

Abstract
A series of isoniazid derivatives bearing a phenolic or heteroaromatic coupled frame were obtained by mechanochemical means. Their pH stability and their structural (conformer/isomer) analysis were checked. The activity of prepared derivatives against Mycobacterium tuberculosis cell growth was evaluated. Some compounds such as phenolic hydrazine 1a and almost all heteroaromatic ones, especially 2, 5 and 7, are more active than isoniazid, and their activity against some M. tuberculosis MDR clinical isolates was determined. Compounds 1a and 7 present a selectivity index >1400 evaluated on MRC5 human fibroblast cells. The mechanism of action of selected hydrazones was demonstrated to block mycolic acid synthesis due to InhA inhibition inside the mycobacterial cell.
Keywords: Mycobacterium tuberculosis, mechanochemistry, hydrazone
1. Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (M.tb), represents an enduring, deadly infectious disease worldwide. According to the World Health Organization (WHO), one third of the global population is infected with M.tb. In comparison with other diseases caused by a single infectious agent, TB is the second leading cause of mortality. It is estimated that in 2015 TB killed 1.8 million people, mainly in underdeveloped countries [1]. New effective drugs for the treatment of TB are necessary firstly to reduce the duration of TB treatment and, secondly, for the treatment of M.tb multidrug-resistant (MDR) [2,3], extensively-drug resistant (XDR) [4], and totally-drug resistant (TDR) strains [5]. In the recent years, two molecules bedaquiline and delamanid (Figure 1) have been approved for the treatment of MDR-TB when an effective treatment regimen is not otherwise available [6,7].
Figure 1.

Antitubercular drug Isoniazid (1952), and Bedaquiline and Delamanid, two new compounds approved for the treatment of MDR-TB.
One of the main known drug targets to fight M.tb growth is the enoyl-ACP reductase or InhA. This well-known enzyme is already the indirect target of Isoniazid (INH), a front-line prodrug used clinically to treat TB. INH remains a key component in all multiple drug treatment regiments recommended by the WHO even if M.tb resistant isolates have been rapidly generated during monotherapy or inappropriate treatment. Hence, improvement of INH molecule by introducing chemical modifications in its core structure in order to enhance biological response (prodrug, increase of bioavailability, and membrane permeability) continues to be an interesting scientific challenge. Recently, for example, compounds containing isonicotinoyl moiety with potential dual inhibition targeting FabG4 and HtdX, were successfully characterized [8].
Hydrazide-hydrazone derivatives have long attracted attention because of their wide range of applications in medicinal chemistry [9,10,11,12]. Hydrazide and hydrazone derivatives showed strong antioxidant and radical scavenging properties, while others ones displayed potent anticancer, antimicrobial, anticonvulsant or anti-inflammatory activities in vitro [9]. Recently, interesting anti-mycobacterial activities were reported about the following derivatives: guanylhydrazones [13], trans-cinnamic acid hydrazides derivatives [14], fluorine containing hydrazones [15], sulfonyl-hydrazones [16], and L-proline derived hydrazones [17].
Moreover, isoniazid-related hydrazones showed similar or better efficiency than the INH [18]. Isonicotinoylhydrazone derivatives were also synthesized and evaluated as anti-mycobacterial agents [19,20]. Interestingly, vanillic acylhydrazones were reported as potential β-keto acyl carrier protein synthase III (FabH) inhibitors [21]. Schiff bases of isoniazid, considered as chemical modification that can block N-acetylation of INH, showed good activity in vitro and in vivo and in some cases low toxicity [22,23]. In 2008, an extended study reported a quantitative structure activity relationships (QSAR) of a large hydrazide family for the developing of antitubercular compounds [24]. Finally, some researchers focused on establishing a predictive QSAR model for different INH derivatives including isonicotinoylhydrazones [25].
Our group is involved both in synthesizing new antitubercular compounds, including cinnamic acid derivatives [26,27,28,29], triazoles [30,31,32,33], pyrrolidines [34,35,36], semicarbazones and hydrazine/hydrazones [37], and in searching for new and innovative synthetic reactions. We have recently reported the solvent-free mechanosynthesis of a series of hydrazones [38,39]. Mechanochemistry has been used for a long time for the chemical and physicochemical transformations of inorganic materials to generate all states of aggregation produced by the effect of mechanical energy [40]. More recently, the mechanical energy has been used to synthesize organic molecules in milling devices [41,42,43,44,45,46,47,48,49,50,51,52].
Considering the pharmaceutical area [53], the mechanical action was used in particular to develop nitrogen-containing heterocycles, well represented in many therapeutic classes. For example, phthalazoles [54], phenazines [55], pyrazoles, pyridazinones [56], and pyrroles [57] were obtained by mechanosynthesis. Non-heterocyclic nitrogen-containing molecules were also synthesized under mechanical solid-state and solvent-free conditions, including imines, azomethines [58,59], azines [56], enamines and hydrazones [60,61,62].
In this work, this methodology was used in order to synthesize hydrazones under solvent-free conditions, in particular the isonicotinoyl ones. Thus, herein we report the synthesis of a series of phenol and hetero aryl isonicotinoylhydrazones through mechanochemistry and the evaluation of their anti-tuberculosis activities.
2. Results and Discussion
2.1. Chemistry
Mechanosynthesis of Isonicotinoyl Hydrazones
The classical methods to synthesize hydrazones are generally carried out at low concentration, and require times from 3 to 24 h or even 48 h under reflux of toluene or ethanol in order to obtain good yields. We previously employed the vibratory mill Pulverisette 0 (P0) (Fritsch, Germany) to synthesize phenolic hydrazones mechanochemically [38]. A comparative study has been therein reported with various hydrazides, among them the isoniazid and phenolic aldehydes leading to compounds already described 1a–d (Figure 2).
Figure 2.

Isonicotinoyl hydrazones synthesized by co-grinding of isoniazide and an aldehyde.
We decided to synthesize a series of isonicotinoylhydrazones derivatives bearing various N-heterocyclic indole, indazole or imidazole moieties using the mechanochemical approach. The corresponding aldehydes were selected because of the importance of these N-heterocyclic fragments in a large number of natural or synthetic biologically active molecules. Indeed, compounds bearing these frames may exhibit various activities, i.e., antibacterial, anticancer, antioxidant, anti-inflammatory, anti-diabetic, antiviral, anti-proliferative, antituberculosis, antispermatogenic or antipsychotic activities [63,64,65,66,67].
The reaction described for phenolic compounds was firstly used with the 4-methyl-5-imidazolecarboxaldehyde as model of heterocyclic aldehyde. However, in spite of the milling was carried out by up to eight hours, TLC and NMR analysis showed incomplete conversion. In that respect, we evaluated the efficiency of the reaction in acidic media.
The reaction was thus carried out in the presence of AlCl3 or of p-toluenesulfonic acid (p-TSA); the latter one showed the best result when using 50% mol of p-TSA. TLC showed the consumption of the reagents and the consequent appearance of the hydrazone, which was confirmed by 1H- and 13C-NMR analysis.
The hydrazones listed in Table 1 were thus obtained with a high transformation ratio in grinding times of 2 h.
Table 1.
Hydrazones 2–11 produced mechanochemically by reacting INH and imidazolic, indazolic or indolic aldehydes. The reaction was catalyzed by p-TSA.
| Aldehydes | Heterocyclic Hydrazones (6) Derived From Isoniazid | |
|---|---|---|
| Imidazole derivatives | 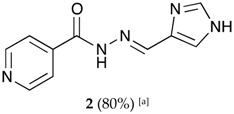 |
 |
| Indazole derivatives |  |
 |
| Indole derivatives | 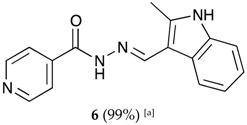 |
 |
 |
 |
|
 |
 |
|
[a] Yields after washing with NaHCO3 aqueous solution to eliminate p-TSA. According to TLC, 1H-NMR and MS, the conversions were quantitative.
It is still important to mention that, differently from the phenolic hydrazones, a melting was produced when the p-TSA was added, and, therefore, the reaction was not fully in solid-state. The formation of a fluid phase is vastly found for solid mixtures (eutectic melting) [68] and surely contributed to reach high conversions in short times of grinding for these hydrazones.
2.2. Structural Analysis of Isonicotinoyl Hydrazones by DFT and NMR: Determination of the Free Activation Energy (ΔG≠) between Conformers of Selected Compounds 1a and 5
The structures of all the hydrazones were identified and fully characterized by 1H- and 13C-NMR, MS and HRMS, FTIR and UV-vis (see Supplementary Materials).
Concerning the NMR data, all isonicotinoyl hydrazones showed the presence of conformers in dimethylsulfoxide (DMSO) solution. NMR spectral and theoretical studies previously demonstrated that acylhydrazones generally exist predominantly or solely as a mixture of isomers [69,70,71]. In theory, N-acylhydrazones may exist with four possible arrangements in respect to (E/Z)-configurational isomers relative to the C=N bond and (E’/Z’)-rotamers caused by inversion of the amide bonds C(O)NH, here named cis/trans amide conformers (Figure 3) [69,72].
Figure 3.

E/Z-configurational isomers and cis/trans amide conformers for N-acylhydrazones.
Although the four forms were considered, E/Z isomerization is generally not observed and the Z geometric isomers are absent or present only in poor part. An exception is for R’ = 2-pyridyl, in which strong intramolecular hydrogen bonds are present in the (Z)-form [70] mainly in less polar solvents.
While all isonicotinoyl hydrazones showed the presence of conformers in DMSO solution, for study purposes, two derivatives were chosen to be further investigated: phenolic hydrazone (1a) and indazole derivative (5). Both of them showed two sets of signals indicating the possibility of equilibrium between rotamers in solution. Theoretical assessment of the existence of the isomers was carried out.
The four structures of Z/E geometrical isomers and cis/trans amide conformers of 1a were modeled by Density Functional theory (DFT), using Gaussian 09, firstly at HF/STO-3G level. The Z conformers were found higher in energy than the E ones, around 6 kcal/mol (see Table S1 in the Supplementary Materials). Thus, only the cisE and transE-isomers were then modeled at the B3LYP/6-31+G(d,p) level and the frequencies calculations were performed on the optimized geometries at 298 K, showing all positive frequencies and allowing evaluation of the Gibbs free energy.
Following Boltzmann distribution, (Pi/Pj = exp((Gj − Gi)/kBT), cisE-isomer was present at 92% and transE at 8% in the gas phase, whereas in DMSO, using the polarizable solvent continuum model (SMD), the ratio was inverted: cis/trans: 6/94 (Table 2).
Table 2.
Geometries and energies of minima and transition state for cis and trans E-isomers of 1a obtained at B3LYP/6-31+G(d,p) level in the gas phase and using the DMSO polarizable continuum model (SMD).

| In the Gas Phase | E (ua) | G (ua) | ΔG (kcal/mol) | % |
| transE_1a | −816.730569 | −816.553707 | 1.47 | 8 |
| cisE_1a | −816.733307 | −816.55605 | 0 | 92 |
| In DMSO | E (ua) | G (ua) | ΔG (kcal/mol) | % |
| transE_1a | −816.763427 | −816.585816 | −1.68 | 94 |
| cisE_1a | −816.761415 | −816.583145 | 0 | 6 |
| TS_1a | −816.765271 | −816.557724 | 17.63 | - |
A transition state (TS-1a) was found between transE and cisE, characterized by its imaginary frequency at −115.79 cm−1. The difference between Gibbs free energy of the transition state and the energy of the cis-E-isomer (ΔG≠) was equal to 17.63 kcal·mol−1.
The NMR chemical shift calculations were then performed at B3LYP/6-311+ (2d,p) level, using the DMSO polarizable continuum model (PCM). Isotropic shielding constants (σ) for 1H and 13C nuclei were transformed in chemical shift (δ) using linear regression procedure proposed by Lodewyk [73]. Calculated values are presented in the Supplementary Materials (Table S2). By comparing calculated values and experimental ones, we unambiguously concluded that the transE-isomer is the major one. This result is also in agreement with the calculated Gibbs free energy values in DMSO, at 298 K, as transE-isomer is −1.68 kcal/mol lower than the cis (ratio cis/trans around 6/94).
This activation energy can also be measured by 1H-NMR analysis, by determining the coalescence temperature Tc.
At the coalescence temperature Tc
kexch = πΔν/√2 = 2.22·Δν (Hz)
kexch = (kB·Tc/h)exp(−ΔG≠/RTc) (Eyringss equation)
ΔG≠ = −RTcLn(kexch·h/kB·Tc)
with kB = 1.38 × 10−23 kJ/K, h = 6.626 × 10−34 J·s, and R = 8.314 J·mol−1·K−1.
The 1H-NMR study at increasing temperatures (from 298 K to 388 K) allowed the determination of coalescence temperatures of the same signals of each isomer (Figure S1). Correlation of the coalescence temperatures Tc with the difference in chemical shift of the signals led to ΔG≠ following the precedent equation described (Supplementary Materials, Table S3). The mean value obtained on several signals led to ΔG≠ = 17.58 kcal·mol−1. This experimental value is in agreement with the calculated one of 17.63 kcal·mol−1 at the B3LYP/6-31+G(d,p) level.
The same study was performed on compound 5, which possesses eight isomers. As previously described, the optimization of geometries on the height isomers at the HF/STO-3G showed that the Z isomers are higher in energy (6–7.5 kcal/mol) (Supplementary Materials, Table S4). Tsshus, the calculations were carried out at the B3LYP/6-31+G(d,p) level on the four isomers: cis and trans E-isomers and the two rotamers, in the gas phase and in the DMSO modeled by the SMD polarizable continuum model. Boltzmann analysis was used to determine the relative distribution of each conformer both in the gas phase and in the DMSO continuum model.
In the gas phase, cis compounds are the major ones, although, in the DMSO continuum, trans isomers are the major ones: transE: 80%, cisE: 20% (Table 3).
Table 3.
Geometries, Gibbs free energies and Boltzmann distribution of the four major conformers of 5 at the B3LYP/6-31+G(d,p) in the gas phase and in the DMSO continuum solvent model (SMD).
| Isomer | Geometry (In Gas Phase) | G (In Gas Phase) | % | G (In DMSO) | % |
|---|---|---|---|---|---|
| transE_5_1 |  |
−888.90736 | 0.34 | −888.94471 | 28.22 |
| transE_5_2 |  |
−888.909926 | 5.09 | −888.94528 | 51.50 |
| cisE_5_1 |  |
−888.910465 | 9.0 | −888.941968 | 1.54 |
| cisE_5_2 |  |
−888.912591 | 85.6 | −888.944326 | 18.72 |
1H- and 13C-NMR chemical shifts were calculated as previously described at the B3LYP/6-311+G(2d,p) using DMSO continuum model (PCM) and taking into account the Boltzmann distribution of the two conformers for each isomer, which means transE-1/transE-2: 35%/65%, and cisE-1/cisE-2: 8%/92%. The comparison of calculated chemical shifts and experimental ones of both 1H and 13C (Supplementary Materials, Figure S2 and Table S5), showed that trans isomer is the major compound in DMSO.
The activation energy was evaluated by 1H-NMR analysis at increasing temperatures (from 298 K to 388 K) and was evaluated at 17.8 kcal/mol (Supplementary Materials, Table S6) in the same order as for compound 1a.
These observations are important as restricted rotations might have an impact on pharmacological properties.
2.3. Physicochemical Studies of Some Isonicotinoyl Hydrazones
2.3.1. Hydrolytic Stability
Stability studies were carried out in order to confirm that biological activities evidenced for isonicotinoyl hydrazones arise from the tested compounds, and not from the hydrolysis of the imine bond. UV-vis spectrophotometry at the λmax of absorbance of the related molecule was used to monitor the stability of the most prominent synthesized compounds. The study was carried out for all compounds; here, we present the results for some of them while all other data are in the Supplementary Materials. Table 4 summarizes the conditions used and the stability results observed for 1a–1d, 5 and 7 compounds.
Table 4.
Stability study of hydrazones.
| Compound | Medium | Conc. (mol/L) | pH | Time | Stability |
|---|---|---|---|---|---|
| 1a | 28% EtOH/H2O | 6.0 × 10−5 | 6.1 | 22 h | stable |
| 1b | 6.3 × 10−5 | 6.5 | 22 h | stable | |
| 1c | 6.9 × 10−5 | 6.3 | 15 h | stable | |
| 1d | 5.9 × 10−5 | 6.2 | 21 h | stable | |
| 5 | 5% EtOH/PIPES buffer (50 mM) | 4.1 × 10−5 | 6.8 | 20 h | 3% Abs. reduction |
| 7 | 3.1 × 10−5 | 6.8 | 20 h | stable | |
| 7 days | 5% Abs. reduction |
As expected, UV-vis spectroscopy demonstrated that all the tested compounds are stable for a prolonged time (up to 22 h–7 days) and no significant decomposition was observed. Only 5 showed a very small reduction (3%) of the absorbance after 20 h. It is known that hydrazones possess greater intrinsic hydrolytic stability than that of imines. In addition to the contribution of the NH-N=C in electron delocalization, the resonance forms in acylhydrazones increase the negative-charge on the C=N and thus, reduce its electrophilicity and the affinity to the nucleophile attack from water [74]. Furthermore, the repulsion of the lone pairs of the NH-N can be relieved in the conjugates [75].
Moreover, the stability of compound 1d was further assessed upon incubation in DMSO, the solvent used for biological assays, by mass spectrometry analysis; in addition, no relevant peaks corresponding to possibly released INH have been detected (Figure S3).
2.3.2. pKa Determination
The acid dissociation constant (Ka, or more commonly expressed by pKa) is a very important physicochemical parameter in a wide range of research areas, including the development of active molecules due to solubility issues. The pKa of the molecules studied for stability could also be determined by using UV-vis spectrometry based on the variation of the absorbance as function of the pH due to the presence of chromophores close to the ionization site of the molecules. The molar absorptivity varies according to the conjugation forms that dynamically change when and where the molecule is charged by the effect of pH and protonation/dissociation. As an example, the different protonation states for the hydrazone 1d are shown in Figure 4.
Figure 4.

Protonation states of the hydrazone 1d as function of pH.
For experimental pKa determination, the molecules were solubilized in hydroalcoholic solutions at ethanol 28% and the ionic strength was maintained by the addition of KCl (0.1 M). The pH was adjusted with concentrated solutions of KOH and HCl. The UV spectra were recorded at each pH value and, at least three wavelengths were monitored. The absorbance variation was plotted as function of the pH. As an example, the variations on the UV-vis spectra for compound 1d and the resulting plot for six wavelength values are shown in Figure 5a,b, respectively (all other spectra are present in Figures S4–S8). The protonation/dissociation of the molecule is accompanied by a variation in the absorbance (Figure 5a) and is graphically represented for six wavelength values as function of pH in Figure 5b.
Figure 5.

The variations on the UV-vis spectra for compound 1d and the resulting plot for six wavelength values. (a) UV-vis spectra obtained for compound 1d as function of pH variation; (b) Plots of absorbance for six wavelength values (λ/nm) as function of pH for compound 1d.
The inflection points correspond to the change in that protonated/dissociated state of the molecule, and, therefore, the pH at that point amounts to the pKa. Thus, by using this method, the pKa values of 1d were determined [76]. The same protocol was applied for the other molecules and the resulted pKa are presented in Table 5, as an average value from three different λ.
Table 5.
pKa values determined for the isonicotinoylhydrazones.
| Compound | 1a | 1b | 1c | 1d | 5 | 7 |
|---|---|---|---|---|---|---|
| pKa1 | nd a | nd a | 3.4 | 3.0 | 3.4 | 3.6 |
| pKa2 | 9.2 ± 0.1 | 7.4 ± 0.1 | 9.1 ± 0.1 | 9.5 ± 0.1 | 10.4 ± 0.1 | 11 |
| pKa3 | - | 8.9 ± 0.1 | - | >11 | - | - |
a nd for not determined.
An inflection point was always observed close to pH 3, which is related with the protonation of 4-pyridinic nitrogen of isoniazid moiety (pKa2 of isoniazid = 3.5), and corresponds to pKa1 of the hydrazones. The pKa2 of the molecules ranges from 7.7 to 11. It means that they are in dissociated form from milder (1b) to stronger (7) basic conditions.
2.4. Biology of Mechanochemically Synthesized Hydrazones
2.4.1. InhA Inhibition Assay
InhA, the NADH-dependent fatty acid biosynthesis (FAS-II) enoylreductase from M. tuberculosis, has emerged as a promising drug target due to its vital role in synthesis of mycolic acids. InhA is the main target of Isoniazid [77,78,79,80]. Recombinant M.tb InhA was expressed in E. coli and subsequently purified.
The selected synthetic compounds, corresponding to the phenolic isonicotinoyl hydrazones 1a–d and the corresponding heterocyclic ones 2–11 series, were evaluated in vitro for the inhibition of M.tb InhA activity at 50 μM by applying a previously described method [81] (Table 6).
Table 6.
Enzyme inhibition values for the INH derivatives. Results are expressed as a percentage of InhA inhibition.
| Compound | % Inhibition at 50 μM (Inhibitor) | Compound | % Inhibition at 50 μM (Inhibitor) |
|---|---|---|---|
| 1a | 45 | 5 | 19 |
| 1b | 54 | 6 | 43 |
| 1c | 48 | 7 | 39 |
| 1d | 64 | 8 | 42 |
| 2 | 54 | 9 | 32 |
| 3 | 3 | 10 | 79 |
| 4 | 33 | 11 | not soluble |
| TCL | >99 |
Considering the four phenolic derivatives 1a–d, the tri-substituted one (1d) presents the better InhA inhibition activity with 64% value at 50 μM. Derivatives 1a–c are less potent with 45–54% InhA inhibition range at 50 μM. Under these conditions, 99% inhibition is obtained for Triclosan (TCL).
Concerning the heterocyclic isonicotinoyl hydrazones, compounds 2, 3 and 11 were difficult to evaluate due to solubility issues at high concentrations. Compound 5 is a very poor inhibitor (19% InhA inhibition), while compounds 4, 6 and 7 may be considered as poor inhibitors with 32%, 43% and 39% values, respectively. Compound 10 is the derivative exhibiting the highest inhibition of InhA enzyme with 79% at 50 μM. In comparing isonicotinoyl derivatives derived from indoles (compounds 6–10), we can notice that those possessing an indol-3-yl frame have the same activities with inhibition values between 32% and 43%. Compounds 6 and 8 differing by one methyl group (position 2 of the indol-3-yl frame) have the same inhibition values. On the contrary, compound 10 possessing an indole-2-yl frame is two-fold more active (79% of inhibition). Concerning the imidazole and indazole derivatives, we might hypothesize that the striking differences in activities could arise by a better positioning of the compounds 2 and 4 guided by their nitrogen atoms on their heterocyclic parts.
2.4.2. Activity of Phenolic Isonicotinoyl Hydrazones (1a–d) against M.tb Cell Growth
The determination of the minimal inhibitory concentration (MIC) was performed using M.tb H37Rv strain and INH as control (Table 7).
Table 7.
Phenolic isonicotinoyl hydrazones tested as inhibitory agents against M.tb growth (H37Rv strain).
| Compound | MW (g/mol) | MIC (μg/mL)/(μM) | LogP | Cpd | MW (g/mol) | MIC (μg/mL)/(μM) | LogP |
|---|---|---|---|---|---|---|---|
| 1a | 241.25 | 0.0125/0.05 | 1.64 | 1c | 271.27 | 0.125/0.46 | 1.51 |
| 1b | 257.24 | 0.125/0.49 | 1.25 | 1d | 301.30 | 0.125/0.41 | 1.38 |
| INH | 137.14 | 0.025/0.18 | −0.64 |
It is noteworthy that a series of phenolic hydrazones bearing the four phenolic frames (a–d), previously synthetized by mechanochemical means were already tested against M.tb. cell growth [38]. Among the different hydrazines (isoniazid, hydralazine, 2-hydrazino-benzothiazole, 3-aminorhodanine, benzyl carbazate and benzhydrazide), used to form the corresponding hydrazones, only the isonicotinoyl derivatives were active against M.tb growth, whilst the other compounds were not effective (MIC > 30 μM; data not shown, but reported in Reference [39]).
Isonicotinoyl derivatives 1a–d showed good anti-mycobacterial activity with 0.0125 or 0.125 μg/mL MIC values. Among these four, those bearing two or more substituents ortho to the phenolic function presented potent activities with a MIC value of 0.125 μg/mL, which is five times higher than that of INH. Compound 1a, synthesized from p-hydroxybenzaldehyde and INH, is the most potent derivative with a MIC value of 0.0125 μg/mL (0.05 μM), which is 2–4 times lower than that of INH (0.025 μg/mL, 0.18 μM).
2.4.3. M.tb H37Rv Growth Inhibition Assays of Nitrogen Heterocyclic Hydrazones (2–11)
The MIC values of the series of hydrazones, bearing the INH moiety coupled with different N-heterocyclic aldehydes, were also determined (Table 8).
Table 8.
MICs of Isoniazid-Nitrogen heterocyclic hydrazones against M.tb H37Rv.
| Compound | MW (g/mol) | MIC (μg/mL)/(μM) | LogP | Cpd | MW (g/mol) | MIC (μg/mL)/(μM) | LogP |
|---|---|---|---|---|---|---|---|
| 2 | 215.21 | 0.03/0.14 | −1.00 | 7 | 265.27 | 0.015/0.056 | 0.24 |
| 3 | 229.24 | 0.03/0.13 | −1.37 | 8 | 264.28 | 0.06/0.23 | 0.86 |
| 4 | 265.27 | 0.06/0.23 | 1.38 | 9 | 343.18 | 0.125/0.36 | 1.69 |
| 5 | 265.27 | 0.03/0.11 | −0.52 | 10 | 264.28 | 0.06/0.23 | 0.52 |
| 6 | 278.31 | 0.25/0.90 | 0.49 | 11 | 309.28 | 0.25/0.81 | 1.39 |
| INH | 137.14 | 0.05/0.36 | −0.64 |
All four imidazole and indazole derivatives (2–5) were 1.5–3 times more effective than isoniazid, while their InhA inhibition activities were much lower to inexistent with activities against M.tb growth, in the range of 0.11–0.23 μM. For indole derivatives, nitro substitution on the aromatic ring (compound 11) or methyl substitution on the indole ring (compound 6), compromised the anti-TB activity. While the bromo derivative 9 had the same activity as INH (0.36 μM), compounds 7, 8 and 10 presented better MIC values than INH. Interestingly, the azaindole derivative 7 showed the highest anti-TB activity in this series and was found as active as the phenolic compound 1a (MIC = 0.015 μg/mL/0.056 μM).
Finally, we can notice that all active compounds here presented, can show different lipophilic values as given by their LogP values, probably supporting the inference that there is no clear relationship between lipophilicity and in vitro activity as pointed also by others [25].
The resistance to the current tested drugs (first- and second-line) remains a very serious problem, mostly resulting from inhA and katG mutations [82] and culminates in the occurrence of M.tb multidrug-resistant (MDR) strains. Owing the good results obtained for the herein studied INH-derived hydrazones, we tested them against a M.tb multidrug-resistant clinical isolate (IC2; resistant to streptomycin, INH, rifampicin, ethambutol, pyrazinamide, ethionamide, and capreomicin) (Table 9). The indazole and indole derivatives were not active against IC2 isolate (MICs > 10 μg/mL), except for the nitro derivative 11. The imidazole containing derivatives 2 and 3 were poor active (MIC = 5–10 μg/mL). By comparing results for compounds 10 and 11, it appears that the nitro substituent improves the activity against IC2 clinical isolate. Interestingly, the phenolic derivatives 1a and 1b presented the best activities against the MDR isolate, with MIC of 2.5 μg/mL (10.36 μM) and 1 μg/mL (3.89 μM), respectively.
Table 9.
MIC of isoniazid derivatives against M.tb MDR isolate IC2.
| Compound | MIC (μg/mL)/(μM) | |
|---|---|---|
| H37Rv | IC2 | |
| 1a | 0.0125/0.05 | 2.5/10.36 |
| 1b | 0.125/0.49 | 1/3.89 |
| 1c | 0.125/0.46 | >2.5/>9.22 |
| 1d | 0.125/0.41 | >2.5/>8.30 |
| 2 | 0.03/0.14 | 5/23.2 |
| 3 | 0.03/0.13 | 5–10/21.8–43.6 |
| 4 | 0.06/0.23 | >10 |
| 5 | 0.03/0.11 | >10 |
| 6 | 0.25/0.90 | >10 |
| 7 | 0.015/0.056 | >10 |
| 8 | 0.06/0.23 | >10 |
| 9 | 0.125/0.36 | >10 |
| 10 | 0.06/0.23 | >10 |
| 11 | 0.25/0.81 | 5–10/18.9–37.8 |
| INH | 0.025/0.18 | >2/>14.58 |
2.4.4. Cytotoxicity and Selectivity Index Determination
Cytotoxicities of all compounds bearing the INH moiety were also evaluated on MRC5 human fibroblast cells. Almost all compounds tested presented LD50 values above 80 μM (Table 10), with the exception of 1b (LD50 = 36.3 μM). The LD50 evaluation is essential to determine the selectivity index (SI), which indicates the best candidates in terms of high biological activity against the target and low cytotoxicity. The SI presented in Table 10 are the ratio between LD50 and the in vitro MIC value against M.tb H37Rv previously obtained.
Table 10.
Cytotoxicity (LD50) and selectivity index (SI) for the most active hydrazones against H37Rv M.tb.
| Compound | LD50 (μM) | SI | Compound | LD50 (μM) | SI |
|---|---|---|---|---|---|
| 1a | >80 | >1600 | 5 | >80 | >727 |
| 1b | 36.3 | 74 | 6 | 129 | 143 |
| 1c | >80 | >173 | 7 | >80 | >1429 |
| 1d | >80 | >195 | 8 | >80 | >364 |
| 2 | >80 | >571 | 9 | >80 | >222 |
| 3 | >80 | >615 | 10 | 71.4 | 310 |
| 4 | >80 | >364 | 11 | 156 | 193 |
| INH | - | - |
Apart from compound 1b, due to its high toxicity, the phenolic hydrazones presented good selectivities higher than 170. A great result was obtained for 1a which has the lowest MIC value (0.0125 μg/mL), LD50 > 80 μM and the highest SI (>1600).
The N-heterocyclic INH derivatives 2–11 presented good SI values, with the exception of 6 and 11, which possessed the highest MIC values. Compounds 4, 8, 9 and 10 conducted to comparable SI. Compound 10 must be considered (MIC = 0.06 μg/mL, LD50 = 71.4 μM and SI = 310), mainly due to its InhA inhibition of 78%. Great SI values closer to 600 are found for 2 and 3 but, nonetheless, some reservation must be taken due to the poor solubility of these molecules. Finally, the SI of compound 5 is higher than 727 and 7 is highlighted with MIC = 0.015 μg/mL and an SI higher than 1469.
2.4.5. In Search for the Molecular Target of Prepared Hydrazones in Mycobacteria
In order to confirm the enoylreductase InhA as a target of synthesized hydrazones, we analyzed the effect of selected compounds with the best MIC values and cytotoxicity scores on synthesis of mycolic acids in avirulent strain M.tb H37Ra. Tested hydrazones, specifically 1a, 3, 5, 7 and 10, as well as INH as control InhA inhibitor, were added to M.tb H37Ra culture when it reached early mid-log phase of growth and, after subsequent 24 h cultivation, 14C acetate was added as a metabolic tracer. TLC analysis of lipid fractions extracted from harvested 14C labeled cells revealed that, similar to INH, all of the tested hydrazones abolish the synthesis of trehalose monomycolates and trehalose dimycolates (Figure 6).
Figure 6.

TLC analysis of lipids extracted from 14C acetate labeled M.tb H37Ra cells treated with compounds 1a, 3, 5, 7 and 10, INH and DMSO as a control. Lipids were separated in chloroform:methanol:water (20:4:0.5) and detected by autoradiography (TDM: trehalose dimycolates; TMM: trehalose monomycolates; PE: phosphatidylethanolamine; CL: cardiolipin).
Analysis of fatty/mycolic acids isolated from whole 14C labeled cells proved that these compounds specifically inhibit synthesis of mycolic acids (Figure 7).
Figure 7.

TLC analysis of methyl esters of fatty (FAME) and mycolic (MAME) acids isolated from 14C acetate labeled M.tb H37Ra cells treated with compounds 1a, 3, 5, 7 and 10, INH and DMSO as a control. Different forms of methyl esters were separated in n-hexane:ethyl acetate (95:5; 3×) and detected by autoradiography. (α, methoxy, and keto refer to forms of MAMEs).
Next, we overproduced InhA protein in M.tb H37Ra and tested sensitivity of overproducer, as well as control strain carrying empty vector against synthesized hydrazones by drop dilution method. Clearly, this testing showed, that MICs of all of tested compounds against M.tb H37Ra pMV261-InhA are 5–10× higher comparing to control strain confirming InhA as molecular target of these inhibitors inside mycobacterial cells (Figure 8).
Figure 8.

Determination of sensitivity of M.tb H37Ra pMV261 and M.tb H37Ra pMV261-InhA against 1a (Left panel) and 3, 5, 7 and 10 (Right panel) by drop dilution method.
3. Materials and Methods
3.1. Material
All chemicals were obtained from Maybridge, TCI, Aldrich or Alfa Aesar, 97–99% and used without further purification. Nuclear magnetic resonance spectra (1H- and 13C-NMR) were recorded on Bruker AC 300, Avance-400 MHz and Avance-500 spectrometers with DMSO-d6 as solvent. Chemical shifts δ were expressed in parts per million (ppm) relative to TMS. Solvent residue signals were used for calibration of spectral data. Mass spectrometry (MS) data were obtained from the “Service Commun de Spectrométrie de masse” of the Plateforme Technique, Institut de Chimie de Toulouse (Toulouse, France). MS were performed using a Waters Quadrupole Time-of-flight mass spectrometer XEVO G2-S QTof. The samples were dissolved in methanol and Electrospray ionization method was used. High-resolution mass spectra (HRMS) were recorded on a ThermoFinnigan MAT 95 XL spectrometer using electrospray ionization (ESI) methods. Melting points were measured using a Kofler heating bench system Heizbank Type WME (Wagner &Munz GmbH, Munich, Germany), with measuring accuracy of ±1 °C in the range of 50–260 °C. If the melting point was higher than 260 °C or if it could not be exactly determined because of an apparent degradation, the DSC analysis was employed. The analysis was performed in a ATG-DSC 111 (Sertaram). The temperature programming was from 20 °C to 200 or 260 °C according to the sample with a constant rate of 5 °C/min under nitrogen atmosphere.
Fourier Transformed Infrared Spectroscopy (FTIR) analysis for identification was performed using KBr pellets on a Thermo Nicolet 5700 spectrometer (Thermo-Nicolet, Madison, WI, USA). The main peaks/bands were identified, especially the –C=N– that is attributed to the hydrazone. FTIR studies with the solid hydrazines as function of temperature were recorded in IN10MX Thermo Scientific FTIR microscope equipped with THMS600 (Linkam Scientific Instruments, Tadworth, Surrey, UK) heating and freezing stage.
UV-vis spectroscopy was performed using a HP (Hewlett Packard, Palo Alto, CA, USA) 8452A diode array spectrophotometer from 200 to 400 nm, with ethanol as a solvent at 20 °C and using quartz cells. The molar absorptivity was determined for the wavelength with the highest absorbance through Lambert–Beer’s law with the molar absorptivity ε in (dm3·mol−1·cm−1) expressed for the λmax of the molecule.
3.2. Chemistry
3.2.1. General Procedure for Phenolic Isonicotinoyl Hydrazones Synthesis
The derivatives 1a–d were synthesized as previously described by us [38]. Compounds 1a–1d have also a CAS number: 1a (840-81-3); 1b (13838-18-1); 1c (149-17-7); 1d (315230-80-9).
3.2.2. General Procedure for Isoniazid Nitrogen-Containing Heterocycles Derivatives 2–11
A mixture the solid reactants, INH (1 equivalent), the aldehyde (1 equivalent) and the catalyst (p-TSA, 0.5 equivalent) were placed in milling device and the reaction proceeded between 1 h–2 h, depending on the aldehyde. The Cryomill (Restch) was used for the screening of catalysts (milling started at the room temperature) at 25 Hz during 1 h. After the choice of the catalyst (p-TSA), all the reactions were carried out in the vibratory ball-mill Pulverisette 0 (Fritsch, Germany) equipped with a single stainless steel ball of 50 mm of diameter and 500 g, in a semi-spherical vessel of 9.5 cm of diameter. The plate vibrates with a frequency of 50 Hz and amplitude of 2.0 mm. The amounts of reactant powder were stoichiometric conditions for reactants totalizing 1 g + the amount of catalyst. The transformation was monitored by TLC. After the reaction time, the powder mixture was washed with a NaHCO3 solution to eliminate the catalyst and the powder was dried under vacuum. 1H-, 13C-NMR spectra and Mass Spectra for all new compounds are included in Supplementary Materials; NMR data show both conformers). NMR data reported below correspond to the major conformer.
(E)-N′-((1H-imidazol-4-yl)methylene)isonicotinohydrazide (2)
 |
m.p.: 296.7 °C (dec.). Rf: 0.1 EtOAc/MeOH (4:1 v/v).
1H-NMR (300 MHz, DMSO-d6) δ ppm: 8.15 (d, J = 1.4 Hz, 1H, H5′), 8.36 (dd, J = 6.6, 1.5 Hz, 2H, H2,6), 8.52 (s, 1H, H-C=N), 9.10 (dd, J = 6.6, 1.5 Hz, 2H, H3,5), 9.21 (d, J = 1.3 Hz, 1H, H3′), 15.67 (s, 2H, N-H).
13C-NMR (75 MHz, DMSO) δ ppm: 122.02 (1C, C5′), 125.10 (2C, C2,6), 128.25 (1C, C1′), 138.37 (1C, C=N), 136,80 (1C, C3′), 144.77 (2C, C3,5), 146.91 (1C, C1), 160.14 (1C, C=O). 126.78 (2C, C2,6), 134,36 (1C, C5′), 143.08 (2C, C3,5).
FTIR (KBr) ν cm−1: 3193.59 (N-H), 3038.08 (C-Har), 1648.96 (C=O), 1626.02 (C=N-N), 1596.86 (C=Car), 1551.04 (Car
 N), 1506.46 (C=N).
N), 1506.46 (C=N).UV (EtOH, 182 μM, 25 °C): λ = 309 nm, ε = 5495.05 dm3·mol−1·cm−1 (very poorly soluble).
MS (ES, TOF, MeOH) m/z: 238.0708 [M + Na+]; 216.0887 [M + H+].
HRMS (ES, TOF) m/z: M + H+ calc. for C10H10N5O: 216.0885. Found: 216.0887.
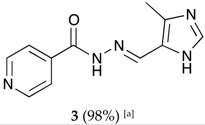
(E)-N′-((4-methyl-1H-imidazol-5-yl)methylene)isonicotinohydrazide (3)
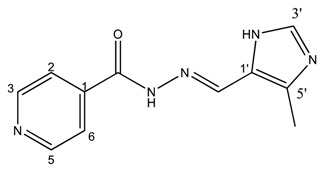 |
m.p.: 299 °C (dec.). Rf: 0.1 EtOAc/MeOH (4:1 v/v).
1H-NMR (300 MHz, DMSO-d6) δ ppm: 2.43 (s, 3H, CH3), 8.42 (dd, J = 5.5, 1.8 Hz, 2H, H2,6), 8.53 (s, 1H, H-C=N), 9.11 (s, 1H, H3′), 9.14 (dd, J = 5.5, 1.8 Hz, 2H, H3,5), 15.98 (br, 2H, N-H).
13C-NMR (75 MHz, DMSO) δ ppm: 9.38 (1C, CH3), 123.60 (1C, C1′) 125.38 (2C, C2,6), 131.78 (1C, C5′), 138.44 (1C, C=N), 142.84 (1C, C3′), 144.03 (2C, C3,5), 147.78 (1C, C1), 159.52 (1C, C=O).
FTIR (KBr) ν cm−1: 3194.75 (N-H), 3097.19 (Car-H), 1660.58 (C=O), 1621.45 (C=N-N), 1602.24 (Car=Car), 1551.49 (Car
N).UV (EtOH, 161.58 μM, 25 °C): λ = 316 nm, ε = 5551.73 dm3·mol−1·cm−1 (very poorly soluble).
MS (ES, TOF, MeOH) m/z: 252.0866 [M + Na+]; 230.1049 [M + H+].
HRMS (ES, TOF) m/z: [M + H+] calc. for C11H12N5O: 230.1042. Found: 230.1049.
(E)-N′-((3a,7a-dihydro-1H-indazol-5-yl)methylene)isonicotinohydrazide (4)
 |
m.p.: 302.5 °C. Rf: 0.45 PE/EtOAc/MeOH (5:5:3 v/v/v).
1H-NMR (300 MHz, DMSO-d6) δ ppm: 7.63 (d, J = 8.7 Hz, 1H, H3′), 7.84 (dd, J = 4.4, 1.6 Hz, 2H, H2,6), 7.90 (dd, J = 8.8, 1.5 Hz, 1H, H2′), 8.03–8.10 (m, 1H, H7′), 8.17 (t, J = 1.2 Hz, 1H, H6′), 8.57 (s, 1H, H-C=N), 8.79 (s, 2H, H3,5), 12.01 (s, 1H, N-H), 13.31 (s, 1H, N-Hind).
13C-NMR (75 MHz, DMSO) δ ppm: 111.39 (1C, C3′), 121.98 (2C, C2,6), 122.73 (1C, C7′), 123.35 (1C, C9′), 124.19 (1C, C2′), 127.23 (1C, C1′), 134.99 (1C, C6′), 141.07 (1C, C1), 141.14 (1C, C8′), 150.31 (1C, C=N), 150.76 (2C, C3,5), 161.87 (1C, C=O).
FTIR (KBr) ν cm−1: 3188.96 (N-H), 3027.37 (Car-H), 1652 (C=O), 1622.47 (C=N-N), 1607.84 (Car=Car), 1549.40 (Car
N).UV (EtOH, 38.37 μM, 25 °C): λ = 234 nm, ε = 22,666.92 dm3·mol−1·cm−1.
MS (ES, TOF, MeOH) m/z: 266.1046 [M + H+].
HRMS (ES, TOF) m/z: [M + H+] calc. for C14H12N5O: 266.1042. Found: 266.1046.
(E)-N′-((2H-indazol-6-yl)methylene)isonicotinohydrazide (5)
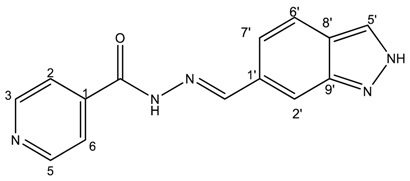 |
m.p.: 295.2 °C. Rf: 0.45 PE/EtOAc/MeOH (5:5:3 v/v/v).
1H-NMR (300 MHz, DMSO-d6) δ ppm: 7.61 (dd, J = 8.5, 1.3 Hz, 1H, H7′), 7.84 (m, 4H, H2,6, H2′, H6′), 8.13 (d, J = 1.3 Hz, 1H, H5′), 8.60 (s, 1H, H-C=N), 8.80 (br, 2H, H3,5), 12.12 (s, 1H, N-H), 13.28 (s, 1H, N-Hind).
13C-NMR (75 MHz, DMSO) δ ppm: 110.75 (1C, C2′), 119.03 (1C, C7′), 121.51 (2C, C2,6), 122.01 (1C, C6′), 124.39 (1C, C8′), 132.43 (1C, C1′), 134.21 (1C, C5′), 140.33 (1C, C1), 140.95 (1C, C9′), 150.03 (1C, C=N), 150.79 (2C, C3,5), 162.08 (1C, C=O).
FTIR (KBr) ν cm−1: 3193.59 (N-H), 3038.08 (C-Har), 1648.96 (C=O), 1626.02 (C=N-N), 1596.86 (C=Car), 1551.04 (Car
N), 1506.46 (C=N).UV (EtOH, 37.39 μM, 25 °C): λ = 313 nm, ε = 25,055.1 dm3·mol−1·cm−1.
MS (ES, TOF, MeOH) m/z: 266.1047 [M + H+].
HRMS (ES, TOF) m/z: [M + H+] calc. for C14H12N5O: 266.1042. Found: 266.1047.
(E)-N′-((2-methyl-1H-indol-3-yl)methylene)isonicotinohydrazide (6)
 |
m.p.: 281.1 °C. Rf: 0.55 PE/EtOAc/MeOH (5:5:3 v/v/v).
1H-NMR (300 MHz, DMSO-d6) δ ppm: 2.54 (s, 3H, CH3), 7.12 (ddt, J = 24.4, 9.3, 1.9, 1.9 Hz, 2H, H5′, H6′), 7.35 (tt, J = 2.4, 1.7, 0.9, 0.8 Hz, 1H, H4′), 7.85 (dd, J = 4.5, 1.8 Hz, 2H, H2,6), 8.16–8.32 (m, 1H, H7′), 8.71 (s, 1H, H-C=N), 8.78 (dd, J = 4.4, 1.6 Hz, 2H, H3,5), 11.55 (d, J = 7.4 Hz, 1H, N-H), 11.67 (s, 1H, N-Hind).
13C-NMR (75 MHz, DMSO) δ ppm: 11.97 (1C, CH3), 107.84 (1C, C1′), 111.34 (1C, C4′), 120.82 (1C, C7′), 121.63(1C, C5′), 121.87 (2C, C2,6), 122.34 (1C, C6′), 125.85 (1C, C9′), 136.19 (1C, C8′), 141.05 (1C, C2′), 141.56 (1C, C1), 146.10 (1C, C=N), 150.69 (2C, C3,5), 161.00 (1C, C=O).
FTIR (KBr) ν cm−1: 3385.07 (N-H), 3209.03 (N-H), 3049.08 (Car-H), 1655.09 (C=O), 1626.02 (C=N-N), 1599.50 (C=Car), 1550.60 (Car
N), 1506.46 (C=N).UV (EtOH, 57.29 μM, 25 °C): λ = 224 nm, ε = 21,909.58 dm3·mol−1·cm−1.
MS (ES, TOF, MeOH) m/z: 279.1246 [M + H+].
HRMS (ES, TOF) m/z: [M + H+] calc. for C16H15N4O: 279.1246. Found: 279.1246.
Compound 6 can also be found (commercial source; CAS No.: 113143-57-0).
(E)-N′-((1H-pyrrolo[2,3-b]pyridin-3-yl)methylene)isonicotinohydrazide (7)
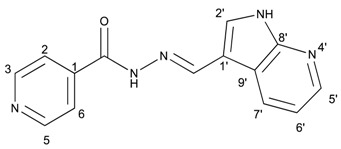 |
m.p.: 323.2 °C (dec.). Rf: 0.34 PE/EtAc/MeOH (5:5:3 v/v).
1H-NMR (300 MHz, DMSO-d6) δ ppm: 77.24 (tt, J = 7.8, 4.7, 4.7 Hz, 1H, H6′), 7.84 (dd, J = 4.2, 1.7 Hz, 2H, H2,6), 8.03 (d, J = 2.3 Hz, 1H, H2′), 8.33 (dd, J = 4.7, 1.7 Hz, 1H, H7′), 8.58 (d, J = 1.6 Hz, 1H, H5′), 8.62 (s, 1H, H-C=N), 8.78 (dd, J = 4.4, 1.7 Hz, 2H, H3,5), 11.86 (s, 1H, N-H), 12.17 (s, 1H, N-Hind).
13C-NMR (75 MHz, DMSO)δ ppm: 110.85 (1C, C1), 117,13 (1C, C9′), 117.44 (1C, C6′), 121.95 (2C, C2,6), 130.56 (1C, C5′), 131.51 (1C, C2′), 141.39 (1C, C1), 144.56 (1C, C7′), 146.09 (1C, C=N), 149.84 (1C, C8′), 150.71 (2C, C3,5), 161.49 (1C, C=O).
FTIR (KBr) ν cm−1: 3454.03 (N-H), 3199.51 (N-H), 3031.09 (Car-H), 1662.68 (C=O), 1611.72 (C=N-N), 1600.48 (C=Car), 1551.26 (Car
N), 1284.78 (C-N).UV (EtOH, 58.08 μM, 25 °C): λ = 200 nm, ε = 18,839.92 dm3·mol−1·cm−1, λ = 218 nm, ε = 17,446.62 dm3·mol−1·cm−1, λ = 322 nm, ε = 17,193.69 dm3·mol−1·cm−1.
MS (ES, TOF, MeOH) m/z: 266.1045 [M + H+].
HRMS (ES, TOF) m/z: [M + H+] calc. for C14H12N5O: 266.1042. Found: 266.1045.
(E)-N′-((1H-indol-3-yl)methylene)isonicotinohydrazide (8)
 |
m.p.: 242 °C. Rf: 0.55 PE/EtAc/MeOH (5:5:3 v/v/v).
1H-NMR (300 MHz, DMSO-d6) δ ppm: δ 7.11–7.28 (m, 2H, H5′,6′), 7.47 (dt, J = 7.9, 0.9 Hz, 1H, H4′), 7.85 (dd, J = 4.4, 1.6 Hz, 2H, H2,6), 7.88 (d, J = 2.8 Hz, 1H, H2′), 8.31 (dd, J = 6.8, 1.5 Hz, 1H, H9′), 8.65 (s, 1H, H-C=N), 8.78 (dd, J = 4.4, 1.6 Hz, 2H, H3, 5), 11.65 (s, 1H, N-H), 11.76 (s, 1H, N-Hind).
13C-NMR (75 MHz, DMSO) δ ppm: 111.96 (1C, C1′), 112.34 (1C, C4′), 121.00 (1C, C5′), 121.95 (2C, C2,6), 122.44 (1C, C7′), 123.17 (1C, C6′), 123.35 (1C, C7′), 124.77 (1C, C9′), 131.36 (1C, C2′), 137.53 (1C, C8′), 141.54 (1C, C1), 146.63 (1C, C=N), 150.69 (2C, C3,5), 161.33 (1C, C=O).
FTIR (KBr) ν cm−1: 3543.66 (N-H), 3395.82 (N-H), 2886.55 (C-Har), 1656.52 (C=O), 1626.02 (C=N-N), 1598.83 (C=Car), 1550.54 (Car
N), 1496.83 (C=N).UV (EtOH, 40.01 μM, 25 °C): λ = 221 nm, ε = 22,519 dm3·mol−1·cm−1.
MS (ES, TOF, MeOH) m/z: 265.1092 [M + H+].
HRMS (ES, TOF) m/z: [M + H+] calc. for C15H13N4O: 265.1089. Found: 265.1092.
Compound 8 can also be found (commercial source; CAS No.: 10245-44-0).
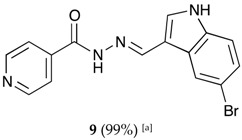
(E)-N′-((5-bromo-1H-indol-3-yl)methylene)isonicotinohydrazide (9)
 |
m.p.: 309.3 °C (dec.). Rf: 0.61 PE/EtAc/MeOH (5:5:3 v/v).
1H-NMR (300 MHz, DMSO-d6) δ ppm: 7.35 (dd, J = 8.7, 2.1 Hz, 1H5′), 7.44 (d, J = 8.5 Hz, 1H, H4′), 7.84 (dd, J = 4.5, 1.9 Hz, 2H, H2,6), 7.95 (d, J = 2.7 Hz, 1H, H9′), 8.48 (s, 1H, H2′), 8.62 (s, 1H, H-C=N), 8.73–8.89 (m, 2H, H3,5), 11.83 (d, J = 6.0 Hz, 2H, N-H).
13C-NMR (75 MHz, DMSO) δ ppm: 111.59 (1C, C1′), 113,70 (1C, C6′), 114.42 (1C, C4′), 121.93 (2C, C2,6), 124.60 (1C, C2′), 125.71 (1C, C5′), 126.42 (1C, C9′), 132.71 (1C, C7′), 136.27 (1C, C8′), 141.38 (1C, C1), 146.13 (1C, C=N), 150.72 (2C, C3,5), 161.40 (1C, C=O).
FTIR (KBr) ν cm−1: 3127.58 (N-Hind), 2891.39 (C-Har), 1662.69 (C=O), 1618.34 (C=N-N), 1538 (C=Car), 1552.13 (Car
N), 1041.12 (Car-Br).UV (EtOH, 52.65 μM, 25 °C): λ = 201 nm, ε = 29,890 dm3·mol−1·cm−1, λ = 226 nm, ε = 25,981 dm3·mol−1·cm−1, λ = 330 nm, ε = 18,196 dm3·mol−1·cm−1.
MS (ES, TOF, MeOH) m/z: 343.0194 [M + H+].
HRMS (ES, TOF) m/z: [M + H+] calc. for C15H12BrN4O: 343.0193. Found: 343.0194.
Compound 9 can also be found (CAS No.: 113143-44-5).
(E)-N′-((1H-indol-2-yl)methylene)isonicotinohydrazide (10)
 |
m.p.: 231 °C (dec.). Rf: 0.82 PE/EtAc/MeOH (5:5:3 v/v/v).
1H-NMR (300 MHz, DMSO-d6) δ ppm: 76.90 (dd, J = 2.1, 0.9 Hz, 1H), 7.02 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 7.18 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 7.47 (dq, J = 8.2, 0.9 Hz, 1H), 7.58 (dd, J = 7.9, 1.1 Hz, 1H), 7.86 (dd, J = 4.4, 1.6 Hz, 2H), 8.51 (s, 1H), 8.81 (dd, J = 4.4, 1.7 Hz, 2H), 11.65 (s, 1H), 12.05 (s, 1H).
13C-NMR (75 MHz, DMSO) δ ppm: 107.98 (1C, C2′), 112.57 (1C, C3′), 120.06 (1C, C4′), 121.31 (1C, C6′), 122.01 (2C, C2,6), 128.05 (1C, C9′), 133.28 (1C, C1′), 138.47 (1C, C8′), 142.16 (1C, C=N), 150.79 (2C, C3, 5), 161.84 (1C, C=O).
FTIR (KBr) ν cm−1: 3250.19 (N-H), 3032.16 (C-Har), 1689.09 (C=O), 1621.50 (C=N-N), 1599.80 (C=Car), 1548.50 (Car
N).UV (EtOH, 52.65 μM, 25 °C): λ = 206 nm, ε = 27,480 dm3·mol−1·cm−1, λ = 350 nm, ε = 33,825 dm3·mol−1·cm−1.
MS (ES, TOF, MeOH) m/z: 265.1090 [M + H+].
HRMS (ES, TOF) m/z: [M + H+] calc. for C15H13N4O: 265.1089. Found: 265.1090.
X-Ray structure of this compound has been recently reported [83].
(E)-N′-((4-nitro-1H-indol-3-yl)methylene)isonicotinohydrazide (11)
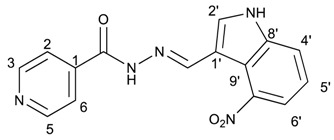 |
m.p.: 317.1 °C (dec.). Rf: 0.70 PE/EtOAc/MeOH (5:5:3 v/v/v).
1H-NMR (300 MHz, DMSO-d6) δ ppm: 7.36 (t, J = 8.0 Hz, 1H, H5′), 7.84 (dd, J = 4.4, 1.7 Hz, 2H, H2,6), 7.94 (dd, J = 11.8, 7.9 Hz, 2H, H4′,6′), 8.28 (d, J = 2.9 Hz, 1H, H2′), 8.78 (dd, J = 4.4, 1.7 Hz, 2H, H3,5), 8.91 (s, 1H, H-C=N), 12.00 (s, 1H, N-H), 12.55 (s, 1H, N-H).
13C-NMR (75 MHz, DMSO) δ ppm: 110.29 (1C, C9′), 117.59 (1C, C1′), 118.94 (1C, C4′), 119.92 (1C, C6′), 121.50 (1C, C5′), 122.01 (2C, C2, 6), 131.31 (1C, C2′), 139.55 (1C, C8′), 141.22 (1C, C1), 141.90 (1C, C-NO2), 146.43 (1C, C=N), 150.66 (2C, C3,5), 161.56 (1C, C=O).
FTIR (KBr) ν cm−1: 3156.34 (N-H), 3137.68 (N-H), 3053.25 (C-Har), 1664.06 (C=O), 1628.34 (C=N-N), 1590.99 (C=Car), 1554.10 (Car
N), 1513.15 (C=N-NO2).UV (EtOH, 52.64 μM, 25 °C): λ = 214 nm, ε = 26,971.88 dm3·mol−1·cm−1, λ = 331 nm, ε = 17,046.35 dm3·mol−1·cm−1.
MS (ES, TOF, MeOH) m/z: 332.0760 [M + Na+]; 310.0940 [M + H+].
HRMS (ES, TOF) m/z: [M + H+] calc. for C15H12N5O3: 310.0937. Found: 310.0940.
3.3. Physicochemical Studies: Hydrolytic Stability and pKa Determination
Physicochemical studies: hydrolytic stability and pKa determination:
UV spectra were recorded with HP8453 (Agilent) temperature controlled spectrophotometer.
The pHs of the solutions were measured at room temperature (temperature probe) with a combined pH electrode with Seven Multi (Mettler Toledo) pHmeter.
3.3.1. Hydrolytic Stability
In the case of compounds 1a, 1b, 1c and 1d, a small quantity (respectively, 1.45 mg, 1.63 mg, 1.87 mg, and 1.79 mg) of each product was weighted and dissolved in 1 mL of EtOH. The aliquots were then placed in 100 mL standard flask with a concentration of 28% EtOH/H2O. Final concentrations of compounds were respectively 1a: 6.0 × 10−5 mol/L, 1b: 6.3 × 10−5 mol/L, 1c: 6.0 × 10−5 mol/L and 1d: 5.9 × 10−5 mol/L.
In the case of compounds 5 and 7, a small quantity (respectively, 1.65 mg and 1.20 mg) of each product was weighted and dissolved in 4 mL of EtOH. A fraction of this solution (respectively 1.30 mL and 1.36 mL) were placed in 50 mL standard flask and mixed with PIPES buffer (50 mM) with a final concentration of 5% EtOH/PIPES.
The solution was stirred at room temperature; pH values and λ max of absorbance of the related compounds were measured for longer than 15 h.
3.3.2. pKa Determination
A small quantity (1–2 mg) of each product was weighted and dissolved in a 1 mL of EtOH. The aliquots were then placed in 100 mL standard flask with a final concentration of 28% EtOH/H2O. The ionic strength was fixed at 0.1 M with potassium chloride. The pH was adjusted to the required value by adding concentrated 0.1 M KOH or 0.1 M HCl. The solution was stirred at room temperature. The absorbance at three different wavelengths was measured. Absorbance vs. pH curves were plotted and the pKa values were evaluated graphically by geometric method [84].
3.3.3. DMSO stability of Compound 1d
The stability of compound 1a in DMSO was assessed at 25 °C. To this purpose, the compound was dissolved in DMSO, at a final concentration of 10 mM, and incubated for 16 h at 25 °C. The compound was then diluted in methanol, and analysed. The mass spectra were recorded in negative ESI resolution mode with a Thermo LTQ-XL mass spectrometer, and compared with those of compound 1a and of INH freshly dissolved in DMSO (10 mM) and diluted in methanol.
3.4. Biological Assays
3.4.1. Inhibition Kinetics in the Presence of InhA
3.4.2. MIC Determination in M.tb
H37Rv strain was used as the reference strain. M.tb H37Rv and IC2 clinical isolate [31] were grown at 37 °C in Middlebrook 7H9 broth (Difco), supplemented with 0.05% Tween 80, or on solid Middlebrook 7H11 medium (Difco) supplemented with oleic acid-albumin-dextrose-catalase (OADC). MICs for the compounds were determined by means of the micro-broth dilution method. Dilutions of M.tb wild-type or mutant strain (about 105–106 cfu/mL) were streaked onto 7H11 solid medium containing a range of drug concentrations (0.125 µg/mL to 40 µg/mL). Plates were incubated at 37 °C for about 21 days and the growth was visually evaluated. The lowest drug dilution at which visible growth failed to occur was taken as the MIC value. Results were expressed as the average of at least three independent determinations.
3.4.3. Determination of LC50
Human primary fibroblast (MRC5 ATCC CCL171) were seeded at 5 k/well in a corning Cell bind 96-well plate and treated with growing concentration of compounds. Seventy-two hours post-treatment, cells were directly stained with Hoechst 33342 and imaged under a Cellomics Array scan HCS microscope using the cell cycle algorithm. Total number of cells were assessed in each condition and normalized over untreated wells. LC50 were determined as concentration inducing 50% reduction in cell number.
3.4.4. Analysis of the Effect of Hydrazones on Lipids and Mycolic Acids of M.tb H37Ra
The modes of actions of selected hydrazones were analyzed by metabolic labeling of M.tb H37Ra strain with 14C acetate as already described [34]. Briefly, M.tb H37Ra culture was grown in Middlebrook 7H9 broth (Difco) supplemented with albumin-dextrose-catalase (Difco) and 0.05% Tween 80 (MP Biomedicals) at 37 °C till OD600 reached 0.24. The culture was then divided into 20 mL aliquots and tested compounds dissolved in DMSO were added in 0.41 μM (0.1 μg/mL) and 2.05 μM (0.5 μg/mL) final concentrations for 1a, 0.44 μM (0.1 μg/mL) and 2.2 μM (0.5 μg/mL) for 3, 0.38 μM (0.1 μg/mL) and 1.9 μM (0.5 μg/mL) for 5, 0.38 μM (0.1 μg/mL) and 1.9 μM (0.5 μg/mL) for 7, 0.38 μM (0.1 μg/mL) and 1.9 μM (0.5 μg/mL) for 10 and 36.5 μM (5 μg/mL) for INH. The final concentration of DMSO was kept at 1% in each experiment. 14C acetate (specific activity 106 mCi/mmol, ARC) in the final concentration of 0.5 μCi/mL was added to each of the cultures after 24 h of cultivation with shaking (120 rpm) and the cells were cultivated for next 24 h.
Lipids were extracted from whole cells harvested from 10 mL culture aliquots as described earlier [34,86], dissolved in chloroform:methanol (2:1)—350 μL per 1 unit of OD600 of harvested cells. Five microliters were loaded on thin-layer chromatography (TLC) silica gel plates F254 (Merck) and the lipids were separated in chloroform:methanol:water (20:4:0.5) and detected by autoradiography. Fatty acid methyl esters (FAME) and mycolic acids methyl esters (MAME) were prepared from whole cells harvested from 10 mL culture aliquots as previously described [34,87]. Dried extracts were dissolved in chloroform:methanol (2:1) and loaded on TLC plates as described for lipid extracts. Different forms of methyl esters were separated by running three times in n-hexane: ethyl acetate (95:5) and detected by autoradiography.
3.4.5. Determination of Sensitivity of M.tb H37Ra Strain Overproducing InhA to Hydrazones
InhA protein was overproduced in M.tb H37Ra using pMV261-InhA construct as already described [88]. Sensitivity of InhA overproducing strain, as well as control strain carrying empty vector to compounds 1a, 3, 5, 7 and 10 was analysed by drop dilution methods. Both cultures grown in 7H9 broth supplemented with albumin-dextrose-catalase and 0.05% Tween 80 were adjusted to OD600 0.5 and 4 µL aliquots of 100, 10−1, 10−2 and 10−3 dilutions were dropped on 7H11 agar supplemented with oleic acid-albumin-dextrose-catalase and incubated 25 days at 37 °C.
4. Conclusions
A series of hydrazones were synthesized through mechanochemistry and evaluated for their ability to inhibit M.tb H37Rv strain growth, the most active being 1a and 7. Imidazole derivative 5 and indazole one 10 are also active against H37Rv with their nitro substituent. Compounds 1a and 1b were the most effective against both M.tb H37Rv strain and drug-resistant IC2 isolate.
The mechanism of anti-mycobacterial activities of selected hydrazones with the best scores regarding the MICs and toxicities was confirmed to be through the cessation of mycolic acid synthesis due to InhA inhibition inside the mycobacterial cell.
Acknowledgments
We thank the CNRS and University Paul Sabatier for financial support. We thank the Minister of Industry for Ph.D. grant (P.F.M.O.); William R. Jacobs for pMV261-InhA construct and grant VEGA 1/0284/15. We thank F. Gaillardo (NeoVirTech, ITAV, Centre P. Potier, Toulouse) for the cytotoxicity evaluation of the hydrazone compounds.
Supplementary Materials
Supplementary Materials are available online.
Author Contributions
P.F.M.O.: Ph.D. student synthetic experiments; B.G., A.C. and M.Baron (co-director of P.F.M.O. thesis): supervised the synthetic part; C.A.B.: NMR and DFT studies; G.M. and B.S.O. performed the MIC experiments; L.R.C. performed MS; M.R.P. directed this part; J.M., J.K.: analysis on lipids and mycolic acids, J.K. directed this part; C.L.: InhA inhibition experiments; C.C.: hydrolytic stability and pKa determination; S.M.: NMR experiments; M.Baltas: conceive and direct the project, co-direct the P.F.M.O. thesis. All authors contributed in writing the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 1a–d, 2–11 are available from the authors.
References
- 1.WHO Gobal Tuberculosis Report 2016. [(accessed on 14 October 2016)];2016 Available online: http://www.who.int/tb/publications/global_report/en/
- 2.Cynamon M.H., Zhang Y., Harpster T., Cheng S., DeStefano M.S. High-dose isoniazid therapy for isoniazid-resistant murine Mycobacterium tuberculosis infection. Antimicrob. Agents Chemother. 1999;43:2922–2924. doi: 10.1128/aac.43.12.2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bemer-Melchior P., Bryskier A., Drugeon H.B. Comparison of the in vitro activities of rifapentine and rifampicin against Mycobacterium tuberculosis complex. J. Antimicrob. Chemother. 2000;46:571–576. doi: 10.1093/jac/46.4.571. [DOI] [PubMed] [Google Scholar]
- 4.Jain A., Mondal R. Extensively drug-resistant tuberculosis: Current challenges and threats. FEMS Immunol. Med. Microbiol. 2008;53:145–150. doi: 10.1111/j.1574-695X.2008.00400.x. [DOI] [PubMed] [Google Scholar]
- 5.Parida S.K., Axelsson-Robertson R., Rao M.V., Singh N., Master I., Lutckii A., Keshavjee S., Andersson J., Zumla A., Maeurer M. Totally drug-resistant tuberculosis and adjunct therapies. J. Intern. Med. 2015;277:388–405. doi: 10.1111/joim.12264. [DOI] [PubMed] [Google Scholar]
- 6.Food and Drug Administration . Silver Spring MD; Food and Drug Administration: 2015. [(accessed on 11 July 2015)]. Sirturo (bedaquiline) Product Insert. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/204384s000lbl.pdf. [Google Scholar]
- 7.European Medicines Agency C for MP for HU . European Medicines Agency C for MP for HU; London, England: 2013. [(accessed on 11 July 2015)]. European Medicines Agency, Assessment Report, Deltyba. Procedure No EMEA/H/C/002552. 2015. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002552/smops/Positive/human_smop_000572.jsp&mid=WC0b01ac058001d127. [Google Scholar]
- 8.Banerjee D.R., Biswas R., Das A.K., Basak A. Design, synthesis and characterization of dual inhibitors against new targets FabG4 and HtdX of Mycobacterium tuberculosis. Eur. J. Med. Chem. 2015;100:223–234. doi: 10.1016/j.ejmech.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Rollas S., Küçükgüzel S.G. Biological Activities of Hydrazone Derivatives. Molecules. 2007;12:1910–1939. doi: 10.3390/12081910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rasras A.J.M., Al-Tel T.H., Al-Aboudi A.F., Al-Qawasmeh R.A. Synthesis and antimicrobial activity of cholic acid hydrazone analogues. Eur. J. Med. Chem. 2010;45:2307–2313. doi: 10.1016/j.ejmech.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Mohareb R.M., Fleita D.H., Sakka O.K. Novel Synthesis of Hydrazide-Hydrazone Derivatives and Their Utilization in the Synthesis of Coumarin, Pyridine, Thiazole and Thiophene Derivatives with Antitumor Activity. Molecules. 2011;16:16–27. doi: 10.3390/molecules16010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asif M. Pharmacologically potentials of hydrazonone containing compounds: A promising scaffold. Int. J. Adv. Chem. 2014;2:85–103. doi: 10.14419/ijac.v2i2.2301. [DOI] [Google Scholar]
- 13.Bairwa R., Kakwani M., Tawari N.R., Lalchandani J., Ray M.K., Rajan M.G.R., Degani M.S. Novel molecular hybrids of cinnamic acids and guanylhydrazones as potential antitubercular agents. Bioorg. Med. Chem. Lett. 2010;20:1623–1625. doi: 10.1016/j.bmcl.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 14.Carvalho S.A., da Silva E.F., de Souza M.V.N., Lourenço M.C.S., Vicente F.R. Synthesis and antimycobacterial evaluation of new trans-cinnamic acid hydrazide derivatives. Bioorg. Med. Chem. Lett. 2008;18:538–541. doi: 10.1016/j.bmcl.2007.11.091. [DOI] [PubMed] [Google Scholar]
- 15.Vavříková E., Polanc S., Kočevar M., Horváti K., Bősze S., Stolaříková J., Vávrová K., Vinšová J. New fluorine-containing hydrazones active against MDR-tuberculosis. Eur. J. Med. Chem. 2011;46:4937–4945. doi: 10.1016/j.ejmech.2011.07.052. [DOI] [PubMed] [Google Scholar]
- 16.Oliveira K.N., Chiaradia L.D., Martins P.G.A., Mascarello A., Cordeiro M.N.S., Guido R.V.C., Andricopulo A.D., Yunes R.A., Nunes R.J., Terenzi J.H. Sulfonyl-hydrazones of cyclic imides derivatives as potent inhibitors of the Mycobacterium tuberculosis protein tyrosine phosphatase B (PtpB) Med. Chem. Comm. 2011;2:500–504. doi: 10.1039/c0md00253d. [DOI] [Google Scholar]
- 17.Reddy K.S., Ramesh M., Srimai V., Chandra K.S., Satyender A. Synthesis, antimycobacterial activity and docking studies of L-proline derived hydrazones. Der Pharma Chemica. 2014;6:267–271. [Google Scholar]
- 18.Maccari R., Ottanà R., Vigorita M.G. In vitro advanced antimycobacterial screening of isoniazid-related hydrazones, hydrazides and cyanoboranes: Part 14. Bioorg. Med. Chem. Lett. 2005;15:2509–2513. doi: 10.1016/j.bmcl.2005.03.065. [DOI] [PubMed] [Google Scholar]
- 19.Naveen Kumar H.S., Parumasivam T., Jumaat F., Ibrahim P., Asmawi M.Z., Sadikun A. Synthesis and evaluation of isonicotinoyl hydrazone derivatives as antimycobacterial and anticancer agents. Med. Chem. Res. 2014;23:269–279. doi: 10.1007/s00044-013-0632-2. [DOI] [Google Scholar]
- 20.Vavříková E., Polanc S., Kočevar M., Košmrlj J., Horváti K., Bősze S., Stolaříková J., Imramovský A., Vinšová J. New series of isoniazid hydrazones linked with electron-withdrawing substituents. Eur. J. Med. Chem. 2011;46:5902–5909. doi: 10.1016/j.ejmech.2011.09.054. [DOI] [PubMed] [Google Scholar]
- 21.Wang X.-L., Zhang Y.-B., Tang J.-F., Yang Y.-S., Chen R.-Q., Zhang F., Zhu H.-L. Design, synthesis and antibacterial activities of vanillic acylhydrazone derivatives as potential β-ketoacyl-acyl carrier protein synthase III (FabH) inhibitors. Eur. J. Med. Chem. 2012;57:373–382. doi: 10.1016/j.ejmech.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Hearn M.J., Cynamon M.H., Chen M.F., Coppins R., Davis J., Joo-On Kang H., Noble A., Tu-Sekine B., Terrot M.S., Trombino D., et al. Preparation andantitubercular activities in vitro and in vivo of novel Schiff bases of isoniazid. Eur. J. Med. Chem. 2009;44:4169–4178. doi: 10.1016/j.ejmech.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hearn M.J., Cynamon M.H. Desing and synthesis of antituberculars: Preparation and evaluation against Mycobacterium tuberculosis of an isoniazid Schiff base. J. Antimicrob. Chemother. 2004;53:185–191. doi: 10.1093/jac/dkh041. [DOI] [PubMed] [Google Scholar]
- 24.Ventura C., Martins F. Application of quantitative structure−activity relationships to the modeling of antitubercular compounds 1. The hydrazide family. J. Med. Chem. 2008;51:612–624. doi: 10.1021/jm701048s. [DOI] [PubMed] [Google Scholar]
- 25.Martins F., Santos S., Ventura C., Elvas-Leitão R., Santos L., Vitorino S., Reis M., Miranda V., Correia H.F., et al. Design, synthesis and biological evaluation of novel isoniazid derivatives with potent antitubercular activity. Eur. J. Med. Chem. 2014;81:119–138. doi: 10.1016/j.ejmech.2014.04.077. [DOI] [PubMed] [Google Scholar]
- 26.De P., Baltas M., Bedos-Belval F. Cinnamic acid derivatives as anticancer agents—A review. Curr. Med. Chem. 2011;18:1672–1703. doi: 10.2174/092986711795471347. [DOI] [PubMed] [Google Scholar]
- 27.De P., Bedos-Belval F., Vanucci-Bacqué C., Baltas M. Cinnamic acid derivatives in tuberculosis, malaria and cardiovascular diseases—A review. Curr. Org. Chem. 2012;16:747–768. doi: 10.2174/138527212799958020. [DOI] [Google Scholar]
- 28.De P., De K., Veau D., Bedos-Belval F., Chassaing S., Baltas M. Recent advances in the development of cinnamic-like derivatives as antituberculosis agents. Expert Opin. Ther. Pat. 2012;22:155–168. doi: 10.1517/13543776.2012.661717. [DOI] [PubMed] [Google Scholar]
- 29.De P., Veau D., Bedos-Belval F., Chassaing S., Baltas M. Cinnamic Derivatives in Tuberculosis. In: Cardona P.-J., editor. Understanding Tuberculosis—New Approaches to Fighting Against Drug Resistance. InTech; Rijeka, Croatia: 2012. pp. 337–362. [DOI] [Google Scholar]
- 30.Menendez C., Chollet A., Rodriguez F., Inard C., Pasca M.R., Lherbet C., Baltas M. Chemical synthesis and biological evaluation of triazole derivatives as inhibitors of InhA and antituberculosis agents. Eur. J. Med. Chem. 2012;52:275–283. doi: 10.1016/j.ejmech.2012.03.029. [DOI] [PubMed] [Google Scholar]
- 31.Menendez C., Rodriguez F., Ribeiro A.L., Zara F., Frongia C., Lobjois V., Saffon N., Pasca M.R., Lherbet C., Baltas M. Synthesis and evaluation of α-ketotriazoles and α,β-diketotriazoles as inhibitors of Mycobacterium tuberculosis. Eur. J. Med. Chem. 2013;69:167–173. doi: 10.1016/j.ejmech.2013.06.042. [DOI] [PubMed] [Google Scholar]
- 32.Menendez C., Mori G., Maillot M., Fabing I. Synthesis and evaluation of β-hydroxytriazoles and related compounds as antitubercular agents. French-Ukrainian J. Chem. 2015;3:82–96. doi: 10.17721/fujcV3I1P82-96. [DOI] [Google Scholar]
- 33.Veau D., Krykun S., Mori G., Orena B.S., Pasca M.R., Frongia C., Lobjois V., Chassaing S., Lherbet C., Baltas M. Triazolophthalazines: Easily accessible compounds with potent antitubercular activity. Chem. Med. Chem. 2016;11:1078–1089. doi: 10.1002/cmdc.201600085. [DOI] [PubMed] [Google Scholar]
- 34.Matviiuk T., Madacki J., Mori G., Orena B.S., Menendez C., Kysil A., André-Barrès C., Rodriguez F., Korduláková J., Mallet-Ladeira S., et al. Pyrrolidinone and pyrrolidine derivatives: Evaluation as inhibitors of InhA and Mycobacterium tuberculosis. Eur. J. Med. Chem. 2016;123:462–475. doi: 10.1016/j.ejmech.2016.07.028. [DOI] [PubMed] [Google Scholar]
- 35.Matviiuk T., Mori G., Lherbet C., Rodriguez F., Pasca M.R., Gorichko M., Guidetti B., Voitenko Z., Baltas M. Synthesis of 3-heteryl substituted pyrrolidine-2,5-diones via catalytic Michael reaction and evaluation of their inhibitory activity against InhA and Mycobacterium tuberculosis. Eur. J. Med. Chem. 2014;71:46–52. doi: 10.1016/j.ejmech.2013.10.069. [DOI] [PubMed] [Google Scholar]
- 36.Matviiuk T., Rodriguez F., Saffon N., Mallet-Ladeira S., Gorichko M., de Jesus Lopes Ribeiro A.L., Pasca M.R., Lherbet C., Voitenko Z., Baltas M. Design, chemical synthesis of 3-(9H-fluoren-9-yl)pyrrolidine-2,5-dione derivatives and biological activity against enoyl-ACP reductase (InhA) and Mycobacterium tuberculosis. Eur. J. Med. Chem. 2013;70:37–48. doi: 10.1016/j.ejmech.2013.09.041. [DOI] [PubMed] [Google Scholar]
- 37.Pavan F.R., da S Maia P.I., Leite S.R.A., Deflon V.M., Batista A.A., Sato D.N., Franzblau S.G., Leite C.Q.F. Thiosemicarbazones, semicarbazones, dithiocarbazates and hydrazide/hydrazones: Anti- Mycobacterium tuberculosis activity and cytotoxicity. Eur. J. Med. Chem. 2010;45:1898–1905. doi: 10.1016/j.ejmech.2010.01.028. [DOI] [PubMed] [Google Scholar]
- 38.Oliveira P.F.M., Baron M., Chamayou A., André-Barrès C., Guidetti B., Baltas M. Solvent-free mechanochemical route for green synthesis of pharmaceutically attractive phenol-hydrazones. RSC Adv. 2014;4:56736–56742. doi: 10.1039/C4RA10489G. [DOI] [Google Scholar]
- 39.Marques de Oliveira P.F. Ph.D. Thesis. Ecole des mines d’Albi-Carmaux; Albi, France: October 2015. Investigation of Mechanochemical Synthesis of Condensed 1,4-diazines and Pharmaceutically Attractive Hydrazones. [Google Scholar]
- 40.Baláž P. Mechanical activation in hydrometallurgy. Int. J. Miner. Process. 2003;72:341–354. doi: 10.1016/S0301-7516(03)00109-1. [DOI] [Google Scholar]
- 41.Balema V.P., Wiench J.W., Pruski M., Pecharsky V.K. Mechanically induced solid-state generation of phosphorus ylides and the solvent-free wittig reaction. J. Am. Chem. Soc. 2002;124:6244–6245. doi: 10.1021/ja017908p. [DOI] [PubMed] [Google Scholar]
- 42.Watanabe H., Hiraoka R., Senna M. A Diels-Alder reaction catalyzed by eutectic complexes autogenously formed from solid state phenols and quinones. Tetrahedron Lett. 2006;47:4481–4484. doi: 10.1016/j.tetlet.2006.04.030. [DOI] [Google Scholar]
- 43.Zhang Z., Peng Z.-W., Hao M.-F., Gao J.-G. Mechanochemical diels-alder cycloaddition reactions for straightforward synthesis of endo-norbornene derivatives. Synlett. 2010;2010:2895–2898. doi: 10.1055/s-0030-1259030. [DOI] [Google Scholar]
- 44.Zhang Z., Dong Y.-W., Wang G.-W., Komatsu K. Highly efficient mechanochemical reactions of 1,3-dicarbonyl compounds with chalcones and azachalcones catalyzed by potassium carbonate. Synlett. 2004:61–64. doi: 10.1002/chin.200421076. [DOI] [Google Scholar]
- 45.Zhang Z., Dong Y.-W., Wang G.-W., Komatsu K. Mechanochemical Michael reactions of chalcones and azachalcones with ethyl acetoacetate catalyzed by K2CO3 under solvent-free conditions. Chem. Lett. 2004;33:168–169. doi: 10.1246/cl.2004.168. [DOI] [Google Scholar]
- 46.Kaupp G., Naimi-Jamal M.R., Schmeyers J. Quantitative reaction cascades of Ninhydrin in the solid state. Chem. Eur. J. 2002;8:594–600. doi: 10.1002/1521-3765(20020201)8:3<594::AID-CHEM594>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 47.Heintz A.S., Gonzales J.E., Fink M.J., Mitchell B.S. Catalyzed self-aldol reaction of valeraldehyde via a mechanochemical method. J. Mol. Catal. A Chem. 2009;304:117–120. doi: 10.1016/j.molcata.2009.01.033. [DOI] [Google Scholar]
- 48.Burmeister C.F., Stolle A., Schmidt R., Jacob K., Breitung-Faes S., Kwade A. Experimental and Computational Investigation of Knoevenagel Condensation in Planetary Ball Mills. Chem. Eng. Technol. 2014;37:857–864. doi: 10.1002/ceat.201300738. [DOI] [Google Scholar]
- 49.Fulmer D.A., Shearouse W.C., Medonza S.T., Mack J. Solvent-free Sonogashira coupling reaction via high speed ball milling. Green Chem. 2009;11:1821. doi: 10.1039/b915669k. [DOI] [Google Scholar]
- 50.Thorwirth R., Stolle A., Ondruschka B. Fast copper-, ligand- and solvent-free Sonogashira coupling in a ball mill. Green Chem. 2010;12:985. doi: 10.1039/c000674b. [DOI] [Google Scholar]
- 51.Schneider F., Ondruschka B. Mechanochemical solid-state Suzuki reactions using an in situ generated base. ChemSusChem. 2008;1:622–625. doi: 10.1002/cssc.200800086. [DOI] [PubMed] [Google Scholar]
- 52.Bernhardt F., Trotzki R., Szuppa T., Stolle A., Ondruschka B. Solvent-free and time-efficient Suzuki-Miyaura reaction in a ball mill: The solid reagent system KF-Al2O3 under inspection. Beilstein J. Org. Chem. 2010;6:30–34. doi: 10.3762/bjoc.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan D., Loots L., Friščić T. Towards medicinal mechanochemistry: Evolution of milling from pharmaceutical solid form screening to the synthesis of active pharmaceutical ingredients (APIs) Chem. Commun. 2016 doi: 10.1039/C6CC02015A. [DOI] [PubMed] [Google Scholar]
- 54.Mikhailenko M.A., Shakhtshneider T.P., Boldyrev V.V. Acylation of sulfathiazole with maleic anhydride under mechanochemical activation. Mendeleev Commun. 2007;17:315–317. doi: 10.1016/j.mencom.2007.11.004. [DOI] [Google Scholar]
- 55.Carlier L., Baron M., Chamayou A., Couarraze G. Use of co-grinding as a solvent-free solid state method to synthesize dibenzophenazines. Tetrahedron Lett. 2011;52:4686–4689. doi: 10.1016/j.tetlet.2011.07.003. [DOI] [Google Scholar]
- 56.Lee B., Kang P., Lee K.H., Cho J., Nam W., Lee W.K., Hur N.H. Solid-state and solvent-free synthesis of azines, pyrazoles, and pyridazinones using solid hydrazine. Tetrahedron Lett. 2013;54:1384–1388. doi: 10.1016/j.tetlet.2012.12.106. [DOI] [Google Scholar]
- 57.Estévez V., Villacampa M., Menéndez J.C. Three-component access to pyrroles promoted by the CAN–silver nitrate system under high-speed vibration milling conditions: A generalization of the Hantzsch pyrrole synthesis. Chem. Commun. 2013;49:591–593. doi: 10.1039/C2CC38099D. [DOI] [PubMed] [Google Scholar]
- 58.Schmeyers J., Toda F., Boy J., Kaupp G. Quantitative solid–solid synthesis of azomethines. J. Chem. Soc. Perk. Trans. 2. 1998;4:989–994. doi: 10.1039/a704633b. [DOI] [Google Scholar]
- 59.Dolotko O., Wiench J.W., Dennis K.W., Pecharsky V.K., Balema V.P. Mechanically induced reactions in organic solids: Liquid eutectics or solid-state processes? New J. Chem. 2010;34:25–28. doi: 10.1039/B9NJ00588A. [DOI] [Google Scholar]
- 60.Kaupp G., Schmeyers J., Boy J. Iminium Salts in Solid-State Syntheses Giving 100% Yield. J. Für Prakt. Chem. 2000;342:269–280. doi: 10.1002/(SICI)1521-3897(200003)342:3<269::AID-PRAC269>3.0.CO;2-0. [DOI] [Google Scholar]
- 61.Mokhtari J., Naimi-Jamal M.R., Hamzeali H., Dekamin M.G., Kaupp G. Kneading Ball-milling and stoichiometric melts for the quantitative derivatization of carbonyl compounds with gas-solid recovery. ChemSusChem. 2009;2:248–254. doi: 10.1002/cssc.200800258. [DOI] [PubMed] [Google Scholar]
- 62.Nun P., Martin C., Martinez J., Lamaty F. Solvent-free synthesis of hydrazones and their subsequent N-alkylation in a Ball-mill. Tetrahedron. 2011;67:8187–8194. doi: 10.1016/j.tet.2011.07.056. [DOI] [Google Scholar]
- 63.Shalini K., Sharma P., Kumar N. Imidazole and its biological activities: A review. Chem. Sin. 2010;1:36–47. [Google Scholar]
- 64.Verma A., Joshi S., Singh D. Imidazole: Having versatile biological activities. J. Chem. 2013;2013:1–12. doi: 10.1155/2013/329412. [DOI] [Google Scholar]
- 65.Thangadurai A., Minu M., Wakode S., Agrawal S., Narasimhan B. Indazole: A medicinally important heterocyclic moiety. Med. Chem. Res. 2012;21:1509–1523. doi: 10.1007/s00044-011-9631-3. [DOI] [Google Scholar]
- 66.De Luca L. Naturally occurring and synthetic imidazoles: Their chemistry and their biological activities. Curr. Med. Chem. 2006;13:1–23. doi: 10.2174/092986706775197971. [DOI] [PubMed] [Google Scholar]
- 67.Sharma V., Kumar P., Pathak D. Biological importance of the indole nucleus in recent years: A comprehensive review. J. Heterocycl. Chem. 2010;47:491–502. doi: 10.1002/jhet.349. [DOI] [Google Scholar]
- 68.Oliveira P.F.M., Haruta N., Chamayou A., Guidetti B., Baltas M., Tanaka K., Sato T., Baron M. Comprehensive experimental investigation of mechanically induced 1,4-diazines synthesis in solid state. Tetrahedron. 2017;73:2305–2310. doi: 10.1016/j.tet.2017.03.014. [DOI] [Google Scholar]
- 69.Palla G., Predieri G., Domiano P., Vignali C., Turner W. Conformational behaviour and/isomerization of -acyl and -aroylhydrazones. Tetrahedron. 1986;42:3649–3654. doi: 10.1016/S0040-4020(01)87332-4. [DOI] [Google Scholar]
- 70.Syakaev V.V., Podyachev S.N., Buzykin B.I., Latypov S.K., Habicher W.D., Konovalov A.I. NMR study of conformation and isomerization of aryl- and heteroarylaldehyde 4-tert-butylphenoxyacetylhydrazones. J. Mol. Struct. 2006;788:55–62. doi: 10.1016/j.molstruc.2005.11.018. [DOI] [Google Scholar]
- 71.Ünsal-Tan O., Özden K., Rauk A., Balkan A. Synthesis and cyclooxygenase inhibitory activities of some N-acylhydrazone derivatives of isoxazolo[4,5-d]pyridazin-4(5H)-ones. Eur. J. Med. Chem. 2010;45:2345–2352. doi: 10.1016/j.ejmech.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 72.Patorski P., Wyrzykiewicz E., Bartkowiak G. Synthesis and conformational assignment of N-(E)-stilbenyloxymethylenecarbonyl-substituted hydrazones of acetone and o- (m-and p-) chloro- (nitro-) benzaldehydes by means of and NMR spectroscopy. J. Spectrosc. 2013:1–12. doi: 10.1155/2013/197475. [DOI] [Google Scholar]
- 73.Lodewyk M.W., Siebert M.R., Tantillo D.J. Computational prediction of 1 H and 13 C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012;112:1839–1862. doi: 10.1021/cr200106v. [DOI] [PubMed] [Google Scholar]
- 74.Kalia J., Raines R.T. Hydrolytic stability of hydrazones and oximes. Angew. Chemie Int. Ed. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wiberg K.B., Glaser R. Resonance interactions in acyclic systems. 4. stereochemistry, energetics, and electron distributions in 3-center-four-.pi.-electron systems A:BC. J. Am. Chem. Soc. 1992;114:841–850. doi: 10.1021/ja00029a006. [DOI] [Google Scholar]
- 76.Doungdee P., Sarel S., Wongvisetsirikul N., Avramovici-Grisaru S. Iron chelators of the pyridoxal 2-pyridyl hydrazone class. Part 4. pKa values of the chelators and their relevance to biological properties. J. Chem. Soc. Perkin Trans. 1995;2:319. doi: 10.1039/p29950000319. [DOI] [Google Scholar]
- 77.Vilchèze C., Jacobs W.R., Jr. The mechanism of isoniazid killing: Clarity through the scope of genetics. Annu. Rev. Microbiol. 2007;61:35–50. doi: 10.1146/annurev.micro.61.111606.122346. [DOI] [PubMed] [Google Scholar]
- 78.Johnsson K., Schultz P.G. Mechanistic studies of the oxidation of isoniazid by the catalase peroxidase from Mycobacterium tuberculosis. J. Am. Chem. Soc. 1994;116:7425–7426. doi: 10.1021/ja00095a063. [DOI] [Google Scholar]
- 79.Rawat R., Whitty A., Tonge P.J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. USA. 2003;100:13881–13886. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lei B., Wei C.J., Tu S.C. Action mechanism of antitubercular isoniazid-activation by Mycobacterium tuberculosis KatG, isolation, and characterization of InhA inhibitor. J. Biol. Chem. 2000;275:2520–2526. doi: 10.1074/jbc.275.4.2520. [DOI] [PubMed] [Google Scholar]
- 81.Menendez C., Gau S., Lherbet C., Rodriguez F., Inard C., Pasca M.R., Baltas M. Synthesis and biological activities of triazole derivatives as inhibitors of InhA and antituberculosis agents. Eur. J. Med. Chem. 2011;46:5524–5531. doi: 10.1016/j.ejmech.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 82.Campbell P.J., Morlock G.P., Sikes R.D., Dalton T.L., Metchock B., Starks A.M., Hooks D.P., Cowan L.S., Plikaytis B.B., Posey J.E. Molecular detection of mutations associated with first- and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011;55:2032–2041. doi: 10.1128/AAC.01550-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Saranaya S., Haribabu J., Bhuvanesh N.S.P., Karvembu R., Gayathri D. Crystal structures of the Schiff base derivatives (E)-N[(1H-indol-3-yl)methylidene]isonicotino-hydrazide ethanol monosolvate and (E)-N-methyl-2-[1-(2-oxo-2H-chromen-3-yl)ethyl-idene]hydrazinecarbothioamide. Pt 4Acta Crystallogr. E Crystallogr. Commun. 2017;73:594–597. doi: 10.1107/S205698901700411X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mochon M. Salicylaldehyde-1-phthalazinohydrazone as an analytical reagent. Talanta. 1986;33:627–630. doi: 10.1016/0039-9140(86)80144-8. [DOI] [PubMed] [Google Scholar]
- 85.Chollet A., Mori G., Menendez C., Rodriguez F., Fabing I., Pasca M.R., Madacki J., Korduláková J., Constant P., Quémard A., et al. Design, synthesis and evaluation of new GEQ derivatives as inhibitors of InhA enzyme and Mycobacterium tuberculosis growth. Eur. J. Med. Chem. 2015;101:218–235. doi: 10.1016/j.ejmech.2015.06.035. [DOI] [PubMed] [Google Scholar]
- 86.Stadthagen G., Korduláková J., Griffin R., Constant P., Bottová I., Barilone N., Gicquel B., Daffé M., Jackson M. p-Hydroxybenzoic acid synthesis in Mycobacterium tuberculosis. J. Biol. Chem. 2005;280:40699–40706. doi: 10.1074/jbc.M508332200. [DOI] [PubMed] [Google Scholar]
- 87.Phetsuksiri B., Baulard A.R., Cooper A.M., Minnikin D.E., Douglas J.D., Besra G.S., Brennan P.J. Antimycobacterial activities of isoxyl and new derivatives through the inhibition of mycolic acid synthesis. Antimicrob. Agents Chemother. 1999;43:1042–1051. doi: 10.1128/aac.43.5.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Larsen M.H., Vilchèze C., Kremer L., Besra G.S., Parsons L., Salfinger M., Heifets L., Hazbon M.H., Alland D., Sacchettini J.C., et al. Overexpression of inhA, but not kasA, confers resistance to isoniazid and ethionamide in Mycobacterium smegmatis, M. bovis BCG and M. tuberculosis. Mol. Microbiol. 2002;46:453–466. doi: 10.1046/j.1365-2958.2002.03162.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.