

Abstract
The use of plant-based compounds derived from traditional medicine to improve human diseases has been gaining momentum, due to their high bioavailability and moderate adverse effects. Sinomenine is one such biomonomer alkali compound derived from Sinomenium acutum and is known for its anti-inflammatory and antitumor effects. However, the molecular mechanism(s) of its antitumor properties are not fully characterized. In the present study, we evaluated the radiosensitizing effects of the water-soluble sinomenine, sinomenine hydrochloride (SH) in human cervical cancer cell line (HeLa). SH sensitized HeLa cells to ionizing radiation (IR) by promoting accumulation of IR-induced DNA double-strand breaks (DSBs) and also by interfering with DNA damage checkpoint activation. We then investigated the molecular mechanisms underlying the SH-mediated cellular sensitization to IR and found that SH inhibited the expression of DNA damage response (DDR) factors Ku80 and Rad51 at the transcription level. Finally, the radiosensitizing activity of SH was confirmed in a cervical cancer mouse xenograft model. The combinatorial treatment of SH and IR significantly slowed the tumor growth rate compared with IR alone. Collectively, our study not only provides molecular insights into the novel role of SH in cellular response to IR, but also suggests a therapeutic potential of SH as a radiosensitizer in cervical cancer therapy.
Keywords: sinomenine hydrochloride, radiosensitizer, DNA damage response, DNA double-strand break repair, cancer therapy
Introduction
Human genomic DNA is subject to multiple endogenous and exogenous insults that include deamination, pyrimidine dimer, mismatches, interstrand crosslink and DNA single or double strand breaks (SSBs or DSBs), induced by free radical species, chemical compounds, UV and ionizing radiation (IR) (1). Among many types of DNA lesions, DSBs are the most lethal form and it is generally believed that a single unrepaired DSB is sufficient to trigger cell death (2). In mammalian cells, DSBs are primarily repaired by non-homologous end joining (NHEJ) and homologous recombination (HR). HR repairs DSBs in an error-free fashion in S/G2 phases when a sister chromatid is available to be used as a repair template. NHEJ could be error prone but plays a central role in repairing DSBs in all cell cycle stages other than the M phase in dividing cells, and is particularly critical for DSB repair (DSBR) in post-mitotic cells such as neurons, in which the HR-mediated repair is not available (3). To ensure that DSBs are repaired prior to initiation of cell division, DSBs induce cell cycle arrest, which is known as DNA damage checkpoint activation. The cellular DNA damage response (DDR) is a kinase-based signal transduction pathway that involves multiple DSB sensor proteins such as an MRN complex, transducer proteins such as ATM and ATR, mediator proteins such as 53BP1 and BRCA1, and effectors such as Chk1 and Chk2, which protect genome stability and integrity against DSB in a coordinated way (4,5). Aberrant expression of DDR proteins has been linked to the initiation and progression of human malignant tumors. For example, overexpression of Rad51 has been linked to pancreatic cancer and deficient BRCA1/BRCA2 has been linked to breast cancer (6,7).
DDR could be a double-edged sword in normal vs. cancer cells. DSB-inducing agents such as IR and radiomimetics is an established therapeutic strategy for cancer, which is known as radiotherapy (RT) and applied in at least 50% of all cancer patients (8). While normal cells rely on efficient DDR to maintain genome fidelity, DDR hampers the RT response in cancer cells. Many cancers display resistance to standardized irradiation due to intrinsic or acquired radioresistance. For example, one of leading malignancies in women worldwide, ~60% of cervical cancer cases are subjected to RT while in an overall 13% of incidences local recurrence is observed due to radioresistance (9). One of the major mechanisms underlying radioresistance is increased DDR activity and DSBR efficiency in those tumors (10). In addition, cancer stem cells that survive fractionated irradiation can also be radioresistant and cause tumor relapse (11). In order to overcome radioresistance, decades of work has been focused on the inhibition of DSB response as a viable strategy, and the link between DDR inhibition and IR-induced cancer cell death has been well demonstrated (12). Inhibitors of DSBR factors and DNA damage checkpoint regulators, and a number of synthetic inhibitors such as BO2 targeting Rad51, AZD7762 targeting Chk1/2, PCI-24781 targeting HDAC, and neutral radiosensitizers extracted from plants such as quercetin targeting ATM, genistein targeting cyclin B, have been evaluated (13,14). However, compared with natural radiosensitizers, synthetic inhibitors have limited improvement on treatment and more side effects in general (14).
Medicinal plant extracts have long been known to be beneficial towards human diseases, and the molecular characterization of plant-based products is critical for the discovery of new drug candidates (15). Sinomenium acutum is such a plant that has been used to treat neuralgia and rheumatoid arthritis in many Asian countries since antiquity (16). As the active ingredient of the plant, alkaloid sinomenine (SIN) was subsequently isolated and the pharmacological effects of SIN on anti-angiogenesis (17), analgesia (18), anti-inflammation (19,20), immunosuppression (19,21) and anti-nociceptive (22) properties were demonstrated by studies in vitro or in vivo. Notably, the anticancer effects of SIN and its water-soluble form, sinomenine hydrochloride (SH) were also characterized recently. Jiang et al found that SIN induced apoptosis of a lung cancer cell line by collapsing the mitochondrial membrane (23); Lv et al found that SIN inhibited the proliferation of gastric cancer cells by suppressing cyclooxygenase-2 expression (24); Lu et al revealed that SH inhibited hepatocellular carcinoma cell growth by involving cell cycle arrest and apoptosis (25); Song et al reported that SIN inhibited breast cancer cell invasion and migration by inactivating NF-κB (26). However, its radiosensitizing function in cancer treatment has never been characterized.
In the present study, we evaluated the sensitizing efficacy of SH on human cervical cancer cell line HeLa to irradiation, and demonstrated its potential as a radiosensitizer on a cellular level and in a mouse model.
Materials and methods
Cell cultures and preparation of SH
Human HeLa cervical cancer cells and SiHa cervical cancer cells were cultured with Dulbecco's modified Eagle's medium (DMEM; HyClone Laboratories; GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; HyClone Laboratories; GE Healthcare Life Sciences), 100 U/ml penicillin and 100 µg/ml streptomycin. Cultures were grown in a 5% CO2 incubator at 37°C. SH (Zhengqing Pharmaceutical Group Co., Ltd., Hunan, China) was dissolved in phosphate-buffered saline (PBS) to a concentration of 100 mmol/l, and stored at −20°C for up to 4 weeks.
Methylthiazoltetrazolium (MTT) assay
HeLa cells were seeded in 96-well plates with a density of 4,000 cells/well in 200 µl culture medium and incubated overnight. SH solutions were prepared with DMEM without serum with final gradient concentrations of 0.5, 1, 1.5, 2 and 5 mmol/l. After the cells were incubated for 24, 48 and 72 h, 20 µl 3-(4,5-dimethylthiazol-2-y1)-2,5-diphenytetrazolium bromide was added to each well and the cell cultures were incubated for an additional 4 h. The colored solution was quantified by a spectrophotometer at an absorbance of 490 nm. The inhibition rate of the cells was then calculated.
Colony forming assay
HeLa and SiHa cells were incubated in 10 cm2 flasks overnight, and then divided into 4 groups: Control, SH (1 mmol/l) alone, radiation alone, and SH combined with radiation. Cells were treated with SH for 48 h, and then irradiated by X-ray linear accelerator. Following IR, the medium containing SH was removed and cells were maintained in normal culture medium. The cell density of groups was: 300 cells for 0 Gy, 1,000 cells for 2 Gy, 2,000 cells for 4 Gy and 4,000 cells for 6 Gy. Fourteen days later, the cells were washed and stained with crystal violet. The colonies containing >50 cells were counted. Cell survival curves were constructed.
Apoptosis and cell cycle assay
Apoptosis was quantitated using the KGI Biotechnology Apoptosis Kit (Nanjing, China) following the manufacturer's instructions. Cells were fixed with 70% ethanol (2 h, 4°C), and stained with propidium iodide and RNase A (30 min, 37°C) for cell cycle analysis. Samples with 10,000 cells/well were used.
Immunofluorescence
We monitored the DNA DSBs and DSB repair capacity of HeLa cells by phospho-H2AX foci immunofluorescence. The cells were incubated in 10 cm2 flasks overnight and then divided into 4 groups as aforementioned. Drugs were added 48 h prior to radiation exposure (6 Gy). After IR, the medium containing SH was removed and the cells were maintained in normal culturing medium. Twenty-four hours later, the cells were fixed with PFA at 37°C for 20 min, treated with 0.02% Triton X-100 for 10 min, and then blocked with a blocking buffer at room temperature for 1 h. Subsequently, the cells were incubated with the phospho-H2AX S139 antibody (cat. no. 9718; Cell Signaling Technology, Inc., Danvers, MA, USA) at a 1:1,000 dilution overnight at 4°C, and then with a goat anti-rabbit IgG fluorescent-conjugated secondary antibody (cat. no. 8889; Cell Signaling Technology, Inc.) at a 1:400 dilution for 2 h at room temperature. Then, DAPI was used for 15 min to stain the nuclei. The images of the γ-H2AX foci were obtained by fluorescence microscopy (Olympus Corp., Tokyo, Japan).
Western blotting and real-time PCR analyses
The cells were lysed with RIPA lysis buffer (Pioneer Biotech, Co., Ltd., Shaanxi, China) and the protein concentrations were quantified using a BCA kit (Roche Applied Science, Penzberg, Germany). The ATM antibody (cat. no. 2873), phospho-ATM Ser1981 antibody (cat. no. 5883), phospho-H2AX S139 antibody (cat. no. 9718), Ku80 antibody (cat. no. 2180), Rad50 antibody (cat. no. 3427), phospho-Chk2 Thr68 antibody (cat. no. 2661), phospho-Chk1 Ser345 antibody (cat. no. 2348) and anti-rabbit IgG HRP-linked antibody (cat. no. 7074) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA) and the dilution used were 1:1,000-1:3,000. Equal amounts of proteins (60 µg per lane) were separated by 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% non-fat milk in Tris-buffered saline with 0.05% Tween-20 at pH 7.5 for 2 h, and then incubated with primary antibodies at 4°C overnight, and secondary antibodies for 2 h at 37°C. The membranes were washed 3 times with Tris-buffered saline with 0.05% Tween-20 and once with Tris-buffered saline. An enhanced chemiluminescence detection kit (cat. no. WBKLS0500; Millipore, Burlington, MA, USA) was used to detect the signals of immunoblotted proteins on an JS-380A automatic gel imaging system (Quantity One Quantitation software; Bio-Rad Laboratories, Inc., Hercules, CA, USA). RNA was isolated from cells using RNAfast200 (Pioneer Biotech, Co., Ltd.). The Ku80 primer/probe sets (F, 5’-ATTTGCTGGAGGACATTGAAAG-3′ and R, 5′-CTGAATCGGCTGCTGAGG-3′); the Rad51 primer/probe sets (F, 5′-CAACACAGACCACCAGACC-3′ and R, 5′-AGAAGCATCCGCAGAAACC-3′); and the GAPDH primer/probe sets (F, 5′-AAGGCTGTGGGCAAGGTCATC-3′ and R, 5′-GCGTCAAAGGTGGAGGAGTGG-3′) were used. PrimeScript™ RT Master Mix and the SYBR® Premix Ex Taq™ II were purchased from Takara Biotechnology, Co., Ltd. (Dalian, China) and used for real-time PCR. The optimal cycling conditions were 30 sec at 95°C, and 40 cycles of 5 sec at 95°C, 30 sec at 60°C. All real-time PCR experiments were performed in triplicate.
Comet assay
HeLa and SiHa cells were incubated in 6 cm2 flasks overnight, and then divided into 4 groups: Control, SH (1 mmol/l) alone, radiation alone, and SH combined with radiation for the neutral comet assay. HeLa cells were also divided into 6 groups: Control, SH, H2O2 (0 min post-H2O2 treatment for 1 h), SH combined with H2O2 (0 min post-H2O2 treatment for 1 h), H2O2 (180 min post-H2O2 treatment for 1 h), SH combined with H2O2 (180 min post-H2O2 treatment for 1 h) for the alkaline comet assay. Comet Assay kits (Trevigen, Inc., Gaithersburg, MD, USA) were used in the present study. Twenty-four hours after IR, or 0 min/180 min after H2O2 treatment, the cells were harvested and mixed with agarose for electrophoresis. The nuclear DNA was stained with SYBR-Green dye for 10 min and the images were obtained by fluorescence microscopy. The olive tail moment was assessed using Comet analysis software (cat. no. 4260-000-CS; Trevigen, Inc.).
Dual-Luciferase reporter assay
The promoter region of the Rad51 gene from 543 bp upstream to 204 bp downstream was cloned (primers used for PCR, 5′-CGGGGTACCGTCTCACTCTGTCATGAGGC-3′ and 5′-CCGCTCGAGGTCTAATTTGGGTCTTGACC-3′) into the pGL-Basic report vector (27). Plasmids pGL-Basic-Rad51-promoter and pRL-TK (as an internal control) were co-transfected into 293T cells seeded in 12-well plates. The luciferase activity was assessed using a Dual-Luciferase Assay Kit (Promega Corp., Madison, WI, USA) 48 h after SH treatment. Reporter luciferase activity was normalized to Renilla luciferase activity.
In vivo test
The animal experiments were approved and supervised by the Laboratory Animal Care Committee of Xi'an Jiaotong University. Twenty-four mice were housed in sterile cages under standard conditions (12 h light/dark cycles at 21±2°C and normal atmosphere) with ad libitum access to disinfected food and water. Twenty-four 4-week-old female BALB/c nude mice were injected s.c. in the back with 1.0×106 HeLa cells. When xenografts reached a volume of 150–200 mm3, they were randomly divided into 4 groups adjusted by initial tumor volume and treated with: i) PBS alone; ii) SH 100 mg/kg by intraperitoneal injection (every day till the mice were sacrificed); iii) irradiation (3 doses of local irradiation at a dose of 4 Gy at 2-day intervals); iv) SH combined with irradiation (IR starting after 2 days of SH injections). Animals were weighed and the tumor volume was assessed every 2 days until the mice were sacrificed 3 weeks later using pentobarbital sodium at a dose of 100 mg/kg. The tumor volume was calculated by V = 1/2 (length × width2).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). Differences between the control and treatment groups were determined by Student's t-test and considered to be significant at P<0.05.
Results
SH sensitizes HeLa cells to irradiation
Although SIN was previously implicated in cancer cell proliferation suppression (23), the radiosensitive effect on cervical cancer cells has not been characterized. To address this question, first, we tested the effect of SH on HeLa cells by performing MTT-based cell viability analysis (Fig. 1A). The cell viability inhibition rate was calculated at 24, 48 and 72 h after SH treatment and as shown in Fig. 1A, cellular survival was significantly inhibited by SH at various concentrations. Concentrations of 1.5, 2 and 5 mM induced severe cell death (~30 to 90%), while 0.5 and 1 mM SH exhibited a similar and relatively moderate effect on cell survival compared with higher concentrations (Fig. 1A). Given that 1 mM was an already established concentration in previously published studies for a number of carcinoma cell lines (24,25,28), we finally chose the 1 mM dose in the following experiments to make it consistent with previous studies, thus enabling the comparison of the SH-induced phenotypes across different cell lines and with other studies. We next examined the radiosensitive effect of SH on HeLa cells by clonogenic assay. HeLa cells were treated with 1 mM SH for 48 h followed by IR with 0, 2, 4 or 6 Gy, and we observed a significant reduction in clonogenic survival of the SH plus IR group compared with IR alone, as illustrated by the survival curves in Fig. 1B, which indicated a sensitization of SH-treated HeLa cells to IR. We also examined whether SH sensitized a squamous cell carcinoma cell line to IR. We performed a clonogenic assay in SiHa cells and observed increased sensitivity of SH-treated SiHa cells to IR (data now shown), which was consistent with the observation in HeLa cells.
Figure 1.
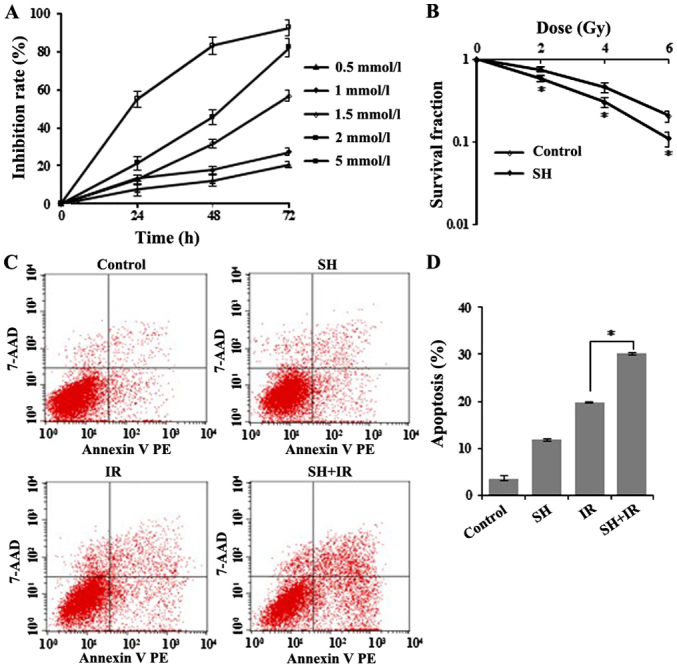
SH sensitizes HeLa cells to irradiation. (A) MTT-based viability analysis of HeLa cells treated with SH. The cells were incubated with SH at indicated concentrations for various time-points. An MTT assay was performedd 24 h after treatment. (B) Clonogenic survival analysis of HeLa cells after SH treatment. Cells were treated with SH (1 mM) for 48 h (*P<0.05). (C) Apoptosis analysis by flow cytometry. HeLa cells were treated with SH (1 mM for 48 h), IR (6 Gy) and SH plus IR, respectively. Untreated cells were indicated as the control. In the SH plus IR group, cells were subjected to IR (6 Gy) 48 h after incubation with 1 mM SH. (D) Quantitation of apoptotic cells in C (*P<0.05). SH, sinomenine hydrochloride; IR, ionizing radiation.
Furthermore, we conducted flow cytometric analysis to test apoptosis in IR-treated HeLa cells incubated with or without SH, and observed significantly increased apoptosis in the group treated by IR in the presence of SH compared with the other groups (Fig. 1C and D). Collectively, these data demonstrated a role of SH in sensitizing HeLa cells to IR.
SH treatment impairs DNA double-strand break response in HeLa cells
Given that IR triggers cellular apoptosis by inducing DNA damage, we tested whether SH enhances IR-induced DSB accumulation. We evaluated DSBs by assessing the level of γ-H2AX. γ-H2AX is a phosphorylated core histone variant H2AX, phosphorylated after DSB induction in an ATM-dependent manner and quickly forms nuclear foci that can be visualized by immunofluorescence (IF) assay (29,30). It has been established that the γ-H2AX-containing foci are correlated directly with the number of DSBs (31). Four groups of cells including one untreated group as a control, and another 3 groups treated with SH, IR or SH plus IR, respectively were fixed for IF following the treatments. Cells with positive γ-H2AX foci were counted by microscopy. The IR-induced DNA damage indicated by γ-H2AX foci was significantly enhanced in the presence of SH (Fig. 2A and B). The results were further confirmed by immunoblotting (IB). In addition to the γ-H2AX, another DSB marker phospho-ATM (32) exhibited a similar pattern (Fig. 2C), indicating a persistent accumulation of IR-induced DSBs in the presence of SH. We further performed neutral single-cell gel electrophoresis assay (comet assay), a method that exclusively visualizes cellular DSBs to confirm the SH-mediated IR-induced DSB accumulation. The higher mean comet tail moment in the SH plus IR group than the IR group, indicated that more IR-induced DSBs were accumulated in HeLa cells when pre-treated with SH (Fig. 2D and E). We noticed that the DSBs were also slightly increased in HeLa cells treated with SH alone, likely caused by the impaired DSBR on endogenous DSBs. Similarly, more IR-induced DSBs were accumulated in the SH pre-treated SiHa cells as well (Fig. 2F and G).
Figure 2.
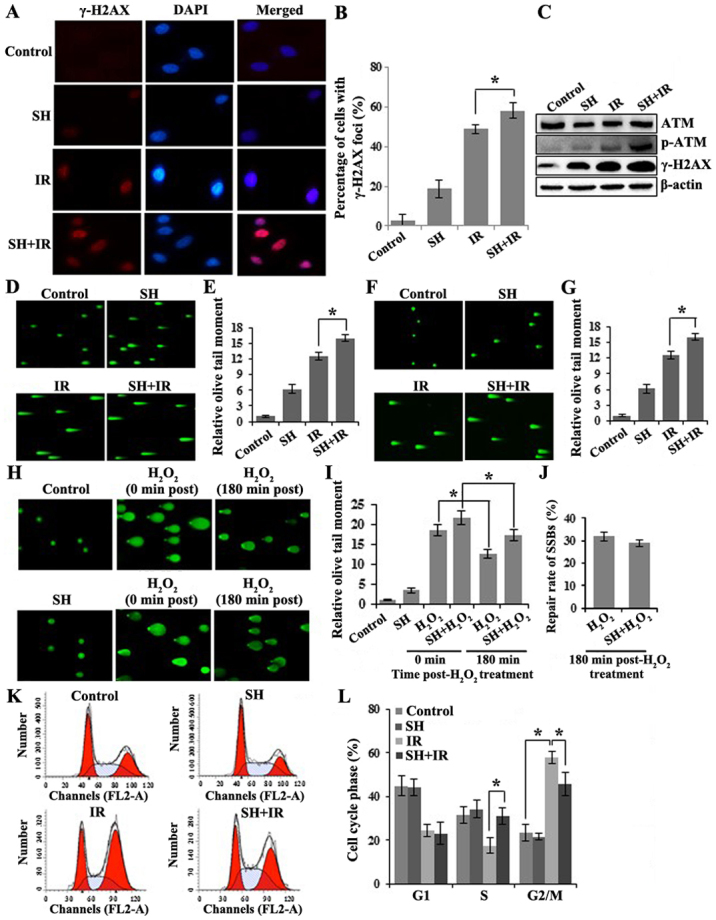
SH treatment impairs DNA damage response in HeLa cells. (A) IF of HeLa cells for γ-H2AX staining (red), nuclei stained with DAPI (blue). (B) Quantitation of HeLa cells with γ-H2AX foci in A (*P<0.05). (C) IB of HeLa cell lysates for ATM, p-ATM and γ-H2AX. β-actin was probed as a loading control. (D and E) Neutral comet assay of HeLa cells with indicated treatments. The quantitation of mean tail moment from 50 randomly selected nuclei is shown in the histogram in E (*P<0.05). (F and G) Neutral comet assay of SiHa cells with indicated treatments. The quantitation of mean tail moment from 50 randomly selected nuclei is shown in the histogram in G (*P<0.05). (H and I) Alkaline comet assay of HeLa cells with indicated treatments. The quantitation of mean tail moment from 50 randomly selected nuclei is shown in the histogram in I (*P<0.05). (J) Histogram revealing the repair rate of SSBs in H2O2- and SH+H2O2-treated HeLa cells. (K and L) Cell cycle analysis by flow cytometry. The distribution of cell cycle phases are revealed in the histogram in L (*P<0.05). SH, sinomenine hydrochloride.
In addition, we investigated whether SH impaired DNA single-strand break repair (SSBR). To reach this aim, we conducted alkaline comet assays, which detect both DSB and SSB. We treated HeLa cells with H2O2 to induce SSBs and performed alkaline comet assays in untreated, SH, H2O2 and SH combined with H2O2-treated HeLa cells. In the SH-treated group, we observed a slightly increased tail moment, likely due to the DSBs that SH caused as we previously demonstrated. H2O2-treated cells exhibited significantly higher accumulation of SSBs as expected and ~30% of damages were repaired after 3-h release from treatment (Fig. 2H and I). Although SH combined with H2O2-treated cells exhibited a higher DNA damage level than the H2O2 group, a comparable repair rate (~30% after 3-h release) was observed in both groups (Fig. 2J), which indicated that the SSBR was not impaired by SH.
Cellular DNA damage activates cell cycle checkpoints. We questioned whether SH alters IR-mediated DNA-damage checkpoint activation. The cell cycle distribution was examined in the 4 groups of HeLa cells by flow cytometry. As shown in Fig. 2K and L, in response to IR treatment, we observed a reduced population in the S phase but a significant enrichment population in the G2/M phase population, indicating the activation of the G2/M checkpoint by IR. We did not observe any changes in cell cycle distribution by SH incubation alone. However, compared with the IR group, the SH plus IR group displayed a markedly decreased cell population arrested in G2/M, which indicated an interfering effect of SH on IR-induced G2/M checkpoint activation. Notably, the IR-induced S phase reduction was rescued by SH, which was comparable with the control (Fig. 2K and L). Overall, our results indicated that SH caused DNA damage accumulation and interfered with cell-cycle checkpoint activation.
SH suppresses expression of DSBR protein Ku80 and Rad51 and enhances the IR-induced activition of Chk1
As aforementioned, IR induces cancer cell death by generating DSBs, which are mainly repaired by HR and NHEJ. As SH sensitizes HeLa cells to IR via accumulation of IR-induced DSBs, we wondered whether SH impairs DSBR pathways. We examined the expression levels of a number of key DSBR factors including ATM, BRCR1, 53BP1, Ku80 and Rad51 by IB, and found that the incubation with SH resulted in a marked reduction in the levels of Ku80 and Rad51 in HeLa cells (Fig. 3A). Furthermore, we examined the mRNA levels of the two proteins by performing RT-PCR and found that the mRNA levels of Ku80 and Rad51 were consistently downregulated in response to SH incubation (Fig. 3B and C), indicating a negative-regulatory role of SH on Ku80 and Rad51 at the transcriptional level. We questioned whether the promotor activity of the genes was affected by SH treatment. Given that the promotor region of the Rad51 gene has been well characterized (27), we cloned Rad51 promoter fragment (from 543 bp upstream to 204 bp downstream) into a pGL-Basic luciferase reporter vector and performed luciferase reporter assays. The results revealed that the promoter activity of the Rad51 gene was significantly inhibited by incubation with SH (data not shown). To explore the underlying mechanism by which pre-treatment with SH promoted IR-dependent S-phase arrest as displayed in Fig. 2K and L, we detected the activation of Chk1 and Chk2, downstream targets of ATR/ATM kinase, which is necessary for DNA damage checkpoint signaling. Upon genotoxic injuries, Chk1 and Chk2 are phosphorylated at Serine 345 (S345) and Threonine 68 (T68) by ATR and ATM, respectively, facilitating G1/S, intra-S and G2/M cell cycle checkpoint activition (33–37). We thus assessed the phosphorylation level of Chk1 S345 and Chk2 T68 by western blotting and found that SH treatment specifically increased IR-induced phosphorylation of Chk1 at S345 (Fig. 3D). Our data indicated that SH may regulate the DSBR pathway by impairing the expression and activition of key DDR proteins.
Figure 3.
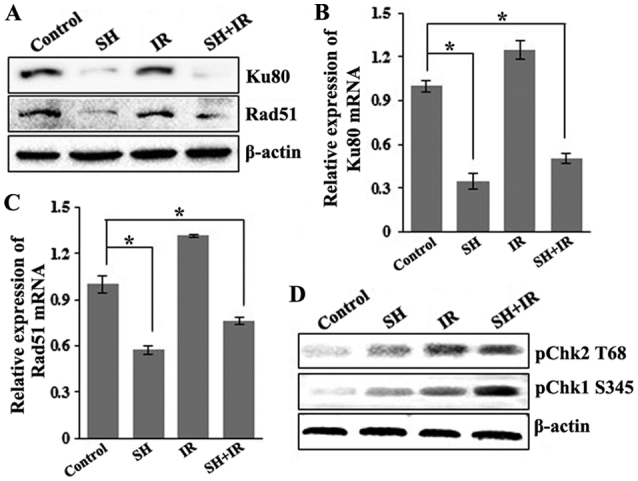
SH suppresses the expression of the DSBR protein Ku80 and Rad51 at the transcriptional level. (A) IB of HeLa cell lysates for Ku80 and Rad51. β-actin was probed as a loading control. (B and C) Relative mRNA level of Ku80 and Rad51 in control cells and cells treated by SH, IR and SH plus IR, respectively. Total RNA was extracted from HeLa cells following various treatments, and the mRNA expression was examined by RT-PCR. Relative levels of Ku80 and Rad51 mRNA were normalized using GAPDH as an internal control (*P<0.05). (D) IB of HeLa cell lysates for pChk2 T68 and pChk1 S345. β-actin was probed as a loading control. SH, sinomenine hydrochloride; DSBR, DSB repair; double-strand break.
Radiosensitizing effect of SH in a cervical cancer xenograft model
Our in-cellulo observations that SH sensitizes HeLa cells and accumulates IR-induced DSBs by impairing DSBR proteins Ku80 and Rad51, led us to evaluate its therapeutic role in RT of cervical cancer, using a nude-mouse tumor xenograft model to investigate SH-mediated radiosensitivity in vivo. HeLa cells (1.0×106) were subcutaneously injected into 4-week old-nude mice, which were randomly divided into 4 groups and treated by PBS injection (control), SH injection, IR and SH injection plus IR, respectively. In the IR and SH plus IR groups, the mice received experimental fractionated radiotherapy (38) when xenografts reached a volume of 150–200 mm3 (39). The mice weight and tumor volume were assessed every 2 days and the mice were sacrificed 3 weeks later. We did not observe any significant difference in the weight of the mice between the control group and SH injection group, while a marked reduced weight was observed in the IR-treated groups compared with the non-irradiated groups (Fig. 4A and B), whereas the SH plus IR group exhibited a similar weight pattern with the IR group (Fig. 4A and B). The tumor volume was assessed every 2 days by vernier calipers according to the formula: Volume = 1/2 (length × width2). As shown in Fig. 4C and D, a large tumor was observed in the control group 3 weeks after subcutaneous injection but the tumor was suppressed by IR, and as anticipated, the suppression was significantly enhanced by SH injection (Fig. 4C and D). Collectively, the results demonstrated that SH injection enhanced IR-induced inhibition of cervical xenograft growth, which indicated that SH could be a potential radiosensitizer to RT for cervical cancer.
Figure 4.

Combination with SH leads to the sensitization of cervical cancer xenograft model to IR. (A) Representative images of mice treated by PBS, SH, IR and SH plus IR, respectively. (B) Body weights of the mice in each group in A. Body weights were measured every 2 days. (C) Representative images of tumors with different sizes isolated from mice of each group. (D) Tumors volumes were measured every 2 days (*P<0.05). SH, sinomenine hydrochloride. IR, ionizing radiation; PBS, phosphate-buffered saline.
Discussion
Cervical cancer is the second leading cause of cancer-related deaths in women worldwide (40). Treatments for cervical cancer include surgery, chemotherapy and RT. Compared to surgery, chemotherapy and RT are mainly used in more advanced stages. RT greatly benefits cervical cancer treatment whereas the survival rate is largely limited by radioresistance developed in patients. Multiple molecular signaling pathways including DDR have been demonstrated to be associated with radioresistance in cervical cancer (41–43). With the use of high throughput screening, independent studies have also indicated DDR gene expression pattern changes in cervical carcinoma tissues or cell lines resisted to IR (41,44,45). For example, radioresistant cervical cancer exhibits upregulated NHEJ proteins (44), which sheds light on the molecular mechanisms underlying DDR-related radioresistance. In addition, recent efforts have been focused on the identification/development and evaluation of radiosensitizers for cervical cancer treatment by targeting DDR factors (46–50).
SIN that is found in the root of the climbing plant Sinomenium acutum is an alkaloid used in traditional herbal medicine in Asian countries, and its antitumor effects in various types of cancers have been subsequently investigated. In the present study, we demonstrated a novel role of SH benefiting cervical cancer treatment as a radiosensitizer. The radiosensitizing effects of SH were examined both in cells and in a xenograft mouse model. In the mouse model, we developed a large tumor through subcutaneous injection of HeLa cells and observed a significant regression by IR. However, IR-induced tumor volume reduction was enhanced by SH pre-treatment, which was consistent with our in vitro experimental results that pre-treatment with SH sensitizes HeLa cells to IR, and indicates it radiosensitizing capacity. Mice treated with SH alone also exhibited a lower tumor volume compared with the control, likely due to its apoptotic effects that were documented in previous publications.
Efficient DSBR is responsible for the RT failure and rapid tumor cell recurrence, and targeting DDR is the key to successful radiosensitization. We also reasoned that SH may impair IR-induced DDR. We observed that HeLa cells pre-treated with SH maintained significantly higher levels of IR-induced DSBs than untreated cells, which was confirmed by either elevated DSB marker levels or long moment tails as revealed by comet assays, likely due to DSBR defects. In addition to HeLa, we also demonstrated the SH-induced sensitivity to IR and DSBR deficiency in squamous cell carcinoma cell line, SiHa. To explore the impairment of SH on DSBR, we screened a number of key DDR proteins and finally, we found that the expression of Rad51 and Ku80 were markedly inhibited by SH incubation at the transcriptional level. Rad51 is a central factor involved in HR-mediated DSBR. When mammalian cells are exposed to genotoxic agents like irradiation, Rad51 is recruited to DSB sites and mediates homologous sequence searching during HR. Rad51 has been established as a radiosensitization target in multiple cancers (51–56). A recent study revealed that Rad51 was highly expressed in glioblastoma stem cells, and inhibition of Rad51 caused significant radiosensitization (51). Notably, the protein level of Rad51 has also been observed to be upregulated ~4 folds in cervical cancer HeLa cells compared with primary cells (57), which indicates its therapeutic applications as a sensitizer target to IR in cervical cancer treatment. Similar to Rad51, Ku80 also plays a crucial role in DSBR but through the NHEJ pathway. Ku80 is one subunit of the Ku80/Ku70 heterodimer, and the Ku heterodimer recruits key DDR proteins like DNA-PK to the DSB sites when DSB occurs and facilitates the DNA damage repair. A number of studies have suggested Ku80 as a molecular target of radiosensitization (58–60). For example, one study demonstrated that inhibition of Ku80 promotes IR combined cisplatin-induced HeLa cell death (58), and another study revealed the relationship between Ku80 suppression and radiosensitivity in human osteosarcoma cells (60). Based on our results, we confirmed the cellular radiosensitization efficacy of inhibiting Rad51 and Ku80 to IR, and identified the potential of SH as a natural radiosensitizer by impairing both HR- and NHEJ-mediated DSBR by inhibiting the two targets. Our observation that SH inhibited promoter activity of the Rad51 gene provided an insight into how SH impairs DSBR. However, why SH selectively regulates the Rad51 and Ku80 genes remain unknown. One of the possibilities is that SH alters histone modification, for example, methylation that specifically regulates transcription of certain genes (61), however further investigation is warranted. In addition to the DSBR defects, SH also disturbed DNA-damage checkpoint activation. Although our data did not reveal any cell cycle disturbance by SH alone, we observed a significantly decreased IR-induced G2/M arrest and increased S-phase arrest by SH pre-incubation, likely due to the elevated phosphorylation level of Chk1 S345, which is important for DNA damage-mediated S-phase checkpoint activation (36).
Overall, our data indicated that SH contributed to radiosensitization through dual pathways, DNA repair and cell cycle checkpoint regulation. A comprehensive investigation of its applications on clinic treatment as radiosensitizer will be a focus of our future studies.
Acknowledgements
The authors thank the members of Han Laboratory in the First Affiliated Hospital of Xi'an Jiaotong University (Xi'an, China) for their technical support.
Glossary
Abbreviations
- SH
sinomenine hydrochloride
- IR
ionizing radiation
- DSBs
double-strand breaks
- DDR
DNA damage response
- NHEJ
non-homologous end joining
- HR
homologous recombination
Funding
The present study was supported by grants from the National Natural Science Foundation of China (nos. 81272488 and 81472795) to SH, (no. 81572968) to HW and NINDS-NIH R01 NS088645 to MLH.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
DZ designed and performed the majority of the experiments in the study and co-wrote the manuscript. YD and CZ contributed to procuring the reagents/materials/analysis tools and analyzed the data. YZ and YQ performed partial experiments. HW and SH designed and supervised the study, and co-wrote and prepared the final manuscript. MLH analyzed and interpreted the data and revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The animal experiments were approved and supervised by the Laboratory Animal Care Committee of Xi'an Jiaotong University (Shaanxi, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15:585–598. doi: 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanungo J. DNA-dependent protein kinase and DNA repair: Relevance to Alzheimer's disease. Alzheimers Res Ther. 2013;5:13. doi: 10.1186/alzrt167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guerrero EN, Wang H, Mitra J, Hegde PM, Stowell SE, Liachko NF, Kraemer BC, Garruto RM, Rao KS, Hegde ML. TDP-43/FUS in motor neuron disease: Complexity and challenges. Prog Neurobiol. 2016;145–146:78–97. doi: 10.1016/j.pneurobio.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freeman AK, Monteiro AN. Phosphatases in the cellular response to DNA damage. Cell Commun Signal. 2010;8:27. doi: 10.1186/1478-811X-8-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srivastava M, Raghavan SC. DNA double-strand break repair inhibitors as cancer therapeutics. Chem Biol. 2015;22:17–29. doi: 10.1016/j.chembiol.2014.11.013. [DOI] [PubMed] [Google Scholar]
- 7.Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517–525. doi: 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. 2005;104:1129–1137. doi: 10.1002/cncr.21324. [DOI] [PubMed] [Google Scholar]
- 9.Zhang B, Chen J, Ren Z, Chen Y, Li J, Miao X, Song Y, Zhao T, Li Y, Shi Y, et al. A specific miRNA signature promotes radioresistance of human cervical cancer cells. Cancer Cell Int. 2013;13:118. doi: 10.1186/1475-2867-13-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239–253. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- 11.Krause M, Dubrovska A, Linge A, Baumann M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv Drug Deliv Rev. 2017;109:63–73. doi: 10.1016/j.addr.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Thoms J, Bristow RG. DNA repair targeting and radiotherapy: A focus on the therapeutic ratio. Semin Radiat Oncol. 2010;20:217–222. doi: 10.1016/j.semradonc.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 13.Mladenova V, Mladenov E, Iliakis G. Novel biological approaches for testing the contributions of single DSBs and DSB clusters to the biological effects of high LET radiation. Front Oncol. 2016;6:163. doi: 10.3389/fonc.2016.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malik A, Sultana M, Qazi A, Qazi MH, Parveen G, Waquar S, Ashraf AB, Rasool M. Role of natural radiosensitizers and cancer cell radioresistance: An update. Anal Cell Pathol. 2016;2016:6146595. doi: 10.1155/2016/6146595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hegde ML. Molecular characterization of neuroprotective activities of plant based products could revive their utilization and lead discovery of new drug candidates for brain diseases. J Pharm Bioallied Sci. 2014;6:63–64. [PMC free article] [PubMed] [Google Scholar]
- 16.Yamasaki H. Pharmacology of sinomenine, an anti-rheumatic alkaloid from sinomenium acutum. Acta Med Okayama. 1976;30:1–20. [PubMed] [Google Scholar]
- 17.Kok TW, Yue PY, Mak NK, Fan TP, Liu L, Wong RN. The anti-angiogenic effect of sinomenine. Angiogenesis. 2005;8:3–12. doi: 10.1007/s10456-005-2892-z. [DOI] [PubMed] [Google Scholar]
- 18.Ju XD, Deng M, Ao YF, Yu CL, Wang JQ, Yu JK, Cui GQ, Hu YL. Protective effect of sinomenine on cartilage degradation and chondrocytes apoptosis. Yakugaku Zasshi. 2010;130:1053–1060. doi: 10.1248/yakushi.130.1053. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q, Li XK. Immunosuppressive and anti-inflammatory activities of sinomenine. Int Immunopharmacol. 2011;11:373–376. doi: 10.1016/j.intimp.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 20.Teng P, Liu HL, Zhang L, Feng LL, Huai Y, Deng ZS, Sun Y, Xu Q, Li JX. Synthesis and biological evaluation of novel sinomenine derivatives as anti-inflammatory agents. Eur J Med Chem. 2012;50:63–74. doi: 10.1016/j.ejmech.2012.01.036. [DOI] [PubMed] [Google Scholar]
- 21.Shu L, Yin W, Zhang J, Tang B, Kang YX, Ding F, Hua ZC. Sinomenine inhibits primary CD4+ T-cell proliferation via apoptosis. Cell Biol Int. 2007;31:784–789. doi: 10.1016/j.cellbi.2007.01.035. [DOI] [PubMed] [Google Scholar]
- 22.Zhu Q, Sun Y, Zhu J, Fang T, Zhang W, Li JX. Antinociceptive effects of sinomenine in a rat model of neuropathic pain. Sci Rep. 2014;4:7270. doi: 10.1038/srep07270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang T, Zhou L, Zhang W, Qu D, Xu X, Yang Y, Li S. Effects of sinomenine on proliferation and apoptosis in human lung cancer cell line NCI-H460 in vitro. Mol Med Rep. 2010;3:51–56. doi: 10.3892/mmr_00000217. [DOI] [PubMed] [Google Scholar]
- 24.Lv Y, Li C, Li S, Hao Z. Sinomenine inhibits proliferation of SGC-7901 gastric adenocarcinoma cells via suppression of cyclooxygenase-2 expression. Oncol Lett. 2011;2:741–745. doi: 10.3892/ol.2011.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu XL, Zeng J, Chen YL, He PM, Wen MX, Ren MD, Hu YN, Lu GF, He SX. Sinomenine hydrochloride inhibits human hepatocellular carcinoma cell growth in vitro and in vivo: Involvement of cell cycle arrest and apoptosis induction. Int J Oncol. 2013;42:229–238. doi: 10.3892/ijo.2012.1704. [DOI] [PubMed] [Google Scholar]
- 26.Song L, Liu D, Zhao Y, He J, Kang H, Dai Z, Wang X, Zhang S, Zan Y. Sinomenine inhibits breast cancer cell invasion and migration by suppressing NF-κB activation mediated by IL-4/miR-324-5p/CUEDC2 axis. Biochem Biophys Res Commun. 2015;464:705–710. doi: 10.1016/j.bbrc.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 27.Hasselbach L, Haase S, Fischer D, Kolberg HC, Sturzbecher HW. Characterisation of the promoter region of the human DNA-repair gene rad51. Eur J Gynaecol Oncol. 2005;26:589–598. [PubMed] [Google Scholar]
- 28.Li X, Wang K, Ren Y, Zhang L, Tang XJ, Zhang HM, Zhao CQ, Liu PJ, Zhang JM, He JJ. MAPK signaling mediates sinomenine hydrochloride-induced human breast cancer cell death via both reactive oxygen species-dependent and -independent pathways: An in vitro and in vivo study. Cell Death Dis. 2014;5:e1356. doi: 10.1038/cddis.2014.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Adhikari S, Butler BE, Pandita TK, Mitra S, Hegde ML. A perspective on chromosomal double strand break markers in mammalian cells. Jacobs J Radiat Oncol. 2014;1:003. [PMC free article] [PubMed] [Google Scholar]
- 31.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Cheng J, Li D, Duan H, Yang H, Bin P, Dai Y, Huang C, Liang X, Leng S, et al. Modulation of DNA repair capacity by ataxia telangiectasia mutated gene polymorphisms among polycyclic aromatic hydrocarbons-exposed studyers. Toxicol Sci. 2011;124:99–108. doi: 10.1093/toxsci/kfr216. [DOI] [PubMed] [Google Scholar]
- 33.Wilson KA, Stern DF. NFBD1/MDC1, 53BP1 and BRCA1 have both redundant and unique roles in the ATM pathway. Cell Cycle. 2008;7:3584–3594. doi: 10.4161/cc.7.22.7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carlessi L, Buscemi G, Fontanella E, Delia D. A protein phosphatase feedback mechanism regulates the basal phosphorylation of Chk2 kinase in the absence of DNA damage. Biochim Biophys Acta. 2010;1803:1213–1223. doi: 10.1016/j.bbamcr.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 35.Choi KS, Kim JY, Lim SK, Choi YW, Kim YH, Kang SY, Park TJ, Lim IK. TIS21/BTG2/PC3 accelerates the repair of DNA double strand breaks by enhancing Mre11 methylation and blocking damage signal transfer to the Chk2T68-p53S20 pathway. DNA Repair (Amst) 2012;11:965–975. doi: 10.1016/j.dnarep.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 36.Sorensen CS, Syljuasen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK, Zhou BB, Bartek J, Lukas J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3:247–258. doi: 10.1016/S1535-6108(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 37.Gamper AM, Rofougaran R, Watkins SC, Greenberger JS, Beumer JH, Bakkenist CJ. ATR kinase activation in G1 phase facilitates the repair of ionizing radiation-induced DNA damage. Nucleic Acids Res. 2013;41:10334–10344. doi: 10.1093/nar/gkt833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang H, Li LW, Shi M, Wang JH, Xiao F, Zhou B, Diao LQ, Long XL, Liu XL, Xu L. In vivo study of breast carcinoma radiosensitization by targeting eIF4E. Biochem Biophys Res Commun. 2012;423:878–883. doi: 10.1016/j.bbrc.2012.06.064. [DOI] [PubMed] [Google Scholar]
- 39.Kim JS, Amorino GP, Pyo H, Cao Q, Price JO, Choy H. The novel taxane analogs, BMS-184476 and BMS-188797, potentiate the effects of radiation therapy in vitro and in vivo against human lung cancer cells. Int J Radiat Oncol Biol Phys. 2001;51:525–534. doi: 10.1016/S0360-3016(01)01658-3. [DOI] [PubMed] [Google Scholar]
- 40.Torre LA, Islami F, Siegel RL, Ward EM, Jemal A. Global cancer in women: Burden and trends. Cancer Epidemiol Biomarkers Prev. 2017;26:444–457. doi: 10.1158/1055-9965.EPI-16-0858. [DOI] [PubMed] [Google Scholar]
- 41.Kitahara O, Katagiri T, Tsunoda T, Harima Y, Nakamura Y. Classification of sensitivity or resistance of cervical cancers to ionizing radiation according to expression profiles of 62 genes selected by cDNA microarray analysis. Neoplasia. 2002;4:295–303. doi: 10.1038/sj.neo.7900251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin C, Chen X, Bai Q, Davis MR, Fang Y. Factors associated with radiosensitivity of cervical cancer. Anticancer Res. 2014;34:4649–4656. [PubMed] [Google Scholar]
- 43.Jeon YT, Song YC, Kim SH, Wu HG, Kim IH, Park IA, Kim JW, Park NH, Kang SB, Lee HP, et al. Influences of cyclooxygenase-1 and −2 expression on the radiosensitivities of human cervical cancer cell lines. Cancer Lett. 2007;256:33–38. doi: 10.1016/j.canlet.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 44.Beskow C, Skikuniene J, Holgersson A, Nilsson B, Lewensohn R, Kanter L, Viktorsson K. Radioresistant cervical cancer shows upregulation of the NHEJ proteins DNA-PKcs, Ku70 and Ku86. Br J Cancer. 2009;101:816–821. doi: 10.1038/sj.bjc.6605201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qing Y, Yang XQ, Zhong ZY, Lei X, Xie JY, Li MX, Xiang DB, Li ZP, Yang ZZ, Wang G, et al. Microarray analysis of DNA damage repair gene expression profiles in cervical cancer cells radioresistant to 252Cf neutron and X-rays. BMC Cancer. 2010;10:71. doi: 10.1186/1471-2407-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kunos CA, Colussi VC, Pink J, Radivoyevitch T, Oleinick NL. Radiosensitization of human cervical cancer cells by inhibiting ribonucleotide reductase: Enhanced radiation response at low-dose rates. Int J Radiat Oncol Biol Phys. 2011;80:1198–1204. doi: 10.1016/j.ijrobp.2011.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wieringa HW, van der Zee AG, de Vries EG, van Vugt MA. Breaking the DNA damage response to improve cervical cancer treatment. Cancer Treat Rev. 2016;42:30–40. doi: 10.1016/j.ctrv.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 48.Mountzios G, Soultati A, Pectasides D, Dimopoulos MA, Papadimitriou CA. Novel approaches for concurrent irradiation in locally advanced cervical cancer: Platinum combinations, non-platinum-containing regimens, and molecular targeted agents. Obstet Gynecol Int. 2013;2013:536765. doi: 10.1155/2013/536765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Candelaria M, Garcia-Arias A, Cetina L, Duenas-Gonzalez A. Radiosensitizers in cervical cancer. Cisplatin and beyond. Radiat Oncol. 2006;1:15. doi: 10.1186/1748-717X-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuhrman CB, Kilgore J, LaCoursiere YD, Lee CM, Milash BA, Soisson AP, Zempolich KA. Radiosensitization of cervical cancer cells via double-strand DNA break repair inhibition. Gynecol Oncol. 2008;110:93–98. doi: 10.1016/j.ygyno.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 51.King HO, Brend T, Payne HL, Wright A, Ward TA, Patel K, Egnuni T, Stead LF, Patel A, Wurdak H, et al. RAD51 is a selective DNA repair target to radiosensitize glioma stem cells. Stem Cell Reports. 2017;8:125–139. doi: 10.1016/j.stemcr.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balbous A, Cortes U, Guilloteau K, Rivet P, Pinel B, Duchesne M, Godet J, Boissonnade O, Wager M, Bensadoun RJ, et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer. 2016;16:604. doi: 10.1186/s12885-016-2647-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen X, Wong P, Radany EH, Stark JM, Laulier C, Wong JY. Suberoylanilide hydroxamic acid as a radiosensitizer through modulation of RAD51 protein and inhibition of homology-directed repair in multiple myeloma. Mol Cancer Res. 2012;10:1052–1064. doi: 10.1158/1541-7786.MCR-11-0587. [DOI] [PubMed] [Google Scholar]
- 54.Kobashigawa S, Morikawa K, Mori H, Kashino G. Gemcitabine induces radiosensitization through inhibition of RAD51-dependent repair for DNA double-strand breaks. Anticancer Res. 2015;35:2731–2737. [PubMed] [Google Scholar]
- 55.Liu Q, Jiang H, Liu Z, Wang Y, Zhao M, Hao C, Feng S, Guo H, Xu B, Yang Q, et al. Berberine radiosensitizes human esophageal cancer cells by downregulating homologous recombination repair protein RAD51. PLoS One. 2011;6:e23427. doi: 10.1371/journal.pone.0023427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gasparini P, Lovat F, Fassan M, Casadei L, Cascione L, Jacob NK, Carasi S, Palmieri D, Costinean S, Shapiro CL, et al. Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc Natl Acad Sci USA. 2014;111:4536–4541. doi: 10.1073/pnas.1402604111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res. 2002;62:219–225. [PubMed] [Google Scholar]
- 58.Zhuang L, Liu F, Peng P, Xiong H, Qiu H, Fu X, Xiao Z, Huang X. Effect of Ku80 on the radiosensitization of cisplatin in the cervical carcinoma cell line HeLa. Oncol Lett. 2018;15:147–154. doi: 10.3892/ol.2017.7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petera J, Sirak I, Beranek M, Vosmik M, Drastikova M, Paulikova S, Soumarova R. Molecular predictive factors of outcome of radiotherapy in cervical cancer. Neoplasma. 2011;58:469–475. doi: 10.4149/neo_2011_06_469. [DOI] [PubMed] [Google Scholar]
- 60.Hu L, Wu QQ, Wang WB, Jiang HG, Yang L, Liu Y, Yu HJ, Xie CH, Zhou YF, Zhou FX. Suppression of Ku80 correlates with radiosensitivity and telomere shortening in the U2OS telomerase-negative osteosarcoma cell line. Asian Pac J Cancer Prev. 2013;14:795–799. doi: 10.7314/APJCP.2013.14.2.795. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y, Reinberg D. Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.