-Wallace K. M., Humphries R. M., Spellberg B., Hujer K. M., Marshall E. K., Rudin S. D., Perez F., Wilson B. M., Wasserman R. B., Chikowski L., Paterson D. L., Vila A. J., Van Duin D., Kreiswirth B. N., Chambers H. F., Fowler V. G., Jacobs M. R., Pulse M. E., Weiss W. J., Bonomo R. A. Antimicrob. Agents Chemother. 2017;61:e02243. doi: 10.1128/AAC.02243-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Kim A., Angela Tanudra M., Harris J. J., McLaughlin R. E., Patey S., O'Donnell J. P., Bradford P. A., Eakin A. E. J. Antimicrob. Chemother. 2015;70:2618–2626. doi: 10.1093/jac/dkv132. [DOI] [PubMed] [Google Scholar]
- Flamm R. K., Farrell D. J., Sader H. S., Jones R. N. J. Antimicrob. Chemother. 2014;69:1589–1598. doi: 10.1093/jac/dku025. [DOI] [PubMed] [Google Scholar]
- Castanheira M., Mills J. C., Costello S. E., Jones R. N., Sader H. S. Antimicrob. Agents Chemother. 2015;59:3509–3517. doi: 10.1128/AAC.00163-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Estabrook M., Jacoby G. A., Nichols W. W., Testa R. T., Bush K. Antimicrob. Agents Chemother. 2015;59:1789–1793. doi: 10.1128/AAC.04191-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry N., Howden B. Expert Rev. Anti-infect. Ther. 2018;16:289–306. doi: 10.1080/14787210.2018.1453807. [DOI] [PubMed] [Google Scholar]
- Winkler M. L., Papp-Wallace K. M., Hujer A. M., Domitrovic T. N., Hujer K. M., Hurless K. N., Tuohy M., Hall G., Bonomo R. A. Antimicrob. Agents Chemother. 2015;59:1020–1029. doi: 10.1128/AAC.04238-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez J. A., González Patzán L. D., Stricklin D., Duttaroy D. D., Kreidly Z., Lipka J., Sable C. Curr. Med. Res. Opin. 2012;28:1921–1931. doi: 10.1185/03007995.2012.748653. [DOI] [PubMed] [Google Scholar]
- Wagenlehner F. M., Sobel J. D., Newell P., Armstrong J., Huang X., Stone G. G., Yates K., Gasink L. B. Clin. Infect. Dis. 2016;63:754–762. doi: 10.1093/cid/ciw378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazuski J. E., Gasink L. B., Armstrong J., Broadhurst H., Stone G. G., Rank D., Llorens L., Newell P., Pachl J. Clin. Infect. Dis. 2016;62:1380–1389. doi: 10.1093/cid/ciw133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeli Y., Armstrong J., Laud P. J., Newell P., Stone G., Wardman A., Gasink L. B. Lancet Infect. Dis. 2016;16:661–673. doi: 10.1016/S1473-3099(16)30004-4. [DOI] [PubMed] [Google Scholar]
- Avycaz webpage, www.avycaz.com.
- Zhanel G. G., Chung P., Adam H., Zelenitsky S., Denisuik A., Schweizer F., Lagac-Wiens P. R. S., Rubinstein E., Gin A. S., Walkty A., Hoban D. J., Lynch J. P., Karlowsky J. A. Drugs. 2014;74:31–51. doi: 10.1007/s40265-013-0168-2. [DOI] [PubMed] [Google Scholar]
- Sucher A. J., Chahine E. B., Cogan P., Fete M. Ann. Pharmacother. 2015;49:1046–1056. doi: 10.1177/1060028015593293. [DOI] [PubMed] [Google Scholar]
- Cho J. C., Fiorenza M. A., Estrada S. J. Pharmacotherapy. 2015;35:701–715. doi: 10.1002/phar.1609. [DOI] [PubMed] [Google Scholar]
- Liscio J. L., Mahoney M. V., Hirsch E. B. Int. J. Antimicrob. Agents. 2015;46:266–271. doi: 10.1016/j.ijantimicag.2015.05.003. [DOI] [PubMed] [Google Scholar]
- Scott L. J. Drugs. 2016;76:231–242. doi: 10.1007/s40265-015-0524-5. [DOI] [PubMed] [Google Scholar]
- Jones J. A., Virga K. G., Gumina G., Hevener K. E. Med. Chem. Commun. 2016;7:1694–1715. doi: 10.1039/C6MD00232C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacobbe D. R., Bassetti M., De Rosa F. G., Del Bono V., Grossi P. A., Menichetti F., Pea F., Rossolini G. M., Tumbarello M., Viale P., Viscoli C. Expert Rev. Anti-infect. Ther. 2018;16:307–320. doi: 10.1080/14787210.2018.1447381. [DOI] [PubMed] [Google Scholar]
- Toda A., Ohki H., Yamanaka T., Murano K., Okuda S., Kawabata K., Hatano K., Matsuda K., Misumi K., Itoh K., Satoh K., Inoue S. Bioorg. Med. Chem. Lett. 2008;18:4849–4852. doi: 10.1016/j.bmcl.2008.07.085. [DOI] [PubMed] [Google Scholar]
- Murano K., Yamanaka T., Toda A., Ohki H., Okuda S., Kawabata K., Hatano K., Takeda S., Akamatsu H., Itoh K., Misumi K., Inoue S., Takagi T. Bioorg. Med. Chem. 2008;16:2261–2275. doi: 10.1016/j.bmc.2007.11.074. [DOI] [PubMed] [Google Scholar]
- Takeda S., Nakai T., Wakai Y., Ikeda F., Hatano K. Antimicrob. Agents Chemother. 2007;51:826–830. doi: 10.1128/AAC.00860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryskier A., Procyk T., Labro M. T. J. Antimicrob. Chemother. 1990;26:1–8. doi: 10.1093/jac/26.suppl_c.1. [DOI] [PubMed] [Google Scholar]
- Livermore D. M., Mushtaq S., Ge Y. J. Antimicrob. Chemother. 2010;65:1972–1974. doi: 10.1093/jac/dkq248. [DOI] [PubMed] [Google Scholar]
- Walkty A., Karlowsky J. A., Adam H., Baxter M., Lagacé-Wiens P., Hoban D. J., Zhanel G. G. Antimicrob. Agents Chemother. 2013;57:5707–5709. doi: 10.1128/AAC.01404-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snydman D. R., McDermott L. A., Jacobus N. V. Antimicrob. Agents Chemother. 2014;58:1218–1223. doi: 10.1128/AAC.02253-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomkin J., Hershberger E., Miller B., Popejoy M., Friedland I., Steenbergen J., Yoon M., Collins S., Yuan G., Barie P. S., Eckmann C. Clin. Infect. Dis. 2015;60:1462–1471. doi: 10.1093/cid/civ097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenlehner F. M., Umeh O., Steenbergen J., Yuan G., Darouiche R. O. Lancet. 2015;385:1949–1956. doi: 10.1016/S0140-6736(14)62220-0. [DOI] [PubMed] [Google Scholar]
- Zerbaxa webpage, https://www.merckconnect.com/zerbaxa/about-zerbaxa.html.
- Crandon J. L., Nicolau D. P. Antimicrob. Agents Chemother. 2015;59:2688–2694. doi: 10.1128/AAC.00033-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandon J., Nicolau D. Pathogens. 2015;4:620–625. doi: 10.3390/pathogens4030620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Akker F., Bonomo R. A. Front. Microbiol. 2018;9:622. doi: 10.3389/fmicb.2018.00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanaik P., Bethel C. R., Hujer A. M., Hujer K. M., Distler A. M., Taracila M., Anderson V. E., Fritsche T. R., Jones R. N., Pagadala S. R. R., Van Den Akker F., Buynak J. D., Bonomo R. A. J. Biol. Chem. 2009;284:945–953. doi: 10.1074/jbc.M806833200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drawz S. M., Bethel C. R., Doppalapudi V. R., Sheri A., Pagadala S. R. R., Hujer A. M., Skalweit M. J., Anderson V. E., Chen S. G., Buynak J. D., Bonomo R. A. Antimicrob. Agents Chemother. 2010;54:1414–1424. doi: 10.1128/AAC.00743-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo J. A., Martínez-Guitián M., Vázquez-Ucha J. C., González-Bello C., Poza M., Buynak J. D., Bethel C. R., Bonomo R. A., Bou G., Beceiro A. J. Antimicrob. Chemother. 2016;71:2171–2180. doi: 10.1093/jac/dkw105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez-Ucha J. C., Maneiro M., Martínez-Guitián M., Buynak J., Bethel C. R., Bonomo R. A., Bou G., Poza M., González-Bello C., Beceiro A. Antimicrob. Agents Chemother. 2017;61:e01172. doi: 10.1128/AAC.01172-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanel G. G., Lawrence C. K., Adam H., Schweizer F., Zelenitsky S., Zhanel M., Lagacé-Wiens P. R. S., Walkty A., Denisuik A., Golden A., Gin A. S., Hoban D. J., Lynch J. P., Karlowsky J. A. Drugs. 2018;78:65–98. doi: 10.1007/s40265-017-0851-9. [DOI] [PubMed] [Google Scholar]
- Lapuebla A., Abdallah M., Olafisoye O., Cortes C., Urban C., Landman D., Quale J. Antimicrob. Agents Chemother. 2015;59:5029–5031. doi: 10.1128/AAC.00830-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lob S. H., Hackel M. A., Kazmierczak K. M., Young K., Motyl M. R., Karlowsky J. A., Sahm D. F. Antimicrob. Agents Chemother. 2017;61:e02209. doi: 10.1128/AAC.02209-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlowsky J. A., Lob S. H., Kazmierczak K. M., Hawser S. P., Magnet S., Young K., Motyl M. R., Sahm D. F. J. Antimicrob. Chemother. 2018;73:1872–1879. doi: 10.1093/jac/dky107. [DOI] [PubMed] [Google Scholar]
- Snydman D. R., Jacobus N. V., McDermott L. A. Antimicrob. Agents Chemother. 2016;60:6393–6397. doi: 10.1128/AAC.01125-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein E. J. C., Citron D. M., Tyrrell K. L., Leoncio E., Merriam C. V. Antimicrob. Agents Chemother. 2018;62:e01992. doi: 10.1128/AAC.01992-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucasti C., Vasile L., Sandesc D., Venskutonis D., McLeroth P., Lala M., Rizk M. L., Brown M. L., Losada M. C., Pedley A., Kartsonis N. A., Paschke A. Antimicrob. Agents Chemother. 2016;60:6234–6243. doi: 10.1128/AAC.00633-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims M., Mariyanovski V., McLeroth P., Akers W., Lee Y. C., Brown M. L., Du J., Pedley A., Kartsonis N. A., Paschke A. J. Antimicrob. Chemother. 2017;72:2616–2626. doi: 10.1093/jac/dkx139. [DOI] [PubMed] [Google Scholar]
- Papp-Wallace K. M., Nguyen N. Q., Jacobs M. R., Bethel C. R., Barnes M. D., Kumar V., Bajaksouzian S., Rudin S. D., Rather P. N., Bhavsar S., Ravikumar T., Deshpande P. K., Patil V., Yeole R., Bhagwat S. S., Patel M. V., van den Akker F., Bonomo R. A. J. Med. Chem. 2018;61:4067–4086. doi: 10.1021/acs.jmedchem.8b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya B., Barcelo I. M., Bhagwat S., Patel M., Bou G., Papp-Wallace K. M., Bonomo R. A., Oliver A. Antimicrob. Agents Chemother. 2017;61:e02529. doi: 10.1128/AAC.01238-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya B., Barcelo I. M., Bhagwat S., Patel M., Bou G., Papp-Wallace K. M., Bonomo R. A., Moya B. Antimicrob. Agents Chemother. 2017;61:e01238. doi: 10.1128/AAC.01238-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sader H. S., Castanheira M., Huband M., Jones R. N., Flamm R. K. Antimicrob. Agents Chemother. 2017;61:e00072. doi: 10.1128/AAC.00072-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sader H. S., Rhomberg P. R., Flamm R. K., Jones R. N., Castanheira M. J. Antimicrob. Chemother. 2017;72:1696–1703. doi: 10.1093/jac/dkx050. [DOI] [PubMed] [Google Scholar]
- Morinaka A., Tsutsumi Y., Yamada M., Suzuki K., Watanabe T., Abe T., Furuuchi T., Inamura S., Sakamaki Y., Mitsuhashi N., Ida T., Livermore D. M. J. Antimicrob. Chemother. 2015;70:2779–2786. doi: 10.1093/jac/dkv166. [DOI] [PubMed] [Google Scholar]
- Morinaka A., Tsutsumi Y., Yamada K., Takayama Y., Sakakibara S., Takata T., Abe T., Furuuchi T., Inamura S., Sakamaki Y., Tsujii N., Ida T. Antimicrob. Agents Chemother. 2016;60:3001–3006. doi: 10.1128/AAC.02704-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinaka A., Tsutsumi Y., Yamada K., Takayama Y., Sakakibara S., Takata T., Abe T., Furuuchi T., Inamura S., Sakamaki Y., Tsujii N., Ida T. J. Antibiot. 2017;70:246–250. doi: 10.1038/ja.2016.150. [DOI] [PubMed] [Google Scholar]
- Livermore D. M., Mushtaq S., Warner M., Woodford N. J. Antimicrob. Chemother. 2015;70:3032–3041. doi: 10.1093/jac/dkv239. [DOI] [PubMed] [Google Scholar]
- Livermore D. M., Warner M., Mushtaq S., Woodford N. Antimicrob. Agents Chemother. 2016;60:554–560. doi: 10.1128/AAC.02184-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand-Réville T. F., Guler S., Comita-Prevoir J., Chen B., Bifulco N., Huynh H., Lahiri S., Shapiro A. B., McLeod S. M., Carter N. M., Moussa S. H., Velez-Vega C., Olivier N. B., McLaughlin R., Gao N., Thresher J., Palmer T., Andrews B., Giacobbe R. A., Newman J. V., Ehmann D. E., De Jonge B., O'Donnell J., Mueller J. P., Tommasi R. A., Miller A. A. Nat. Microbiol. 2017;2:17104. doi: 10.1038/nmicrobiol.2017.104. [DOI] [PubMed] [Google Scholar]
- Tommasi R., Iyer R., Miller A. A. ACS Infect. Dis. 2018;4:686–695. doi: 10.1021/acsinfecdis.8b00027. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B., Gao N., Jahić H., Carter N. M., Chen A., Miller A. A. ACS Infect. Dis. 2017;3:833–844. doi: 10.1021/acsinfecdis.7b00113. [DOI] [PubMed] [Google Scholar]
- Iyer R., Moussa S. H., Durand-Réville T. F., Tommasi R., Miller A. ACS Infect. Dis. 2018;4:373–381. doi: 10.1021/acsinfecdis.7b00168. [DOI] [PubMed] [Google Scholar]
- Chang H. K., Baek S. Y., Kim M. J., Oh K. M., Choi J. S., Ha S. B., Kim S. M., Chung C.-W., Kang D. H., Kwon H. J., Cho Y. L. and Kim Y. Z., US Pat., 20170096430A1, 2017.
- Gu Y. G., He Y., Yin N., Alexander D. C., Cross J. B. and Metcalf III C. A., US Pat., 20140315876A1, 2014.
- Maiti S. N., Ganguli B., Nguyen D. Q., Khan J., Ling R., Ha C. M. and Khlebnikov V., WO Pat., 2014141132A1, 2014.
- Basarab G. S., Moss B., Comita-Prevoir J., Durand-Reville T. F., Gauthier Lise, O'donnell J., Romero J., Tommasi R., Verheijen J. C., Wu F., Wu X. and Zhang J., WO Pat., 2018053215A1, 2018.
- Ke W., Sampson J. M., Ori C., Prati F., Drawz S. M., Bethel C. R., Bonomo R. A., Van Den Akker F. Antimicrob. Agents Chemother. 2011;55:174–183. doi: 10.1128/AAC.00930-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trippier P. C., McGuigan C. MedChemComm. 2010;1:183–198. [Google Scholar]
- Powers R. A., Swanson H. C., Taracila M. A., Florek N. W., Romagnoli C., Caselli E., Prati F., Bonomo R. A., Wallar B. J. Biochemistry. 2014;53:7670–7679. doi: 10.1021/bi500887n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen N. Q., Krishnan N. P., Rojas L. J., Prati F., Caselli E., Romagnoli C., Bonomo R. A., Van Den Akker F. Antimicrob. Agents Chemother. 2016;60:1760–1766. doi: 10.1128/AAC.02643-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouza A. A., Swanson H. C., Smolen K. A., Vandine A. L., Taracila M. A., Romagnoli C., Caselli E., Prati F., Bonomo R. A., Powers R. A., Wallar B. J. ACS Infect. Dis. 2018;4:325–336. doi: 10.1021/acsinfecdis.7b00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner J. P., Mitchell J. M., Taracila M. A., Bonomo R. A., Powers R. A. Protein Sci. 2017;26:515–526. doi: 10.1002/pro.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem J., Cain R., Cahill S., McDonough M. A., Clifton I. J., Jiménez-Castellanos J. C., Avison M. B., Spencer J., Fishwick C. W. G., Schofield C. J. Nat. Commun. 2016;7:12406. doi: 10.1038/ncomms12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill S. T., Cain R., Wang D. Y., Lohans C. T., Wareham D. W., Oswin H. P., Mohammed J., Spencer J., Fishwick C. W. G., McDonough M. A., Schofield C. J., Brema J. Antimicrob. Agents Chemother. 2017;61:e02260. doi: 10.1128/AAC.02260-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomo R., Prati F., Caselli E. and Romagnoli C., US Pat., 20170065626A1, 2017.
- Reddy R. K., Glinka T., Totrov M., Hecker S. and Rodny O., WO Pat., 2016003929A1, 2016.
- Reddy R., Glinka T., Totrov M. and Hecker S., WO Pat., 2014107536A1, 2014.
- Burns C. J., Liu B., Yao J., Daigle D. and Boyd S. A., US Pat., 20170073360A1, 2017.
- Burns C. J., Daigle D., Liu B., Mcgarry D., Pevear D. C. and Lee Trout R. E., WO Pat., 2014151958A1, 2014.
- Burns C. J., Daigle D., Liu B., Jackson R. W., Hamrick J., McGarry D., Pevear D. C. and Lee Trout R. E., US Pat., 20160264598A1, 2016.
- Burns C. J., Daigle D., Liu B., McGarry D., Pevear D. C., Lee Trout R. E. and Jackson R. W., US Pat., US20140194386A1, 2014.
- Somboro A. M., Osei Sekyere J., Amoako D. G., Essack S. Y., Bester L. A. Appl. Environ. Microbiol. 2018 doi: 10.1128/AEM.00698-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King D. T., Strynadka N. C. J. Future Med. Chem. 2013;5:1243–1263. doi: 10.4155/fmc.13.55. [DOI] [PubMed] [Google Scholar]
- Fast W., Sutton L. D. Biochim. Biophys. Acta, Proteins Proteomics. 2013;1834:1648–1659. doi: 10.1016/j.bbapap.2013.04.024. [DOI] [PubMed] [Google Scholar]
- Ju L.-C., Cheng Z., Fast W., Bonomo R. A., Crowder M. W. Trends Pharmacol. Sci. 2018;39:635–647. doi: 10.1016/j.tips.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groundwater P. W., Xu S., Lai F., Váradi L., Tan J., Perry J. D., Hibbs D. E. Future Med. Chem. 2016;8:993–1012. doi: 10.4155/fmc-2016-0015. [DOI] [PubMed] [Google Scholar]
- Rotondo C. M., Wright G. D. Curr. Opin. Microbiol. 2017;39:96–105. doi: 10.1016/j.mib.2017.10.026. [DOI] [PubMed] [Google Scholar]
- McGeary R. P., Tan D. T. C., Gerhard S. Future Med. Chem. 2017;9:673–691. doi: 10.4155/fmc-2017-0007. [DOI] [PubMed] [Google Scholar]
- Klingler F. M., Wichelhaus T. A., Frank D., Cuesta-Bernal J., El-Delik J., Müller H. F., Sjuts H., Göttig S., Koenigs A., Pos K. M., Pogoryelov D., Proschak E. J. Med. Chem. 2015;58:3626–3630. doi: 10.1021/jm501844d. [DOI] [PubMed] [Google Scholar]
- Li N., Xu Y., Xia Q., Bai C., Wang T., Wang L., He D., Xie N., Li L., Wang J., Zhou H. G., Xu F., Yang C., Zhang Q., Yin Z., Guo Y., Chen Y. Bioorg. Med. Chem. Lett. 2014;24:386–389. doi: 10.1016/j.bmcl.2013.10.068. [DOI] [PubMed] [Google Scholar]
- Liu S., Jing L., Yu Z.-J., Wu C., Zheng Y., Zhang E., Chen Q., Yu Y., Guo L., Wu Y., Li G.-B. Eur. J. Med. Chem. 2018;145:649–660. doi: 10.1016/j.ejmech.2018.01.032. [DOI] [PubMed] [Google Scholar]
- Büttner D., Kramer J. S., Klingler F. M., Wittmann S. K., Hartmann M. R., Kurz C. G., Kohnhäuser D., Weizel L., Brüggerhoff A., Frank D., Steinhilber D., Wichelhaus T. A., Pogoryelov D., Proschak E. ACS Infect. Dis. 2018;4:360–372. doi: 10.1021/acsinfecdis.7b00129. [DOI] [PubMed] [Google Scholar]
- Li G.-B., Brem J., Lesniak R., Abboud M. I., Lohans C. T., Clifton I. J., Yang S.-Y., Jiménez-Castellanos J.-C., Avison M. B., Spencer J., McDonough M. A., Schofield C. J. Chem. Commun. 2017;53:5806–5809. doi: 10.1039/c7cc02394d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusof Y., Tan D. T. C., Arjomandi O. K., Schenk G., McGeary R. P. Bioorg. Med. Chem. Lett. 2016;26:1589–1593. doi: 10.1016/j.bmcl.2016.02.007. [DOI] [PubMed] [Google Scholar]
- Brem J., Van Berkel S. S., Zollman D., Lee S. Y., Gileadi O., McHugh P. J., Walsh T. R., McDonough M. A., Schofield C. J. Antimicrob. Agents Chemother. 2016;60:142–150. doi: 10.1128/AAC.01335-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. L., Shi Y., Kang J. S., Oelschlaeger P., Yang K. W. ACS Med. Chem. Lett. 2015;6:660–664. doi: 10.1021/acsmedchemlett.5b00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. L., Yang K. W., Zhang Y. J., Ge Y., Xiang Y., Chang Y. N., Oelschlaeger P. Bioorg. Med. Chem. Lett. 2016;26:4698–4701. doi: 10.1016/j.bmcl.2016.08.048. [DOI] [PubMed] [Google Scholar]
- Arjomandi O. K., Hussein W. M., Vella P., Yusof Y., Sidjabat H. E., Schenk G., McGeary R. P. Eur. J. Med. Chem. 2016;114:318–327. doi: 10.1016/j.ejmech.2016.03.017. [DOI] [PubMed] [Google Scholar]
- Chang Y.-N., Xiang Y., Zhang Y.-J., Wang W.-M., Chen C., Oelschlaeger P., Yang K.-W. ACS Med. Chem. Lett. 2017;8:527–532. doi: 10.1021/acsmedchemlett.7b00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. L., Yang K. W., Zhou Y. J., LaCuran A. E., Oelschlaeger P., Crowder M. W. ChemMedChem. 2014;9:2445–2448. doi: 10.1002/cmdc.201402249. [DOI] [PubMed] [Google Scholar]
- Yang S.-K., Kang J. S., Oelschlaeger P., Yang K.-W. ACS Med. Chem. Lett. 2015;6:455–460. doi: 10.1021/ml500534c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai L., Zhang Y. L., Kang J. S., Oelschlaeger P., Xiao L., Nie S. S., Yang K. W. ACS Med. Chem. Lett. 2016;7:413–417. doi: 10.1021/acsmedchemlett.5b00495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Chang Y.-N., Ge Y., Kang J. S., Zhang Y.-L., Liu X.-L., Oelschlaeger P., Yang K.-W. Bioorg. Med. Chem. Lett. 2017;27:5225–5229. doi: 10.1016/j.bmcl.2017.10.038. [DOI] [PubMed] [Google Scholar]
- Cain R., Brem J., Zollman D., McDonough M. A., Johnson R. M., Spencer J., Makena A., Abboud M. I., Cahill S., Lee S. Y., McHugh P. J., Schofield C. J., Fishwick C. W. G. J. Med. Chem. 2018;61:1255–1260. doi: 10.1021/acs.jmedchem.7b01728. [DOI] [PubMed] [Google Scholar]
- González M. M., Kosmopoulou M., Mojica M. F., Castillo V., Hinchliffe P., Pettinati I., Brem J., Schofield C. J., Mahler G., Bonomo R. A., Llarrull L. I., Spencer J., Vila A. J. ACS Infect. Dis. 2016;1:544–554. doi: 10.1021/acsinfecdis.5b00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchliffe P., González M. M., Mojica M. F., González J. M., Castillo V., Saiz C. Proc. Natl. Acad. Sci. U. S. A. 2016:E3745–E3754. doi: 10.1073/pnas.1601368113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem J., van Berkel S. S., Aik W., Rydzik A. M., Avison M. B., Pettinati I., Umland K.-D., Kawamura A., Spencer J., Claridge T. D. W., McDonough M. A., Schofield C. J. Nat. Chem. 2014;6:1084–1090. doi: 10.1038/nchem.2110. [DOI] [PubMed] [Google Scholar]
- Xiang Y., Chen C., Wang W. M., Xu L. W., Yang K. W., Oelschlaeger P., He Y. ACS Med. Chem. Lett. 2018;9:359–364. doi: 10.1021/acsmedchemlett.7b00548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Markoulides M. S., Stepanovs D., Rydzik A. M., El-Hussein A., Bon C., Kamps J. J. A. G., Umland K. D., Collins P. M., Cahill S. T., Wang D. Y., von Delft F., Brem J., McDonough M. A., Schofield C. J. Bioorg. Med. Chem. 2018;26:2928–2936. doi: 10.1016/j.bmc.2018.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts J. W., Phee L. M., Abdul Momin M. H. F., Umland K.-D., Brem J., Schofield C. J., Wareham D. W. Med. Chem. Commun. 2016;7:190–193. [Google Scholar]
- Xiao J., Fang M., Shi Y., Chen H., Shen B., Chen J., Lao X., Xu H., Zheng H. Mol. Inf. 2015;34:559–567. doi: 10.1002/minf.201400178. [DOI] [PubMed] [Google Scholar]
- Shen B., Zhu C., Gao X., Liu G., Song J., Yu Y. PLoS One. 2017;12:e0177293. doi: 10.1371/journal.pone.0177293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skagseth S., Akhter S., Paulsen M. H., Muhammad Z., Lauksund S., Samuelsen Ø., Leiros H.-K. S., Bayer A. Eur. J. Med. Chem. 2017;135:159–173. doi: 10.1016/j.ejmech.2017.04.035. [DOI] [PubMed] [Google Scholar]
- Schlesinger S. R., Bruner B., Farmer P. J., Kim S. K. J. Enzyme Inhib. Med. Chem. 2013;28:137–142. doi: 10.3109/14756366.2011.640632. [DOI] [PubMed] [Google Scholar]
- Shin W. S., Bergstrom A., Bonomo R. A., Crowder M. W., Muthyala R., Sham Y. Y. ChemMedChem. 2017;12:845–849. doi: 10.1002/cmdc.201700182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevaille L., Gavara L., Bebrone C., De Luca F., Nauton L., Achard M., Mercuri P., Tanfoni S., Borgianni L., Guyon C., Lonjon P., Turan-Zitouni G., Dzieciolowski J., Becker K., Bénard L., Condon C., Maillard L., Martinez J., Frère J.-M., Dideberg O., Galleni M., Docquier J.-D., Hernandez J.-F. ChemMedChem. 2017;12:972–985. doi: 10.1002/cmdc.201700186. [DOI] [PubMed] [Google Scholar]
- Song G. Q., Wang W. M., Li Z. S., Wang Y., Wang J. G. Chin. Chem. Lett. 2018;29:899–902. [Google Scholar]
- Mollard C., Moali C., Papamicael C., Damblon C., Vessilier S., Amicosante G., Schofield C. J., Galleni M., Frère J. M., Roberts G. C. K. J. Biol. Chem. 2001;276:45015–45023. doi: 10.1074/jbc.M107054200. [DOI] [PubMed] [Google Scholar]
- Tehrani K. H. M. E., Martin N. I. ACS Infect. Dis. 2017;3:711–717. doi: 10.1021/acsinfecdis.7b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsfall L. E., Garau G., Liénard B. M. R., Dideberg O., Schofield C. J., Frère J. M., Galleni M. Antimicrob. Agents Chemother. 2007;51:2136–2142. doi: 10.1128/AAC.00866-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin K. S., Son B. R., Hong S. B., Kim J. Diagn. Microbiol. Infect. Dis. 2008;62:102–105. doi: 10.1016/j.diagmicrobio.2008.04.015. [DOI] [PubMed] [Google Scholar]
- Kimura S., Ishii Y., Yamaguchi K. Diagn. Microbiol. Infect. Dis. 2005;53:241–244. doi: 10.1016/j.diagmicrobio.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Pasteran F., Veliz O., Faccone D., Guerriero L., Rapoport M., Mendez T., Corso A. Clin. Microbiol. Infect. 2011;17:1438–1441. doi: 10.1111/j.1469-0691.2011.03585.x. [DOI] [PubMed] [Google Scholar]
- Yong D., Lee Y., Jeong S. H., Lee K., Chong Y. J. Clin. Microbiol. 2012;50:3227–3232. doi: 10.1128/JCM.00818-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. Y., Thomas P. W., Stewart A. C., Bergstrom A., Cheng Z., Miller C., Bethel C. R., Marshall S. H., Credille C. V., Riley C. L., Page R. C., Bonomo R. A., Crowder M. W., Tierney D. L., Fast W., Cohen S. M. J. Med. Chem. 2017;60:7267–7283. doi: 10.1021/acs.jmedchem.7b00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchliffe P., Tanner C. A., Krismanich A. P., Labbé G., Goodfellow V. J., Marrone L., Desoky A. Y., Calvopiña K., Whittle E. E., Zeng F., Avison M. B., Bols N. C., Siemann S., Spencer J., Dmitrienko G. I. Biochemistry. 2018;57:1880–1892. doi: 10.1021/acs.biochem.7b01299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett M., Sprynski N., Coelho A., Castandet J., Bayet M., Bougnon J., Lozano C., Davies D. T., Leiris S., Zalacain M., Morrissey I., Magnet S., Holden K., Warn P., De Luca F., Docquier J.-D., Lemonnier M. Antimicrob. Agents Chemother. 2018;62:e00074. doi: 10.1128/AAC.00074-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraiwa Y., Saito J., Watanabe T., Yamada M., Morinaka A., Fukushima T., Kudo T. Bioorg. Med. Chem. Lett. 2014;24:4891–4894. doi: 10.1016/j.bmcl.2014.08.039. [DOI] [PubMed] [Google Scholar]
- Feng L., Yang K. W., Zhou L. S., Xiao J. M., Yang X., Zhai L., Zhang Y. L., Crowder M. W. Bioorg. Med. Chem. Lett. 2012;22:5185–5189. doi: 10.1016/j.bmcl.2012.06.074. [DOI] [PubMed] [Google Scholar]
- Tang H., Yang S.-W., Mandal M., Su J., Li G., Pan W., Tang H., DeJesus R., Pan J., Hagmann W., Ding F.-X., Xiao L., Pasternak A., Huang Y., Dong S. and Yang D., US Pat., 20170173035A1, 2017.
- King A. M., Reid-Yu S. A., Wang W., King D. T., De Pascale G., Strynadka N. C., Walsh T. R., Coombes B. K., Wright G. D. Nature. 2014;510:503–506. doi: 10.1038/nature13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D., Yang S., Wang J., Zhang J., Hong B., Wu F., Lei X. Angew. Chem., Int. Ed. 2016;55:4291–4295. doi: 10.1002/anie.201509960. [DOI] [PubMed] [Google Scholar]
- Koteva K., King A. M., Capretta A., Wright G. D. Angew. Chem., Int. Ed. 2016;55:2210–2212. doi: 10.1002/anie.201510057. [DOI] [PubMed] [Google Scholar]
- Albu S. A., Koteva K., King A. M., Al-Karmi S., Wright G. D., Capretta A. Angew. Chem., Int. Ed. 2016;55:13259–13262. doi: 10.1002/anie.201606657. [DOI] [PubMed] [Google Scholar]
- Zhang J., Wang S., Bai Y., Guo Q., Zhou J., Lei X. J. Org. Chem. 2017;82:13643–13648. doi: 10.1021/acs.joc.7b02342. [DOI] [PubMed] [Google Scholar]
- Fu H., Zhang J., Saifuddin M., Cruiming G., Tepper P. G., Poelarends G. J. Nat. Catal. 2018;1:186–191. [Google Scholar]
- Bergstrom A., Katko A., Adkins Z., Hill J., Cheng Z., Burnett M., Yang H., Aitha M., Mehaffey M. R., Brodbelt J. S., Tehrani K. H. M. E., Martin N. I., Bonomo R. A., Page R. C., Tierney D. L., Fast W., Wright G. D., Crowder M. W. ACS Infect. Dis. 2018;4:135–145. doi: 10.1021/acsinfecdis.7b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchettini J. C., Mire J. A., Thurman C. C., Zhou N. W., Joachimiak A., Babnigg G. and Youngchang Kim, WO Pat., 2015157618A1, 2015.
- Somboro A. M., Tiwari D., Bester L. A., Parboosing R., Chonco L., Kruger H. G., Arvidsson P. I., Govender T., Naicker T., Essack S. Y. J. Antimicrob. Chemother. 2014;70:1594–1596. doi: 10.1093/jac/dku538. [DOI] [PubMed] [Google Scholar]
- Zhang E., Wang M. M., Huang S. C., Xu S. M., Cui D. Y., Bo Y. L., Bai P. Y., Hua Y. G., Xiao C. L., Qin S. Bioorg. Med. Chem. Lett. 2018;28:214–221. doi: 10.1016/j.bmcl.2017.10.074. [DOI] [PubMed] [Google Scholar]
- Azumah R., Dutta J., Somboro A. M., Ramtahal M., Chonco L., Parboosing R., Bester L. A., Kruger H. G., Naicker T., Essack S. Y., Govender T. J. Appl. Microbiol. 2016;120:860–867. doi: 10.1111/jam.13085. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V., Sarkar P., Samaddar S., Manjunath G. B., Das Mitra S., Paramanandham K., Shome B. R., Haldar J. ACS Infect. Dis. 2018;4:1093–1101. doi: 10.1021/acsinfecdis.8b00011. [DOI] [PubMed] [Google Scholar]
- Brem J., Rydzik A. M., McDonough M. A., Schofield C. J., Morrison A., Hewitt J., Pannifer A. and Jones P., WO Pat., 2017093727A1, 2017.
- Mandal M., Tang H., Xiao L., Su J., Li G., Yang S.-W., Pan W., Tang H., DeJesus R., Hicks J., Lombardo M., Chu H., Hagmann W., Pasternak A., Gu X., Jiang J., Dong S., Ding F.-X., London C., Biswas D., Young K., Hunter D. N., Zhao Z. and Yang D., US Pat., 20160333021A1, 2016.
- Bennett F., Jiang J., Pasternak A., Dong S., Gu X., Scott J. D., Tang H., Zhao Z., Huang Y., Hunter D., Yang D., Zhang Z., Fu J., Bai Y., Zheng Z., Zhang X., Young K. and Xiao L., WO Pat., 2016206101A1, 2016.
- Becker D. P., Reidl C., Moore M., Heath T. K. and Fast W., WO Pat., 2017011408A1, 2017.
- Thomas P. W., Cammarata M., Brodbelt J. S., Fast W. ChemBioChem. 2014;15:2541–2548. doi: 10.1002/cbic.201402268. [DOI] [PubMed] [Google Scholar]
- Christopeit T., Albert A., Leiros H. K. S. Bioorg. Med. Chem. 2016;24:2947–2953. doi: 10.1016/j.bmc.2016.04.064. [DOI] [PubMed] [Google Scholar]
- Chiou J., Wan S., Chan K.-F., So P.-K., He D., Chan E. W., Chan T., Wong K., Tao J., Chen S. Chem. Commun. 2015;51:9543–9546. doi: 10.1039/c5cc02594j. [DOI] [PubMed] [Google Scholar]
- Brindisi M., Brogi S., Giovani S., Gemma S., Lamponi S., De Luca F., Novellino E., Campiani G., Docquier J. D., Butini S. J. Enzyme Inhib. Med. Chem. 2016;31:98–109. doi: 10.3109/14756366.2016.1172575. [DOI] [PubMed] [Google Scholar]
- Zhang Y. L., Zhang Y. J., Wang W. M., Yang K. W. Phosphorus, Sulfur Silicon Relat. Elem. 2017;192:14–18. [Google Scholar]
- Li G.-B., Abboud M. I., Brem J., Someya H., Lohans C. T., Yang S.-Y., Spencer J., Wareham D. W., McDonough M. A., Schofield C. J. Chem. Sci. 2017;8:928–937. doi: 10.1039/c6sc04524c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeary R. P., Tan D. T. C., Selleck C., Monteiro Pedroso M., Sidjabat H. E., Schenk G. Eur. J. Med. Chem. 2017;137:351–364. doi: 10.1016/j.ejmech.2017.05.061. [DOI] [PubMed] [Google Scholar]
- Wang R., Lai T. P., Gao P., Zhang H., Ho P. L., Woo P. C. Y., Ma G., Kao R. Y. T., Li H., Sun H. Nat. Commun. 2018;9:439. doi: 10.1038/s41467-018-02828-6. [DOI] [PMC free article] [PubMed] [Google Scholar]