

Abstract
Multiple variant alleles of the human arylamine N-acetyltransferase genes, NAT1* and NAT2*, alter the capacity of individuals to metabolize arylamines by N-acetylation. Although biochemical and genetic studies have improved our understanding of the molecular basis of the acetylation polymorphism in humans and other mammals, regulation of NAT* gene expression is not understood. In the present study, a segment of the 5′-untranslated region of mouse Nat2* was sequenced and characterized. Primer extension analysis and RNase protection assays exposed multiple transcription initiation sites located 112 to 151 bases upstream of the translational start site. Computer sequence analysis revealed a promoter-like region located within the region 530 bases upstream of the translational start site consisting of TATA boxes, upstream promoter elements such as a CAAT box and Spl binding site, regulatory elements such as a palindromic hormone response element (HRE), and enhancer regions such as an AP-1 transcription factor binding site. Transient expression of CAT reporter constructs of the mouse Nat2*-palindromic HRE demonstrated positive regulation of the HSV-thymidine kinase 1 (tkl) promoter and induced the expression of chloramphenicol acetyltransferase (CAT). This induction was initiated by the addition of hormones such as 5α-dihydrotestosterone (DHT) or dexamethasone and was entirely dependent on the presence of androgen or glucocorticoid receptors, respectively. Together with recent discoveries regarding the effects of testosterone on the expression of Nat2* in mouse kidney during development, the findings reported in this article suggest that the HRE found in the promoter region of Nat2* is a potential candidate for the mediation of androgenic regulation of Nat2* in mouse kidney.
Keywords: N-Acetyltransferase, Promoter, Hormone response element, Androgens, glucocorticoids
FOR the past five decades the study of arylamine Af-acetyltransferases has been focused on genetic variations or polymorphisms, the effects of those variations on the N-acetylation of therapeutic and carcinogenic compounds, and the toxicological consequences of such genetic differences. Two N-acetyltransferases, NAT1 and NAT2, have been identified in humans and both show extensive genetic variability. At least 6 and 15 alleles have been described for NAT1* and NAT2*, respectively (10,29). The hereditary basis of the acetylation polymorphism has also been characterized in several animal models including rabbits (4,25), Syrian hamsters (12,13,21), rats (9), and mice.
In mice the Nat2* gene locus is responsible for the interstrain polymorphism in N-acetylation of p-aminobenzoic acid (PABA) and certain other arylamine substrates. The coding region of this gene has been studied extensively and only a single base difference (A296T) has been found between rapid and slow acetylator strains (8,18,19). Although biochemical and genetic studies have vastly improved our knowledge of the acetylation polymorphisms in humans, mice, and other animal models, the expression and regulation of the NAT* genes are not understood.
Our laboratory has identified an androgen-dependent difference in mouse kidney PABA-NAT activity between males and females. Kidney N-acetylation of PABA measured in A/J and C57BL/6J mice of both sexes during development (10–215 days of age) revealed significant gender differences as early as 20 days of age coinciding with sexual maturation and elevation of testosterone. Castrated and tfm/y mice showed similar Nat activities to females, and treatment of castrated animals with testosterone increased the N-acetylation of PABA to that of intact males (27). A second study examined the gender influences on the developmental pattern of kidney and liver Nat2 activity with the Nat2-selective substrate, PABA, in CD1 mice from birth to 80 days of age (11). The liver Nat2 activity showed no gender-specific differences. Unlike the liver, kidney Nat2 catalytic activity revealed statistically significant differences between males and females that are associated with an increase in the Nat protein levels. This study implies not only gender-specific but tissue-specific regulation of Nat2* gene expression. Studies in Harderian gland NAT activity in Syrian hamsters have also suggested that the sex difference observed in NAT activity is under androgenic control (20).
In the present study, we begin to investigate the sexual dimorphism in mouse kidney Nat activity by describing the structural organization of the 5′-flanking region of mouse Nat2* gene. We have sequenced 1720 bases upstream of the transitional start codon and determined the location of transcription start sites, TATA and CAAT boxes, Sp1 binding sites, a hormone response element (HRE), and AP-1 transcription factor binding sites. We also provide a preliminary functional analysis of the HRE and explore its potential role in the modulation of Nat2* expression in mouse kidney.
MATERIALS AND METHODS
Primer Extension Analysis
Synthetic oligonucleotide primers (Nat2-A and Nat2-B) complementary to a region within the 5′ UTR of mouse Nat2* transcript (Fig. 1) were labeled at their 5′ ends with [γ-32P]ATP by T4 polynucleotide kinase. The labeled primers were purified on NuClean D25 columns (IBI Corp). Labeled primer was mixed with 150 μg of total RNA isolated from C57BL/6J mouse liver or kidney and precipitated with ethanol. The pellet was dissolved in 30 μl of hybridization buffer (40 mM PIPES, pH 6.4, 1 mM EDTA, pH 8.0, 400 μM NaCl, 80% formamide) at 65°C for 10 min, denatured at 85°C for 10 min, and transferred to 55°C for overnight hybridization. Twenty microliters of 5× transcriptase buffer containing 1 mM of each dNTP, 1 U/μl Rnasin, 50 μg/ml actinomycin, and 50 U of Superscript reverse transcriptase were added to the primer-RNA duplex (Gibco, BRL). Extension was performed at 37°C for 2 h; elongated products were extracted with phenol/chloroform, ethanol precipitated, and fractionated through a 6% polyacrylamide gel containing 8 M urea.
FIG. 1.
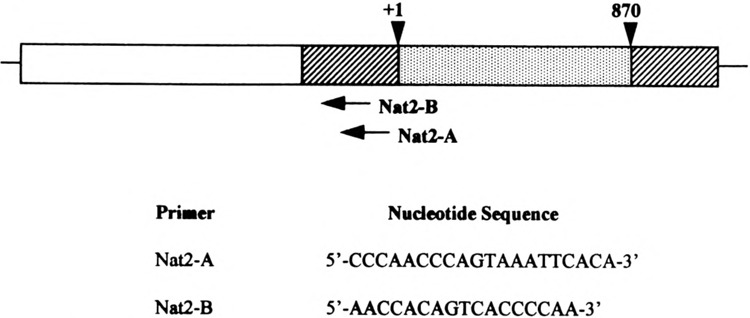
Position and sequence of oligonucleotide primers. Primers Nat2-A and Nat2-B were employed in the primer extension analysis of mouse Nat2*. The gene structure of Nat2* is as depicted by Martell et al. (19). The 5′-flanking region (open bar), 5′- and 3′-untranslated regions (hatched bars), and coding exon (dotted bar).
RNase Protection Assay
A 171-bp 5′ UTR Nat2* probe was generated by PCR. The nucleotide sequence of the SP6 promoter was appended to the 5′ end of the antisense primer (5′ -GCGCGCCACGTAGTGATTTAGGTGACACTATAGGATATCTGTGAATTT ACTGGGT-3′). The sense primer was designed as follows: 5′ -GATATCTCATCCCAGGTCACTT-3′. The cRNA probe was synthesized with SP6 phage RNA polymerase according to the supplier’s specifications (Ambion) in the presence of 50 μCi [α-32P]dUTP (800 μCi/mmol), purified by polyacrylamide gel electrophoresis, and evaluated for full-length extension. The band was excised and extracted in 0.5 M ammonium acetate, 1 mM EDTA, and 0.2% SDS overnight at 37°C. Hybridization was performed by mixing 1 × 105 cpm of eluted probe with 10 μl of HybSpeed hybridization (Ambion) containing 85 or 65 μg of ethanol-precipitated total RNA from slow acetylator congenic (B6.A) mouse liver or kidney, respectively. The solution was heated at 95°C for 3 min, immediately transferred to a water bath maintained at 68°C, and incubated for 10 min. After hybridization, 350 μl of HybSpeed RNase digestion buffer (Ambion) containing 1:50 RNase A and 1:100 RNase T1 dilutions was added and the mixture was incubated for 30 min at 37°C. Reactions were inactivated and ethanol precipitated by adding 150 μl of HybSpeed inactivation/precipitation mix (Ambion). The protected fragments were fractionated by electrophoresis through a 6% polyacrylamide gel containing 8 M urea.
Identification of Promoter and Regulatory Elements
The 5′-flanking region (1720 bp) of mouse Nat2* was sequenced from a previously isolated genomic clone from B6 mouse strain (19) and from PCR-generated fragment for the CD1 strain. Conditions for sequencing of PCR-generated fragments were as described by Vatsis et al. (28). TATA boxes and various response elements were searched by the FINDPATTERN command in the GCG program (Version 7.0, Genetics Computing Group, WI). The CAAT box, Spl region, and other promoter elements were identified in the TRANSFAC web site (transfac@gnf-braunsch weig.ed) and TFSEARCH version 1.3.
Cell Culture
CV1 cells, originally obtained from Dr. D. M. Robins, were cultured in DMEM and supplemented with 10% fetal calf serum (Hyclone, Logan, UT) and penicillin-streptomycin (Gibco, BRL). For transfections, cells were grown in DMEM without phenol red supplemented with 10% steroid-depleted fetal calf serum. For examination of hormone effect on gene expression, 5α-dihydrotestosterone (DHT) (Sigma, St. Louis, MO) was added to a final concentration of 340 nM. In indicated cases, dexamethasone or β-estradiol dipropionate (Sigma) were added to a final concentration of 100 nM.
Construction of CAT Plasmids
The reporter vector pUTKAT3 (22) provided by Dr. M. A. Burgos-Trinidad (The University of Michigan, Ann Arbor) contains the HSV-tk1 promoter from −109 to −1 before the chloramphenicol acetyltransferase (CAT) gene. For HRE plasmids, complementary 25-bp oligonucleotides of the HRE sequence (AGCTCAttcTGTTCT) with BamHI termini were ligated into BamHI-digested pUTKAT3. The oligonucleotides were designed as follows: sense primer, 5′-GATCCACAGCTCATTCTGTTCTCTG-3′, and antisense primer, 5′-GATCCAGAGAACAGAATGAGCTGTG-3′. Cloned HRE nucleotide sequences were verified by direct sequencing (Sequencing Core, The University of Michigan, Ann Arbor). The pG3tk-CATF-3X HRE3 construct, which contains three Slp enhancer sequences of HRE3, was a gift from Dr. D. M. Robins (the University of Michigan, Ann Arbor) and was designed as described previously (2). The mouse androgen receptor (mAR) and the rat glucocorticoid expression vectors (rGR) were also kindly provided by Dr. D. M. Robins (The University of Michigan, Ann Arbor) (1).
Transfections
CV1 cells were transfected with lipofectamine reagent following the manufacturer’s specifications (Life Technologies BRL, Bethesda, MD). Briefly, CV1 cells were grown in 35-mm tissue culture plates at 37°C in a 5% C02 incubator until they reached 70% confluency. For each transfection, 100 ng of mAR or rGR, 2 ng of target DNA, and 25 μl lipofectamine reagent were combined and incubated at room temperature for 45 min. After the DNA-lipofectamine complexes were allowed to form, 0.8 ml of serum-free medium (OPTI-MEM, Life Technologies BRL) was added to the solution and overlaid onto rinsed cells. The transfection was allowed to continue for 5 h at 37°C in a 5% C02 incubator. Following incubation, the transfection mixture was removed and replaced with DMEM media without phenol red containing 10% steroid-depleted fetal calf serum, antibiotics, and the corresponding hormone treatment. Cell extracts were assayed 72 h after the start of transfection.
CAT Assay
Cells were washed in phosphate buffer solution (PBS) once and incubated in hypotonic buffer (25 mM Trizma, pH 7.5, 2 mM magnesium chloride) for 5 min at room temperature. The cells were lysed in 20 μ1 of hypotonic Tris-Triton buffer (25 mM Trizma, pH 7.5, 2 mM magnesium chloride, 0.5% Triton X-100), scraped, and the supernatant was heated at 70°C for 15 min. Cytosol in duplicate samples was assayed for CAT activity by addition of 10 mmol (0.2 μCi) [3H]chloramphenicol and 25 μg butyryl-CoA (Sigma). The reaction mixtures were incubated overnight (15–17 h) at 37°C. The next day the reactions were extracted with xylene to separate the butylated chloramphenicol (26). The xylene layer was analyzed in a liquid scintillation counter.
RESULTS
Mapping of the Transcription Start Site(s) of Mouse Nat2* Gene
To identify the cap site(s) of mouse Nat2*, we performed two types of experiments: primer extension and ribonuclease protection assays. Primer extension of mouse Nat2* was performed with radiolabeled oligonucleotides, Nat2-A and Nat2-B (Figs. 1 and 4), and total RNA isolated from C57BL/6J mouse liver and kidney. The extended products were electrophoresed in parallel with the sequencing reaction performed with each oligonucleotide, which served as size markers (Fig. 2, lanes a, c, g, and t). Multiple mouse Nat2* transcription initiation sites were identified with primer Nat2-A (Fig. 2A) in mouse liver (lane 1) and kidney (lane 2). The sizes of the extended fragments generated by primer Nat2-A were 45, 46, 51, 57, 68, and 84 bp, which places the cap sites at positions −112, −113, −118, −124, −135, and −151, respectively. A single extended product located 135 bases upstream of the translation start site was clearly identified with oligonucleotide Nat2-B (Fig. 2B) in mouse liver (lane 1) and kidney (lane 2). This band was detected with both oligonucleotides, which may indicate that the cap site at −135 nt is a major transcription initiation site.
FIG. 4.

Nucleotide sequence of the 5′-flanking and untranslated regions of C57BL/6J mouse Nat2*. The sequence was determined for genomic clone 5H, previously isolated and mapped by Martell et al. (19). Translation start site (ATG) is indicated by a double arrow (+1) and the identified transcription initiation sites are indicated by asterisks (*). TATA boxes (underlined) and response elements (boxes) were identified as described in Materials and Methods. HRE, hormone response element; AP-1, activator protein 1 binding site; CCAAT Box and Sp1, upstream promoter elements. Also shown are the locations of primers Nat2-A and Nat2-B employed in primer extension analysis.
FIG. 2.
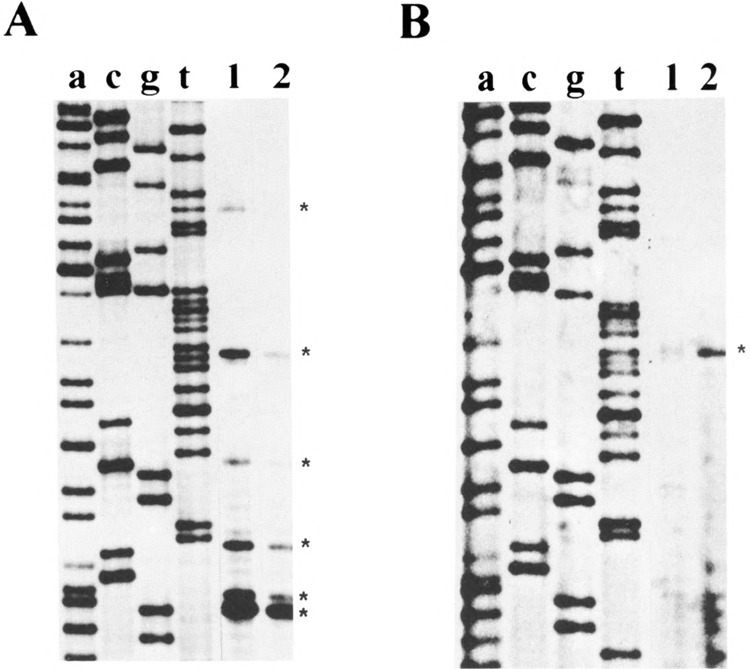
Determination of the transcription start sites of mouse Nat2* by primer extension assay. 32P-labeled primer Nat2-A (A) and primer Nat2-B (B) were hybridized with 150 μg of total RNA from C57BL/6J mouse liver (lane 1) and kidney (lane 2). Lanes a, c, g, and t are the reference dideoxy-sequencing reactions with each corresponding primer. Extended fragments are indicated by asterisks (*).
We used the ribonuclease protection assay to complement the above results. A 171-bp antisense radiolabeled RNA probe (Fig. 3, lane 5) was hybridyzed to total RNA from B6.A mouse liver and kidney and subjected to RNase digestion as described in Materials and Methods. Six protected fragments were observed with probe A (Fig. 3) in both mouse liver (lane 2) and kidney (lane 3), which confirms the presence of multiple initiation sites in the region −51 to −221 bases upstream of the translation start site. The exact location of these fragments is not clearly identified by the ribonuclease protection assay because the expected sizes of the protected fragments do not match the results obtained. No fragments were protected when the probe was hybridized to yeast tRNA (lane 4).
FIG. 3.
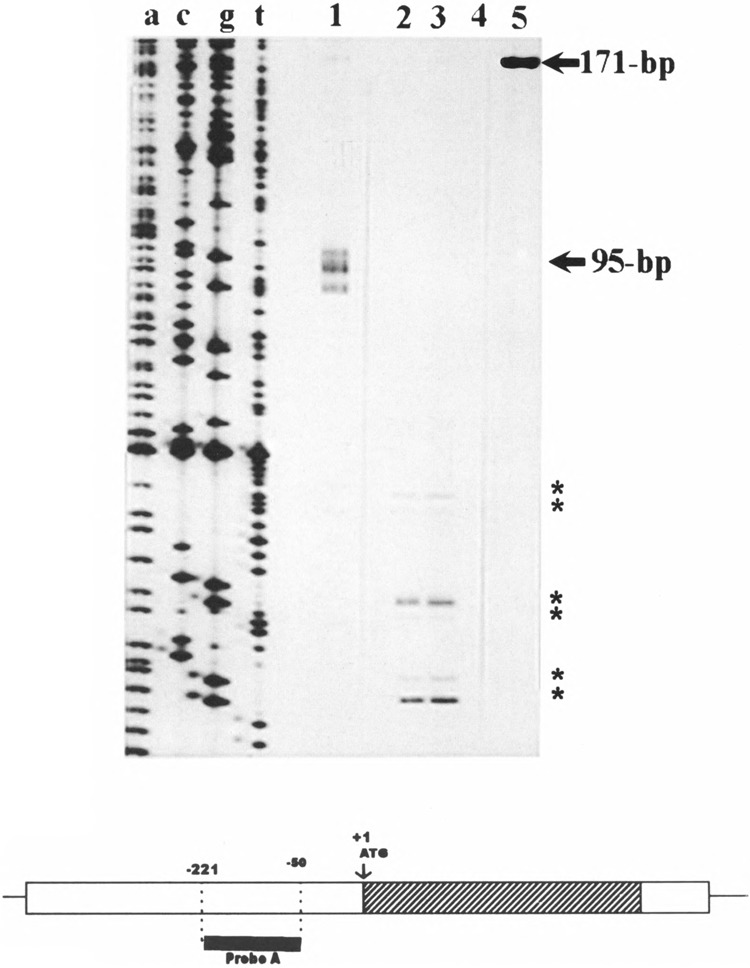
Determination of the transcription start sites of mouse Nat2* by ribonuclease protection assay. Total RNA (85 μg) from B6.A mouse liver (lane 2), 65 μg kidney (lane 3), or 50 μg yeast (lane 4) was hybridized with 171-bp cRNA probe (lane 5). After treatment with RNase A/T, the protected fragments were analyzed on a 6% polyacrylamide/8 M urea gel. Sizes of the protected fragments were determined by comparison to the sequencing reaction on lane a, c, g, and t and to RNA markers in lane 1 (95 bp). The scheme (bottom) depicts the location of the probe (probe A) relative to the coding region (hatched bar) and 5′- and 3′-flanking regions (open bars) of mouse Nat2*.
Analysis of Mouse Nat2* Primary Structure
A PCR-generated fragment including 1720 bases upstream of the translational start site was sequenced to identify potential cis elements that may be involved in the regulation of mouse Nat2* expression (Fig. 4). The sequence analysis of the region flanking the transcription initiation sites revealed that the Nat2* promoter is an AT-rich region, containing 62% A + T. Analysis of DNA sequences (GCG Version 7.0, TRANSFAC, and TFSEARCH) revealed numerous putative elements proximal to the transcription initiation sites. Among those, multiple TATA boxes were identified as well as a CAAT box and an Sp1 binding site at positions −534 and −525, respectively. A number of potential regulatory and enhancer elements were identified by comparison to the consensus sequences (17). In mouse Nat2*,one AP-1 enhancer site at position −553 nt was identified. In addition, the computer analysis disclosed a palindromic HRE located −543 to −576 bases upstream of the translational start site.
Reporter Gene Expression by a Putative Promoter Region of the Nat2* Gene
Studies on the expression of Nat2 during the development of the mouse reveal that the N-acetylating activity of this isoenzyme increases significantly 30 days after birth in the kidney of male C57BL/6J and A/J mouse inbred strains (27) and in CD1 outbred mouse strain 21 days postnatally (11). The observed increase in PABA-NAT activity in males persists throughout maturity in all strains. The kidney Nat activity of females, however, remains unchanged. The increase in N-acetylation of PABA by Nat2 is associated with an increase in Nat protein level (11). These findings suggest that the increase of kidney Nat activity in male mice during development is gender specific and under the control of sex hormones.
To explore the potential role of the palindromic HRE sequence found within the Nat2* promoter in androgen response, we constructed various reporter constructs containing the palindromic Nat2*-HRE segment. Figure 5 illustrates the fusion reporter constructs made with one or two copies of the Nat2*-HRE and the HSV-tk1 promoter-driven CAT gene. The vector, pUTKAT3 (22), with no copies of the HRE (0 × HRE) serves as a negative control whereas the positive control (3 × HRE3) contains three copies of the HRE found in the Slp gene (2). Fusion constructs containing one (1 × HRE) or two copies (2 × HRE) of the Nat2*-HRE were used to test whether this putative element can regulate the HSV-tk1 promoter and, subsequently, the expression of CAT. We co-transfected the various pUTKAT3 constructs (Fig. 5) and mAR expression vector into CV1 cells, which lack endogenous steroid responses (Fig. 6). In the presence of DHT, cells transfected with 0 × HRE construct showed a low level of CAT activity, which was not altered with DHT treatment. This indicates that the HSV-tk promoter is a weak promoter under the present conditions. One copy of the Nat2*-HRE (1 × HRE) elevated basal CAT expression in CV1 cells in the presence or absence of DHT. On the other hand, the 2×HRE construct and the positive control (3 × HRE3) conferred 10- and 14-fold induction, respectively, relative to cells without hormone treatment (Fig. 6A). The CAT activities were determined to be linear relative to amount of total cytosolic protein and the time the assay was allowed to proceed.
FIG. 5.
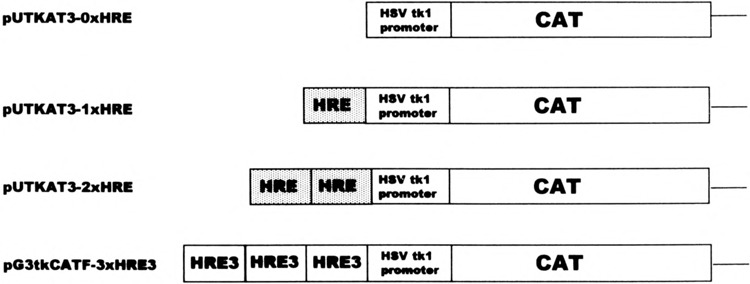
Diagram constructs of CAT reporter genes containing multiple copies of hormone response elements (HRE). A 25-bp double-stranded oligonucleotide corresponding to the −579 to −560 nucleotide relative to the mouse Nat2* translation start site (+l), and flanked by BamHI sites, was synthesized and inserted into pUTKAT3 vector as one and two copies, as shown (dotted bars), upstream of the HSV-tk1 promoter element fused to the bacterial chloramphenicol acetyltransferase gene (open bars). The positive control construct (pG3tkCATF-3×HRE3) contains three HRE copies of the mouse Slp gene whereas the negative control (0 × HRE) has no copies of the HRE.
FIG. 6.
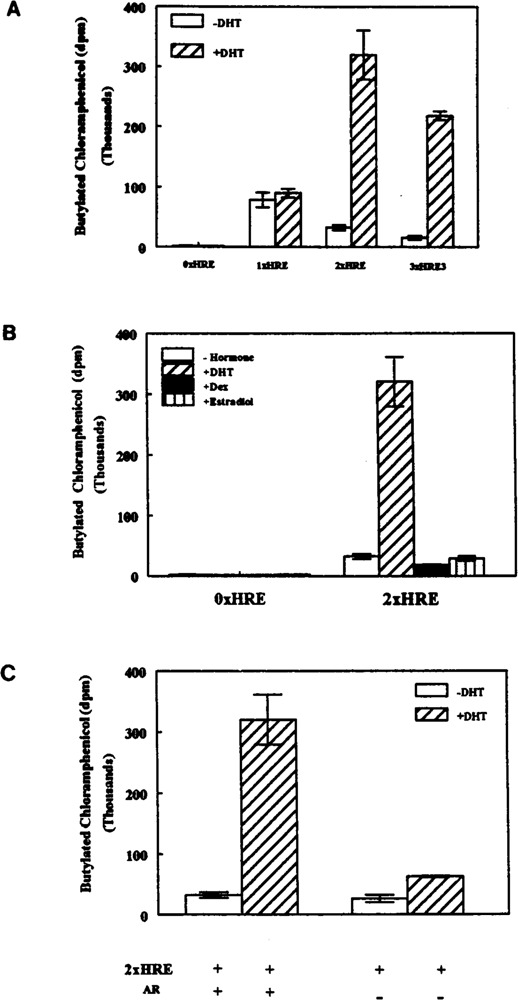
The mouse Nat2* HRE displays positive gene regulation upon androgen stimulation. CV1 cells were cotransfected with 100 ng of mouse androgen receptor expression vector and 2 μg of the indicated CAT reporter construct (0× HRE, 1×HRE, 2 × HRE, or 3 × HRE3) shown in Fig. 5. After transfection, cells were incubated for 72 h with fresh media containing (A) no hormone or 340 nM of dihydrotestosterone (DHT), (B) no hormone or 100 nM dexamethasone (Dex), 340 nM DHT, or 100 nM of β-estradiol as indicated. (C) Cells were also transfected with 2×HRE in the presence or absence of mAR and incubated with or without DHT. Cells were harvested and heat-treated extracts were subjected to the nonchromatographic CAT assay described in Materials and Methods. CAT activities were normalized to total amount of protein (mg) and are expressed as average (dpm) of triplicate samples ± SEM.
To determine the hormone specificity of the regulation of tkl promoter in vitro, cells cotransfected with Ox HRE or 2xHRE constructs and mAR were treated with DHT, dexamethasone, or β-estradiol (Fig. 6B). Under these conditions, only DHT significantly elevated the CAT activity of CV1 cells transfected with 2×HRE target DNA. This result was expected because negligible endogenous glucocorticoid or estrogen receptors are present in CV1 cells. To establish that the positive regulation of CAT gene expression was dependent on the presence of androgen receptor, we cotransfected the cells with 2×HRE reporter gene construct in the presence or absence of exogenous receptor (Fig. 6C). Only cells cotransfected with both target DNA (2×HRE) and mAR expression vector activate promoter function, demonstrating that the androgenic effect on the HSV-tk1 promoter regulation conferred by Nat2*-HRE is entirely dependent on the presence of androgen receptor.
It is a well-studied event that the HRE, also known as glucocorticoid response element (GRE), can not only mediate induction of gene expression by androgen receptor but also by glucocorticoids, progesterone, and mineralocorticoid receptors (3). Therefore, we asked whether the palindromic HRE found in the Nat2* promoter might also elicit a response to glucocorticoids by cotransfection of CAT reporter constructs (Fig. 5) and rat glucocorticoid receptor into receptor-deficient CV1 cells. As expected, 2×HRE and 3×HRE3 constructs conferred 11- and 12-fold glucocorticoid response upon the HSV-tk1 promoter, respectively (Table 1). The effect of Nat2*-HRE on CAT expression is dependent on glucocortcoid hormone (dexamethasone) and glucocorticoid receptor (Table 1). Thus, the Nat2*-HRE confers receptor- and hormone-dependent positive regulation upon HSV-tk promoter function in vitro.
TABLE 1.
GLUCOCORTICOIDS RESPONSIVENESS OF THE Nat2*-HRE TRANSFECTED INTO CV1 CELLS
| CAT Constructs | rGR | Butylated Chloramphenicol (× 104 dpm) ± SEM | |||
|---|---|---|---|---|---|
| Hormone Treatment | |||||
| No Hormone | Dexamethasone | DHT | β-Estradiol | ||
| 0 × HRE | + | 0.63 ± 0.03 | 0.31 ± 0.08 | 0.52 ± 0.08 | 0.78 ± 0.07 |
| 1 × HRE | + | 8.70 ± 0.44 | 4.21 ± 0.30 | nd | nd |
| 2 × HRE | − | 6.42 ± 0.42 | 8.52 ± 0.76 | nd | nd |
| 2 × HRE | + | 3.86 ± 0.58 | 41.37 ± 2.94 | 3.98 ± 0.16 | 5.06 ± 0.51 |
| 3 × HRE3 | + | 9.41 ± 0.63 | 111.00 ± 5.27 | nd | nd |
Cells were cotransfected with 100 ng of rat glucocorticoid receptor (GR) and 2 μg of target DNA (0 × HRE, 1 × HRE, 2 × HRE, or 3 × HRE3). Subsequent treatment was for 72 h with no hormone, 340 nM DHT, 100 nM dexamethasone, or 100 nM β-estradiol. Cells were also transfected in the presence or absence of GR. CAT activities measured as the formation of butylated chloramphenicol were normalized to total amount of protein (mg) and are expressed as average (× 104 dpm) of triplicate samples ± SEM. nd, not determined.
DISCUSSION
This report describes the structural organization of the 5′ region of the mouse Nat2* gene and provides a preliminary functional analysis of the HRE activity. Multiple transcription initiation sites for the Nat2* gene were identified in mouse kidney and liver by primer extension analysis in the region spanning 112 to 151 bases upstream of the translational start site (ATG). The initiation site at position −135 (A) may be a potential major cap site as two distinct oligonucleotides yielded extended fragments, which identify this nucleotide as the cap site. Also, comparative analyses of 5′ UTR and promoter regions in an array of unrelated eukaryotic genes and mRNAs with precisely mapped 5′ ends have disclosed that there is a strong preference for purines at the cap site. This is especially true of adenosine, which occurs at mRNA 5′ ends with a frequency of 76–95% (6,15). It is also known that the bases on either side of the cap nucleotide are predominantly pyrimidines, preferentially a cytosine (5–7,15). In the case of mouse Nat2* the purine at position −135 is indeed adjacent to a pyrimidine (T). Although one of the advantages of the primer extension analysis is the mapping of the location of transcription initiation sites, the possibility of identifying false cap sites due to premature pauses by the reverse transcriptase is a limitation of this assay.
To exclude the possibility that the cap sites identified by the primer extension assay were merely artifacts, we performed a ribonuclease protection assay. The designed probe covered the region from −50 to −221, which contains the initiation sites previously identified. Multiple protected fragments were obtained by the RNase protection assay, confirming the primer extension results: the mouse Nat2* promoter contains multiple transcription initiation sites. Mapping the exact nucleotide location of the transcription initiation sites identified by ribonuclease protection assays is complicated because the sizes of the bands obtained do not coincide with the sizes predicted. Such discrepancies may be explained by the presence of a stem-and-loop secondary structure of the 5′ UTR region of mouse Nat2* as predicted by GCG version 7.0 (FOLD command) (14).
Examination of the promoter region of mouse Nat2*, a highly AT-rich region, revealed a typical eukaryotic gene consisting of four distinct transcriptional control elements as reviewed by Latchman (16). Multiple TATA boxes are present in the region spanning 100–450 bases upstream of the translation start site. Seventeen and 20 bases upstream of the transcription initiation sites identified by primer extension analysis at positions −112 and −134, respectively, we find typical consensus sequences of TATA boxes. Twenty to 30 bases is the preferred positional distribution of TATA boxes with respect to RNA polymerase II initiation sites in vertebrates (6). In addition, the following elements are located within the promoter region of mouse Nat2*: upstream promoter elements such as CAAT and Sp1 boxes, which are required for efficient transcription; regulatory elements such as HRE, which activate the gene in particular tissues or in response to particular stimuli; and enhancer elements such as AP-1, which increase gene activity in all tissues or in a regulated manner. We also discovered a variety of other regulatory elements in the 5′-flanking region of the mouse Nat2* by computer analysis. However, the element reported in this study (HRE) is of particular interest in view of its potential connection to mouse Nat2* tissue- and gender-specific gene expression and regulation during postnatal development. We investigated the potential role of the HRE found within the mouse Nat2* gene on the androgenic effect observed on the expression of this gene in mouse kidney during development. The results of this study demontrate that Nat2*-HRE can positively regulate the HSV-tk1 promoter in vitro. This activation of promoter function was shown to be dependent on the presence of both androgen receptor and hormone, which agrees with the recognized steroid molecular mechanism of action on gene regulation. The direct effect of the Nat2*-HRE and androgens on the native promoter and the regulation of Nat2* expression remains to be established.
Our laboratory has previously demonstrated a clear androgenic influence on development of PABA-NAT activity in kidney of male, which was not observed in female C57BL/6J and A/J inbred mice (27). These studies argue that androgens play a significant role in the regulation of mouse arylamine N-acetyltransferase 2 gene expression in kidney during development. Our more recent study (11) extended the finding to tissue- and gender-specific expression of Nat2 in CD1 outbred mouse strain. The fact that androgen regulation occurs in kidney but not mouse liver is an intriguing finding, because it may provide the opportunity to study interplay between an androgen response element and other tissue-specific elements in the promoter of mouse Nat2*.
A basic question of steroid hormone regulation is how specific transcription takes place in vivo when glucocorticoids, androgen, progesterone, and mineralocorticoid receptors recognize the same DNA sequence, HRE (1–3,24). The HRE consensus sequence, a palindrome of TGTTCT half-sites separated by three bases, is required for induction of gene expression by androgens; however, this simple motif is not sufficient to confer androgen specificity. The enhancer of the mouse sex-limited protein (Slp) gene, for example, is activated by androgens but not by glucocorticoids in Robins et al. (24). The specific induction of Slp gene by androgens requires both the core HRE and multiple auxiliary elements within 120 bp proximate to the HRE (1,2,24). In this report, we demonstrated the response of the Nat2*-HRE to androgens. As expected, the core Nat2*-HRE sequence also activates the HSV-tk1 promoter function in vitro upon glucocorticoid stimulation, an effect that is entirely dependent on the addition of exogenous glucocorticoid receptor. This finding may be significant to the study of NATs as several studies report the induction of liver N-acetylation activity by glucocorticoids in rats (30) and rabbits (23); however, the action of glucocorticoids on the expression of mouse Nat enzymes in vivo is yet to be investigated.
ACKNOWLEDGMENTS
The authors are very grateful to Dr. Diane M. Robins and laboratory members for providing the CV1 cells and the mAR, rGR, and 3 × HRE3 plasmids and for her valuable advice and expert assistance. We also wish to thank Dr. Audrey F. Seasholtz for helpful discussions and recommendations and Dr. Maria A. Burgos-Trinidad for the pUTKAT3 vector and the technical advice provided. This work was supported by NIH grants GM 44965 and CA 39018.
REFERENCES
- 1. Adler A. J.; Danielsen M.; Robins D. M. Androgen-specific gene activation via a consensus glucocorticoid response element is determined by interaction with nonreceptor factors. Proc. Natl. Acad. Sci. USA 89:11660–11663; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adler A. J.; Scheller A.; Hoffman Y.; Robins D. M. Multiple components of a complex androgen-dependent enhancer. Mol. Endocrinol. 5:1587–1596; 1991. [DOI] [PubMed] [Google Scholar]
- 3. Beato M. Regulation of gene expression by steroid hormones. In: Kahn P., et al. , eds. Oncogenes and growth control. New York: Springer-Verlag; 1986:219–225. [Google Scholar]
- 4. Blum M.; Grant D. M.; Demierre A.; Meyer U. A. N-acetylation pharmacogenetics: A gene deletion causes absence of arylamine N-acetyltransferase in liver of slow acetylator rabbits. Proc. Natl. Acad. Sci. USA 86:9554–9557; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Breathnach R.; Chambon P. Organization and expression of eucaryotic split genes coding for proteins. Annu. Rev. Biochem. 50:349–383; 1981. [DOI] [PubMed] [Google Scholar]
- 6. Bucher P. Weight matrix descriptions of four eukaryotic RNA polymerase II promoter elements derived from 502 unrelated promoter sequences. J. Mol. Biol. 212:563–578; 1990. [DOI] [PubMed] [Google Scholar]
- 7. Corden J.; Wasylyk B.; Buchwalder A.; Sassone-Corsi P.; Kedinger C.; Chambon P. Promoter sequences of eukaryotic protein-coding genes. Science 209:1406–1414; 1980. [DOI] [PubMed] [Google Scholar]
- 8. De Leon J. H.; Martell K. J.; Vatsis K. P.; Weber W. W. Slow acetylation in mice is caused by a labile and catalytically impaired mutant N-acetyltransferase (NAT2 9). Drug Metab. Dispos. 23:1354–1361; 1995. [PubMed] [Google Scholar]
- 9. Doll M. A.; Hein D. W. Cloning, sequencing and expression of NAT1 and NAT2 encoding genes from rapid and slow acetylator inbred rats. Pharmacogenetics 5:247–251; 1995. [DOI] [PubMed] [Google Scholar]
- 10. Doll M. A.; Jiang W.; Deitz A. C.; Rustan T. D.; Hein D. W. Identification of a novel allele at the human NAT1 acetyltransferase locus. Biochem. Biophys. Res. Commun. 233:584–591; 1997. [DOI] [PubMed] [Google Scholar]
- 11. Estrada-Rodgers L.; Levy G. N.; Weber W. W. Expression of arylamine N-acetyltransferase, NAT2, during development of CD1 mice. Pharmacologist 39:22; 1997. [Google Scholar]
- 12. Ferguson R. J.; Doll M. A.; Baumstark B. R.; Hein D. W. Polymorphic arylamine N-acetyltransferase encoding gene (NAT2) from homozygous rapid and slow acetylator congenic Syrian hamsters. Gene 140:247–249; 1994. [DOI] [PubMed] [Google Scholar]
- 13. Ferguson R. J.; Doll M. A.; Rustan T. D.; Baumstark B. R.; Hein D. W. Syrian hamster monomorphic N-acetyltransferase (NAT1) alleles: Amplification, cloning, sequencing, and expression in E. coli. . Pharmacogenetics 4:82–90; 1994. [PubMed] [Google Scholar]
- 14. Jbilo O.; Toutant J. P.; Vatsis K. P.; Chatonnet A.; Lockridge O. Promoter and transcription start site of human and rabbit butyrylcholinesterase genes. J. Biol. Chem. 269:20829–20837; 1994. [PubMed] [Google Scholar]
- 15. Kozak M. Compilation and analysis of sequences upstream from the translational start site in eukaryotic mRNAs. Nucleic Acids Res. 12:857–872; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Latchman D. S. Eukaryotic transcription factors. 2nd ed. San Diego: Academic Press Limited; 1995. [Google Scholar]
- 17. Lewin B. Genes V. 5th ed. New York: Oxford University Press Inc.; 1994. [Google Scholar]
- 18. Martell K. J.; Levy G. N.; Weber W. W. Cloned mouse N-acetyltransferases: Enzymatic properties of expressed Nat-1 and Nat-2 gene products. Mol. Pharmacol. 42:265–272; 1992. [PubMed] [Google Scholar]
- 19. Martell K. J.; Vatsis K. P.; Weber W. W. Molecular genetic basis of rapid and slow acetylation in mice. Mol. Pharmacol. 40:218–227; 1991. [PubMed] [Google Scholar]
- 20. Menendez-Pelaez A.; Marrufo B.; Reiter R. J.; Santana C.; Guerrero J. M. Testosterone increases N-acetyltransferase activity in both male and female Syrian hamster harderian glands. Biochem. Int. 19:115–121; 1989. [PubMed] [Google Scholar]
- 21. Nagata K.; Ozawa S.; Miyata M.; Shimada M.; Yamazoe Y.; Kato R. Primary structure and molecular basis of polymorphic appearance of an acetyltransferase (AT-II)* in hamsters. Pharmacogenetics 4:91–100; 1994. [DOI] [PubMed] [Google Scholar]
- 22. Prost E.; Moore D. D. CAT vectors for analysis of eukaryotic promoters and enhancers. Gene 45:107–111; 1986. [DOI] [PubMed] [Google Scholar]
- 23. Reeves P. T.; Minchin R. F.; Ilett K. F. In vivo mechanisms for the enhanced acetylation of sulfamethazine in the rabbit after hydrocortisone treatment. J. Pharmacol. Exp. Ther. 248:348–352; 1989. [PubMed] [Google Scholar]
- 24. Robins D. M.; Scheller A.; Adler A. J. Specific steroid response from a nonspecific DNA element. J. Steroid Biochem. Mol. Biol. 49:251–255; 1994. [DOI] [PubMed] [Google Scholar]
- 25. Sasaki Y.; Ohsako S.; Deguchi T. Molecular and genetic analyses of arylamine N-acetyltransferase polymorphism of rabbit liver. J. Biol. Chem. 266:13243–13250; 1991. [PubMed] [Google Scholar]
- 26. Seed B.; Sheen J. Y. A simple phase-extraction assay for chloramphenicol acyltransferase activity. Gene 67:271–277; 1988. [DOI] [PubMed] [Google Scholar]
- 27. Smolen T. N.; Brewer J. A.; Weber W. W. Testosterone modulation of N-acetylation in mouse kidney. J. Pharmacol. Exp. Ther. 264:854–858; 1993. [PubMed] [Google Scholar]
- 28. Vatsis K. P.; Weber W. W. Structural heterogeneity of Caucasian N-acetyltransferase at the NAT1 gene locus. Arch. Biochem. Biophys. 301:71–76; 1993. [DOI] [PubMed] [Google Scholar]
- 29. Vatsis K. P.; Weber W. W.; Bell D. A.; Dupret J. M.; Evans D. A.; Grant D. M.; Hein D. W.; Lin H. J.; Meyer U. A.; Relling M. V.; et al. Nomenclature for N-acetyltransferases. Pharmacogenetics 5:1–17; 1995. [DOI] [PubMed] [Google Scholar]
- 30. Zaher H.; Svensson C. K. Glucocorticoid induction of hepatic acetyl CoA:arylamine N-acetyltransferase activity in the rat. Res. Commun. Chem. Pathol. Pharmacol. 83:195–208; 1994. [PubMed] [Google Scholar]