

Abstract
Expression of vascular endothelial growth factor (VEGF), an angiogenic factor and endothelial cell-specific mitogen, is induced by hypoxia in various cell lines as well as in solid tumors. In this study, we report that cell density has a profound effect on the expression of VEGF in human glioblastoma cells (U87) and human fibrosarcoma cells (HT1080), an effect that is independent of hypoxia. Northern blot analysis revealed that VEGF mRNA levels were four- to eightfold higher in cells seeded at high density compared to cells seeded at low density. This upregulation of VEGF message in response to seeding at high density was not seen with other mRNAs such as those for TGF-β1 or GAPDH. Conditioned medium switch experiments between sparse and dense cells suggested that soluble factor(s) may not account for the observed changes in VEGF expression. Incubation with genistein, a protein tyrosine kinase inhibitor, for 3 h following seeding resulted in the reduction of the VEGF mRNA levels in highly confluent cultures but not in sparse cultures. To identify protein tyrosine kinases involved in the upregulation of the steady-state levels of VEGF mRNA in highly dense cultures, we analyzed the phosphorylation state of the c-src tyrosine kinase, in high versus low confluency cultures of U87 and HT1080 cells. Interestingly, an increased phosphorylation at Tyr416 of c-src was noted in high compared to low confluency, suggesting the activation of c-src in highly confluent cultures. Because extracellular signal-regulated kinases (ERKs) such as MAP kinase have been shown to be activated by extracellular stimuli and act downstream of c-src, we examined their possible involvement in this process. We found that the tyrosine phosphorylation level of MAP kinase is higher in dense compared to sparse cultures and, moreover, 6-thioguanine (6-TG), a potent inhibitor of ERKs, reduced VEGF mRNA levels in high but not low confluency. Furthermore, reintroduction of wild-type, but not mutant, von Hippel-Lindau (VHL) gene product in 786-0 cells (a renal carcinoma cell line) specifically abrogated the induction of VEGF mRNA due to high cell density. Taken together, these data suggest that VEGF gene expression is regulated by cell density, and the proto-oncogene c-src and the tumor-suppresser VHL are modulators of this regulation.
Keywords: VEGF, Cell density, Protein tyrosine phosphorylation, Cancer, Gene regulation, von Hippel-Lindau (VHL)
ANGIOGENESIS, the development of a micro-vascular bed, plays an important role in physiological as well as pathological conditions: in normal growth and development, in the female reproductive cycle, in wound healing, in diabetic retinopathy, in solid tumor growth, and in the expansion of metastatic colonies (1,8–10,21,25,26). New capillaries provide nourishment to a growing tumor and also allow the tumor cells to establish continuity with the vasculature of the host. Angiogenesis is a complex process, involving the interaction of cells, soluble factors, and extracellular matrix components (8). Several factors, both peptide and nonpeptide, have been shown to induce angiogenesis in vivo. Recently, an endothelial cell-specific mitogen, known as vascular endothelial growth factor (VEGF) or vascular permeability factor (VPF), has been characterized (2,17). Overexpression of VEGF by gene transfection in xenografted tumor lines enhances tumor growth and vascular density (28). Moreover, systemic administration of antibodies to VEGF has been shown to inhibit tumor growth in vivo (13). Furthermore, the dominant-negative mutant of Flk-1 (which is specifically expressed in endothelial cells and acts as a receptor for VEGF) can prevent tumor growth (20).
Hypoxia can induce VEGF in several cell types in vitro as well as in solid tumors leading to ischemia-mediated angiogenesis (23,24). Recently, we have characterized an intracellular signaling pathway by which hypoxia induces VEGF expression. In brief, we defined VEGF as a new downstream target for c-src and suggested a role for Src in promoting angiogenesis (22). During the course of these studies, we noted that cell density also regulates VEGF expression, which is independent of hypoxia-mediated signaling. Here, we describe how cell density influences VEGF expression and define part of the signaling pathway involved.
MATERIALS AND METHODS
Cell Culture
Human glioblastoma-astrocytoma cells U-87 MG (U87; ATCC HTB-14) and human fibrosarcoma cells (HT1080; ATCC CCL 121) were maintained in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum (HyClone Laboratories) and 1 mM sodium pyruvate (only for U87 cells). 786-0 renal carcinoma subclones stably transfected with pRC/CMV, pRC/CMV-VHL (wt-VHL), and pRC/CMV-VHL (1–115) (ΔVHL) plasmids were grown as described elsewhere (12). These cell lines are a generous gift from Dr. W. G. Kaelin. Cells were grown to 50–60% confluence, trypsinized, and seeded at different densities as described, and incubated for 3–4 h or as described. Cells were harvested for total cellular RNA samples or protein samples as necessary.
Northern Blot Analysis
RNA, isolated by the single-step acid phenol extraction method (4), was separated on a formaldehyde-agarose gel, transferred to GeneScreen (DuPont) membrane using 10 × SSC, and probed with random primer labeled cDNAs in a solution containing 0.5 M sodium phosphate (pH 7.2), 7% SDS, 1% bovine serum albumin (BSA), 1 mM EDTA, and sonicated herring sperm DNA (50 μg/ml) at 68°C. Blots were washed three times with a solution containing 40 mM sodium phosphate (pH 7.2), 0.5% SDS, 0.5% BSA, 1 mM EDTA at 68°C, and then exposed to Kodak X-OMAT film.
Western Blot Analysis
Cells were plated at high or low densities for different time points (as indicated). Cells were washed twice with 10 ml cold buffered saline (PBS), lysed with ice-cold lysis buffer (50 mM Tris, pH 7.5, 1% NP-40, 150 mM NaCl, 1 mM Na3VO4, 2 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 0.5% aprotinin, 2 mM pepstatin A), incubated for 10 min on ice, and centrifuged for 10 min at 4°C. Immunoprecipitations were carried out using 0.5 mg of total protein with c-src (Upstate Biotechnology Inc.) or ERK (Santa Cruz Co.) monoclonal antibody (4 μg) at antibody excess. Immunocomplex capture was accomplished by protein A agarose beads (BioRad) suspended in a buffered solution containing 200 mM HEPES (pH 7.0), 10% (v/v) glycerol, and 0.1% NP-40. Western blot analysis with antiphosphotyrosine 416 Src antibody, c-src monoclonal antibody, or antiphosphotyrosine monoclonal antibody (ICN) and ERK monoclonal antibody was done on split samples. The antiphosphotyrosine 416 Src antibody was a kind gift from Andy Laudano. To generate this antiserum, the peptide arginine-leucine-isoleucine-glutamic acid-aspartic acid-asparagine-glutamic acid-phosphotyrosine-threonine-alanine-arginine-glutamine-glycine-alanine-lysine (sequence derived from the Src catalytic domain) was coupled to bovine serum albumin and injected into rabbits. The resulting polyvalent serum was adsorbed sequentially with a nonphosphorylated version of the peptide and with free phosphotyrosine. Subsequently it was purified by binding to the above phosphorylated peptide using an Affigel column. This antibody reacts against Src protein phosphorylated on tyrosine 416 (22).
RESULTS AND DISCUSSION
Regulation of VEGF by Cell Density
It is known that hypoxia upregulates VEGF in solid tumors as well as in cell culture (23,24). Here we examined whether cell density can influence VEGF expression. U87 cells were grown in T-225 flasks at close to 90% confluent. The cells were then seeded at low (L) or high (H) densities as described in Materials and Methods and in the figure legends. Figure 1A shows that the higher density cells expressed sevenfold more VEGF mRNA as determined by Northern blot analysis compared to cells grown at lower density. To examine whether this effect was a result of a hypoxic microenvironment surrounding highly dense cells, we subjected both highly densed and sparse cultures to hypoxic conditions, in parallel. Figure 1a shows the effect of hypoxia on the expression of VEGF in high and low confluent cultures. Hypoxia produced an additional increase of the VEGF mRNA levels in highly confluent cultures, whereas the mRNA levels of GAPDH (Fig. 1a) or TGF-β1 (data not shown) were unaffected. These results suggest that the upregulation of VEGF mRNA levels in densed cultures is independent of hypoxia. Next we examined the expression of VEGF mRNA at various cell densities. Figure 1b shows that VEGF message was cell density dependent. By increasing the number of cells plated, VEGF expression levels increased in a sigmoidal fashion, with a major inflection occurring between 6 and 30 × 105 cells per 60-mm dish and corresponding visually to tight packing of cells at the higher density. These data suggest that different levels of cell-cell contact (high versus low con-fluency) may affect VEGF mRNA levels.
FIG. 1.
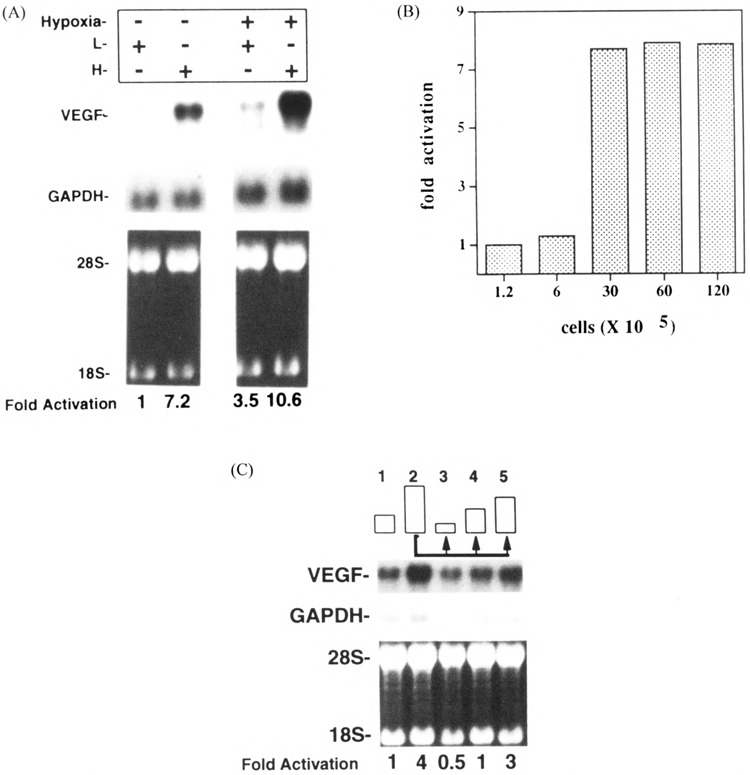
Effect of cell density on the steady-state levels of VEGF mRNA. (A) Total RNA (5 μg) was extracted from low confluency (L, 5 × 105 cells in a 100-mm TC plate) and high confluency (H, 3 × 106 cells in a 60-mm TC plate) U87 cells incubated in normoxic or hypoxic (95% N2 and 5% CO2) conditions (3 h). The blot was hybridized to human VEGF and GAPDH (for normalization) cDNAs. Lower panels show ethidium bromide-stained RNA samples before transfer; ribosomal RNA markers (18S and 28S) are indicated. (B) U87 cells were seeded to different densities in 60-mm plates. Cells were grown for 3 h and then total RNA was extracted and VEGF mRNA levels were determined by Northern blotting. Total RNA (5 μg) was loaded per lane, transferred to nylon membranes, and hybridized to VEGF or GAPDH cDNA probes. Autoradiograms were quantified by scanning densitometry and data were presented as fold activation over levels of VEGF mRNA measured at the lowest confluency. (C) U87 cells were seeded at low (4 × 105 cells/60-mm plate) or high (H, 3 × 106 cells/60-mm plate), incubated for 3 h, and total RNA was extracted and probed with a 32P-labeled VEGF probe by Northern analysis (lanes 1 and 2). Lanes 3–5 represent the results obtained by Northern analysis of RNA extracted from 2 × 105, 4 × 105, or 1.5 × 106 cells/60-mm plate, respectively. All three subcultures were derived from a highly confluent culture (H, 3 × 106 cells/60-mm plate) similar to the one represented in lane 2. Similar results were obtained in two independent experiments.
To examine whether the effect of cell density on the steady-state levels of VEGF mRNA was reversible, highly densed cell cultures (3 × 106 cells/60-mm plate) were split 3 h following seeding, to different cell densities (2 and 4 × 105 and 1.5 × 106 cells/60-mm plate). These subcultures were grown for 3 h, cells were harvested, and VEGF mRNA levels were assayed by Northern blotting. Figure 1c shows that the level of VEGF mRNA seen at 3 × 106 cells/60-mm plate was reduced to 12.5% when these cells were split and then seeded at 2 × 105/60-mm plate (Fig. 1c, lanes 2 and 3). However, VEGF mRNA level was linearly increased as increasing number of cells were plated (Fig. 1c, lanes 3–5). These results show that the regulation of VEGF expression by cell density is reversible and may not involve long-term changes of cellular phenotypes.
Possible Mechanisms of the Cell Density Effect
To investigate the mechanisms by which cell density affects VEGF gene expression, we considered three distinct possibilities. First, soluble factors) may be differentially secreted in high versus low confluency. Second, placing of cells at high versus low confluency may result in changes of cell shape that could be responsible for the different VEGF mRNA levels seen in high versus low confluent cultures and, finally, cell-cell contact directly or indirectly may regulate VEGF message. A direct regulation of VEGF by cell-cell contact may be a result of the activation of a signaling pathway triggered by a cell adhesion molecule. An indirect regulation could occur when cells packed together and cell surface molecules come in close proximity. Such ligand-receptor interactions may signal VEGF mRNA upregulation. To test the first hypothesis, we added conditioned medium from higher density cells (following a 3-h incubation) to lower density cells and vice versa. Figure 2 shows that there was no significant change in VEGF expression levels as a result of the conditioned medium switch, suggesting that soluble factors) were not sufficient to account for the cell density effect. To examine whether changes in cell shape were responsible for the regulation of VEGF mRNA levels in high versus low confluency, we seeded the cells in bacterial dishes versus tissue culture dishes at low density. By plating cells onto bacterial plates, cells assume a spherical morphology that is distinct from the regular shape of adherent cells grown as monolayers. No significant changes of VEGF mRNA levels were seen when U87 or HT1080 cells were plated either on bacterial or tissue culture dishes (Fig. 3).
FIG. 2.
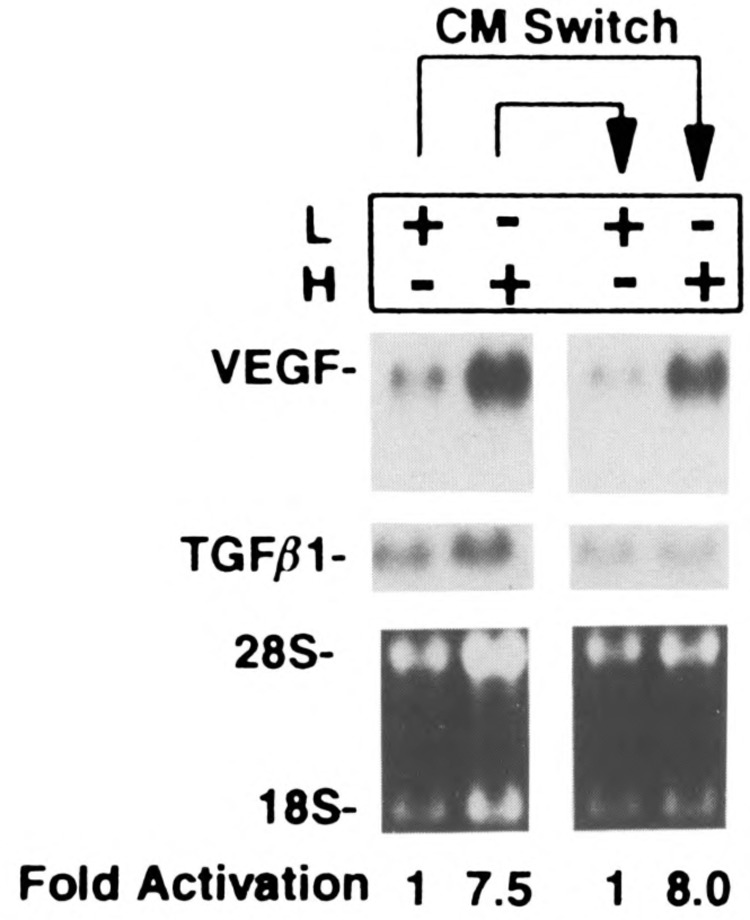
Conditioned medium switch experiment. U87 cells were seeded at low confluence (L, 5 × 105 cells in a 100-mm TC plate) or high confluence (H, 3 × 106 cells in 60-mm TC plate), incubated for 3 h; total RNA was extracted and analyzed by Northern blot (lanes 1 and 2). Conditioned media (3 h) from these cells were added to a new set of L and H plates. The L plate received conditioned medium from the previously harvested H culture and vice versa. L and H plates were incubated with the conditioned media for 3 h and levels of VEGF mRNA were determined by Northern blotting. The blot was hybridized to VEGF and TGF-β1 (for normalization) cDNAs, and quantified as described previously.
FIG. 3.
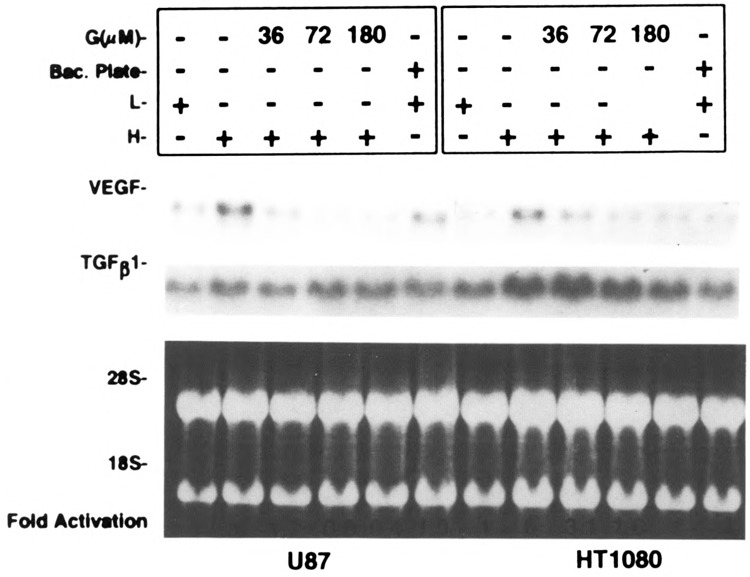
Effect of a bacterial plate and a tyrosine kinase inhibitor on cell density-mediated VEGF expression. U87 and HT1080 cells were seeded at low and high confluences as described. The cells at lower confluence were plated on a bacterial plate to assess the role of cell shape in VEGF induction. Different amounts of genistein were added to high confluence cells for 3 h. Total RNA were extracted and analyzed by Northern blot hybridization. Blots were hybridized to VEGF and TGF-β1 cDNAs and quantified as described. The results shown here were repeated and similar data obtained.
Because the production and secretion of soluble factors and/or changes of cell shape may be considered long-term effects, our results—showing that soluble factors or changes of cell shape are not sufficient to account for increased VEGF mRNA in high density cultures—are in good agreement with our previous data showing that the cell density effect on VEGF mRNA is reversible and not the result of long-term changes of cellular phenotypes. However, the possibility of a combined effect of cell shape, soluble factor(s), and cell-cell contact cannot be excluded.
Involvement of Protein Kinases
To dissect the signaling events responsible for high cell density-mediated VEGF induction, we utilized genistein, a tyrosine kinase-specific inhibitor. Figure 3 shows that, in both U87 cells and HT1080 cells, genistein blocked VEGF upregulation seen at high cell density in a dose-dependent manner. Our previous studies have shown hypoxia induces c-src activation, which results in VEGF induction, and this c-src activation by hypoxia appeared to correlate with an increased phosphorylation of Tyr416. Thus, we used a specific antibody that recognizes phosphorylated Tyr416 to compare the phosphorylation status of Tyr416 in dense versus sparse cultures. Figure 4 shows that the phosphorylation at Tyr416 is increased in high compared to low density cultures without a significant increase Src protein. Because there is a correlation between the phosphorylation of the 416Y residue of c-src and c-src activation, these data indicate a role for c-src in high cell density-mediated VEGF induction.
FIG. 4.
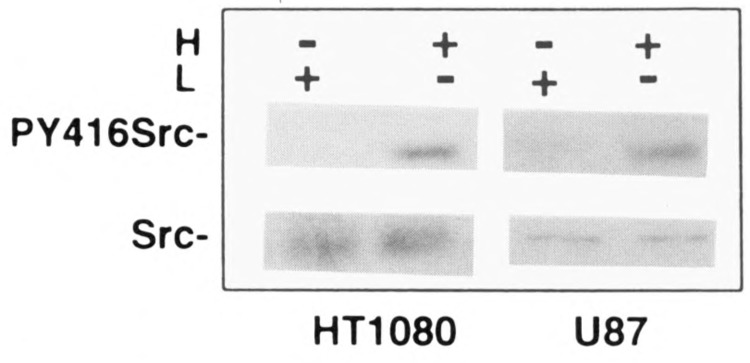
Phosphorylation of Src 416Y by cell density. U87 cells and HT1080 cells were seeded at low and high cell density as described. After 3 h cells were lysed with cell (VPS) as described in Materials and Methods. Western blot analysis with antiphosphotyrosine 416 Src antibody and c-src monoclonal antibody as determined on split samples.
ERKs belong to a family of kinases that have been shown to respond to a variety of extracellular stimuli and may act downstream of Src. Because our studies suggest a potential activation of c-src in high cell density, we further investigated whether ERKs have a role in this signaling cascade. First, we examine the tyrosine phosphorylation status of MAP kinase, a member of the ERK family, in high versus low confluency. MAP kinase was immunoprecipitated from dense or sparse cultures of U87 cells and then its phosphorylation status was determined by immunoblotting using a monoclonal antiphosphotyrosine antibody. Figure 5a shows an increase in the tyrosine phosphorlyation level of MAP kinase in high compared to low confluency cells extracts. This increase in the tyrosine phosphorylation level does not reflect an increase at the protein level (Fig. 5A, lower panel). To correlate this potential activation of ERKs by high confluency with the upregulation of VEGF observed in high confluency, we used 6-thioguanine (6TG), a potent inhibitor of ERKs. Incubation of high confluency cultures of U87 cells with 100 μM 6TG for 3 h resulted in 70% reduction of VEGF induction, which suggests a potential role of ERKs in this signaling pathway (Fig. 5B).
FIG. 5.
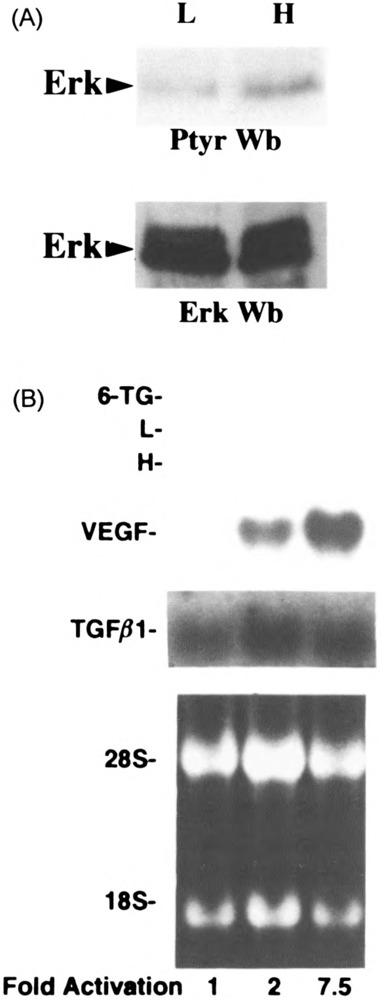
Effect of ERKs in cell density-mediated VEGF induction. U87 cells were seeded at low (L, 5 × 105 cells in a 100-mm TC plate) and high (H, 3 × 106 cells in a 60-mm TC plate) cell densities. (a) Upper panel: ERK was immunoprecipated as described in Materials and Methods, and protein tyrosine phosphorylation was evaluated by antiphosphotyrosine immunoblotting. Lower panel: Western blot analysis with ERK antibody as determined by split samples. (b) Total RNA (5 μg per lane) was extracted from the low, high, and high confluence cells treated with 6-thioguanine (100 μM) for 3 h, the blot hybridized with VEGF and TGF-β1 cDNAs, and quantified as described.
Recently, it has been shown that germline mutations of the von Hippel-Lindau (VHL) gene suppresser exhibit a heredity cancer syndrome, which is characterized by the development of highly vascularized tumors such as renal cell carcinomas (RCC), hemangioblastomas, and pheochromocytomas (3,5,11,15,19). These highly vascularized tumors overproduce the angiogenic cytokine, VEGF (27). It is also noteworthy to mention that the localization of VHL gene product appears to be at the nuclear versus the cytoplasm depending upon cell density (16). Because the VEGF mRNA level is upregulated in high cell density, where VHL is expected to be mainly accumulated in the cytoplasm, it tempted us to examine the status of VEGF expression in the wt-VHL-containing cell line at higher cell density. For this purpose, we utilized 786-O cell lines, which are stably transfected with wt-VHL, mutant VHL (a naturally occurring deletion mutation), and empty vector cassette, and seeded those cell lines in low and high cell density as described earlier. Northern blot analysis was performed using VEGF cDNA as a probe and fold activation was measured by densitometry using GAPDH mRNA expression as a normalization control. Interestingly, the cell line containing wt-VHL transfectants could not induce VEGF mRNA level due to high cell density, whereas, neo-cassette and mutant VHL-containing cell lines clearly showed seven- to eightfold induction of VEGF mRNA (Fig. 6). These data suggest VHL is one of the key components of high cell density-mediated VEGF induction in RCC cell lines. Of importance, VHL could not block VEGF induction due to hypoxia (data not shown).
FIG. 6.
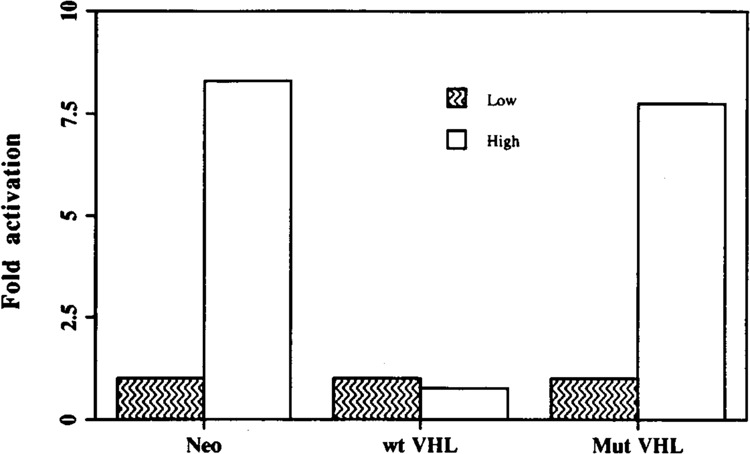
Wild-type VHL gene product inhibits high cell density-mediated VEGF induction in stably transfected renal cell carcinoma lines. RCC lines were grown in low (5 × 105 cells in a 100-mm TC plate) and high (3 × 106 cells in a 60-mm TC plate) cell densities. Total RNA (5 μg) were extracted from the cells and Northern blot analysis was done using VEGF and GAPDH cDNAs as probes. Fold activation over levels of VEGF mRNA measured at low confluency was assayed by scanning densitometry using GAPDH expression as a normalization control. The results shown here were repeated an additional time with similar data.
In large solid tumors, hypoxia-mediated VEGF expression plays a critical role in promoting angiogenesis (23,24). It is not yet clear how angiogenesis is induced in smaller tumors or early metastatic colonies where hypoxia is less likely to be the critical instigating event. Tumors less than 1–2 mm in diameter can receive nutrients by diffusion but further growth depends on an adequate blood supply established through angiogenesis (8–10). Of importance, it has been shown recently that human colon cancer cells with metastatic potential expressed higher levels of VEGF compared to nonmetastatic cells (6). The cell density effect on VEGF expression described here may serve as a model system for angiogenesis in a “small” meta-static colony that occurs in the absence of hypoxia (7,18). Moreover, because the cell density and hypoxia effects on VEGF expression are additive, it is possible that the cell density effect also plays a role in larger tumors, where cell density may vary somewhat within the tumor mass.
Note: In the process of revision of this manuscript, Koura et al. also showed that regulation of VEGF expression is cell density dependent (14).
ACKNOWLEDGMENTS
We thank J. Abraham for the VEGF cDNA, A. Laudano for rabbit polyvalent serum against Src phospho Y416, and S. Bossone and A. G. Nicolson for critical reading of the manuscript. This work was supported in part by NIH DK44921 to V.P.S.
REFERENCES
- 1. Adamis A. P.; Miller J. W.; Bernal M. T.; D’Amico D. J.; Folkman J.; Yeo T. K.; Yeo K. T. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am. J. Ophthalmol. 118:445–450; 1994. [DOI] [PubMed] [Google Scholar]
- 2. Berse B.; Brown L. F.; Van De Water L.; Dvorak H. F.; Senger D. R. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol. Biol. Cell 3:211–220; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen F.; Kishida T.; Duh F-M.; Renbaum P.; Orcutt M. L.; Schmidt L.; Zbar B. Suppression of growth of renal carcinoma cells by the von Hippel-Lindau tumor suppressor gene. Cancer Res. 55:4804–4807; 1995. [PubMed] [Google Scholar]
- 4. Chomczynski P.; Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocynatephenol-chloroform extraction. Anal. Biochem. 162:156–159; 1987. [DOI] [PubMed] [Google Scholar]
- 5. Crossey P. A.; Richards F. M.; Foster K.; Green J. S.; Prowse A.; Latif F.; Lerman M. I.; Zbar B.; Affara N. A.; Ferguson-Smith M. A.; Maher E. R. Identification of intragenic mutations in the von Hippel-Lindau disease tumor suppressor gene and correlation with disease phenotype. Hum. Mol. Genet. 3:1303–1308; 1994. [DOI] [PubMed] [Google Scholar]
- 6. Ellis L. M.; Liu W. Vascular endothelial growth factor (VEGF) expression and alternate splice in non-metastatic and metastatic human colon cancer cell lines. Proc. Ann. Meet. Am. Cancer Res. 36:A524; 1995. [Google Scholar]
- 7. Fidler I. J.; Ellis L. M. The implications of angiogenesis for the biology and therapy of cancer metastasis. Cell 79:185–188; 1994. [DOI] [PubMed] [Google Scholar]
- 8. Folkman J. Fighting cancer by attacking its blood supply. Sci. Am. 275:150–4; 1996. [DOI] [PubMed] [Google Scholar]
- 9. Folkman J. The role of angiogenesis in tumor growth. Semin. Cancer Biol. 3:65–71; 1992. [PubMed] [Google Scholar]
- 10. Folkman J.; Shing Y. Angiogenesis. J. Biol. Chem. 267:10931–10934; 1992. [PubMed] [Google Scholar]
- 11. Gnarra J. R.; Tory K.; Weng Y.; Schmidt L.; Wei M. H.; Li H.; Latif F.; Liu S.; Chen F.; Duh F-M.; Lubensky I.; Duan D. R.; Florence C.; Pozzatti R.; Walther M. M.; Bander N. H.; Grossman H. B.; Brauch H.; Pomer S.; Brooks J. D.; Isaacs W. B.; Lerman M. I.; Zbar B.; Linehan W. M. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nature Genet. 7:85–90; 1994. [DOI] [PubMed] [Google Scholar]
- 12. Kibel A.; Iliopoulos O.; DeCaprio J. A.; Kaelin W. G. J. Binding of the von Hippel-Lindau tumor suppressor protein to elongin B and C. Science 269:1444–1446; 1995. [DOI] [PubMed] [Google Scholar]
- 13. Kim K. J.; Li B.; Winer J.; Armanini M.; Gillett N.; Phillips H. S.; Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 362:841–844; 1993. [DOI] [PubMed] [Google Scholar]
- 14. Koura A. N.; Liu W.; Kitadai Y.; Singh R. K.; Radinsky R.; Ellis L. M. Regulation of vascular endothelial growth factor expression in human colon carcinoma cells by cell density. Cancer Res. 56:3891–3894; 1996. [PubMed] [Google Scholar]
- 15. Latif F.; Tory K.; Gnarra J.; Yao M.; Duh F-M.; Orcutt M. L.; Stackhouse T.; Kuzmin I.; Modi W.; Geil L.; Schmidt L.; Zhou F.; Li H.; Wei M. H.; Chen F.; Glenn G.; Choyke P.; Walther M. M.; Weng Y.; Duan D-S. R.; Dean M.; Glavac D.; Richards F. M.; Crossey P. A.; Ferguson-Smith M. A.; Paslier D. L.; Chumakov L.; Cohen D.; Chinault A. C.; Maher E. R.; Linehan W. M.; Zbar B.; Lerman M. I. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260:1317–1320; 1993. [DOI] [PubMed] [Google Scholar]
- 16. Lee S.; Chen D. Y. T.; Humphrey J. S.; Gnarra J. R.; Linehan W. M.; Klausner R. D. Nuclear/cytoplasmic localization of the von Hippel-Lindau tumor suppressor gene product is determined by cell density. Proc. Natl. Acad. Sci. USA 93:1770–1775; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leung D. W.; Cachianes G.; Kuang W-J.; Goeddel D. V.; Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246:1306–1309; 1989. [DOI] [PubMed] [Google Scholar]
- 18. Liotta L. A.; Steeg P. S.; Stetler-Stevenson W. G. Cancer metastasis and angiogenesis: An imbalance of positive and negative regulation. Cell 64:327–336; 1991. [DOI] [PubMed] [Google Scholar]
- 19. Maher E. R.; Yates J. R.; Harries R.; Benjamin C.; Harris R.; Moore A. T.; Ferguson-Smith M. A. Clinical features and natural history of von Hippel-Lindau disease. Q. J. Med. 77:1151–1163; 1990. [DOI] [PubMed] [Google Scholar]
- 20. Millauer B.; Shawver L.; Plate K.; Risau W.; Ullrich A. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature 367:576–579; 1994. [DOI] [PubMed] [Google Scholar]
- 21. Miller J. W.; Adamis A. P.; Shima D. T.; D’Amore P. A.; Moulton R. S.; O’Reilly M. S.; Folkman J.; Dvorak H. F.; Brown L. F.; Berse B.; Yeo T-K.; Yeo K-T. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am. J. Pathol. 145:574–584; 1994. [PMC free article] [PubMed] [Google Scholar]
- 22. Mukhopadhyay D.; Tsiokas L.; Zhou X. M.; Foster D.; Brugge J. S.; Sukhatme V. P. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature 375:577–81; 1995. [DOI] [PubMed] [Google Scholar]
- 23. Plate K. H.; Breier G.; Weich H. T.; Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo . Nature 359:845–848; 1992. [DOI] [PubMed] [Google Scholar]
- 24. Shweiki D.; Itin A.; Soffer D.; Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 359:843–845; 1992. [DOI] [PubMed] [Google Scholar]
- 25. Weidner N.; Carroll P. R.; Flax J.; Blumenfeld W.; Folkman J. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am. J. Pathol. 143:401–409; 1993. [PMC free article] [PubMed] [Google Scholar]
- 26. Weidner N.; Semple J. P.; Welch W. R.; Folkman J. Tumor angiogenesis and metastasis—correlation in invasive breast carcinoma. N. Engl. J. Med. 324:1–8; 1991. [DOI] [PubMed] [Google Scholar]
- 27. Wizigmann-Voos S.; Breier G.; Risau W.; Plate K. H. Up-regulation of vascular endothelial growth factor and its receptors in von Hippel-Lindau disease-associated and sporadic hemangioblastomas. Cancer Res. 55:1358–1364; 1995. [PubMed] [Google Scholar]
- 28. Zhang H. T.; Craft P.; Scott P. A.; Ziche M.; Weich H. A.; Harris A. L. Enhancement of tumor growth and vascular density by transfection of vascular endothelial cell growth factor into MCF-7 human breast carcinoma cells. J. Natl. Cancer Inst. 87:213–219; 1995. [DOI] [PubMed] [Google Scholar]