

Abstract
Given our possible future dependence on carbon fiber reinforced composites, the introduction of a renewable matrix might be advantageous for the vision of a sustainable world. Cellulose is a superior green candidate and provides exceptional freedom in composite design as the free OH groups can be conveniently functionalized to give tailor‐made materials. To obtain a high‐performing carbon fiber reinforced cellulose propionate composite, we accurately tailored the interfacial adhesion by invoking click chemistry. The synthetic strategy involved grafting of a phenylacetylene structure onto the carbon fiber surface, onto which O‐acylated 6‐azido‐6‐deoxycellulose and a number of aromatic azides could be covalently attached. Single‐fiber fragmentation tests indicated that the lipophilicity and size of the substituent on the deposited structure played a crucial role in determining molecular entanglement and mechanical interlocking effects, as penetration into the cellulose propionate matrix was of utmost importance. Enhanced interfacial shear strength was obtained for the carbon fiber covalently functionalized with the cellulose derivative. Nevertheless, the greatest increase was observed for the derivative substituted with a compact and highly lipophilic CF3 substituent. In a broader sense, our study provides a synthetic platform to bind cellulose derivatives to graphitic surfaces and paves the ways towards the preparation of innovative cellulose‐based carbonaceous materials.
Keywords: carbon fibers, cellulose, click chemistry, interfaces, surface chemistry
1. Introduction
As the demand for carbon fiber reinforced polymers increases at a tremendous pace, environmental issues are coming into focus to decrease the environmental footprint of these energy‐intensive materials.1 The most commonly applied thermosetting epoxy matrix provides a composite with excellent parameters; however, there are many concerns surrounding its recyclability and relatively long production time.2, 3 From this point of view, changing the matrix to a thermoplastic polymer [e.g. poly(phthalazinone ether ketone), polypropylene, poly(phenylene sulfide)] is gaining attention, as these polymers overcome several of the disadvantages of thermosetting resins (e.g. epoxy matrix).4, 5, 6, 7, 8 In an attempt to introduce cellulose‐based carbon fiber reinforced polymers as renewable green alternatives, we chose commercially available cellulose propionate as a promising candidate for the matrix, because it features some advantageous characteristics owing to its high molecular weight (processability at high temperature, enhanced tensile strength). To obtain a composite with high performance, interfacial adhesion between the relatively inert graphitic surface of the carbon fiber and the polymer chains of cellulose propionate needs to be intensified. This study is about to fill this knowledge gap by tailoring the surface of carbon fiber by applying click chemistry. Click reactions refer to a group of reactions making a connection between two molecular building blocks through an efficient and fast process (e.g. CuI‐catalyzed azide–alkyne cycloaddition).
While the matrix is responsible for the efficient transfer of the load to the reinforcing fiber, the final performance of the composite is highly dependent on the interactions taking place at the fiber–matrix interphase.9 This area of research has experienced a boost in the last decades, giving rise to a wide range of surface‐modification strategies.4, 5, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 In the course of these studies, main concepts about molecular design to tailor the interfacial adhesion between the fiber and the matrix have also been revealed,11, 15, 22, 23 and these concepts were recently supported by molecular dynamics calculations.24 As it turns out, mechanical interlocking, interfacial cross‐links, and the structural properties of the grafted molecule are crucial factors that intensify interfacial adhesion in a carbon fiber–epoxy composite system. However, the interfacial behavior of thermoplastic composites is not well understood, and the implementation of previously discovered rules is not straightforward; therefore, there is much to be done in this field.
Although several peculiar reactions have been applied on the carbon fiber surface, many of them will probably never reach industrial application on a large scale. Presumably, the most promising method to date involves grafting aromatic structures via diazonium species.5, 17, 18, 19, 21, 22, 23 Its potentiality resides in the fact that these species can also be electrochemically reduced on the carbon fiber surface to provide a robust technology, which is already in application for the production of carbon fibers.16, 22 Furthermore, the reaction does not affect key single‐fiber performance parameters, and the aromatic structures can be decorated with several functionalities that are able to take part in additional reactions, which expands the versatility of chemistries applied on the surface.18, 23 In our previous study, we covalently immobilized tosylated cellulose propionate on the carbon fiber surface by a nucleophilic displacement reaction involving surface‐grafted amine functions.25 However, we also envisioned another, powerful catalytic synthetic procedure for binding a cellulose derivative to the surface, and we intend to report on our novel synthetic approach herein.
Recently, advances in the successful modification of carbon fibers have been achieved by applying the powerful CuI‐catalyzed azide–alkyne cycloaddition (click reaction) after functionalization of the carbon fiber surface with a phenylacetylene moiety via the corresponding diazonium species.23, 26 Inspired by this approach, we decided to apply a similar strategy for the interphase engineering of a cellulose‐based carbon fiber reinforced composite. As click reactions are also compatible with cellulose chemistry,27 we proposed a synthetic route to bind the propionyl ester of cellulose azide to the carbon fiber surface by aromatic structures serving as molecular bridges. Furthermore, to have a deeper understanding of the interfacial effects, we also deposited a range of molecules with different side chains by applying similar solid‐phase click chemistry. Our study provides a novel strategy to bind cellulosic derivatives covalently to a largely graphitic surface.
2. Results and Discussion
In an attempt to increase the interfacial adhesion between the relatively inert carbon fiber surface and cellulose propionate, we decided to bind the polymer covalently to the surface. For this purpose, we found a previously elaborated functionalization procedure involving the use of diazonium species to be especially attractive,5, 17, 18, 19, 21, 22, 23 as the grafted aromatic structures with appropriate substituents could serve as molecular bridges, which would make the surface compatible with cellulose chemistry. These molecular bridges are thought to make the carbon fiber surface not only chemically available but also sterically available for cellulose derivatives, as steric effects are known to play a crucial role in surface functionalization.18 Besides recent advances in solid‐phase click chemistry (CuI‐catalyzed azide–alkyne cycloaddition) on carbon fiber surfaces,23, 26 this highly efficient synthetic approach was previously also successfully introduced to cellulose chemistry.27 Therefore, we propose a synthetic strategy to bind a cellulose derivative to the carbon fiber surface on the basis of a click reaction (Figure 1).
Figure 1.
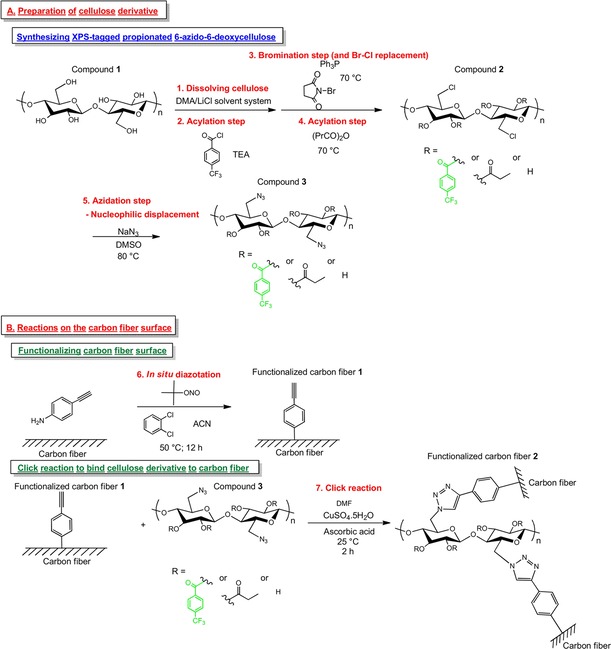
Synthetic strategy to bind cellulose derivatives to the carbon fiber surface. TEA=triethylamine; ACN=acetonitrile.
The synthetic approach necessitated the initial preparation of O‐acylated 6‐azido‐6‐deoxycellulose (compound 3, Figure 1) and carbon fiber samples functionalized with a phenylacetylene moiety (functionalized carbon fiber 1, Figure 1), followed by the CuI‐catalyzed azide–alkyne cycloaddition by using these stable intermediates (step 7, click reaction; Figure 1). The greatest obstacle was encountered in the synthesis of the cellulose derivative as a result of solubility issues, but this was eventually solved by a convenient and facile synthetic route based on previous studies (one‐pot procedure including bromination and acylation steps).28, 29
2.1. Synthesis of O‐Acylated 6‐Azido‐6‐deoxycellulose
Upon considering possible reaction paths to obtain O‐acylated azidodeoxycellulose 3 (Figure 1), commercially available cellulose propionate was discarded as a starting compound due to its high degree of substitution (DS) value (DS=2.76, i.e. 92% of the free OH groups of cellulose are substituted) and total saturation at the C6 position according to our quantitative 13C NMR spectroscopy measurements.25 In particular, we intended to apply a regioselective synthetic route for azidation at the C6 position due to its putative beneficial effects on sterically promoted surface modification. As a result, we decided to use microcrystalline cellulose (Avicel PH 101, compound 1; Figure 1) as the starting compound and dimethylacetamide (DMA)/LiCl as the solvent system (some considerations about the solvent system are given below).
Given that it is challenging to detect molecules on a heterogeneous carbon fiber surface without doubt by using X‐ray photoelectron spectroscopy (carbon fiber is made out of the same elements as the grafted entity), an XPS‐detectable molecule needed to be incorporated into the deposited structure to ease characterization. For this purpose, the trifluoromethyl group was shown to be a promising entity according to previous studies.16, 18, 19 Therefore, cellulose was initially treated with 4‐(trifluoromethyl)benzoyl chloride to afford the corresponding ester, with the fluorine atom being an XPS‐detectable element, not native to the surface (step 2, acylation; Figure 1). At the same time, we also had to consider that this substituent should not interfere with any subsequent steps in the reaction sequence (Figure 1). For this reason, we tried to keep the degree of substitution as low as possible by applying only 0.2 equivalents of the reagent. As a result, we obtained a DS of 0.03 for the XPS tag (Figure S1 in the Supporting Information).
Azidation of cellulose can be performed via halodeoxycelluloses or tosylated derivatives by exploiting their facile nucleophilic displacement reaction with NaN3.30, 31 We were especially interested in regioselective azidation at the C6 position, as it was expected to give further conformational freedom to the reaction system (less sterically hindered), which would facilitate the click reaction on the surface. The most straightforward way to achieve our goal was bromination of cellulose, as it occurred exclusively at the C6 position;32 this was followed by facile nucleophilic displacement of the Br atom with the N3 moiety (step 5, azidation; Figure 1).
Furthermore, to obtain the corresponding azidodeoxycellulose ester, the brominated/chlorinated derivative needed to be acylated before the azidation step, and at this point, we needed to face some problems arising from the poor solubility of brominated/chlorinated cellulose in organic solvents. A one‐pot reaction was recently developed to overcome this limitation by preparing the acylated product after the bromination step without isolating the intermediate.29 We decided to adopt this procedure, however, with some modifications by using the DMA/LiCl system (instead of LiBr). We performed the reaction (step 3, bromination, followed by step 4, acylation; Figure 1) with this slight modification, and in this case, our product was identified as O‐acyl 6‐chloro‐6‐deoxycellulose 2 (Figure 1; NMR spectra are shown in Figures S1 and S2). The facile displacement of bromide with chloride is in line with the literature.32 Decisive evidence for this displacement reaction came from 13C NMR spectroscopy experiments, as instead of a typical signal at δ=32 ppm for the Br‐substituted C6 atom, a signal at δ=43 ppm characteristic of a Cl‐substituted C6 atom was found (Figure S2).28, 33 Furthermore, in the FTIR spectrum, characteristic absorption bands appeared at =730 and 758 cm−1, which could be assigned to the νC−Cl vibration (Figure S3)34 instead of the band at about =550 cm−1 for the νC−Br vibration.28, 35 The DS values were determined to be 2.16 with respect to the propionate units (determined from Figure S1) and 0.81 for the chlorine substituent (determined indirectly, as the benzoylation method indicated substitution of all hydroxy groups36).
Following this experiment, the azidation procedure was executed (step 5, azidation; Figure 1) to afford O‐acylated 6‐azido‐6‐deoxycellulose 3 (Figure 1) with excellent performance (1H NMR, 13C NMR,19F NMR, and FTIR spectra are shown in Figures S4, S5, S6, and S3, respectively), and this yielded a derivative with DS=0.77 according to elemental analysis. Furthermore, for the mechanical test, O‐acylated 6‐azido‐6‐deoxycellulose was synthesized without the XPS tag, and in this case, complete regioselective azidation was achieved with a DS of 1.
Regarding the application of a modified procedure for the bromination reaction, the following issues were considered. According to the original method, cellulose is dissolved in the DMA/LiBr solvent system to avoid nucleophilic displacement of Br with Cl in a DMA/LiCl system. Bromodeoxycellulose is preferred over the chlorinated derivative, as bromide is a better leaving group (for the next SN2 reaction with NaN3). However, on the basis of the reaction rate constants reported for a model displacement reaction comparing chloro‐ and bromodeoxycellulose,35 we did not expect to have any difference on the timescale of the reactions applied in the literature.29 Upon using LiBr, huge amount of the salt needs to be added to the system (at least nine times the amount of cellulose, according to reported procedures), and a higher temperature (160 °C) needs to be applied to dissolve cellulose.28, 29, 30, 32 From this point of view, the use of LiCl is beneficial, as a smaller amount (three times that of cellulose) and a lower temperature (130 °C) are needed.
2.2. Functionalization of the Carbon Fiber Surface using Diazonium Species
Functionalization of the carbon fiber surface was performed by applying a previously elaborated procedure to generate a phenylacetylene moiety on the surface (step 6, in situ diazotation; Figure 1).18, 23, 37 This method exploits the instability of diazonium species, which upon heating generate radicals that are able to be covalently grafted onto the surface.16 The method was proved to be applicable to the carbon fiber surface, as it did not detrimentally affect key single‐fiber performance parameters (e.g. single‐fiber tensile strength).18, 23
Evidence for successful grafting came from high‐resolution C 1s spectral analysis (Figure 2), which clearly showed the presence of new types of C−C bonds on the surface relative to the control carbon fiber (Figure 2, inset). A control sample was prepared from the same carbon fiber by using the same experimental conditions without reagents (only solvents). The main broad C 1s band obtained for the control sample (Figure 2, inset) arises from highly delocalized and localized C−C bonds,14, 15 whereas the band at 288.88 eV can be attributed to defect sites with −COOH and N components and π–π* shake‐up transitions.38
Figure 2.
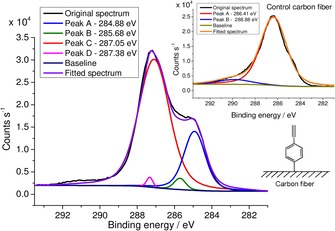
High‐resolution C 1s spectrum of the phenylacetylene‐functionalized carbon fiber (functionalized carbon fiber 1). Inset shows the high‐resolution C 1s spectrum of a control fiber.
Relative to the main band of the control, that of the functionalized sample is slightly shifted to higher energy and has lower full width at half maximum (FWHM); furthermore, in the spectrum of the functionalized sample, a second, appreciably intense band is observable at 284.88 eV, and it can be assigned to sp‐hybridized C≡C bonds, according to a previous study.39 Furthermore, a smaller band can be observed at 286.58 eV, and it can be assigned to sp2 carbon atoms present on the carbon fiber surface. The band at 287.38 eV is still expected to arise from C−C bonds with C(sp2) character, and these bonds are probably in close proximity to the C≡C bonds. The broad band peaking at 291 eV arises from π–π* shake‐up bands of sp‐hybridized carbon atoms; however, interference due to defect sites and π–π* bands of the bare carbon fiber surface cannot be excluded.38
2.3. Binding of O‐Acylated 6‐Azido‐6‐deoxycellulose to the Carbon Fiber through Click Reaction
The click reaction between the functionalized carbon fiber (functionalized carbon fiber 1, Figure 1) sample and azidodeoxycellulose derivative 3 (Figure 1) was performed by using l‐ascorbic acid and CuSO4 ⋅5 H2O to generate a catalytic amount of CuI in situ according to previous studies that applied click chemistry on a carbon fiber surface.23, 26 Despite our efforts to incorporate the XPS tag into the cellulose structure (see above), XPS analysis did not show the presence of fluorine attached to the fiber. Nevertheless, the high‐resolution C 1s and N 1s spectra gave clear evidence for successful establishment of a covalent bond between the polymer and the carbon fiber surface (Figure 3). Besides the main band (at 284.99 eV) belonging to delocalized and localized carbon bonds,14, 15, 38 a band with appreciable intensity centered at 286.48 eV could be assigned to C−N bonds in the triazole ring.40 Furthermore, the band peaking at 288.08 eV was indicative of the presence of C−O linkages originating from cellulose.41 The band at 289.18 eV could be attributed partly to defect sites with −COOH and N components38 and presumably to an sp3‐hybridized carbon atom bound to a nitrogen atom. Moreover, the N 1s spectra undoubtedly substantiated the presence of triazole structures on the carbon fiber surface (Figure 3, inset). According to the survey spectrum in Figure S7, no nitrogen could be detected in the control sample, whereas the nitrogen content amounted to 7.73±0.85 % in the case of the functionalized sample. In conclusion, successful covalent functionalization of the carbon fiber surface was achieved, as outlined in Figure 1.
Figure 3.
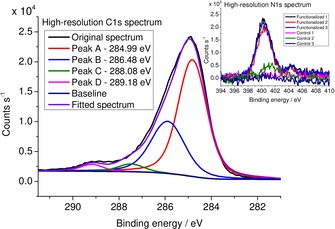
High‐resolution C 1s spectrum of the carbon fiber sample functionalized with the cellulose derivative by a click reaction (functionalized carbon fiber 2). Inset shows the high‐resolution N 1s spectrum of the same sample.
In rationalizing the unsuccessful detection of fluorine, we suggest that the substituent with low DS (DS=0.03) might have been hydrolyzed during the processes with the carbon fiber (this low DS value appeared to be applicable for XPS detection on the basis of our previous study25).
2.4. Functionalization of the Carbon Fiber Surface using Diazonium Species
To gain deeper insight into the effects that determine surface adhesion at a molecular level, a range of entities were deposited on the surface by taking molecular length, rigidity, and polarity of the structure into account. For this purpose, the grafted phenylacetylene moiety served as a scaffold, to which tailor‐made structures could be attached by click chemistry. Upon choosing the synthetic approach, we adapted a previously elaborated, especially effective one‐pot synthetic procedure that was shown to be applicable for electron‐deficient and electron‐rich anilines, in addition to sterically demanding anilines42 (the experimental procedure is shown in the Supporting Information, page S4). We also performed the click reaction with an aminomethylaniline derivative, and in this case, the amino group was protected with a tert‐butoxycarbonyl (Boc) group. After the click reaction, a deprotection step was performed according to a previously elaborated procedure with carbon fiber.18 This reaction was shown to preserve the mechanical properties of the single carbon fiber (experimental procedure is shown in the Supporting Information, page S5).
Being different from the synthetic approach applied for azidodeoxycellulose, we realized that validation of the new strategy needed to be accomplished. In doing so, we chose 4‐(trifluoromethyl)aniline as a model compound. Unfortunately, the fluorine signal expected to arise from the CF3 substituent of the grafted structure could not be obtained in this case either (see above).
The high‐resolution C 1s spectrum (Figure S8) could be resolved to the characteristic component centered at 285.52 eV, representing delocalized and localized C−C bonds.14, 15, 38 From our point of view, spectral components peaking at 286.38 and 287.08 eV are of high interest, as they can be assigned to C−N bonds of the triazole ring and probably to sp2 carbon atoms of the benzene ring bound to nitrogen atoms, respectively.40 Furthermore, the relatively small band at higher binding energies could be attributed to defect sites on the carbon fiber surface (−COOH and N components).38 In conclusion, the high‐resolution C 1s spectrum indicated a successful functionalization process, and the unsuccessful detection of fluorine in this case might be attributed to low functionalization density and the lower sensitivity of XPS experiments to detect fluorine on the surface.
2.5. Surface Morphologies of Carbon Fibers
It was previously shown that single‐fiber mechanical parameters (tensile strength) are not affected negatively during the grafting procedure and the click reaction.18, 23 To confirm that we did not damage the fibers physically during the processing, FE‐SEM images were recorded and chemical mapping was performed (these results are shown in Figures S9–S17). SEM images were also recorded (Figure 4) by using the Au/Pd sputtering technique. The scanning electron micrograph of the control sample shows only slight surface roughness (Figure 4 a), but this surface roughness is slightly increased for the phenylacetylene‐functionalized derivative (Figure 4 b).
Figure 4.

Surface morphologies of a) the control carbon fiber and carbon fiber samples functionalized with b) a phenylacetylene moiety, c) the cellulose derivative, and d) the structure depicted in Figure S8 d.
Furthermore, Figure 4 c indicates that a smooth coating might be present on the surface of the carbon fiber sample functionalized with the cellulose derivative. Similarly, a smooth surface with a slight morphological change can be seen in Figure 4 d for the derivative that was modified by the click reaction by using 4‐(trifluoromethyl)aniline. Moreover, chemical mapping was also performed to observe the integrity of the fiber structure penetrating into deeper layers (during the EDS measurements, we could obtain signals from a few micrometers depth). The EDS images also support the theory that the fibers preserve their structural integrity, and this implies a benign functionalization procedure (Figures S9–S12). Data addressing the chemical mapping are provided in the Supporting Information for all of the functionalized derivatives synthesized in this study (Figures S13–S17).
2.6. Interfacial Shear Strength (IFSS) Determination
Interfacial shear strength, as an ultimate measure of the degree of interfacial adhesion, was determined by using the fragmentation test according to previous studies.16, 18, 19, 20, 22, 23, 43 The theory and calculation of the IFSS can be found in the Experimental Section. As our matrix is a thermoplastic polymer, the composites were prepared by means of a hot‐pressing technique.
As seen in Figure 5, the IFSS increases slightly as the originally purchased carbon fiber is thoroughly cleaned, as this process removes the sizing agent from the surface. This phenomenon can be attributed to the polar groups of the sizing agent, as they do not favor secondary interactions with the aliphatic side chains of the cellulose propionate matrix. A significant increase in the IFSS was obtained for the phenylacetylene‐functionalized carbon fiber sample, and this highlights the importance of the lipophilicity of the grafted structure. Such a gain in the IFSS can be clearly assigned to mechanical interlocking and molecular entanglement effects that can be put in force if the grafted structure can efficiently penetrate into the matrix. For such an event to occur, the grafted structure should be compatible with the matrix so that it is able to establish secondary interactions with the matrix constituents.
Figure 5.
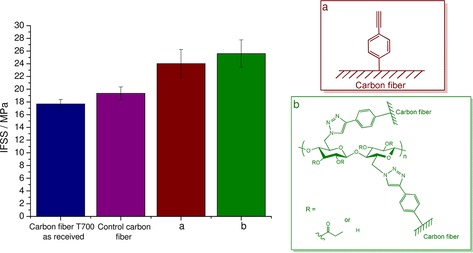
Interfacial shear strength determination of single‐fiber composites: Effect of binding cellulose derivative to the carbon fiber surface.
Interestingly, binding O‐acylated 6‐azido‐6‐deoxycellulose to the surface did not give rise to a significant improvement in the IFSS relative to that of the simple phenylacetylene moiety (Figure 5). By covalently attaching the cellulose derivative to the carbon fiber, we practically make a coating on the surface. Therefore, we are still limited by secondary interactions between the bulk matrix and the coating. In light of other studies in the literature,18, 23 a large increase could only be achieved if there were cross‐links in the matrix that were able to transfer the load within the matrix efficiently to the inherently stronger fiber. Clearly, this is not the case for thermoplastic composites, for which a limit in the IFSS is anticipated to be obtained, and further development is not expected to depend on the interface anymore but on the matrix structure.
To unravel the effects that govern interfacial adhesion, the structures of the deposited entities were tailored to obtain a range of derivatives with different lipophilicities and chain lengths (Figure 6). An enhanced IFSS was obtained for the carbon fiber sample functionalized by the click reaction by using aniline (Figure 6, column c); however, this increase was still lower than that for the phenylacetylene‐functionalized sample (Figure 5). A further improvement could be noticed in the interfacial adhesion if an aminomethyl substituent was attached to the aromatic side chain (Figure 6, column d). This effect was attributed to possible hydrogen‐bonding interactions between the carbonyl oxygen atom of the propionate units of the cellulose derivative and the amino group of the grafted entity. Similarly, a high IFSS was achieved if a propyl side chain was present on the aromatic structure (Figure 6, column e). It is also clear from Figure 6 that increasing the chain length of the substituent on the aromatic structure did not lead to any further increase in the IFSS (columns e–g), probably due to steric reasons. Nevertheless, lipophilicity has a crucial impact on interfacial adhesion, as the highest IFSS was reached if a highly lipophilic and yet compact trifluoromethyl substituent was attached to the aromatic structure (Figure 6, column h). It follows that penetration into the matrix might be the best in this case.
Figure 6.
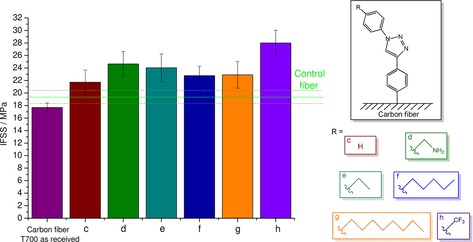
Interfacial shear strength determination of single‐fiber composites: Effect of the structure of the grafted molecule.
2.7. Electrochemical Grafting
An electrochemical grafting experiment was performed to confirm the applicability of the surface modification targeting an industrially relevant robust grafting procedure able to be integrated into the manufacturing train of carbon fibers.22, 23 The initial electrochemical reduction process of the diazonium salt was elaborated by Servinis et al.,23 and we basically followed this well‐established electrochemical method optimized for our experimental setup. In the cyclic voltammetry experiment, a long carbon fiber (≈20 cm) was applied as the working electrode, and it was immersed in 10 mm diazonium salt solution in acetonitrile, also containing 0.1 m tetrabutylammonium hexafluorophosphate as the electrolyte. The cyclic voltammogram obtained for reductive deposition of a phenylacetylene moiety on the carbon fiber surface is shown in Figure S18. The first sweep indicates a successful reduction process typical for diazonium species (reduction peak lies between −0.4 and −0.8 V).22, 23 In the next cycles, a redox process takes place only to a small extent due to the partial saturation of the working electrode during the first sweep (Figure S18). Therefore, to make functionalized carbon fibers for the fragmentation test, only one electrochemical cycle was applied, and this was followed by the click reaction as discussed above. The results of the fragmentation test for the most promising candidates (from Figures 5 and 6) are shown in Figure 7.
Figure 7.
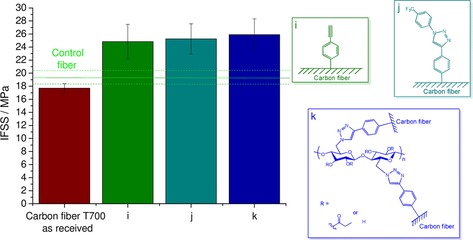
Interfacial shear strength determination of single‐fiber composites: Carbon fibers functionalized by an initial electrochemical grafting procedure.
The IFSS values obtained for the electrochemical process are very close to the values presented in Figures 5 and 6 (purely chemical functionalization), which clearly indicates the potential of an electrochemical method to be applied as a robust surface‐modification technique to intensify the interfacial adhesion in a cellulose‐based carbon fiber reinforced composite.
2.8. Future Outlook
Although the reagents used herein might, in fact, not be considered to be environmentally friendly, through our synthetic procedure many interesting findings have shed light on the molecular design of a carbon fiber surface leading to a green carbon fiber reinforced composite with improved properties. Basic understanding of the effects that govern interfacial interactions in the case of a cellulose derivative as a matrix reinforced with carbon fiber is crucial to develop additional, possibly more environmentally friendly functionalization processes. Furthermore, the authors believe that a process agreeing well with green‐chemistry issues can indeed be elaborated in the future by a facile electrochemical process, which was shown to lead to similar results in this work.
Upon comparing the IFSS values of our study with those from other works in the literature, one needs to keep in mind the crucial role of the matrix in determining the IFSS. Whereas in the case of the commonly used epoxy matrix we have a cross‐linked polymer network that is able to transfer the load to the fiber efficiently, in case of thermoplastic resins, we are mainly restricted to secondary interactions between polymer chains. Therefore, it is not surprising that the highest IFSS values are indeed reported for epoxy resins, particularly for cases in which cross‐links can also connect the fiber with the matrix (e.g. an IFSS of ≈58 MPa was recently reported for functionalized carbon fiber possessing a free amine function23). Nevertheless, the highest IFSS value reported in our study (≈28 MPa) is substantially higher than that for an unfunctionalized fiber in an epoxy matrix (≈18 MPa23). As these materials are already used in applications, the future potential of cellulose‐based carbon fiber reinforced plastic (cellulose‐based CFRP) is indisputable. Furthermore, in the case of the thermoplastic polypropylene matrix, a substantial increase in the IFSS was reported for a carbon fiber functionalized with amine‐capped cross‐linked polyphosphazenes (IFSS≈32 MPa4). Our result is comparable to this value, and the cellulose matrix is still superior with respect to sustainability issues. Therefore, the authors believe that the results obtained in this study hold promise for the successful introduction of cellulose‐based CFRPs as nature‐derived future materials.
3. Conclusions
The successful introduction of cellulose‐based carbon fiber reinforced composites as future green alternatives of their conventionally used, entirely manmade counterparts necessitates tailoring of the interfacial interactions to achieve a material with high performance.
By depositing a phenylacetylene moiety on the carbon fiber surface as a molecular bridge for a click reaction, we successfully functionalized the carbon fiber covalently by using O‐acylated 6‐azido‐6‐deoxycellulose and a range of aromatic azides. As a result of the interfacial shear strength determination, it appeared that lipophilicity and size of the grafted structure were very important for effective penetration into the cellulose propionate matrix and had a profound impact on mechanical interlocking and molecular entanglement effects. An enhanced IFSS, increased by roughly 32 % relative to that of the control sample, was obtained if a covalently bound cellulose propionate coating was present on the surface. Slightly higher interfacial adhesion, increased by about 45 % relative to that of the control sample, was achieved with a small molecule possessing a highly lipophilic and compact −CF3 substituent on the pendant rigid aromatic structure. The potential of the functionalization procedure for industrial application was testified by involving a robust electrochemical treatment in the reaction sequence, which led to similar results.
Thus, a synthetic strategy to bind cellulose derivatives to a largely graphitic surface was provided, and this method should pave the way towards the preparation of innovative cellulose‐based carbonaceous materials.
Experimental Section
Materials
Avicel PH 101, aniline, 4‐pentylaniline, 4‐octylaniline, 4‐[(N‐Boc)aminomethyl]aniline, 4‐trifluoromethyl aniline, anhydrous LiCl, and anhydrous N,N‐dimethylacetamide were obtained from Sigma–Aldrich (St. Louis, MO, USA). ortho‐Dichlorobenzene, tert‐butyl nitrite (isoamyl nitrite), 1,4‐dioxane, 4‐ethylaniline, 4‐ethynylaniline, l‐ascorbic acid, N,N‐dimethylformamide, trimethylsilyl azide, ethylenediaminetetraacetic acid, N‐bromosuccinimide, triphenylphosphine, propionic anhydride, sodium azide, and tetrabutylammonium hexafluorophosphate were supplied by Tokyo Chemicals Industry Co., Ltd. (Tokyo, Japan). Acetonitrile and hydrochloric acid were purchased from Naclai Tesque, Inc. (Kyoto, Japan). All other chemicals were from Kanto Chemical (Tokyo, Japan).
Carbon fiber T700SC‐12000‐50C was provided by Toray Industries (Tokyo, Japan) with the following specifications: diameter, 7 μm; tensile strength, 4900 MPa; sizing agent, 1 % of the total mass. The fibers were cleaned before the experiments to remove the sizing agent and impurities from the surface according to our previous report.25 The cleaning procedure included ultrasonic washing in acetone (2×) for 20 min at 40 °C, followed by rinsing with methanol and water, and ending with drying in a vacuum oven (ADP 200, Yamato Scientific Co., Ltd, Tokyo, Japan) at 150 °C for 3 h. The carbon fibers were stored in sealed vials, and before use, they were cleaned in an ultrasonic bath with acetone (5×) for 10 min; the solvent was changed after each step. The removal of the sizing agent was confirmed by TGA, XPS, and AFM measurements in our previous study.25
Cellulose propionate (M w≈200000 g mol−1) was supplied by Scientific Polymer Products, Inc. (Ontario, NY, USA) and was used as received for the matrix of the composite. The degree of substitution (DS) value was determined to be 2.76 by using the benzoylation method according to a previous study.36
Characterization of Synthesized Cellulose Derivatives
1H NMR and 13C NMR spectra were recorded with JEOL 400 and 600 MHz FT‐NMR spectrometers (Tokyo, Japan) by using [D6]DMSO or CDCl3 as the solvent. The DS value with respect to the propionate unit in the chlorinated sample was calculated from the 1H NMR spectrum by integrating the signal areas of the proton signals of the cellulose backbone (I 3.4–5.5 ppm) and the proton signals of the propionate methyl group (I 0.5‐1.3 ppm) as follows [Eq. (1)]:
| (1) |
The DS value with respect to the azido group was determined from elemental analysis performed with a Micro Order JM10 Organic Elemental Micro Analyzer (J‐Science Lab Co., Ltd., Kyoto, Japan).
FTIR spectra were obtained by using a Thermo Scientific Nicolet iS10 FTIR spectrometer (Waltham, MA, USA) and a GladiATR accessory (Pike Technologies, Madison, WI, USA). An average of 128 scans was taken with a resolution of 2 cm−1.
Surface Analysis of Carbon Fiber Samples
XPS analysis was performed by using a Thermo Scientific K‐Alpha X‐ray Photoelectron Spectrometer System with an AlKα (1486.6 eV) monochromated X‐ray source (12 kV×3 mA) (Waltham, MA, USA). Carbon fiber filaments were fixed at both ends to the sample holder by using a carbon tape, and the target spot was chosen to cover the carbon fibers only (400 μm X‐ray spot size). The elemental composition of the surface was discovered by recording the survey spectrum with a resolution of 1 eV. High‐resolution C 1s, N 1s, O 1s, and F 1s spectra were obtained with a resolution of 0.1 eV by using a pass energy of 20 eV. Data processing was performed with Thermo Scientific Avantage Software version 5.89 (Waltham, MA, USA).
Surface topography of the samples was investigated by using a Hitachi S4500 scanning electron microscope by applying an accelerating voltage of 15 kV; the Au/Pd coating was deposited with a Hitachi E1030 ion sputter for 40 s (Hitachi, Ltd., Tokyo, Japan). Field‐emission scanning electron microscopy (FE‐SEM) experiment was also performed with a JSM‐7610F and an accelerating voltage of 15 kV (JEOL, Tokyo, Japan). The FE‐SEM system was coupled with a JEOL EX‐230**BU EX‐37001 energy‐dispersive X‐ray analyzer, which enabled us to record the energy‐dispersive X‐ray spectrum and to carry out chemical mapping experiments.
Interfacial Shear Strength Determination
The IFSS was determined by using the single‐fiber fragmentation method. A hydraulic hot‐press machine (Type MH‐10, Imoto Machinery Co., Kyoto, Japan) was applied to prepare the single carbon fiber–cellulose propionate composite according to our previous study.25 At least ten specimens were prepared from each type of carbon fiber samples. The specimens were strained by using a Shimadzu Autograph AG‐X Plus 5 kN tensile tester (Kyoto, Japan) with a crosshead speed of 0.5 mm min−1 until saturation in the fiber‐fragmentation process occurred. The fragmentation test was followed in real time with a high‐resolution digital camera (N.O.W.‐D2X3Z‐KSH, Nihonkouki, Aoki, Japan). The fragment size was measured by means of an AUSB‐K version 14.4 program (Nihonkouki, Aoki, Japan) calibrated with an objective micrometer (Shibuya Optical Co., Ltd., Wako, Japan). The apparent IFSS (τ) was calculated from the average fragment size (l) according to the Kelly–Tyson model [Eqs. (2), (3), (4)]:44, 45
| (2) |
| (3) |
| (4) |
in which l c is the critical fragment size, σ fu is the strength of the fiber at the critical length, and d is the fiber diameter. The value of σ fu can be calculated by using the average tensile strength (σl) of the fiber according to Eq. (4), in which ll is the gauge length applied in the single‐fiber tensile test experiment and m is the Weibull modulus representing the data spread of the single‐fiber tensile strength.45, 46
Significant differences between interfacial shear strength values were analyzed (multiple t‐test, α=0.05) by using GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA).
Synthetic procedures for preparing the cellulose derivatives and functionalized carbon fibers can be found in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors gratefully acknowledge the COI program “Construction of next‐generation infrastructure using innovative materials—Realization of a safe and secure society that can coexist with the Earth for centuries” supported by Ministry of Education, Culture, Sports, Science and Technology (MEXT) and Japan Science and Technology Agency (JST). This study was also supported in part by the Advanced Low Carbon Technology Research and Development Program (ALCA) of the JST and the Cross‐ministerial Strategic Innovation Promotion Program (SIP) from the JST.
L. Szabó, S. Imanishi, N. Kawashima, R. Hoshino, D. Hirose, T. Tsukegi, K. Ninomiya, K. Takahashi, ChemistryOpen 2018, 7, 720.
Contributor Information
Dr. László Szabó, Email: szabo-laszlo@se.kanazawa-u.ac.jp.
Prof. Kenji Takahashi, Email: ktkenji@staff.kanazawa-u.ac.jp.
References
- 1. Witik R. A., Teuscher R., Michaud V., Ludwig C., Månson J. A. E., Composites Part A 2013, 49, 89. [Google Scholar]
- 2.S. Das, J. Warren, D. West, S. M. Schexnayder, Global Carbon Fiber Composites Supply Chain Competitiveness Analysis, Technical Report ORNL/SR-2016/100-NREL/TP-6A50-66071, Clean Energy Manufacturing Analysis Center, Denver, USA, 2016.
- 3. Henshaw J. M., Han W. J., Owens A. D., J. Thermoplast. Compos. Mater. 1996, 9, 4. [Google Scholar]
- 4. Zhang X., Xu H., Fan X., RSC Adv. 2014, 4, 12198. [Google Scholar]
- 5. Li N., Wu Z., Huo L., Zong L., Guo Y., Wang J., Jiang X., RSC Adv. 2016, 6, 70704. [Google Scholar]
- 6. Lee H. S., Kim S. Y., Noh Y. J., Kim S. Y., Composites Part B 2014, 60, 621. [Google Scholar]
- 7. Liu D., Zhu Y., Ding J., Lin X., Fan X., Composites Part B 2015, 77, 363. [Google Scholar]
- 8. Zhang K., Zhang G., Liu B., Wang X., Long S., Yang J., Compos. Sci. Technol. 2014, 98, 57. [Google Scholar]
- 9. Karger-Kocsis J., Mahmood H., Pegoretti A., Prog. Mater. Sci. 2015, 73, 1. [Google Scholar]
- 10. Sharma M., Gao S., Mäder E., Sharma H., Wei L. Y., Bijwe J., Compos. Sci. Technol. 2014, 102, 35. [Google Scholar]
- 11. He J., Huang Y., Meng L., Cao H., Gu H., J. Appl. Polym. Sci. 2009, 112, 3380. [Google Scholar]
- 12. Peng Q., Li Y., He X., Lv H., Hu P., Shang Y., Wang C., Wang R., Sritharan T., Du S., Compos. Sci. Technol. 2013, 74, 37. [Google Scholar]
- 13. Servinis L., Henderson L. C., Gengenbach T. R., Kafi A. A., Huson M. G., Fox B. L., Carbon 2013, 54, 378. [Google Scholar]
- 14. Ma L., Meng L., Fan D., He J., Yu J., Qi M., Chen Z., Huang Y., Appl. Surf. Sci. 2014, 296, 61. [Google Scholar]
- 15. Ma L., Meng L., Wang Y., Wu G., Fan D., Yu J., Qi M., Huang Y., RSC Adv. 2014, 4, 39156. [Google Scholar]
- 16.L. Servinis, Ph.D. Thesis, Deakin University, Geelong, Australia, 2014.
- 17. Wang Y., Meng L., Fan L., Ma L., Qi M., Yu J., Huang Y., Appl. Surf. Sci. 2014, 316, 366. [Google Scholar]
- 18. Servinis L., Henderson L. C., Andrighetto L. M., Huson M. G., Gengenbach T. R., Fox B. L., J. Mater. Chem. A 2015, 3, 3360. [Google Scholar]
- 19. Beggs K. M., Servinis L., Gengenbach T. R., Huson M. G., Fox B. L., Henderson L. C., Compos. Sci. Technol. 2015, 118, 31. [Google Scholar]
- 20. Servinis L., Gengenbach T. R., Huson M. G., Henderson L. C., Fox B. L., Aust. J. Chem. 2015, 68, 335. [Google Scholar]
- 21. Wang Y., Meng L., Fan L., Wu G., Ma L., Zhao M., Huang Y., Appl. Surf. Sci. 2016, 362, 341. [Google Scholar]
- 22. Servinis L., Beggs K. M., Scheffler C., Wölfel E., Randall J. D., Gegenbach T. R., Demir B., Walsh T. R., Doeven E. H., Francis P. S., Henderson L. C., Carbon 2017, 118, 393. [Google Scholar]
- 23. Servinis L., Beggs K. M., Gengenbach T. R., Doeven E. H., Francis P. S., Fox B. L., Pringle J. M., Pozo-Gonzalo C., Walsh T. R., Henderson L. C., J. Mater. Chem. A 2017, 5, 11204. [Google Scholar]
- 24. Demir B., Henderson L. C., Walsh T. R., ACS Appl. Mater. Interfaces 2017, 9, 11846. [DOI] [PubMed] [Google Scholar]
- 25. Szabó L., Imanishi S., Kawashima N., Hoshino R., Takada K., Hirose D., Tsukegi T., Ninomiya K., Takahashi K., RSC Adv. 2018, 8, 22729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang Y., Ibrahim A. A., Stockdill J. L., Hashemi P., Anal. Methods 2015, 7, 7352. [Google Scholar]
- 27. Negishi K., Mashiko Y., Yamashita E., Otsuka A., Hasegawa T., Polymers 2011, 3, 489. [Google Scholar]
- 28. Fox S. C., Edgar K. J., Cellulose 2011, 18, 1305. [Google Scholar]
- 29. Fox S. C., Edgar K. J., Biomacromolecules 2012, 13, 992. [DOI] [PubMed] [Google Scholar]
- 30. Matsui Y., Ishikawa J., Kamitakahara H., Takano T., Nakatsubo F., Carbohydr. Res. 2005, 340, 1403. [DOI] [PubMed] [Google Scholar]
- 31. Baumann H., Liu C., Faust V., Cellulose 2003, 10, 65. [Google Scholar]
- 32. Furuhata K., Koganei K., Chang H. S., Aoki N., Sakamoto M., Carbohydr. Res. 1992, 230, 165. [DOI] [PubMed] [Google Scholar]
- 33. Cimecioglu A. L., Ball D. H., Kaplan D. L., Huang S. H., Macromolecules 1994, 27, 2917. [Google Scholar]
- 34. Furuhata K., Chang H. S., Aoki N., Sakamoto M., Carbohydr. Res. 1992, 230, 151. [DOI] [PubMed] [Google Scholar]
- 35. Aoki N., Sakamoto M., Furuhata K. in Cellulose Derivatives: Modification, Characterization, and Nanostructures (ACS Symposium Series), (Eds.: T. J. Heinze, W. G. Glasser), Washington, DC, 1998, Vol. 688, Ch. 6. [Google Scholar]
- 36. Kakuchi R., Ito R., Nomura S., Abroshan H., Ninomiya K., Ikai T., Maeda K., Kim H. J., Takahashi K., RSC Adv. 2017, 7, 9423. [Google Scholar]
- 37. Bahr J., Tour J. M., Chem. Mater. 2001, 13, 3823. [Google Scholar]
- 38. Zhang G., Sun S., Yang D., Dodelet J. P., Sacher E., Carbon 2008, 46, 196. [Google Scholar]
- 39. Kang X., Zuckerman N. B., Konopelski J. P., Chen S., Angew. Chem. Int. Ed. 2010, 122, 9786; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9686. [Google Scholar]
- 40. Ciampi S., Böcking T., Kilian K. A., James M., Harper J. B., Gooding J. J., Langmuir 2007, 23, 9320. [DOI] [PubMed] [Google Scholar]
- 41. Johansson L. S., Campbell J. M., Surf. Interface Anal. 2004, 36, 1018. [Google Scholar]
- 42. Barral K., Moorhouse A. D., Moses J. E., Org. Lett. 2007, 9, 1809. [DOI] [PubMed] [Google Scholar]
- 43. Lopattananon N., Kettle A. P., Tripathi D., Beck A. J., Duval E., France R. M., Short R. D., Jones F. R., Composites Part A 1999, 30, 49. [Google Scholar]
- 44. Kelly A., Tyson W. R., J. Mech. Phys. Solids 1965, 13, 329. [Google Scholar]
- 45. Kettle A. P., Beck A. J., O'Toole L., Jones F. R., Short R. D., Compos. Sci. Technol. 1997, 57, 1023. [Google Scholar]
- 46.K. Naito in Improvement of Resin Impregnation Property and Reliability Evaluation of CFRP (carbon fiber reinforced plastic), Technical Information Institute Co., Ltd., Tokyo, Japan 2010, Ch. 4, pp. 34–46; in Japanese.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary