

Abstract
Topochemical engineering is a method of designing the fractionation (disassembly) and fabrication (assembly) of highly engineered functional materials using a combination of molecular and supramolecular techniques. Cellulose is one of the naturally occurring biopolymers, currently considered to be an important raw material for the design and development of sustainable products and processes. This feature article deals with new insights into how cellulose can be processed and functionalized using topochemical engineering in order to create functional fibers, enhance biopolymer dissolution in water-based solvents, and control the shaping of porous materials. Subsequently, topochemical engineering of cellulose offers a variety of morphological structures such as highly engineered fibers, functional cellulose beads, and reactive powders that find relevant applications in pulp bleaching, enzyme and antimicrobial drug carriers, ion exchange resins, photoluminescent materials, waterproof materials, fluorescent materials, flame retardants, and template materials for inorganic synthesis. The topochemical engineering of biopolymers and biohybrids is an exciting and emerging area of research that can boost the design of new bioproducts with novel functionalities and technological advancements for biobased industries.
Introduction
Wood, one of the most abundant natural materials in this world, has been used as a thermal energy resource, construction material, and raw material for pulp fibers for the large-scale production of paper. Recently, electronic materials revolutionized the medium of communication and caused a reduced usage of printing paper. However, new applications of pulp fiber in personal care and packaging keep increasing with the growing population and demands for a high-quality life. The annual production of pulp exceeds 160 million tonnes, and emerging trends in utilizing biomass in refineries for transportation fuels and chemicals have caused forest industries to expand their focus on pulp and biomass valorization.
Pulp is a lignocellulosic material containing cellulose fibers, hemicelluloses, and lignin. Pulp fibers are extracted from wood using mechanical, chemo-mechanical, and chemical methods and are bleached prior to paper- and board-making applications. Pulping and bleaching techniques have huge influences on the quality of the resulting pulp, which gives rise to a variety of pulp grades suitable for different purposes depending on the residual amounts of lignin and hemicelluloses present in the material. Mechanical and chemo-mechanical fibers contain greater amounts of lignin and hemicelluloses than do chemical pulps. All pulps contain anionic groups (AGs) that are of high relevance to interfacial interactions with polyelectrolytes and other cationic chemicals used in the manufacture of fiber-based materials. The quantity of AGs on fibers depends on the macromolecular properties and quantity of hemicelluloses of wood raw material and the dissolution and reaction of biopolymers during pulping and bleaching.1
Kraft and sulfite pulping are the dominant chemical pulping techniques for the production of fibers. The bleaching of chemical pulp fibers is currently performed using elemental chlorine free (ECF) or total chlorine free (TCF) sequences. Fibers produced using a combination of Kraft pulping and ECF bleaching dominate the market of chemical pulps and are used in numerous applications including paper, packaging, tissue products, and absorbent materials in diapers. Prehydrolysis Kraft pulping or sulfite pulping combined with TCF bleaching is used for the extensive removal of hemicellulose and lignin for the production of pulps suitable for cellulose dissolution and derivatization to cellulose products. For many years, cellulose has been seen as a pulp and paper source material. However, the scenario changed very fast due to the invention of potential applications of cellulose fibers in thin films, textiles, and personal care. These new concepts are the driving force in studying its chemical and physical properties in order to convert/value-add cellulose to novel applied materials. Cellulose is a polysaccharide in which anhydroglucose units are repeated. These units have primary hydroxyl groups at the C6 position and secondary hydroxyl groups at the C2 and C3 positions. These hydroxyl functionalities pave the way to modify cellulose chemically, for example, in the preparation of cellulose esters and ethers. Oxidation is another modification process in which the primary −OH groups at position C6 can be oxidized to −COOH and secondary −OH groups at positions C2 and C3 and to −CHO and −COOH.2,3 These treatments are aimed to modify cellulose from neutral to anionic or cationic, to change its hydrophilic nature to hydrophobic character, and to increase its mechanical strength and chemical reactivity. Overall, cellulose has a unique structure, which results in properties such as hydrophilicity, crystallinity, multichirality, stereoregularity, thermal and mechanical stability, biocompatibility, and sustainability. Our current research focuses on the topochemical engineering of cellulose into well-defined objects such as sponges, fibers, film or sheets, and spherical beads having the desired geometry and functionality. The challenge pertaining to these new morphological formations include the disassembly of inter/intramolecular hydrogen bonding in cellulose fibers to separate them as independent cellulose molecules, the choice of solvents and reagents, and the process of extrusion and drying. When these new morphologies are functionalized or assembled with applied molecules possessing the desired properties, they unveil new surfaces and interfaces carrying targeted functionalities suitable for a variety of functional material applications.
Topochemical Engineering of Cellulose
A topochemical reaction is one in which both the nature and properties of the products of the reaction are governed by the three-dimensional topological environment of molecules or atoms.4 Photochemical reactions in organic crystals5 are examples of topochemical reactions that have been extensively studied by organic chemists. Langmuir also first noted the kinetic consequences of reaction zones in a topological space6 (i.e., the rate of a topochemical reaction is not proportional to the total amount of unreacted material but rather to the amount of material present in the reaction zone). The field of topochemistry is fascinating, and different definitions of topochemical reactions have been suggested.7 However, in our view, the relevance of topochemistry has been overlooked in many scientific fields and sometimes even confused with surface engineering or supramolecular chemistry. Reactions in three-dimensional confined spaces are extremely relevant to cell metabolism, protein biosynthesis, and numerous biological interactions and materials. Topochemistry influences the structure–function relationship of natural and man-made systems. Topochemical engineering, in our view, is a method of directed assembly or directed disassembly of functional materials. Directed assembly and disassembly are controlled via the design of molecular and intermolecular interactions in a topological space. The directed assembly uses electrostatic interactions, hydrogen bonding, hydrophobic interactions, or solid-state cross-linking reactions of components to create functional shapes and interfaces. The directed disassembly of a component from a multicomponent system is designed on the basis of the controlled cleavage of molecular and intermolecular bonds and the formation of new molecular and intermolecular interactions that enhance separation and selected fractionation. This feature article focuses on the topochemical assembly of functional cellulose fibers and the shaping of functional cellulose beads. The degradation of fiber primary wall layers to enhance cellulose dissolution in water-based solvents, the removal of lignin to enhance the purification of cellulose, and enzymatic hydrolysis to glucose are examples of the topochemical disassembly presented here.
Topochemical Engineering of Fiber Surfaces with Biomolecules
Cellulose fibers in their natural form are chemically inactive with low reactivity for the modification of −OH groups under multiphase conditions. However, fibers contain carboxyl groups that are ionized in water and in combination with hydroxyl groups form an excellent template for intermolecular interactions such as electrostatic and hydrogen bonds. The topochemical engineering of fiber surfaces is an attractive strategy for large-scale application because it can be performed in water using intermolecular interactions between fibers and organic/inorganic molecules/complexes. Furthermore, this approach can also be applied to make fibers more reactive for subsequent processing steps, reducing processing time and energy input. In addition, new functional bioproducts can be designed and tuned with targeted properties.
Cellulose Fiber Surfaces Modified with Xylan Biopolyelectrolytes
Cellulose fiber surfaces can be treated with a hemicellulose derivative in order to make them more reactive for chemical conversions and other charged pulp applications. The adsorption of hemicelluloses on pulp for fiber modification was started in the early 1950s.8−11 However, the main disadvantage of hemicelluloses was their laborious isolation step followed by obtaining a homogeneous composite material with cellulose. Alternatively, we used pressurized hot water for xylan extraction from birch wood. From xylan, a group of hemicelluloses can be derivatized to carboxymethyl xylan (CMX), xylan sulfate (XS), and xylan-4-[N,N,N-trimethylammonium] butyrate chloride (XTMAB) through functional modification (Figure 1a). These new functionalities acted as polyelectrolytes (PEs), and they were able to adsorb on cellulose surfaces (pine Kraft pulp) due to their charge. The adsorbed PEs were determined by wet chemical titrations. The results suggested that the cellulose surface had more affinity for the cationic xylan derivative (XTMAB) than for anionic derivatives XS and CMX. The surface anionic groups (SAGs) value calculated by XPS showed that the number of anionic groups at the pulp surface matched the maximum number of cationic groups that could be adsorbed onto the fiber surfaces. This revealed that the sorption of XTMAB onto the fiber surfaces was mainly driven by electrostatic forces. Thus, charge interactions overruled hydrogen bond interactions. The adsorption of XTMAB followed the Langmuir model of the sorption isotherm (Figure 1b). ToF-SIMS (time-of-flight secondary ion mass spectrometry) imaging revealed that the distribution of XTMAB was even on fiber surfaces, and thus XTMAB turned out to be a efficient fiber-modifying agent.12 Since XTMAB is renewable, it can be used as a biopolyelectrolyte in pulp and papermaking processes to enhance the mechanical strength and sustainability.
Figure 1.

(a) Schemes for the synthesis of ionic xylan derivatives illustrated using an idealized xylan. (b) Sorption isotherm showing the amount of xylan-4-[N,N,N-trimethylammonium]butyrate chloride (XTMAB) per 100 mg of bleached pine Kraft pulp versus meq free representing the amount of XTMAB still in solution after sorption. Reprinted from ref (12) with permission from Elsevier.
Reactive Fiber Interfaces Using Multifunctional Cellulose Derivatives
Reactive fibers carrying special functional groups can be obtained from various physical, chemical, and enzymatic treatments. However, these treatments should not degrade cellulose with the inherent loss of its mechanical properties.13−18 The disadvantages with many other methods were the elevated operating temperatures and uncontrolled assembly of guest molecules on the fiber surface. To counter this, pulp fibers were treated with novel water-soluble derivative of cellulose in order to produce a charge-induced reactive interface at room temperature. The ionic groups of the cellulose derivative triggered the solubility in water and allowed charge-mediated self-assembly. Here, a cellulosic −OH group was converted to an azide functionality (a (3-carboxypropyl)trimethylammonium chloride ester of 6-deoxyazidocellulose) and then this derivative underwent charge-directed self-assembly on the bleached Kraft pine pulp fibers in an aqueous environment at 25 °C. It was found that the cationic group of N3-cell+ electrostatically attached to the anionic counterpart of the pulp fibers and remained intact. Furthermore, these azide groups were available for covalent bond formation with any alkyne molecule in the presence of a transition-metal catalyst (e.g., Cu1+) for cycloaddition. The (3-carboxypropyl)trimethylammonium chloride ester of 6-deoxyazidocellulose (N3-cell+)-decorated cellulose fibers was applied in the copper(I)-catalyzed azide–alkyne Huisgen cycloaddition (CuAAc) reaction (Figure 2a) to prove the reactivity, and the final products were amino (−NH2) and photoactive cellulose (Figure 2b,c). N3-cell+ was also prepared from an organic solvent medium, and it was found to have no influence on the surface morphology of the fibers.19 These functionalized fibers can be applied in the production of photoluminescent tapes and labels for authentication purposes.
Figure 2.

(a) Synthesis scheme for the preparation of cellulose fibers decorated with photoactive molecules (photofibers) and amino functional groups (amino fibers) prepared from cellulose fibers decorated with azide functions (reactive fibers). Cu(II)-TBTA, Cu(II)-tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine complex solution; AAc, ascorbic acid; TMA, triethylammonium acetate buffer. (b) Section (xy plane) of scanned xyz volume of photofibers obtained with TPM (left). A 3D rendering of a cross-section of an individual fiber at the position marked as “ROI2” on the left figure (right). The figures illustrate the dense labeling of the fibers and the preserved 3D shape during the activation and labeling. (c) Image of a photofiber observed with an Olympus BX60 epi-fluorescence microscope at 10× (left) and 40× magnifications (right). Excitation filter, 330–385 nm; dichroic mirror, 400 nm; and barrier filter, >420 nm. Reprinted from ref (19) with permission from Elsevier.
Biopolyelectrolyte Complexes for Engineering Cellulose Fiber Surfaces
Assembling novel polysaccharide derivatives functioning as polyelectrolytes or polyelectrolyte complexes (PEC) on the charge-directed sites of pulp fibers is another way of functionalizing cellulose (Figure 3a). Previous studies focused on adsorbing polysaccharide-based polyelectrolyte multilayer systems on pulp to improve paper quality.20−27 The present study28 used never-dried bleached sulfate (Kraft) pine pulp as a substrate and cellulose (3-carboxypropyl)trimethylammonium chloride ester (CN+), carboxymethyl xylan (CMX–), and xylan sulfate (XS–) as assemblies or modifying agents (Figure 3b). The assemblies were made by reacting CMX– and XS– with CN+, which in turn produced [CN+CMX–] and [CN+XS–] complexes. These complexes were made to bind with anionic cellulose fibers electrostatically for functional modification. Thus, polycations acted as carriers for valuable polyanions.
Figure 3.

(a) Graphical representation of the functionalizing cellulose with PEC. (b) Cellulose (3-carboxypropyl)trimethylammonium chloride ester (CN+), carboxymethylxylan (CMX–), and xylan sulfate (XS–). (c) Sorption isotherms obtained from polyelectrolyte titrations of [CN+CMX–] (a system composed of carboxymethylxylan (CMX–) and cellulose(3-carboxypropyl)trimethylammonium chloride ester (CN+)), [CN+XS–] (a system composed of xylan sulfate (XS–)), and CN+. S, amount of adsorbed charge (μmol/g); C, equilibrium concentration in the solution (μmol/L). Reprinted from ref (28) with permission from the American Chemical Society.
The adsorption of a single polymer of high charge density, CN+ and PECs ([CN+CMX–], [CN+XS–]) on the pulp fibers was determined by elemental analysis and electrochemical titrations. The fibers treated with PECs (neutral) had double the amount of adsorption compared to CN+-treated fibers. Thus, it was evident that the higher charge density on CN+ lessened its adsorption, and it was sufficient for the modification. However, these experiments showed that the maximum amount of net charge developed on the fiber surface was the same irrespective of the modifying agent used, proving that the adsorption was purely electrostatic in nature (Figure 3c). The PECs were chemically stable during the course of the time after the adsorption. The distribution of fiber-modifying agents was measured by a surface-sensitive technique, ToF-SIMS. The results revealed that the mechanical properties must be different for the modified fibers on their charged interfaces than for the unmodified fibers.28 The newly introduced charged species on the fiber surfaces can be utilized in strengthening paper, the immune detection of viruses, organic syntheses, ion exchange resins, and charge transfer reactions.
Topochemical Activation of Pulp Fibers Prior to Bleaching
The activation of the cellulose surface by the employment of precursors such as peracids of mono and disaccharides before peroxide bleaching was a new attempt where these activators promoted high brightness and whiteness in mechanical pulps. Activators used were weak acid esters and a weak base such as lactose octaacetate (LOA, colloidal particles, and low water solubility), sucrose octaacetate (SOA, partially soluble in water), and tetraacetylethylenediamine (TAED, water-soluble). TAED was the first reported activator in hydrogen peroxide bleaching for enhanced laundry performance and was later applied in mechanical and chemical pulps.29−37 The mechanism behind the activators was that they generate active oxygen from hydrogen peroxide (the bleaching agent) via peroxyacid intermediates. These peroxyacid anions were formed in situ by the hydrolysis of esters or amides. The activators introduced carboxylic and amino groups to the fiber surfaces at different depths, yielding enhanced mechanical strength for the pulp.38 The results showed that LOA was very effective in topochemical activation even at low concentrations as the brightness increased during bleaching and the surface coverage of lignin was minimal (Table 1, Figure 4). The important observation made here was that the aqueous solubility of the activators negatively influences the surface specificity. This pretreatment approach can be applied in pulp bleaching to achieve a reduced coverage of lignin on fibers and enhanced brightness. The disadvantage with this method could be the toxic wastes produced in the process, such as unreacted TAED and H2O2.
Table 1. Brightness Parameters on Bleached TMP with Different Surface Activatorsa.
| pH |
residual (kg/ton) |
||||||
|---|---|---|---|---|---|---|---|
| sample | initial | final | peroxide | alkali | brightness % ISO | whiteness % | yellowness % |
| unbleached (reference) | 4.5 | 4.5 | 58.0 ± 0.1 | 1.3 ± 0.1 | 27.9 ± 0.1 | ||
| H2O2 | 11.4 | 9.0 | 1.42 | 0.01 | 65.6 ± 0.1 | 19.1 ± 0.2 | 22.8 ± 0.1 |
| TAED | 11.3 | 8.2 | 2.41 | 0.02 | 66.9 ± 0.2 | 22.6 ± 0.2 | 21.6 ± 0.1 |
| SOA | 11.5 | 8.2 | 2.58 | 0.02 | 67.1 ± 0.1 | 23.1 ± 0.2 | 21.4 ± 0.1 |
| LOA | 10.7 | 7.4 | 1.14 | 0.01 | 68.2 ± 0.2 | 24.4 ± 0.3 | 21.3 ± 0.1 |
Reprinted from ref (38) with permission from Springer Nature.
Figure 4.

ToF-SIMS positive mode image of the lignin distribution before and after the bleaching of thermomechanical pulp (TMP) with and without activators. The size of the images is 100 μm × 100 μm. Reprinted from ref (38) with permission from Springer Nature.
Photoresponsive Cellulose Fibers
Light responsive cellulose fibers were developed by incorporating novel biobased cellulose derivatives on the eucalyptus pulp fibers. Light-induced chemical conversions such as cross-linking, dissociation, or isomerization were previously reported to cause changes in the physical properties of soft materials.39−41 In our work, cellulose was converted to its multifunctional derivative in order to have both cationic and photoactive groups.42 The cationic group introduced on the maiden cellulose was (3-carboxypropyl)trimethylammonium chloride ester moieties, and the photoactive groups were 2-[(4-methyl-2-oxo-2H-chromen-7-yl)oxy]acetate substituents (Figure 5a,b, Table 2). The adsorption of cellulose derivative on the pulp fibers was driven by electrostatic interaction and followed the Freundlich model. Hydrophobic interactions leading to multilayers were also suggested. When UV light was shined on the modified material, there was fast photo-cross-linking as measured by the change in the light absorption and fluorescence of the pulp fibers. This type of light-controlled cross-linking also increased mechanical properties such as the tensile strength of the pulp fibers.42 Hence, these functionalized fibers will find application in photoluminescent textiles, films, composites, and absorbent substrates for personal care.
Figure 5.

(a) Schematic presentation of the structure of the photoactive cationic cellulose derivative (PCCD). (b) Schematic illustration of the adsorption of the photoactive cationic cellulose derivative (PCCD) onto the pulp fibers. Reprinted from ref (42) with permission from Elsevier.
Table 2. Adsorption Data of Photoactive Cationic Cellulose Derivatives (PCCDs) Treated with Langmuir and Freundlich Equationsa.
| adsorption
model | |||||||
|---|---|---|---|---|---|---|---|
| Langmuir |
Freundlich |
||||||
| derivative | ns | KL | R2 | R2 | n | 1/n | A |
| PCCD-1b | 36.76 | 0.007 | 0.8980 | 0.9606 | 2.86 | 0.35 | 3.0 |
| PCCD-2b | 30.30 | 0.011 | 0.9478 | 0.9915 | 2.56 | 0.39 | 2.4 |
Reprinted from ref (42) with permission from Elsevier.
The DS (degree of substitution) of the cationic group is 0.34, and the DS values of the photoactive group are 0.11 and 0.37 for PCCD-1 and PCCD-2, respectively.
Photocontrolled Formation of Fiber-to-Fiber Bonds Using Polysaccharide Derivatives
Directed assembly of polysaccharide derivatives such as photoactive cationic cellulose on pulp fibers can lead to changes in its mechanical properties, such as the tensile strength of fiber networks and the stiffness of individual fibers.43,44 These cationic cellulose derivatives underwent a (2π + 2π) cycloaddition reaction under UV-light exposure (320 nm), leading to fast photo-cross-linking of the covalent bonds between the photoactive groups. These groups were 2-[(4-methyl-2-oxo-2H-chromen-7-yl)oxy]acetate (coumarin) and (3-carboxypropyl)trimethylammonium chloride ester moieties. These moieties adsorbed on the bleached eucalyptus Kraft pulp surface via electrostatic interactions and in the presence of UV light facilitated the creation of new fiber-to-fiber bonds (Figure 6a,b). Dynamic mechanical analysis (DMA) and Z-directional tensile testing characterized the outcome of this fiber modification. This testing revealed that there was an increase in the unidirectional stiffness of fibers by 60% and in the strength of the fiber network from 81 to 84%.44 Thus, the mechanical properties of both individual fibers and fiber networks can be engineering to improve the performance in smart packaging, composites, membranes, and tissue paper.
Figure 6.

(a) Schematic drawing of the structure of the photoactive cationic cellulose derivative and its interaction with pulp fibers. (b) Visualization of the 2π + 2π cycloaddition reaction creating covalent bonds between pulp fibers and along the fibrils of the fiber. Reprinted from ref (44) with permission from John Wiley and Sons.
Fluorescent Cellulose Fibers
Polysaccharide materials exhibiting photoluminescence were composed of pulp fibers under aqueous conditions in order to make them fluoresce under given conditions. The polysaccharide derivative studied was N-(3-propanoic acid)- and N-(4-butanoic acid)-1,8-naphthalimide esters of cellulose with (3-carboxypropyl)trimethylammonium chloride cationic moieties (Figure 7). The adsorption of a fluorescent multifunctional cellulose derivative (FMCD) on the pulp fibers was facilitated by an ion-exchange mechanism,45 and it was also dependent on the length of the aliphatic chain connecting the naphthalimide group to cellulose fibers. When the aliphatic chain length increased, the adsorption decreased as shown by propanoic and butanoic naphthalimide moieties. The modified material fluoresced in the dark and distinguished itself from the reference unmodified fibers.
Figure 7.

Synthesis scheme of N-(3-propanoic acid)-1,8-naphthalimide and N-(4-butanoic acid)-1,8-naphthalimide esters of cellulose and the corresponding mixed naphthalimide (3-carboxypropyl)trimethylammonium chloride esters of cellulose via in situ activation of N-(3-propanoic acid)-1,8-naphthalimide, N-(4-butanoic acid)-1,8-naphthalimide, and (3-carboxypropyl)trimethylammonium chloride4 with N,N-carbonyldiimidazole (CDI) in N,N-dimethylacetamide/LiCl (DMA/LiCl). Reprinted from ref (46) with permission from the American Chemical Society.
The light absorption measurements were carried out using a UV–vis spectrometer in which reference fibers exhibited absorption at 230 and 275 nm originating from residual hexenuronic acid and lignin, whereas modified fibers showed strong absorption at 340 nm which corresponded to the naphthalimide chromophore. The fluorescent properties of the modified fibers were studied using a spectrofluorimeter, and the emission band at 393–398 nm was attributed to naphthalimides. Also, the modified fibers were scrutinized by epifluoro microscopy under UV and white light exposure. In the case of UV, the fibers were excited at 330–385 nm and the emitted light was collected at λ > 420 nm. The morphology and microstructural elements looked similar in both lighting environments (Figure 8a,b). Figure 8c showed the fluorescence illumination of hand sheets made up of modified fibers obtained from propanoic and butanoic naphthalimide moieties. Though the butanoic naphthalimide moiety had lower adsorption on the fibers compared than did the propanoic counterpart, it showed a high degree of luminescence. The fluorescent behavior of the modified fibers can be utilized for identity or authenticity checks in packaging, fluorescent labeling, and detector applications.46 A few other reports also dealt with cellulose modification with the use of fluorescent whitening agents (FWA) on the fibers to make them fluoresce, but they suffered from a low affinity of FWA for the fiber. Hence, they needed additional fixing agents/salts for that purpose.47,48
Figure 8.

Visualization of fluorescent pulp fibers by an epifluorescence microscope under UV light exposure (a) and white light (b). The fibers were modified with 5b, and the dosage was 2% (w/w). N-(4-Butanoic acid)-1,8-naphthalimide-(3-carboxypropyl)trimethylammonium chloride ester of cellulose (5b, DSphoto 0.22, DScat 0.33). (c) Picture of fiber hand-sheets under black light illumination. The quadrates and the background are made of treated FMCDs and reference fibers, respectively. N-(3-Propanoic acid)-1,8-naphthalimide-(3-carboxypropyl)trimethylammonium chloride ester of cellulose (4a, DSphoto 0.07, DScat 0.31) and N-(4-butanoic acid)-1,8-naphthalimide-(3-carboxypropyl)trimethylammonium chloride esters of cellulose (5a, DSphoto 0.11, DScat0.32; 5b, DSphoto 0.22, DScat 0.33). Reprinted from ref (46) with permission from the American Chemical Society.
Topochemical Immobilization of Enzymes on Fiber Surfaces
Cellulose fiber surfaces have been explored for the immobilization of enzymes49 because this could open up new possibilities in bioprocessing systems employing renewable materials. The glucose oxidase (G*) enzyme obtained from Aspergillus niger was a cross-linked enzyme studied for its immobilization on bleached Kraft pulp fibers containing amino groups (Figure 9a). The amino groups were introduced into cellulose by derivatizing it to the (3-carboxypropyl)trimethylammonium chloride ester of 6-deoxyazidocellulose first, followed by reacting it with propargyl amine and the copper(II)-tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine complex solution in the presence of ascorbic acid. The reactants undergo a copper(I)-catalyzed azide–alkyne Huisgen cycloaddition (CuAAc) reaction to form amino-substituted fibers. The concentration of amino groups in the amino fibers (CNH2, μmol/g of fibers) was calculated according to
| 1 |
where A485 is the absorbance at 485 nm, V is the total volume of the supernatant at pH 3 (L), ε is the molar absorptivity (L mol–1 cm–1) at 485 nm, b is the path length of the cuvette in which the sample is contained (cm), and m is the mass of amino fibers (g). In eq 1, the factor of 106 was used to convert moles of amino groups per gram of amino fibers in mol/g.
Figure 9.

(a) Enzyme activity shown by biocatalytic 673 fibers FG*. STED image of a biocatalytic fiber cross-section bearing free enzymes labeled with Abberior STAR 635 (b) and the corresponding transmission image (c). Dye/enzyme ratio of 1.4. Picture dimensions: 32.53 μm × 32.53 μm (b, c). Field emission scanning electron microscope (FESEM) pictures of the reference fibers (d, e) and the fibers obtained after chemical reaction between the functional fibers (F), the immobilized enzymes (G*), and GA (f, g). All images are at 25K magnification. Reprinted from ref (50) with permission from the American Chemical Society.
These amino fibers subsequently attracted enzyme moieties due to their charge-directed self-assembling behavior in the presence of glutaraldehyde (GA).50 Unmodified cellulose without amino groups could attract only a very small quantity of enzymes. These amino groups were suitable for anchoring both original and cross-linked enzymes (Figure 9b–g). Thus, these biocatalytic fibers (FG*) can be tuned and utilized to immobilize a variety of enzymes for potential applications in biotechnology, biosensors, and biomedicine.51,52 There are several other types of enzyme immobilization on a support, but in those works immobilization either was propelled by weak van der Waals forces or required strong ionic interaction. Sometimes these supports were detrimental, especially when the loading of enzyme was high.
Cellulose–Shellac Biocomposite for Reinforced Fibers
Shellac is a polyester type of resin derived from polyhydro carboxylic acids by intra- and intermolecular esterification. It is used as a coating material as it adheres to many smooth surfaces due to its hydroxyl acid groups. Shellac-coated material surfaces provide a glossy look, UV resistance, and hardness.53,54 The presence of −OH and −COOH groups in this resin make it suitable for synthesizing a composite with cellulose as its −OH groups would form hydrogen bonds easily. In a typical preparation, cellulose and shellac were mixed in a ratio of 1:2 with alcohol additives for 20 min and spread in a hot mold for 10 min followed by pressing and cooling. For the biocomposite, refined cellulose fibers and cellulose acetate butyrate particles were used for comparison. The refined fibers showed better mechanical strength over cellulose derivative particles even though they had a uniform dispersion. In addition, the coating was better with the fibers as they possessed a great adhesion for shellac, over cellulose particles. A cellulose–shellac composite containing a low content of cellulose with a high concentration of ethanol and poly(ethylene glycol) (PEG) additives, exhibited high elasticity and a low Young’s modulus, whereas a high content of cellulose with a low concentration of additives showed high stress resistance and a high Young’s modulus (E). PEG acted as a plasticizer and improved the interfacial interaction between cellulose and shellac by its C–O–C- and O–H-initiated dipolar interaction and hydrogen bonding. On the basis of the characterization of cellulose–shellac composites, they can be classified from hard material (E = 1731 ± 300 MPa) to soft (E = 0.40 ± 10 MPa).55 These composite materials will potentially find a place in reinforced fiber applications such as building components, packaging, and fabrics. The disadvantage associated with this material is that it cannot be used in applications where the temperature is above 80 °C as shellac might start to melt if it is used in its dewaxed form.
Topochemical Engineering of Fibers with Organic–Inorganic Hybrids
Cellulose fiber surfaces and interfaces for chemical modification and value addition can be carried out in a systematic way. Hybridization is one such way in which inorganic clay minerals are composed with organic cellulosic fibers.56 Layered double hydroxide (LDH) is one such inorganic clay material having the formula [M1–xIIMxIII(OH)2]x+[Ax/nn–·mH2O], where MII and MIII denote the divalent (Mg2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, etc.) and trivalent metal cations (Ti3+, Cr3+, Fe3+, Al3+, etc.). An– is the intercalated anion (CO32–, SO42–, NO3–, Cl–, etc.) located in the hydrated galleries.57−59 A layered double hydroxide (LDH) is a class of ionic solids with a layered structure in which cationic layers are interconnected through anionic layers by the compensation of charge. The hybridization with organic moieties is mainly propelled by the charged sites in the mineral structures.
Cellulose Modification with Layered Double Hydroxides
When pulp fibers were modified with LDH nanoparticles, the LDH acted as a contact point on the fiber surface for the incoming bleaching and brightening agents in the pulp process. The functionalization of cellulose by LDH occurred as a result of electrostatic interaction between metal cations of LDH and anionic cellulose groups. Haartman et al. have shown that LDH can enhance the ISO brightness by up to 10% and decrease the κ number by 2 units in the Kraft pulp oxygen delignification process. In addition, LDH increased the selectivity of oxygen delignification, thus lignin removal was more efficient. However, modified LDH composed of terephthalate anions decreased the consumption of the hydrogen peroxide (H2O2) bleaching agent and enhanced the opacity of fibers by 3 units in the thermomechanical pulp (TMP). Moreover, the retention of an optical brightening agent (OBA) was found to be improved with LDH-modified pulp fibers.60 The mechanism of bleaching was not clear, hence control over bleaching was quite challenging. Otherwise, this modification increased the loading of OBA on the fibers and protected the leaching of OBA into wastewater. Thus, LDH-modified cellulose fibers can be applied in the production of paper and cardboard having high brightness.
Fiber Functionalization with LDH-Interfaced Stearic Acid Nanohybrids
Cellulose, being inherently hydrophilic in nature due to its primary and secondary −OH groups, is prone to undergo hybridization with inorganic solid-state materials having charge centers by the same −OH functionalities. When cellulose was hybridized with LDH, it not only improved the fiber quality in terms of optical brightness but also acted as a linker/sandwich material for anchoring other specialty guest molecules such as fatty acids by the use of its leftover charges. Fatty acids are carboxylic acids with long aliphatic chain having 4–28 carbons derived from animal or vegetable fats.61 Each of these long carbon chains has a negatively polarizable functional group which is susceptible to undergoing neutralization with positively charged metal cations. The fatty acid-coated LDH–cellulose hybrid fibers became oleophilic and hydrophobic in character and thus changed cellulose’s hydrophilic nature in a cost-effective and ecofriendly way. This unique property can be extrapolated to a variety of other hydrophilic target materials to produce various customized waterproof/oil-loving material applications. Hence, this hybrid material will potentially lead to innovations benefiting sustainable processes for hydrophobicity. This approach differed from previously reported methods where hydrophobic organic moieties were directly incorporated on cellulose to bring about hydrophobicity without the use of any linker material, which was also achieved only on regenerated cellulose. The LDH–cellulose hybrid fibers (LDH–CEL) allows native cellulose fibers (natural pulp) even in their wet state to undergo hydrophobization with stearic acid (SA) right away (Figure 10).
Figure 10.

(a) Schematic representation showing the formation of hybrid fibers. (b) XRD patterns of the pristine and modified fibers.*Peaks from the copper sample holder (to be ignored). (c) Influence of pH on the water contact angle as demonstrated with SA-LDH-CEL hybrid fibers. Reprinted from ref (62) with permission from Elsevier.
The experimental data on the structural configuration/mechanism revealed that cellulose and stearic acid self-assembled on LDH through its cationic (brucite) layers on either side to make SA–LDH–CEL, a hybrid composite material. The negative charges obtained from cellulosic −OH and stearic −COOH were attracted toward the positively charged brucite layers of LDH particles via electrostatic interaction by keeping the nonpolar aliphatic carbon chain tailing out of the surface, which eventually introduced hydrophobicity into pulp fibers. The XRD pattern showed that stearic acid did not occupy (e.g., an intercalated/anion-exchanged carbonate anion) the interlayer galleries of LDH, thus it anchored only on the brucite sites of LDH (Figure 10b). The SA-LDH-CEL materials synthesized from varying concentrations of LDH metal precursor and stearic acid showed different degrees of hydrophobicity as identified from water contact angle (CA) measurements. Also, water solutions having different pH values were drop cast on SA-LDH-CEL material to study the influence of pH on contact angle values (Figure 10c) The SA-LDH-CEL composite material surpassed all other materials that have been reported in the literature in terms of its high water contact angle (150°) (directly proportional to water repellence), which is a mandatory requirement for the production of waterproof materials.62
The idea of utilizing the charge centers of LDH can be effectively materialized to conjugate hydrophobic fatty acid and hydrophilic cellulose simultaneously. This produced an entirely new kind of hybrid material which offered not only hydrophobicity but also superhydrophobicity to the cellulose network. With the employment of LDH, a minimal amount of a hydrophobic moiety led to a high order of hydrophobicity in cellulose and reduced the processing time of about 5 days, compared to that in other reported works.63 The development of superhydrophobic cellulose material with LDH as the interface can be extrapolated to incorporate long-chain fatty acids such as palmitic, arachidic, and lignoceric acids (having 16, 20, and 24 carbons, respectively) to achieve the desired tensile strength and water-repellence. This LDH-sandwiched cellulose–stearic acid composite will find application in waterproof packaging materials, films, oil adsorbents, and cleaning sponges. The drawback for this material could come from its tensile strength, which was weaker than unmodified cellulose fiber.
In Situ Hybridization of Cellulose Fibers by LDH Material
Layered double hydroxides (LDH) were synthesized in the presence of pulp fibers and behaved slightly different due to their change in properties as the LDH crystal growth occurred on the cellulose surface from its nascent state.64,65 For this, pine pulp was used as a substrate to grow Mg- and Al-containing LDH in an aqueous medium. The growth was initiated by the precipitation of Mg and Al precursor solutions under basic conditions (pH ≥9). The nature of the base used to alter the pH influenced the properties of LDH particles, causing a wide range of sizes, from micrometer to submicrometer to nanometer. The high and low super saturated solution (hss and lss) medium obtained by the use of sodium carbonate yielded 100–200 and 70 nm respectively, whereas the use of urea for pH adjustment caused an increase in particle size (2–5 μm). Even though urea hydrolysis increased the fiber densification, it decreased the fiber compliance by 50%. This indicated that LDH formation happened not only outside but inside the fiber walls as well. The choice of a basic medium also affected the cellulose depolymerization in the order of urea > lss > hss. The charge centers on the fibers were quantified by methylene blue (MB) and metanil yellow (MY) adsorption measurements. The results suggested that LDH-hybridized cellulose prepared from a low supersaturated solution medium retains most of the original charge on cellulose and provided a higher loading of anionic MY (10 μmol/g), indicating the development of positive charge on LDH-modified cellulose fibers. In the thermochemistry point of view, LDH incorporation into cellulose reduced its exothermic heat emitted, making it suitable for flame retardant applications.66 There were few limitations pertaining to this method: (1) hss formed LDH particles with low crystallinity; (2) the size of the LDH particles was changed after synthetic hydrothermal treatment; and (3) elevated temperature in urea hydrolysis produced very large LDH particles due to cation migration in the crystalline structure.
Fire-Resistive Cellulosic Fiber Foams by Nanoengineering
Modified lightweight fibrous foam (lwFF) with various LDH loadings was optimized under oxidative pyrolysis conditions, and a 34% (w/w) LDH composition was found to be a fitting candidate for flame resistance applications.67,68 The nanoengineering of hybridized cellulose foam was carried out under urea alkalined in situ LDH nucleation conditions, producing nanometer to micrometer-sized particles. This carbonate (CO32–)-containing LDH nucleation was favored in the sites of fibril and flake structures over fibers. These coprecipitated LDH particles surrounded the cellulose fibers and protected it from the external heat by reducing the rate of oxidation. Thermogravimetric analysis showed that incorporation of LDH into lwFF suppressed the ignition and the amount of exothermic energy release. The oxidative pyrolysis experimental results expressed that the amount of soot liberation and the peak heat release rate (PHRR) were reduced. Thus, lwFF contributed to a 60% reduction in the CO2 formation rate.69 Therefore, LDH offers an inexpensive and sustainable solution in place of traditional polyphosphate or a halogen-based fire retardant, where the toxicity level is high and the smoke carries heavy particulate matter. However, the material cannot be reused without performing a reduction because the LDH gets oxidized to a mixed metal oxide under the combustion conditions.
Topochemical Engineering for Biomass Deconstruction and Cellulose Dissolution
Topochemical Pretreatment of Cellulose Fibers for Sugar Production
Cellulose can be depolymerized to produce mono- and disaccharides through enzymatic hydrolysis. The rate of enzyme action was dependent upon the chemical composition surrounding the cellulose fibers in wood biomass because these protective layers inhibited the enzymatic action on cellulose directly.70,71 When the wood fibers were treated with solvents such as hot water and ionic liquids, their chemical composition changed due to the partial removal of lignin. Surface coverage by lignin on the fiber surface slowed down the hydrolysis of cellulose into a monosaccharide sugar such as glucose.72 Hence, its removal by eco-friendly methods was highly demanded for downstream/cut-down applications. Ionic liquids (ILs) are a class of molten salts which can be used as polar solvents to dissolve organic substrates. In this case, 1-ethyl-3-methylimidazolium acetate (EmimAC) and 1-butyl-3-methylimidazolium chloride (BmimCl) IL were used to treat birch and pine wood. Also, hydrothermal or hydrotropic methods were employed for lignin removal in separate experiments followed by enzymatic hydrolysis.73 The results suggested that a topochemical treatment using sodium xylene sulfonate (SXS)–water solution removed lignin efficiently whereas a hydrothermal method using hot water and ILs showed no removal of lignin but ILs swelled the fiber and subsequently increased its the surface area. Lignin removal by the SXS–water system could increase the yield of glucose by up to 80% after enzymatic hydrolysis of the birch wood fibers. This result was achieved by the topochemical disassembly of lignin from fiber surfaces and the exposure of cellulose to the enzymatic reaction.
Topochemical Disassembly of Cellulose and Lignin with Hydrotropes
Lignin is the most abundant natural aromatic polymer, and its separation from cellulose from wood was performed using topochemical extraction. Gabov and co-workers extracted lignin from birch wood by an eco-friendly method in which almost 67% of the lignin was recovered.74 They adopted a modified hydrotropic method in which the structure of lignin underwent significant changes such as an increase in the number of phenolic hydroxyl groups and a decrease in aliphatic hydroxyl functionalities. The modified hydrotropic method employed formic acid and hydrogen peroxide reagents in addition to hydrotropic agent sodium xylenesulfonate for the breakdown of wood content at pH 3.5. This method can be used to purify cellulose fibers from lignin and change the properties of lignin.
Topochemical Pretreatment of Cellulose Fibers for Dissolution in Water-Based Solvents
The dissolution of cellulose using ecofriendly solvent systems is of high importance due to the environmental impact. A water-based solvent system such as NaOH–urea–water has been a most desirable dissolution medium. However, this multisolvent system requires energy-intensive pretreatments such as milling and refining. To solve this problem, an inexpensive pretreatment method was used in which an ethanol–hydrochloric acid mixture was applied to pulp fibers at 65 °C to enhance the disruption of the remnant primary wall fiber layer and decrease the degree of polymerization.75 In forest product industries, raw pulp fibers undergo hydrolysis under aqueous mineral acid conditions to yield various biopolymers. Obtaining cellulose polymer with the desired molecular weight is a challenge, and it can be achieved by performing the controlled hydrolysis of pulp. Trygg et al. demonstrated that the addition of ethanol (0–96%) to an acid solution medium (pKa 10–4.7) can control the level of depolymerization to a specific value. The presence of ethanol in the acid solvent system preserved the cellulose content at the original level with a decrease in the viscosity-averaged degree of polymerization (DPv) (Figure 11), whereas the absence of ethanol decreased both DPv and the relative cellulose content. In their observation, weak (organic) acid treatment showed a much higher DPv as the outer cell wall layers were still intact. On the contrary, strong (mineral) acids caused a disruption in cell walls and reduced the DPv by 75–80%.76 The removal of a remnant primary wall of fiber surfaces significantly affected the mechanism of fiber dissolution in a cellulose solvent (Figure 12).
Figure 11.

DPv profile against ethanol concentrations on pulp fibers. Reproduced by permission of the Romanian Academy Publishing House, the owner of the publishing rights (ref (76)).
Figure 12.
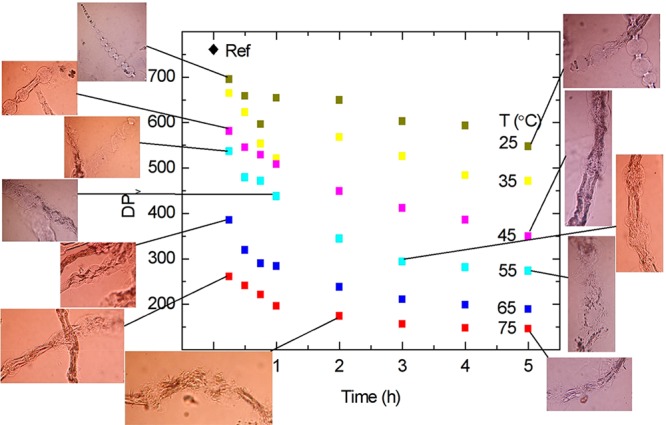
Viscosity-average degree of polymerization (DPv) of ethanol–acid-treated dissolving pulp at various temperatures as a function of time.
Topochemical Engineering of Functional Cellulose Beads
Cellulose obtained as chemical pulp fibers is usually dissolved in complex multisolvent systems/reagents and then converted to different physical forms such as films, fibers, and particles. Beads are another form in which cellulose can be processed as spheres with diameter ranging from micrometer to millimeter. There are numerous ways to produce these beads through chemical or physicochemical routes. Those methods involve dissolution and regeneration, reagent treatment, flow techniques, and milling equipment. Our approach has the advantage of dissolving topochemically pretreated cellulose fibers in water-based solutions and thus increasing the application potential of beads for different biomaterials. Moreover, the beads can be engineered by adding functional groups or via the adsorption of new active groups on their surfaces, facilitating applications across many fields for catalysis and the chromatographic separation of biomolecules,77 drug delivery,78 bioconjugation, wastewater and radioactive waste treatment,79,80 blood purification, enzyme immobilization, and the separation and filtration of heavy metals.81−85
Mesoporous Neutral Cellulose Beads for Drug Entrapment and Delivery
Cellulose beads (CBs) carrying neutral charge were prepared by dissolving pulp pretreated fibers in an environmentally friendly solvent mixture of NaOH–H2O–urea followed by precipitating it from a HNO3 solution under the following conditions: T1 = 0.5 M/25 °C, T2 = 2 M/25 °C, and T3 = 2 M/50 °C. The size (diameter) of the beads was 2 to 3 mm. The concentration of the acid medium and the temperature influenced physical parameters such as the porosity, diameter, and specific surface area (Table 3). This change in parameters influenced the drug loading capacity and release in real time drug delivery applications.
Table 3. Physical Properties of Beads Prepared from Three Different Conditions: T1, T2, and T3a.
| batch no. | coagulation bath molarity and temperature | specific surface area (m2 g–1/bead) | water swollen volume (mm3/bead) | water swollen weight (mg/bead) | porosity (%/bead) |
|---|---|---|---|---|---|
| T1 | 0.5 M, 25 °C | 470 | 11.2 | 14.1 | 93.6 |
| T2 | 2 M, 25 °C | 447 | 14.9 | 17.0 | 93.8 |
| T3 | 2 M, 50 °C | 381 | 17.2 | 17.8 | 94.7 |
Reprinted from ref (86) with permission from Elsevier.
Three water-soluble model drug molecules, namely, (a) anhydrous theophylline (Thp), (b) riboflavin 5-phosphate sodium (RSP), and (c) lidocaine hydrochloride monohydrate (LiHCl), were employed to evaluate the drug delivery performance and in vitro toxicity properties of the cellulose bead carrier material. The solubility levels of these drugs were 8, 112, and 147 mg/mL, respectively, in water. These drugs were loaded into CBs prepared in various environments (T1, T2, and T3) by immersing in 20 mg/mL RSP, 20 mg/mL LiHCl, and 4 mg/mL Thp under magnetic stirring for 24 h. Furthermore, these beads were dried for at least 24 h at room temperature until there was no water left inside the beads. The size of the drug-loaded dried beads was around 1 mm.
Surface morphology investigations using scanning electron microscopy revealed that the external morphology had no significant difference for the drug-loaded and unloaded beads, whereas the internal surface for loaded beads showed the presence of drug against the unloaded beads (Figure 13). Thus, drug molecules diffused into the pores of beads and were absorbed. The higher the porosity and dissolution of the drug, the higher the drug loading on the beads, as observed from UV–vis spectra. The drug entrapment was determined spectrophotometrically by crushing the beads mechanically and soaking in a water solution for 24 h prior to measuring the wavelength (λmax) at 267 nm (RSP), 218 nm (LiHCl), and 272 nm (Thp), respectively (Table 4). Cell viability experiments were carried out to determine the compatibility of the carrier cellulose material with the human epithelial cells. For this, a mechanically crushed bead solution of 10 mg/mL in cell culture medium was incubated, and then fluorescent signals were measured. The results showed that the cellulose bead material did not affect the cell viability under the given conditions and the value stayed at 95% as against 100% for nontreated cells.
Figure 13.

Internal morphology of an unloaded bead (a) and a loaded bead (L 20 T1 (LiHCl 20 mg/mL, condition T1) (b). Reprinted from ref (86) with permission from Elsevier.
Table 4. Drug Loading Shown as the Amount of Drug Incorporated per Bead as Well as Drug Loading Presented as the Mass Percentagea,b.
| loaded beads | incorporated drug amount in one bead (mg) | drug amount percentage per one dry bead (%) |
|---|---|---|
| T_4_T3 | 0.072 ± 0.002 | 5.0 |
| T_4_T2 | 0.052 ± 0.002 | 4.2 |
| T_4_T1 | 0.049 ± 0.004 | 3.7 |
| L_20_T3 | 0.396 ± 0.032 | 27.3 |
| L_20_T2 | 0.333 ± 0.050 | 26.6 |
| L_20_T1 | 0.313 ± 0.052 | 23.3 |
| L_4_T3 | 0.118 ± 0.022 | 8.1 |
| L_4_T2 | 0.095 ± 0.036 | 7.6 |
| L_4_T1 | 0.065 ± 0.008 | 4.8 |
| R_20_T3 | 0.207 ± 0.005 | 14.3 |
| R_20_T2 | 0.163 ± 0.015 | 13.0 |
| R_20_T1 | 0.172 ± 0.038 | 12.7 |
| R_4_T3 | 0.081 ± 0.005 | 5.6 |
| R_4_T2 | 0.063 ± 0.002 | 5.1 |
| R_4_T1 | 0.068 ± 0.002 | 5.0 |
Average weights of a bead for T1, T2, and T3 beads are 1.35, 1.25, and 1.45 mg, respectively. T.4, Thp 4 mg/mL; L.20 and L.4, LiHCl 20 and 4 mg/mL; R.20 and R.4, RSP 20 and 4 mg/mL.
Reprinted from ref (86) with permission from Elsevier.
The in vitro drug release was measured using the USP paddle method, where 4–20 beads dissolved in 500 mL of 0.1 N HCl buffer (pH 1) at 37 ± 0.5 °C. The resultant solution was checked for λmax and quantified. The dissolution data showed a low solubility of RSP (2 mg/mL) in the chosen medium, leading to a slower release of RSP from the beads compared to Thp and LiHCl. The order of dissolution was LiHCl > Thp > RSP, and it followed the same trend in releasing constant values. This can be explained by the fact that the cellulose beads irreversibly contracted and became rigid upon drying, causing a tight matrix around encapsulated drug entities. Thus, the drug delivery kinetics was highly dependent on the solubility of drug-loaded carrier materials and the loading degree rather than how these beads were prepared.86 This suggested that nascent cellulose beads in their wet state can be applied straightaway for drug encapsulation and delivery. However, these beads can be used only for water-soluble drugs, which narrowed their application.
Anionic Cellulose Beads for Drug Encapsulation and Release
Neutral cellulose beads were first prepared from an ecofriendly water-based solvent system, as there is a serious focus on dissolving cellulose in an environmentally friendly way without producing any toxic waste or byproducts.87−89 Neutral cellulose beads (Figure 14a) were formed by dissolving 5% cellulose solution in 7% NaOH–12% urea–H2O solvent followed by dropwise extrusion using a 0.8 mm needle in 2 M HNO3 at room temperature.90
Figure 14.
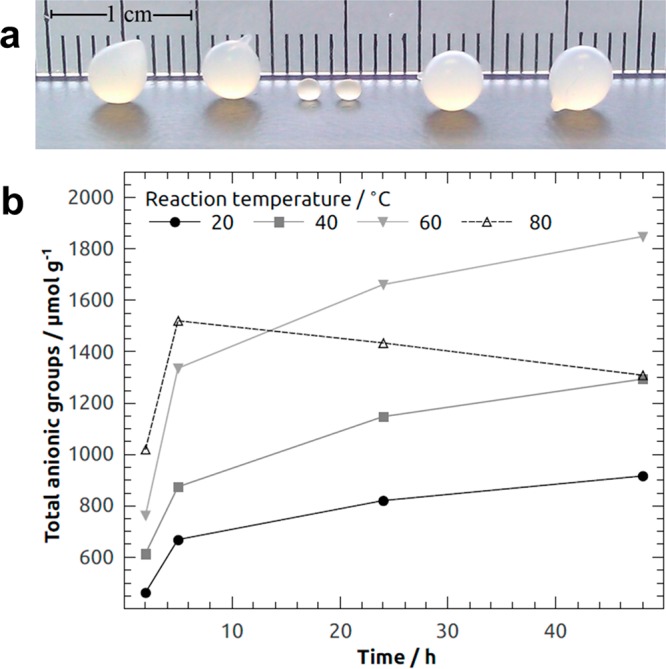
(a) (Left) Cellulose beads prepared from a 5% cellulose–7% NaOH–12% urea–water solution and coagulated in nitric acid and (right) oxidized with Anelli’s reaction at 60 °C for 48 h. (Middle) Beads after drying at room temperature. (b) Total anionic groups in CBs oxidized for 2–48 h at 20–80 °C. Reprinted from ref (90) with permission from Springer Nature.
These beads exhibited a high surface area with an ordered pore network. Such morphology was highly beneficial for the incorporation of drugs and its delivery. The shape and size of the beads offered technological benefits over conventional powders such as the improvement in flowability, the reduction of dust during processing, and the uniformity of the particle size.
Drugs of any charge or without charge can be loaded on cellulose beads for their applications in pharmaceutics. The loading of water-soluble drugs was done by immersing these beads under constant agitation for at least 24 h or until there was no water residue remaining inside the beads. For charged drugs, anionic groups (AG) were introduced on neutral cellulose beads (CB) by a modified Anelli’s reaction at 20–80 °C for 2–48 h. In detail, the beads were oxidized in TEMPO (2,2,6,6-tetramethylpiperidin-1-oxyl)/NaClO2/NaClO (0.1/10/1) with 1.2 moles of NaClO2 for each anhydroglucose unit (AGU). Oxidation at 80 °C yielded a lower level of anionization, which resulted in hypochloric acid-induced nonspecific oxidation instead of specific oxidation of the C6 hydroxyl group by the N-oxoammonium ion. This caused the degradation of cellulose.91 The maximum amount of AG introduced on CB was ∼1850 μmol g–1 (corresponding to a degree of substitution of 0.31) (Figure 14b). The amount of AG on the oxidized cellulose beads (OCB) can be tailored as a function of reaction time and temperature. This contributed to new properties such as swelling, water retention, and pore size distribution which were the key factors in using these anionic beads for drug encapsulation and delivery. The pore size distributions of the beads were measured using a solute exclusion technique with dextran molecules as probes.90,92 The modified Anelli’s reaction decreased the number of macropores (≥560 Å) while increasing the number of mesopores (39–139 Å). The total porosity was about 95%. The swelling capability of beads can be studied by measuring water retention values (WRVs) (Table 5) after immersing the cellulose beads in a sodium phosphate-buffered water solution at pH 7.4. The WRV was calculated with eq 2:
| 2 |
m1 is the mass of centrifuged beads, and m2 is the dry mass before swelling. The data suggested that there was an increase of 40% in swellability and 500% in water retention. The swelling and water retention values increased with increasing temperature of the oxidation reaction.
Table 5. Swelling Ratios of Minimum Diameters Compared to Never-Dried Beads and Water Retention Values (WRV) of Dried Beads Oxidized at 20, 40, and 60 °Ca.
| swelling ratio % | WRV | |
|---|---|---|
| CB | 48 | 0.22 |
| OCB20 | 73 | 2.50 |
| OCB40 | 84 | 4.01 |
| OCB60 | 88 | 5.10 |
Reprinted from ref (93) with permission from Springer Nature.
Anionic cellulose beads were studied for applications related to drugs such as ranitidine, an antacid that cures stomach illnesses. In a typical drug encapsulation experiment, anionic cellulose beads (A-CB) were immersed in ranitidine HCl solution (20 mg/mL) at a concentration of 2 beads/mL of solution followed by gradual drying for 2 days. The dried beads were further investigated for the controlled release of ranitidine according to the USP paddle method (United States Pharmacopeia, 35th ed.). This method employed a smart dissolution tester (Sotax AT7) wherein the drug adsorbants (A-CB) were treated with sodium phosphate buffer solution (1 bead/100 mL) at 37 °C. The pH of the buffer was maintained at 7.4. The released drug was determined by UV–vis spectroscopy using its characteristic peak at 288 nm. The in vitro drug release profile showed that the oxidized or anionized beads (A-CB) released twice the amount of ranitidine when compared to unoxidized beads (CB) even though the amount of accessible water inside the pores did not change much between these beads. This can be explained by the fact that the oxidation process increased small meso and micropores where ranitidine HCl could diffuse and get absorbed. The dimensions of the rantidine HCl crystal lattices (a, b, and c) were 12, 7, and 22 Å, respectively, and this was sufficient enough to enter into the newly formed micropores of A-CB. This increased the amount of encapsulated drug. Also, the e-folding time measurement (drug retention) suggested that there was no stronger interaction between the cationic drug and anionic surfaces of beads as the time difference between CB and A-CB was very low. The weaker interaction between drug and beads was also supported by the molar ratio of the released ranitidine and AG present on the beads, which was 110 μmol g–1. This is an advantageous property required in every drug delivery system. Nevertheless, the release profile showed the model of exponential decay, indicating a diffusion-controlled mechanism93 that could be a barrier for a higher loading of the drug and its delivery. Besides drug delivery, A-CB can be utilized in ion exchange chromatography to purify water, ad/absorbents, nutrient capsules for soil fertility, and intermediates/supports in biorefinery and catalysis.
Zwitterionic Cellulose Beads
Zwitterionic cellulose beads can be synthesized by a spin drop atomization/sol–gel transition technique followed by the introduction of carboxylic anion (−COO–) and cationic quaternary ammonium (−N+) groups on the bead surface. To prepare these beads, the dissolution of pretreated cellulose pulp (viscosity 124 mL/g) was carried out in a NaOH–urea–H2O solvent mixture followed by shaping into spherical beads by spin drop atomization and regeneration in 10% H2SO4.75 The beads formed with this technique ranged in diameter from 250 μm to 4 mm (Figure 15a,b).
Figure 15.

Images of cellulose beads of different sizes: (a) microbeads and (b) millibeads. (c) Scheme for the synthesis of zwitterionic beads. Cross sections of reference (d) and zwitterionic (e, f) cellulose beads. Reprinted from ref (94) with permission from Springer.
In a typical zwitterionization on the beads, −COOH and −C=O groups were generated using a NaClO2/TEMPO-mediated oxidation system under heterogeneous reaction conditions. The N+R3 group was incorporated via a coupling reaction between −C=O and Girard’s reagent T (carboxymethyl trimethylammonium chloride hydrazide) in a water medium (Figure 15c), which was also a step toward green chemistry. NaClO2/TEMPO was a stoichiometric oxidation system in which a catalytic amount of NaClO oxidized TEMPO to the N-oxoammonium ion at 60 °C, which in turn oxidized primary cellulosic −OH groups to −CHO and formed a byproduct, hydroxylamine. In a consecutive oxidation step, −CHO got converted to −COOH by NaClO2, generating NaClO, which then reoxidized hydroxylamine to the N-oxoammonium ion.95,96 The carbonyl groups (−CHO) formed during the oxidation process were further coupled with hydrazides containing quaternary ammonium ions through the click chemistry approach.97−101 This was similar to a coupling reaction between an aldehyde and a primary amine, which led to the formation of Schiff’s base in a weakly acidic environment.101 In our case, a stable hydrazone was formed as supported by the literature,102−104 thus beads were made into zwitterions in nature.
When these beads were subjected to conductometric titrations, they revealed the quantitative amount of −COOH present on them. To determine −C=O oximation, nitrogen elemental analysis was employed. The reaction between carbonyl present in oxidized cellulose beads and carboxymethyl trimethylammonium chloride hydrazide was monitored by nitrogen elemental analysis. SEM morphological analysis showed that zwitterionic beads have well-defined macropores with thicker walls than nonfunctionalized cellulose beads having thin walls around pores (Figure 15d–f). The increase in pore size was attributed to the induced hydrophilicity of beads caused by the introduction of ionic moieties. These macropores provide an opportunity to carry bulk biomolecules and encapsulate drugs in theranostics applications. Thus, zwitterionization proved to be an effective way to tune the pore dimension in cellulose beads.94
These doubly charged hydrophilic cellulose beads meet the requirements of future applications such as the separation of biomolecules, the immobilization of proteins or enzymes, and the encapsulation of zwitterionic drugs.105 In this way, zwitterionic exchange resins with both anionic and cationic groups together become a potential replacement for nonrenewable organic polymers, which are widely dominant in ion exchange/carrier applications.
Hybrid Beads of Cellulose-Synthetic Polymer for Antimicrobial Applications
Bead-shaped cellulose fibers was also studied for interaction with microorganisms such as Gram-positive bacteria like Micrococcus luteus when they were coated with bactericidal agents for QPVP-C5. Cellulose beads (CB) and oxidized cellulose beads (OCB) of 3 mm size were studied for the adsorption of QPVP-C5 followed by their bactericidal action. Biocidal action was dependent on the amount of interaction exerted by the biocide carrier’s surface on the cell membrane of the microorganism.106−109 CB by its nature could incorporate biocide agents either covalently or by adsorption.
CB were prepared from a dripping method where 5–7 wt % solutions of cellulose were dissolved in ecofriendly 7% NaOH–12% urea–H2O solvent and then spheronized from 2 M HCl coagulation bath. In another set of experiments, CB was oxidized by TEMPO reagent in order to introduce carboxylic groups (−COOH) on bead surfaces. Young moduli experiments showed that CBs prepared from 7 wt % solution (CB7%) possessed very good stress–strain compression values. Hence, those samples were chosen for further investigation. CB (7%) and oxidized cellulose beads (OCB7%) having a charge density of 0.77 mmol/g were studied for the adsorption of poly(4-vinyl-N-pentyl pyridinium)bromide (QPVP-C5). In a typical experiment, 30 mg of CB and OCB were dispersed in a glass flask containing 4 mL of QPVP-C5 solutions at concentrations of 0.5, 2.5, 5.0, 12.5, or 17.5 g/L. These mixtures were sealed and stirred magnetically at 55 °C for 15 h. The amount of QPVP-C5 agent adsorbed was measured through the absorption spectra of the pyridinium cation intermediate at 256.5 nm before and after adsorption on the beads. Similarly, the desorption of the agent from the beads was also measured by UV–vis after washing and soaking the beads in 4 mL of distilled water for 24 h under orbital shaking (Figure 16a,b). FT-IR experiments showed that the adsorbed amount of QPVP-C5 (absorption band at 1640 cm–1 assigned to C=N stretching of pyridinium group) increased with increasing concentration of the dispersion medium. OCB adsorbed more QPVP-C5 than the CB suggesting that newly introduced anions on the beads electrostatically interacts more with the cationic counterpart of the bactericide agent. This attraction also reduced the desorption to less than 5%.
Figure 16.

Amount of adsorbed QPVP-C5 (ΓQPVP-C5) onto (a) CB7% and (b) OCB7% as a function of the QPVP-C5 bulk concentration. Red and blue columns stand for the amounts of adsorbed and desorbed polycations, respectively. SEM images of freeze-dried (c) OCB7%, (d) OCB7%/QPVP-C5-5, and (e) OCB7%/QPVP-C5-12.5 surfaces after 24 h of contact with M. luteus suspensions. The scale bars correspond to 1 μm, and the circles highlight the presence of M. luteus or bacterial debris. Schematic representation of the interaction between bacteria (red solid circles) and beads after 24 h. (f) Partial adhesion of bacteria on OCB7%, (g) adsorption of bacteria and weakly charged OCB7%/QPVP-C5-5 without bacteria disruption, and (h) adhesion of bacteria onto highly charged OCB7%/QPVP-C5-12.5, followed by bacterial disruption. Reprinted from ref (110) with permission from the American Chemical Society.
The antibiotic action of QPVP-C5 coated/adsorbed CBs (prepared from 7% solution) and OCBs (7%) was assessed by allowing 3 mL of a 2.75 g/L aqueous dispersion of Micrococcus luteus to interact with 35 mg of beads for 24 h at pH 6. Dispersions of microorganism in water were turbid due to the micrometric size of bacteria. However, when the contact took place with the biocidal material the size decreased due to cell disruption (Figure 16c–h). The magnitude of interaction was correlated with the amount of turbidity originating from the dispersion using spectrophotometry. The turbidity of the bacterial dispersion was measured at 650 nm at room temperature before (τi) and after (τf) its contact with the beads. The efficiency of antibiotic action was calculated from the relative decrease in turbidity (Δτ) which corresponded to bacterial disruption. The larger the Δτ, the stronger the bactericide.
| 3 |
The blank or control bacterial dispersion showed the relative turbidity to be 23%, whereas OCB7%-QPVP-C5-5,12.5 samples exhibited 85 and 99%, suggesting that the material was efficient in disrupting the bacterial cells. The results conveyed that the anionic charges developed on the OCB surface first adsorbed positively charged QPVP-C5, and in turn negatively charged phospholipids on the bacteria envelope were attracted to cationic pyridinium groups, causing cell collapse.110 Thus, antimicrobial-agent-incorporated OCB can be a potential drinking water sterilizer because the water-soluble CB might release the antibiotic agent in water, which will affect human health eventually. This type of binding and disinfectant nature of cellulose-bead-supported sterilizing agents can be extended to antifungal and antiviral materials. Furthermore, the introduction of negative charge reduced the compressive strength and density of beads. Hence, the oxidation of cellulose beads must be carefully done without the loss of their physical properties.
Hybrid Cellulose–Lignin Biocomposite Beads
Lignin is an abundant cell wall material in plants, and its chemical composition consists of complex cross-linked phenolic polymers. When lignin was composed of biopolymer cellulose in a controlled amount, its association caused them to fight against the growth of microorganisms such as bacteria.111,112 This property was not shown by them when they were associated naturally in the plant cells. Lignin composed of cellulose beads was prepared from a 7% NaOH–12% urea aqueous solution of dissolving grade pulp and hydrotropic lignin together, and it was mixed and then extruded into beads. These beads were either air dried or wiped with paper towels (never dried). The never-dried beads possessed advantageous characteristics (increased porosity and lignin uptake) over air-dried beads; hence, they exhibited improved antimicrobial activity. Lignin’s introduction into the cellulose macromolecule network caused a change in the physical properties of beads, such as shape, porosity, microstructure, and swelling in water. The bonding between lignin and cellulose was established by intermolecular hydrogen bonding as evidenced by the shift of the −OH stretching band to higher wavenumbers in IR characterization experiments. The lignin–cellulose composite material was studied for the inhibition of the growth of Gram-negative Escherichia coli and Gram-positive Staphylococcus aureus bacteria.113 The composite beads (Figure 17) did not inhibit the growth of E. coli, but they exerted resistance to the growth of S. aureus when the concentration of lignin on the beads was 40%. However, pure cellulose beads had no antimicrobial activity, and the amount of original hydrotropic lignin corresponding to 40% was less efficient in inhibiting bacterial growth on its own (Figure 18). This suggested that hydrotropic lignin can be immobilized on cellulose beads in order to tune its antibiotic properties. Thus, the carrier behavior of cellulose for drugs, catalysts, and enzymes can be further explored.
Figure 17.

Distribution of lignin in cellulose (C)–lignin (L) beads as examined by confocal fluorescence microscopy. Cross section of the beads in the reflection channel: (a) 90C10L, (b) 75C25L, and (c) 60C40L. Surface of the beads in the reflection channel: (g) 90C10L, (h) 75C25L, and (i) 60C40L. Cross sections in the fluorescence channel: (d) 90C10L, (e) 75C25L, and (f) 60C40L. Surface in the fluorescence channel: (j) 90C10L, (k) 75C25L, and (l) 60C40L. The colors of the images are artificial. The parameters for all the fluorescent images were adjusted in the same way during processing. Reprinted from ref (113) with permission from Springer.
Figure 18.

Concentration of S. aureus (a, b) in the broth with different beads and hydrotropic lignin after incubation for 24 h at 37 C. Inhibition of S. aureus growth calculated on the basis of the bacterial concentrations (c, only positive values are presented). (a and b) Runs were performed on two different days. Load of the beads and the dosages of lignin were the same as in the tests with E. coli. Initial concentrations of bacteria were 6.38 and 6.26 log(CFU/mL) for runs a and b, respectively. 90C10L denotes 90% cellulose, 10% lignin. Reprinted from ref (113) with permission from Springer Nature.
Chitosan–Cellulose Multifunctional Hydrogel Beads
Chitosan is another naturally occurring polysaccharide material, and its structure closely resembles the structure of cellulose, with a difference of an −OH group at the C2 position replaced by an amino group. Trivedi et al. composed chitosan with cellulose in order to produce a new functional material in the form of hydrogel beads aiming at cellular uptake applications. They used a NaOH–urea–water solvent system to coagulate the polysaccharides under weakly and strongly acidic conditions. It was found that a weakly acidic condition (CH3COOH) yielded higher chitosan retention in cellulose comparatively and did not have any impact on the chitosan molecular structure, whereas in a strongly acidic medium (HCl, H2SO4), the amino (−NH2) functional group of chitosan was protonated. The chitosan–cellulose composite hydrogel beads were tested for cyto-compatibility in breast adenocarcinoma cells (Figure 19), where acetic acid (weak)-treated beads showed more compatibility than did the strong-acid-treated beads. This suggested that the chemical structure of chitosan must be preserved in the hydrogel cellulose composite so that it will not have any negative influence on the biological system. The surface morphology of hydrogel beads had larger pore sizes after treatment with chitosan and acids (Figure 20).114 The acetic acid-coagulated chitosan–cellulose composite was also tested for compatibility in osteoblast cells (Figure 21). The sample treated with acetic acid at pH 5 showed enhanced cell viability after 144 h of experiment compared to the control sample. Therefore, the chitosan–cellulose composite will have a huge scope as a carrier material in bone regeneration and cancer therapy.
Figure 19.

(a) Surface and (b) core morphology of 0A, 0B, 0C, 70A, 70B, and 70C chitosan–cellulose hydrogel beads with a scale bar of 1 μm. (A) Acetic acid, (B) HCl, and (C) H2SO4. 0 refers to 0% initial chitosan content, and 70 refers to 70% chitosan content in the composite.
Figure 20.
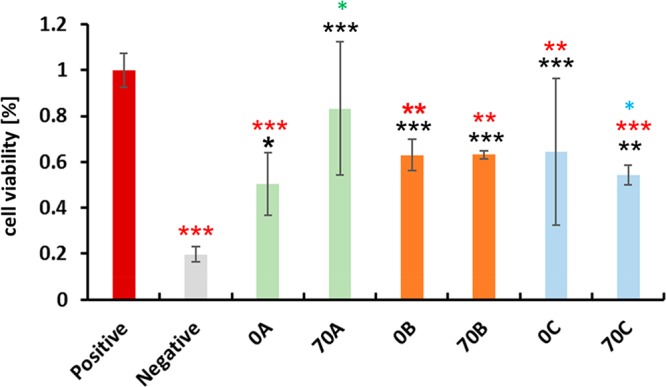
Human breast adenocarcinoma (MDA-MB-231). Biocompatibility with 0A, 70A, 0B, and 70B and 0C and 70C chitosan–cellulose hydrogel beads over 48 h. Values are expressed as percentage of the means ± SD (n = 4). The statistical significance was defined as *P < 0.05, **P < 0.01,***P < 0.005 compared to control samples (ANOVA test). Black * and red * indicate sample comparisons to negative and positive controls, respectively. Green * shows comparisons among 0A–70A, 0B–70B, and 0C-70C, and blue * shows comparisons among 70A–70B, 70A–70C, and 70B–70C. A, Acetic acid; B, HCl; and C, H2SO4. 0 refers to 0% initial chitosan content, and 70 refers to 70% chitosan content in the composite.
Figure 21.
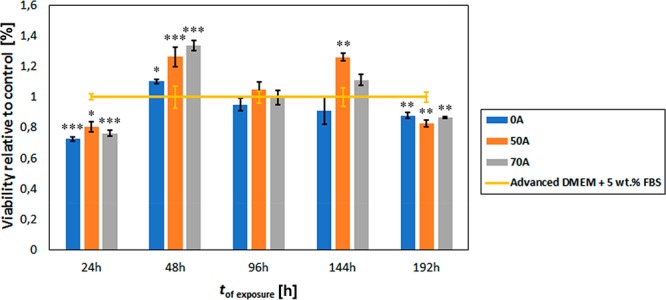
Osteoblast biocompatibility (proliferation behavior) with 0A, 50A, and 70A chitosan–cellulose hydrogel beads based on the MTT assay over 192 h. Each value is expressed as a percentage of the mean ± SD (n = 4). Statistical significance was defined as *P < 0.05, **P < 0.005, ***P < 0.001 compared to control sample (ANOVA test). A, Acetic acid; B, HCl; and C, H2SO4. 0 refers to 0% initial chitosan content, and 70 refers to 70% chitosan content in the composite. DMEM is Dulbecco’s modified Eagle’s medium, and FBS is fetal bovine serum.
Cellulose as a Biotemplate for the Topochemical Engineering of 3D Inorganic Clay Nanoarchitectures
Cellulose can even be used in a destructive technique such as carbonization for templating the desired architectural growth of inorganic solids. Microcrystalline cellulose as a carbonizable template in inorganic material synthesis is not well explored so far, and it will be useful for the sustainable synthesis routes because of its renewability and low toxicity. When cellulose was used as removable skeleton while synthesizing ionic solids, it offered porous structure to the product material with increased surface area. Apart from the regular use of cellulose in pulp and papermaking, it was also used as a biotemplate for the synthesis of inorganic nanomaterial namely, layered double hydroxides (LDH). Geraud and co-workers reported the use of polystyrene beads as organic templates by the inverse opal method for the synthesis of the MgAl-LDH macroporous framework.115 However, the biodegradable and renewable property of cellulose made it an attractive alternative to that of conventionally used templates such as vesicles, micelles, microemulsions, peptides, and polymers.116 Microcrystalline cellulose (MCC), a purified form of plant cellulose, was used as the template. MCC was inert and stable with efficient emulsion and foam stabilizing abilities, which was taken into account for the formation of conceivable LDH networks.117 MCC acted like a skeleton to yield reproducible structures and prevent unorganized LDH nanoparticle growth with different functions and interfaces.118 Therefore, cellulose as a biobased template is an interesting area where scientists have more to explore because it is a cheap, abundant precursor and can be removed under relatively mild conditions (300 °C).119
Briefly, the templating procedure can be explained as follows: MCC was allowed to swell under basic conditions, and the swollen MCC was added dropwise to a beaker containing mixed metal solution (M2+/M3+ = 3:1). The slurry was subjected to microwave hydrothermal treatment. The particles were then washed until neutral conditions were achieved and freeze dried to yield the final product LDH. The template was removed by calcination. The removal of the MCC from the particles displayed the successful production of the network structure of LDH (Figure 22).
Figure 22.

Schematic synthesis of LDH networks using cellulose biotemplates. Reprinted from ref (120) with permission from Elsevier.
The outcome of using cellulose as a biotemplate resulted in the formation of a material with the specific surface area and pore volume increasing from 36 to 152 m2/g and 0.083 to 0.417 cm3/g, respectively. The increase in the pore volume might be due to the expulsion of gases produced during the burning of the template. LDH also maintained its structural integrity during these processes supported by powder X-ray diffraction analysis. It provided information that there existed no chemical interaction between MCC and metal cations during the synthesis, whereas a weak van der Waals force of attraction existed and that facilitated the growth of LDH particles on the biotemplate. There originated an interaction between the electron-rich oxygen atoms of the polar hydroxyl groups of organic cellulose and the inorganic phases of electron-deficient transition-metal cations. The most important observation included size reduction with the fibril nature of the particles (Figure 23). This caused an increase in the surface area and allowed the study of its adsorption properties. Methyl orange (orange II dye), a potential water pollutant from the dyeing industries, was used as a model molecule, and its adsorption on LDH was studied. The adsorption was found to be directly proportional to its surface area.120 By virtue of the unique structure offered by cellulose, this composite material could become a strong platform for diverse applications in the areas of drug delivery, catalysis, catalyst support, flame retardants, and adsorbents in environmental applications.
Figure 23.

SEM images of as-synthesized LDH particles: (a, b) MCC, (c, d) LDH, (e, f) LDH-0.5% MCC, (g, h) LDH-1.5% MCC, (i, j) LDH-0.5% MCC removed, and (k, l) LDH-1.5% MCC removed. Reprinted from ref (120) with permission from Elsevier.
Conclusions
Topochemical engineering is an exciting field for the design and development of processes and products. The assembly and disassembly of biomass components can be creatively combined to generate biobased functional materials that are highly engineered at the molecular and nanoarchitectural levels. Among the various biomass resources, wood fibers/cellulose occupy a distinct candidate for functionalized material applications due to the potential to drive interactions of electrostatic, hydrophilic, and hydrophobic forces with guest molecules and macromolecules. Topochemical engineering has been applied to advance the design and applications of cellulose-based materials such as drug/enzyme carriers and chemo-, bio-, and photoresponsive functional materials. We were able to design hybrid templates or scaffolds for the fabrication of functional nanoarchitectures by using their fantastic three-dimensionally cross-linked porous structures, micro-to-nano hierarchical morphologies, and preformed shapes. This advanced approach to creating functional biopolymer and biohybrid materials can be integrated in process and product design leading to potential innovations in bioeconomy and environmental benefits.
Impact and Future Direction
We are living in the infancy of topochemical engineering, and we foresee exciting future developments in many fields from drug discovery and tissue engineering to the creation of smart materials and processes. It emphasizes the control and direction of assembly and disassembly, and it can be applied to material sciences, bioengineering, biomedicine, and new emerging technologies for additive manufacture. The disassembly of biocomponents from biomass or bioprocess products is a crucial unit operation that affects purity, crystallinity, structure–property relationships, reproducibility, and the quality and cost of biopolymers, pharmaceuticals, and biomolecules. Generally, the design of separation and purification methods is done via a trial-and-error approach, being specific for targeted applications and difficult to reproduce in other value chains. Topochemical engineering concepts applied to disassembly and down-streaming processing of biocomponents will advance the creation of highly engineered bioproducts with targeted macromolecular properties, the structure–function relationship, and the designed biological function. The assembly of bio- and synthetic components in engineered nanostructures is essential to the design and shape of advanced materials. Topochemical engineering concepts applied to the assembly and shaping of materials can combine methods of molecular and supramolecular chemistry to design highly engineered architectures suitable for advances in tissue engineering, targeted drug delivery, energy storage, catalysis, and stimuli-responsive applications. Topochemical engineering of functional biopolymer-based materials also has the great potential to advance the creation of processes and products of the emerging bioeconomy.
Biographies

LijiSobhana S. Sobhanadhas is a postdoctoral researcher in the Laboratory of Fibre and Cellulose Technology at Åbo Akademi University. She received her Ph.D. degree in chemistry-nanotechnology (2013) from CLRI, University of Madras, India. Her research interest lies in the functionalization of pulp fibers/surface treatment using inorganic materials (layered double hydroxides), organic (fatty) acids, surfactants, resin acids, and acrylates to produce advanced cellulose hybrid materials.

Lokesh Kesavan is TCSM (Turku Collegium for Science and Medicine) fellow carrying out his research in hybrid materials for energy, chemicals, and engineering materials at the University of Turku, Finland. He obtained his Ph.D. from Cardiff University, United Kingdom in the field of heterogeneous catalysis under eminent physical chemist Graham Hutchings FRS, followed by postdoctoral research positions held at Aalto University (Finland) and Trinity University (USA). During his career, he collaborated with Åbo Akademi University for the development of functional cellulose materials.

Pedro Fardim is affiliated with the University of Leuven in Belgium and Åbo Akademi University in Finland. He obtained his Doctor of Science in chemistry at State University of Campinas, Brazil and his Habilitation in Chemical Engineering at Åbo Akademi University in Finland. He is a Fellow of the Royal Society of Chemistry and the International Academy of Wood Science, vice president of the European Polysaccharide Network of Excellence (EPNOE), and a member of the American Chemical Society. His research interest lies in engineering biopolymers and biohybrids for health and care and topochemical engineering for process and bioproduct design.
The authors declare no competing financial interest.
References
- Fardim P.; Holmbom B.; Ivaska A.; Karhu J.; Mortha G.; Laine J. Critical comparison and validation of methods for determination of anionic groups in pulp fibres. Nord. Pulp Pap. Res. J. 2002, 17, 346–351. 10.3183/NPPRJ-2002-17-03-p346-351. [DOI] [Google Scholar]
- de Nooy A. E. J.; Besemer A. C.; van Bekkum H. On the use of stable organic nitroxyl radicals for the oxidation of primary and secondary alcohols. Synthesis 1996, 10, 1153–1176. 10.1055/s-1996-4369. [DOI] [Google Scholar]
- Saito T.; Isogai A. TEMPO-mediated oxidation of native cellulose. The effect of oxidation conditions on chemical and crystal structures of the water-insoluble fractions. Biomacromolecules 2004, 5, 1983–1989. 10.1021/bm0497769. [DOI] [PubMed] [Google Scholar]
- Kohlschutter V.; Haenni P. Zur Kenntnis des Graphitischen Kohlenstoffs und der Graphitsäure. Z. Anorg. Allg. Chem. 1919, 105, 121–144. 10.1002/zaac.19191050109. [DOI] [Google Scholar]
- Ramamurthy V.; Venkatesan K. Photochemical reactions of organic crystals. Chem. Rev. 1987, 87, 433–481. 10.1021/cr00078a009. [DOI] [Google Scholar]
- Langmuir I. The constitution and fundamental properties of solids and liquids. Part I. Solids. J. Am. Chem. Soc. 1916, 38, 2221–2295. 10.1021/ja02268a002. [DOI] [Google Scholar]
- Boldyrev V. V. Topochemistry and topochemical reactions. React. Solids 1990, 8, 231–246. 10.1016/0168-7336(90)80023-D. [DOI] [Google Scholar]
- Yllner S.; Enstrom B. Studies of the adsorption of xylan on cellulose fibres during the sulphate cook, Part 1. Svensk Papperstidning 1956, 59, 229–232. [Google Scholar]
- Yllner S.; Enstrom B. Studies of the adsorption of xylan on cellulose fibres during the sulphate cook, Part 2. Svensk Papperstidning 1957, 60, 549–554. [Google Scholar]
- Linder A.; Bergman R.; Bodin A.; Gatenholm P. Mechanism of assembly of xylan onto cellulose surfaces. Langmuir 2003, 19, 5072–5077. 10.1021/la0341355. [DOI] [Google Scholar]
- Esker A.; Becker U.; Jamin S.; Beppu S.; Renneckar S.; Glasser W.. Self-Assembly Behavior of Some Co- and Heteropolysaccharides Related to Hemicelluloses. In Hemicelluloses: Science and Technology; International Symposium on Xylans, Mannans and Other Hemicelluloses; Gatenholm P., Tenkanen M., Eds.; American Chemical Society: Washington, DC, 2002; pp 198–219.
- Vega B.; Petzold-Welcke K.; Fardim P.; Heinze T. Studies on the fibre surfaces modified with xylan polyelectrolytes. Carbohydr. Polym. 2012, 89, 768–776. 10.1016/j.carbpol.2012.04.006. [DOI] [PubMed] [Google Scholar]
- Heinze T.; Koschella A.; Brackhagen M.; Engelhardt J.; Nachtkamp K. Studies on non-natural deoxyammonium cellulose. Macromol. Symp. 2006, 244 (1), 74–82. 10.1002/masy.200651206. [DOI] [Google Scholar]
- Elegir G.; Kindl A.; Sadocco P.; Orlandi M. Development of antimicrobial cellulose packaging through laccase-mediated grafting of phenolic compounds. Enzyme Microb. Technol. 2008, 43, 84–92. 10.1016/j.enzmictec.2007.10.003. [DOI] [Google Scholar]
- Kim S.; Zille A.; Murkovic M.; Güebitz G.; Cavaco-Paulo A. Enzymatic poly-merization on the surface of functionalized cellulose fibers. Enzyme Microb. Technol. 2007, 40, 1782–1787. 10.1016/j.enzmictec.2007.01.001. [DOI] [Google Scholar]
- Li X.; Tabil L. G.; Panigrahi S. Chemical treatments of natural fiber foruse in natural fiber-reinforced composites: A review. J. Polym. Environ. 2007, 15 (1), 25–33. 10.1007/s10924-006-0042-3. [DOI] [Google Scholar]
- Persson P.Strategies for Cellulose Fiber Modification. Ph.D. Thesis, KTH, STFI-Packforsk AB, Stockholm, Sweden, April 2004. [Google Scholar]
- Qiu X.; Hu S. Smart” materials based on cellulose: A Review of the preparations, properties, and applications. Materials 2013, 6 (3), 738–781. 10.3390/ma6030738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega B.; Holger W.; Bretschneider L.; Näreoja T.; Fardim P.; Heinze T. Preparation of reactive fibre interfaces using multifunctional cellulose derivatives. Carbohydr. Polym. 2015, 132, 261–273. 10.1016/j.carbpol.2015.05.048. [DOI] [PubMed] [Google Scholar]
- Köhnke T.Adsorption of Xylans on Cellulosic Fibres. Influence of Xylan Composition on Adsorption Characteristics and Kraft Pulp Properties. Ph.D. Thesis, Chalmers University of Technology, 2010. [Google Scholar]
- Larsson P.; Puttaswamaiah S.; Ly C.; Vanerek A.; Hall C.; Drolet F. Filtration, Adsorption and Immunodetection of Virus Using Polyelectrolyte Multilayer-Modified Paper. Colloids Surf., B 2013, 101, 205–209. 10.1016/j.colsurfb.2012.06.023. [DOI] [PubMed] [Google Scholar]
- Ankerfors C.; Lingström R.; Wågberg L. A Comparison of Polyelectrolytes Complexes and Multilayers: Their Adsorption Behavior and Use for Enhancing Tensile Strength of Paper. Nord. Pulp Pap. Res. J. 2009, 24 (1), 77–86. 10.3183/NPPRJ-2009-24-01-p077-086. [DOI] [Google Scholar]
- Gernandt R.; Wågberg L.; Gärdlund L.; Dautzenberg H. Polyelectrolyte Complexes for Surface Modification of Wood Fibres. I. Preparation and Characterisation of Complexes for Dry and Wet Strength Improvement of Paper. Colloids Surf., A 2003, 213, 15–25. 10.1016/S0927-7757(02)00335-7. [DOI] [Google Scholar]
- Kikuchi Y.; Noda A. Polyelectrolyte Complexes of Heparin with Chitosan. J. Appl. Polym. Sci. 1976, 20, 2561–2563. 10.1002/app.1976.070200924. [DOI] [Google Scholar]
- Fukuda H. Polyelectrolyte Complexes of Chitosan with Sodium Carboxymethylcellulose. Bull. Chem. Soc. Jpn. 1980, 53, 837–840. 10.1246/bcsj.53.837. [DOI] [Google Scholar]
- Hara M.; Nakajima A. Formation of Polyelectrolyte Complex of Heparin with Aminoacetalized poly(vinyl alcohol). Polym. J. 1978, 10 (1), 37–44. 10.1295/polymj.10.37. [DOI] [Google Scholar]
- Dumitriu S.; Chornet E. Inclusion and Release of Proteins from Polysaccharide-Based Polyion Complexes. Adv. Drug Delivery Rev. 1998, 31, 223–246. 10.1016/S0169-409X(97)00120-8. [DOI] [PubMed] [Google Scholar]
- Vega B.; Holger W.; Zarth C. S. P.; Heikkila E.; Fardim P.; Heinze T. Charge-Directed Fiber Surface Modification by Molecular Assemblies of Functional Polysaccharides. Langmuir 2013, 29, 13388–13395. 10.1021/la402116j. [DOI] [PubMed] [Google Scholar]
- Dence C. W.Chemistry of Mechanical Pulp Bleaching. In Pulp Bleaching: Principles and Practice; Dence C. W., Reeve D. W., Eds.; TAPPI Press: Atlanta, 1994; pp 161–181. [Google Scholar]
- Pan G. X.; Spencer L.; Leary G. J. A comparative study on reactions of hydrogen peroxide and peracetic acid with lignin chromophores. Part 1. The reaction of coniferaldehyde model compounds. Holzforschung 2000, 54 (2), 144–152. 10.1515/HF.2000.025. [DOI] [Google Scholar]
- Plesnicar B.Oxidation with Peroxy Acids and Other Peroxides. In Oxidation in Organic Chemistry, Part C; Trahanovsky W. S., Ed.; Academic Press: New York, 1978; p 211. [Google Scholar]
- Anderson J. R.; Amini B.. Hydrogen Peroxide Bleaching. In Pulp Bleaching: Principles and Practice; Dence C. W., Reeve D. W., Eds.; TAPPI Press: Atlanta, 1994; pp 411–442. [Google Scholar]
- James A. P.; Mackirdy I. S. The chemistry of peroxygen bleaching. Chem. Ind. (London) 1990, 20, 641–645. [Google Scholar]
- Coucharriere C.; Mortha G.; Lachenal D.; Briois S.; Larnicol P. Rationalization of the use of TAED during activated peroxide bleaching. Part II: Bleaching optimization. J. Pulp. Pap. Sci. 2004, 30 (2), 35–41. [Google Scholar]
- Zeinaly F.; Shakhes J.; Zeinali N. Multi stage peroxide and activated peroxide bleaching of kenaf bast pulp. Carbohydr. Polym. 2013, 92 (2), 976–981. 10.1016/j.carbpol.2012.10.043. [DOI] [PubMed] [Google Scholar]
- Turner N. A.; Mathews A. J.. Enhanced Delignification and Bleaching Using TAED Activated Peroxide. TAPPI Pulping Conference; Montréal, Québec, Canada, 1998, Proceedings Book 3, pp 1269–1276.
- Zhao Q.; Sun D.; Wang Z.; Pu J.; Jin X.; Xing M. Effects of different activation processes on H2O2/TAED bleaching of Populus nigra chemi-thermomechanical pulp. BioResources 2012, 7 (4), 4889–4901. 10.15376/biores.7.4.4889-4901. [DOI] [Google Scholar]
- Iamazaki E. T.; Orblin E.; Fardim P. Topochemical activation of pulp fibres. Cellulose 2013, 20, 2615–2624. 10.1007/s10570-013-0016-2. [DOI] [Google Scholar]
- Irie M. Properties and applications of photoresponsive polymers. Pure Appl. Chem. 1990, 62, 1495–1502. 10.1351/pac199062081495. [DOI] [Google Scholar]
- Wondraczek H.; Kotiaho A.; Fardim P.; Heinze T. Photoactive polysaccharides. Carbohydr. Polym. 2011, 83, 1048–1061. 10.1016/j.carbpol.2010.10.014. [DOI] [Google Scholar]
- Wondraczek H.; Pfeifer A.; Heinze T. Synthetic photocrosslinkable polysaccharide sulfates. Eur. Polym. J. 2010, 46, 1688–1695. 10.1016/j.eurpolymj.2010.06.005. [DOI] [Google Scholar]
- Grigoray O.; Wondraczek H.; Heikkila E.; Fardim P.; Heinze T. Photoresponsive Cellulose Fibers by Surface Modification with Multifunctional Cellulose Derivatives. Carbohydr. Polym. 2014, 111, 280–287. 10.1016/j.carbpol.2014.04.089. [DOI] [PubMed] [Google Scholar]
- Wondraczek H.; Pfeifer A.; Heinze T. Water soluble photoactive cellulose derivatives: synthesis and characterization of mixed 2-[(4-methyl-2-oxo-2H-chromen-7-yl)oxy]acetic acid–(3-carboxypropyl)trimethylammonium chloride esters of cellulose. Cellulose 2012, 19, 1327–1335. 10.1007/s10570-012-9708-2. [DOI] [Google Scholar]
- Grigoray O.; Holger W.; Stephan D.; Katrin K.; Seyed K.; Latifi; Pooya S.; Pedro F.; Pasi K.; Heinze T. Photocontrol of Mechanical Properties of Pulp Fibers and Fiber-to-Fiber Bonds via Self- Assembled Polysaccharide Derivatives. Macromol. Mater. Eng. 2015, 300 (3), 277–282. 10.1002/mame.201400286. [DOI] [Google Scholar]
- Wagberg L. Polyelectrolyte Adsorption onto Cellulose Fibres - A Review. Nord. Pulp Pap. Res. J. 2000, 15 (5), 586–597. 10.3183/NPPRJ-2000-15-05-p586-597. [DOI] [Google Scholar]
- Grigoray O.; Holger W.; Annett P.; Fardim P.; Heinze T. Fluorescent multifunctional polysaccharides for sustainable supramolecular functionalization of fibers in water. ACS Sustainable Chem. Eng. 2017, 5 (2), 1794–1803. 10.1021/acssuschemeng.6b02539. [DOI] [Google Scholar]
- Kurrle F. L.; Parks C. J.. Process of Manufacturing Authenticatable Paper Products. U.S. Patent 6,054,021, 2000.
- Foster J. J.; Mulcaahy L. T.. Process for Making and Detecting Anti-Counterfeit Paper. U.S. Patent 6,045,656, 2000.
- de Souza Bezerra T. M.; Bassan J. C.; Tabosa de Oliveira Santos V.; Ferraz A.; Monti R. Covalent immobilization of laccase in green coconut fiber and use in clarification of apple juice. Process Biochem. 2015, 50, 417–423. 10.1016/j.procbio.2014.12.009. [DOI] [Google Scholar]
- Erramuspe I. B. V.; Fazeli E.; Nareoja T.; Trygg J.; Hänninen P.; Heinze T.; Fardim P. Advanced cellulose fibers for efficient immobilization of enzymes. Biomacromolecules 2016, 17 (10), 3188–3197. 10.1021/acs.biomac.6b00865. [DOI] [PubMed] [Google Scholar]
- Marconi W. Immobilized enzymes: their catalytic behaviour and their industrial and analytical applications. React. Polym. 1989, 11, 1–19. 10.1016/0923-1137(89)90078-X. [DOI] [Google Scholar]
- Liu Y.; Chen Y. Enzyme immobilization on cellulose matrixes. J. Bioact. Compat. Polym. 2016, 31 (6), 553–567. 10.1177/0883911516637377. [DOI] [Google Scholar]
- Schaeffer B.; Gardner H. Nature and constitution of shellac separation of the constituent acids. Ind. Eng. Chem. 1938, 30 (3), 333–336. 10.1021/ie50339a024. [DOI] [Google Scholar]
- Sharma S. K.; Shukla S. K.; Vaid D. N. Shellac-structure, characteristics & modification. Def. Sci. J. 1983, 33 (3), 261–271. 10.14429/dsj.33.6181. [DOI] [Google Scholar]
- Obradovic J.; Petibon F.; Fardim P. Preparation and characterisation of cellulose-shellac biocomposites. BioResources 2016, 12 (1), 1943–1959. 10.15376/biores.12.1.1943-1959. [DOI] [Google Scholar]
- Wang Q.; O’Hare D. Recent advances in the synthesis and application of layered double hydroxide (LDH) nanosheets. Chem. Rev. 2012, 112 (7), 4124–4155. 10.1021/cr200434v. [DOI] [PubMed] [Google Scholar]
- Dou Y.; Xu S.; Liu X.; Han J.; Yan H.; Wei M.; Evans D. G.; Duan X. Transparent, Flexible Films Based on Layered Double Hydroxide/Cellulose Acetate with Excellent Oxygen Barrier Property. Adv. Funct. Mater. 2014, 24, 514–521. 10.1002/adfm.201301775. [DOI] [Google Scholar]
- Liji Sobhana S. S.; Mehedi R.; Mika M.; Petriina P.; Mika L.; Marinela M. D.; Garcia Y.; Fardim P. Heteronuclear nanoparticles supported hydrotalcites containing Ni(II) and Fe(III) stable photocatalysts for Orange II degradation. Appl. Clay Sci. 2016, 132–133, 641–649. 10.1016/j.clay.2016.08.016. [DOI] [Google Scholar]
- Sobhana L.; Mohamed S.; Prevot V.; Fardim P. Layered double hydroxides decorated with Au-Pd nanoparticles to photodegradate Orange II from water. Appl. Clay Sci. 2016, 134, 120–127. 10.1016/j.clay.2016.06.019. [DOI] [Google Scholar]
- von Hartman S.; Heikkila E.; Lange C.; Fardim P. Potential applications of hybrid layered double hydroxide particles in pulp and paper production. BioResources 2014, 9 (2), 2274–2288. 10.15376/biores.9.2.2274-2288. [DOI] [Google Scholar]
- Jung Y. C.; Bhushan B. Mechanically Durable Carbon Nanotube Composite Hierarchical Structures with Superhydrophobicity, Self-Cleaning, and Low-Drag. ACS Nano 2009, 3 (12), 4155–4163. 10.1021/nn901509r. [DOI] [PubMed] [Google Scholar]
- Liji Sobhana S. S.; Xue Z.; Lokesh K.; Pirkko L.; Fardim P. Layered double hydroxide interfaced stearic acid - Cellulose fibres: A new class of super-hydrophobic hybrid materials. Colloids Surf., A 2017, 522, 416–424. 10.1016/j.colsurfa.2017.03.025. [DOI] [Google Scholar]
- Shu W.; Zhaojun T.; Zengfu J.; Zelong Y.; Lijuan W. Preparation and characterization of hydrophobic cotton fibre for water/oil separation by electroless plating combined with chemical corrosion. Int. J. Environ. Res. Publ. Health. 2015, 2 (10), 144–150. 10.15739/irjpeh0.32. [DOI] [Google Scholar]
- Lange C.; Lundin T.; Fardim P. Hydrophobisation of mechanical pulp fibres with sodium dodecyl sulphate functionalised layered double hydroxide particles. Holzforschung 2012, 66, 433–441. 10.1515/hf.2011.168. [DOI] [Google Scholar]
- Lange C.; Touaiti F.; Fardim P. Hybrid clay functionalized biofibres for composite applications. Composites, Part B 2013, 47, 260–266. 10.1016/j.compositesb.2012.10.032. [DOI] [Google Scholar]
- Lange C.; Lastusaari M.; Reza M.; Latifi S.; Kallio P.; Fardim P. In Situ Hybridization of Pulp Fibers Using Mg-Al Layered Double Hydroxides. Fibers 2015, 3, 103–133. 10.3390/fib3020103. [DOI] [Google Scholar]
- Wang S.; Huang J.; Chen F. Study on Mg-Al hydrotalcites in flameretardant paper preparation. BioResources 2012, 7 (1), 997–1007. [Google Scholar]
- Li Y. C.; Mannen S.; Cain A. C.; Grunlan J.. Layer-by-Layer Assembly of Layered Double Hydroxides on Cotton Fabric for Anti-Flammability. Conference Proceedings of the 241st ACS National Meeting & Exposition; Anaheim, CA, USA, 2011, pp 27–31.
- Lange C.; Eriksson J.; Lehmonen J.; Tuominen M.; Ek P.; Fardim P. Nanoengineering of Hybrid Lightweight Cellulosic Fibre Foams for better Flame Resistance. J. Nanosci. Adv. Technol. 2016, 1 (3), 1–13. 10.24218/jnat.2015.11. [DOI] [Google Scholar]
- Blanch H. W.; Wilke C. R. Sugars and chemicals from cellulose. Rev. Chem. Eng. 1983, 1, 71–119. 10.1515/revce-1983-0103. [DOI] [Google Scholar]
- Mooney C. A.; Mansfield S. D.; Touhy M. G.; Saddler J. N. The effect of initial pore volume and lignin content on the enzymatic hydrolysis of softwoods. Bioresour. Technol. 1998, 64, 113–119. 10.1016/S0960-8524(97)00181-8. [DOI] [Google Scholar]
- Rahikainen J.; Mikander S.; Marjamaa K.; Tamminen T.; Lappas A.; Viikari L.; Kruus K. Inhibition of enzymatic hydrolysis by residual lignins from softwood – study of enzyme binding and inactivation on lignin-rich surface. Biotechnol. Bioeng. 2011, 108, 2823–2834. 10.1002/bit.23242. [DOI] [PubMed] [Google Scholar]
- Mou H.; Elina O.; Kruus K.; Fardim P. Topochemical pretreatment of wood biomass to enhance enzymatic hydrolysis of polysaccharides to sugars. Bioresour. Technol. 2013, 142, 540–545. 10.1016/j.biortech.2013.05.046. [DOI] [PubMed] [Google Scholar]
- Gabov K.; Gosselink R. J. A.; Smeds A. I.; Fardim P. Characterization of Lignin Extracted from Birch Wood by a Modified Hydrotropic Process. J. Agric. Food Chem. 2014, 62 (44), 10759–10767. 10.1021/jf5037728. [DOI] [PubMed] [Google Scholar]
- Trygg J.; Fardim P. Enhancement of cellulose dissolution in water-based solvent via ethanol–hydrochloric acid pretreatment. Cellulose 2011, 18, 987–994. 10.1007/s10570-011-9550-y. [DOI] [Google Scholar]
- Trygg J.; Poonam T.; Fardim P. Controlled depolymerisation of cellulose to a given degree of polymerisation. Cellulose Chem. Technol. 2016, 50 (5–6), 557–567. [Google Scholar]
- Guile G. R.; Wong S. Y. C.; Dwek R. A. Analytical and preparative separation of anionic oligosaccharides by weak anion-exchange high-performance liquid chromatography on an inert polymer column. Anal. Biochem. 1994, 222, 231–235. 10.1006/abio.1994.1478. [DOI] [PubMed] [Google Scholar]
- Bilandi A.; Mishra A. K. Ion exchange resins: an approach towards taste making of bitter drugs and sustained release formulations with their patents. Int. Res. J. Pharm. 2013, 4, 65–74. 10.7897/2230-8407.04811. [DOI] [Google Scholar]
- Sheldon A.; Newman S. Technical reports: The "life-line" test. Soc. Sci. Med. 1968, 1, 441–444. 10.1016/0037-7856(68)90046-2. [DOI] [Google Scholar]
- Agency IAE, Vienna (1967). The plutonium-oxygen and uranium-plutonium-oxygen systems: a thermochemical assessment. Technical Reports Series No. 79. Report of a Panel on Thermodynamics of Plutonium Oxides. Vienna, Austria, 24–28 Oct 1966.
- Ettenauer M.; Loth F.; Thümmler K.; Fischer S.; Weber V.; Falkenhagen D. Characterization and functionalization of cellulose microbeads for extraporeal blood purification. Cellulose 2011, 18, 1257–1263. 10.1007/s10570-011-9567-2. [DOI] [Google Scholar]
- Guo X.; Du Y.; Chen F.; Park H.-S.; Xie Y. Mechanism of removal of arsenic by bead cellulose loaded with iron oxyhydroxide (β-FeOOH). J. Colloid Interface Sci. 2007, 314, 427–433. 10.1016/j.jcis.2007.05.071. [DOI] [PubMed] [Google Scholar]
- Štamberg J.; Peška J.; Dautzenberg H.; Phillip B.; Gribnau T. C. J.; Visser J.; Nivard R. J. F., Eds.; Affinity Chromatography and Related Techniques; Elsevier Science Publishing Co.: Amsterdam, 1982; pp 131–141. [Google Scholar]
- Weber V.; Linsberger I.; Ettenauer M.; Loth F.; Höyhtyä M.; Falkenhagen D. Development of specific adsorbents for human tumor necrosis factor-alpha: influence of antibody immobilization on performance and biocompatibility. Biomacromolecules 2005, 6, 1864–1870. 10.1021/bm040074t. [DOI] [PubMed] [Google Scholar]
- Zhou D.; Zhang L.; Guo S. Mechanisms of lead biosorption on cellulose/chitin beads. Water Res. 2005, 39, 3755–3762. 10.1016/j.watres.2005.06.033. [DOI] [PubMed] [Google Scholar]
- Yildir E.; Kolakovic R.; Genina N.; Trygg J.; Gericke M.; Hanski L.; Ehlers H.; Rantanen J.; Tenho M.; Vuorela P.; Fardim P.; Sandler N. Tailored beads made of dissolved cellulose—Investigation of their drug release properties. Int. J. Pharm. 2013, 456 (2), 417–423. 10.1016/j.ijpharm.2013.08.047. [DOI] [PubMed] [Google Scholar]
- Isogai A.; Atalla R. Dissolution of cellulose in aqueous NaOH solutions. Cellulose 1998, 5, 309–319. 10.1023/A:1009272632367. [DOI] [Google Scholar]
- Liu W.; Budtova T.; Navard P. Influence of ZnO on the properties of dilute and semi-dilute cellulose-NaOH-water solutions. Cellulose 2011, 18, 911–920. 10.1007/s10570-011-9552-9. [DOI] [Google Scholar]
- Qi H.; Chang C.; Zhang L. Effects of temperature and molecular weight on dissolution of cellulose in NaOH/urea aqueous solution. Cellulose 2008, 15, 779–787. 10.1007/s10570-008-9230-8. [DOI] [Google Scholar]
- Trygg J.; Fardim P.; Gericke M.; Mkil E.; Salonen J. Physicochemical design of the morphology and ultrastructure of cellulose beads. Carbohydr. Polym. 2013, 93, 291–299. 10.1016/j.carbpol.2012.03.085. [DOI] [PubMed] [Google Scholar]
- De Nooy A. E.; Besemer A. C.; van Bekkum H. Highly selective nitroxyl radical-mediated oxidation of primary alcohol groups in water-soluble glucans. Carbohydr. Res. 1995, 269, 89–98. 10.1016/0008-6215(94)00343-E. [DOI] [Google Scholar]
- Stone J.; Scallan A. A structural model for the cell wall of water-swollen wood pulp fibres based on their accessibility to macromolecules. Cell. Chem. Technol. 1968, 2, 343–358. [Google Scholar]
- Trygg J.; Yildir E.; Kolakovic R.; Sandler N.; Fardim P. Anionic cellulose beads for drug encapsulation and release. Cellulose 2014, 21, 1945–1955. 10.1007/s10570-014-0253-z. [DOI] [Google Scholar]
- Trivedi P.; Trygg J.; Saloranta T.; Fardim P. Synthesis of novel zwitterionic cellulose beads by oxidation and coupling chemistry in water. Cellulose 2016, 23, 1751–1761. 10.1007/s10570-016-0939-5. [DOI] [Google Scholar]
- Hirota M.; Tamura N.; Saito T.; Isogai A. Oxidation of regenerated cellulose with NaClO2 catalyzed by TEMPO and NaClO under acid-neutral conditions. Carbohydr. Polym. 2009, 78, 330–335. 10.1016/j.carbpol.2009.04.012. [DOI] [Google Scholar]
- Saito T.; Hirota M.; Tamura N. Individualization of nanosized plant cellulose fibrils by direct surface carboxylation using TEMPO catalyst under neutral conditions. Biomacromolecules 2009, 10, 1992–1996. 10.1021/bm900414t. [DOI] [PubMed] [Google Scholar]
- Ganguly T.; Kasten B. B.; Bučar D.-K. The hydrazide/hydrazone click reaction as a biomolecule labeling strategy for M(CO)3 (M = Re, 99mTc) radiopharmaceuticals. Chem. Commun. 2011, 47, 12846–12848. 10.1039/c1cc15451f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donovan L.; De Bank P. A. A hydrazide-anchored dendron scaffold for chemoselective ligation strategies. Org. Biomol. Chem. 2014, 12, 7290–7296. 10.1039/C4OB00870G. [DOI] [PubMed] [Google Scholar]
- O’Shannessy D. J.; Wilchek M. Immobilization of glycoconjugates by their oligosaccharides: use of hydrazidoderivatized matrices. Anal. Biochem. 1990, 191, 1–8. 10.1016/0003-2697(90)90377-L. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Li X.; Martin D. B.; Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 2003, 21, 660–666. 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- Raddatz S. Hydrazide oligonucleotides: new chemical modification for chip array attachment and conjugation. Nucleic Acids Res. 2002, 30, 4793–4802. 10.1093/nar/gkf594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne H.; Neubert P.; Kuhn K. Carbonyl-reactive tandem mass tags for the proteome-wide quantification of N-linked glycans. Anal. Chem. 2012, 84, 3716–3724. 10.1021/ac300197c. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Cao Z.; Bai T. Zwitterionic hydrogels implanted in mice resist the foreign-body reaction. Nat. Biotechnol. 2013, 31, 553–556. 10.1038/nbt.2580. [DOI] [PubMed] [Google Scholar]
- Jia Y.; Jarrett H. W. Method for trapping affinity chromatography of transcription factors using aldehyde–hydrazide coupling to agarose. Anal. Biochem. 2015, 482, 1–6. 10.1016/j.ab.2015.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gericke M.; Trygg J.; Fardim P. Functional cellulose beads: preparation, characterization, and applications. Chem. Rev. 2013, 113, 4812–4836. 10.1021/cr300242j. [DOI] [PubMed] [Google Scholar]
- Kenawy E. R.; Abdel-Hay F. I.; El-Shanshoury A. E. R.; El-Newehy M. H. Biologically active polymers: synthesis and antimicrobial activity of modified glycidyl methacrylate polymers having a quaternary ammonium and phosphonium groups. J. Controlled Release 1998, 50, 145–152. 10.1016/S0168-3659(97)00126-0. [DOI] [PubMed] [Google Scholar]
- Kugler R.; Bouloussa O.; Rondelez F. Evidence of a charge-density threshold for optimum efficiency of biocidal cationic surfaces. Microbiology 2005, 151, 1341–1348. 10.1099/mic.0.27526-0. [DOI] [PubMed] [Google Scholar]
- Liang T.; Zhang Y.; Li S.; Nguyen T. T. H. Synthesis, characterization, and bioactivity of rosin quaternary ammonium salt derivatives. BioResources 2013, 8 (1), 735–742. 10.15376/biores.8.1.735-742. [DOI] [Google Scholar]
- Nurdin N.; Helary G.; Sauvet G. Biocidal polymers active by contact. III. Ageing of biocidal polyurethane coatings in water. J. Appl. Polym. Sci. 1993, 50 (4), 671–678. 10.1002/app.1993.070500412. [DOI] [Google Scholar]
- Blachechen L. S.; Fardim P. Denise Freitas Siqueira Petri, Multifunctional Cellulose Beads and Their Interaction with Gram Positive Bacteria. Biomacromolecules 2014, 15 (9), 3440–3448. 10.1021/bm5009876. [DOI] [PubMed] [Google Scholar]
- Nada A. M. A.; El-Diwany A. I.; Elshafei A. M. Infrared and antimicrobial studies on different lignins. Acta Biotechnol. 1989, 9, 295–298. 10.1002/abio.370090322. [DOI] [Google Scholar]
- Nelson J. L.; Alexander J. W.; Gianotti L.; Chalk C. L.; Pyles T. Influence of dietary fiber on microbial growth in vitro and bacterial translocation after burn injury in mice. Nutrition 1994, 10, 32–36. [PubMed] [Google Scholar]
- Gabov K.; Oja T.; Deguchi T.; Fallarero A.; Fardim P. Preparation, characterization and antimicrobial application of hybrid cellulose-lignin beads. Cellulose 2017, 24, 641–658. 10.1007/s10570-016-1172-y. [DOI] [Google Scholar]
- Trivedi P.; Saloranta T.; Uroš M.; Lidija G.; Neeraj P.; Jan-Henrik S.; Tamilselvan M.; Martin G.; Heinze T.; Fardim P. Chitosan–Cellulose Multifunctional Hydrogel Beads: Design, Characterization and Evaluation of Cytocompatibility with Breast Adenocarcinoma and Osteoblast Cells. Bioengineering 2018, 5 (1), 3. 10.3390/bioengineering5010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraud E.; Prevot V.; Leroux F. Synthesis and characterization of macroporous MgAl LDH using polystyrene spheres as template. J. Phys. Chem. Solids 2006, 67 (5–6), 903–908. 10.1016/j.jpcs.2006.01.002. [DOI] [Google Scholar]
- Mann S.; Archibaid D. D.; Didymus J. M.; Douglas T.; Heywood B. R.; Meldrum F. C.; Reeves N. J. Crystallization at Inorganic-organic Interfaces: Biominerals and Biomimetic Synthesis. Science 1993, 261 (5126), 1286–1292. 10.1126/science.261.5126.1286. [DOI] [PubMed] [Google Scholar]
- Alan P. I. In Thickening and Gelling Agents for Food, 2nd ed.; Imeson A., Ed.; Blackie Academic & Professional: New York, 1999; p 188. [Google Scholar]
- Liu S.; Tao D. Cellulose scaffold: A green template for the controlling synthesis of magnetic inorganic nanoparticles. Powder Technol. 2012, 217, 502–509. 10.1016/j.powtec.2011.11.010. [DOI] [Google Scholar]
- Liu Y.; Goebl J.; Yin Y. Templated synthesis of nanostructured materials. Chem. Soc. Rev. 2013, 42 (7), 2610–2653. 10.1039/C2CS35369E. [DOI] [PubMed] [Google Scholar]
- Liji Sobhana S. S.; Raj Bogati D.; Reza M.; Gustafsson J.; Fardim P. Cellulose biotemplates for layered double hydroxides networks. Microporous Mesoporous Mater. 2016, 225, 66–73. 10.1016/j.micromeso.2015.12.009. [DOI] [Google Scholar]