

Abstract
Recently, we demonstrated that the natural cytokinin nucleosides N6-isopentenyladenosine (iPR) and N6-benzyladenosine (BAPR) exert a potent and selective antiviral effect on the replication of human enterovirus 71. In order to further characterize the antiviral profile of this class of compounds, we generated a series of fluorinated derivatives of BAPR and evaluated their activity on the replication of human enterovirus 71 in a cytopathic effect (CPE) reduction assay. The monofluorination of the BAPR-phenyl group changed the selectivity index (SI) slightly because of the concomitant high cell toxicity. Interestingly, the incorporation of a second fluorine atom resulted in a dramatic improvement of selectivity. Moreover, N6-trifluoromethylbenzyladenosine derivatives (9–11) exhibited also a very interesting profile, with low cytotoxicity observed. In particular, the analogue N6-(3-trifluoromethylbenzyl)-adenosine (10) with a four-fold gain in potency as compared to BAPR and the best SI in the class represents a promising candidate for further development.
Keywords: fluorinated N6-benzyladenosines, synthesis and antiviral activity, SAR, enterovirus 71
1. Introduction
For many years, natural products (NPs) have been a leading source for the majority of the approved drugs, and their structures are a valuable source of inspiration for medicinal chemists [1]. Interestingly, only 36% of the new chemical entities discovered between 1981 and 2010 were developed without inspiration from a natural product [2].
Among natural products, the development of nucleosides is by far the most fruitful field of investigation. About one hundred drugs derive from nucleoside structures: the vast majority of them were developed as antiviral drugs, and a consistent proportion as antitumor drugs. Natural nucleosides are isolated from DNA, RNA, nucleotides, and coenzymes of various natural sources. Nowadays, the nucleoside library consists of about 550 compounds, and is a promising pool for the development of new biologically active compounds [3,4,5].
N6-Modified purine nucleosides (cytokinin nucleosides) are an important group of biologically active natural compounds with a unique spectrum of biological activities [3]. Cytokinin nucleosides contain a hydrophilic ribofuranose moiety and a purine heterocyclic scaffold modified with a hydrophobic residue at the N6 position. tRNA contains N6-isopentenyladenosine and some related nucleosides [6,7]. N6-Substituted adenosines are naturally present in plants [8,9,10] and bacteria [11].
In 2008, Arita and co-workers found that N6-benzyladenosine (BAPR) exhibited a pronounced antiviral activity against the replication of human enterovirus 71 (EV71) [12]. EV71 is a non-enveloped, single-stranded, positive-sense RNA virus belonging to the Enterovirus genus within the Picornaviridae family. EV71 commonly causes hand-, foot-, and mouth disease (HFMD), a mild and self-limiting illness mostly affecting children under the age of five. In some patients, EV71 has been associated with severe neurological complications including encephalitis, aseptic meningitis, and acute flaccid paralysis [13,14,15]. EV71 is prevalent worldwide, but most of the large outbreaks of neurotropic EV71 have occurred in the Pacific-Asia area [15,16,17]. However, in recent years, such epidemic tracts have been reported also in America and in Europe [15,17]. The World Health Organization has placed EV71 as one of the next biggest worldwide threats to public health, especially to young children, due to the lack of effective antiviral treatments [18,19].
Recently, we showed that, similarly to BAPR, two other naturally occurring plant cytokinin nucleosides, namely N6-isopentenyladenosine and N6-furfuryladenosine (kinetin riboside), possessed a potent and selective antiviral effect on EV71 [20,21]. Unfortunately, these compounds were rather cytotoxic, with CC50 values in the low micromolar range (4–8 μM). We were able to improve the selectivity of this group of compounds by modifying the size and the nature of the linker. In particular, a modified BAPR with a two-to-three atom-long linker had a very pronounced antiviral activity, and a 50-fold improvement of the selectivity index (SI) as result of a lower cytotoxicity [21].
The introduction of fluorine in order to improve the pharmacological properties of a drug is a modern trend in medicinal chemistry. Currently, there are about 200 fluorinated drugs on the market (~20% of all pharmaceuticals), with even higher figures for agrochemicals (up to 30%) [22,23]. Therefore, in the present study, we report on the modification of natural BAPR by the substitution in the phenyl ring with fluoro-, difluoro-, and trifluorometyl groups to evaluate the eventual improvement in the antiviral profile of these fluorinated compounds in the context of EV71 replication (Figure 1).
Figure 1.

Strategy of modification of natural cytokinin nucleoside BAPR.
2. Results and Discussion
2.1. Chemistry
Recently, we have developed a new useful and versatile approach for the preparation of N6-modified adenosine derivatives by the regioselective N6-alkylation of N6-acetyl-2′,3′,5′-tri-O-acetyladenosine with alcohols under Mitsunobu reaction conditions or with alkyl halides promoted by a base [20,21,24,25]. The main advantage of our method is the possibility to use both alkyl halides and alcohols for N6-modification. This is important, especially in the case when an amine is not stable or is hardly available. Using this methodology, several hundred N6-substituted adenosines have been synthesized in one of our laboratories.
The traditional approach for the preparation of N6-alkylated or N6-arylated adenosines is the substitution of the chlorine atom in commercially available 6-Chloropurine riboside with alkyl- or arylamines [26,27]. 6-hloropurine riboside can be readily prepared by the deacetylation of 2′,3′,5′-tri-O-acetyl-6-chloropurine riboside [28].
To simplify the separation procedure, we used 2′,3′,5′-tri-O-acetyl-6-chloropurine riboside directly in the substitution reactions. The acetyl groups are completely preserved in the reaction with aniline, and the protected intermediate can be isolated by silica gel chromatography and characterized. After the removal of the acetyl groups by ammonolysis, N6-phenyladenosine was obtained in overall high yield [21]. On the other hand, the reaction of 2′,3′,5′-tri-O-acetyl-6-chloropurine riboside with benzylamines was accompanied by the formation of by-products due to the partial removal of the protective groups, which complicated the chromatographic control of the reaction, and required a large excess of amines for the full conversion of the starting compound. Therefore, we decided to study the stability of different O-acyl groups to select the one optimal for our purposes. The results of the O-deacylation experiments of 2′,3′,5′-tri-O-acylinosine are summarized in Table 1.
Table 1.
Stability of O-acyl protecting groups under different deblocking conditions.
| Substrate | O-Deacylation Conditions, 20 °C a | t½. h | Complete O-Deacylation, h |
|---|---|---|---|
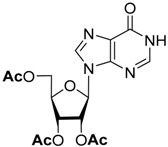 |
CH3NH2/C2H5OH (4M) | 0.25 | 2 |
| NH3/MeOH (4M) | 1 | 5 | |
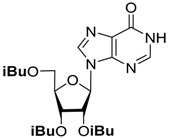 |
CH3NH2/C2H5OH (4M) | 3 | 15 |
| NH3/MeOH (4M) | 15 | 75 | |
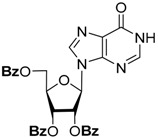 |
CH3NH2/C2H5OH (4M) | 6 | 26 |
| NH3/MeOH (4M) | 19 | 96 |
a The reagent was used in at least 400-fold excess.
According to the data in Table 1, the acetyl group is rather labile under basic conditions, and the benzoyl group is the most stable. The properties of the iso-butyroyl group exhibited the best behavior for our aims, since it is fairly resistant to the action of benzylamines, and its synthesis is more straightforward than that of the benzoyl derivatives. The compound 2′,3′,5′-tri-O-isobutyroyl-6-chloropurine riboside (4) has then been used as the starting substrate in the reactions with a small excess of benzylamines with fluoro- and trifluoromethyl groups (Scheme 1). The protective groups were removed in the presence of MeNH2/EtOH at room temperature with the subsequent chromatographic purification of the resulting products. Compounds 5–11 were obtained in overall good yield (50–98%). It should be mentioned that some of these compounds were previously prepared starting from 6-chloropurine riboside [26].
Scheme 1.

Synthesis of N6-alkyladenosines by the substitution of the chlorine atom in 2′,3′,5′-tri-O-isobutyroyl-6-chloropurineriboside. Reagents and conditions: (i) RNH2, DIPEA, MeCN, 70 °C, 10–24 h; (ii) MeNH2/EtOH, room temperature., 24 h, 50–98% (overall yields); (The structure of R is given in Table 1).
The structure of the obtained compounds was confirmed by NMR and mass spectroscopy. The presence of fluorine atoms in the phenyl residue was confirmed by spin-spin coupling constants between 19F and 1H in 1H-NMR spectra (JH-F) and between 19F and 13C in 13C-NMR spectra (JC-F). The 1H-NMR spectra of the fluorinated N6-benzyladenosine analogues (5–8) in the low field region were complicated by the presence of 19F-1H couplings: 3JH-F–8.0–9.0 Hz, 4JH-F–6.7–5.5 Hz, and 5JH-F ˂ 2.0 Hz. In the 13C-NMR spectra, three types of coupling constants JC-F were present, which are characteristic of fluorinated aromatic compounds [29]: 1JC-F–240–248 Hz, 2JC-F–12–24 Hz, and 3JC-F–7.5 Hz. The presence of trifluoromethyl residue in nucleosidic derivatives (9–11) was confirmed by low-intensive quartet with a coupling constant of ~30 Hz in 13C-NMR spectra. This constant was consistent with the literature data for trifluoromethylated aromatic compounds [29]. Despite the majority of the synthesized compounds having been characterized by NMR earlier, their detailed analysis and the assignment of all chemical shifts and coupling constants has not been presented. Therefore, we provided in the Supplementary section a detailed NMR analysis for each analogue produced.
2.2. Biological Activity on EV71 and Structure-Activity Relationship (SAR)
We have shown earlier that three natural cytokinin nucleosides (compound 1–3) exerted a potent antiviral effect on the replication of EV71 with an EC50 of 0.3–1.4 μM, but exhibited also a rather high cytotoxicity [20,21] (Table 2). As previously mentioned, modifications of the N6-substituent (linker) of the BAPR scaffold led to a remarkable increase of selectivity [21]. Here, we produced a series of BAPR analogues to evaluate the effect of the fluorination of BAPR on the replication of EV71. A cytopathic effect (CPE) reduction assay was performed with the newly synthetized analogues (compounds 5–11) in rhabdomyosarcoma (RD) cells. BAPR, N6-isopenthenyladenosine, and N6-furfuryladenosine were included in the screening, and the toxicity of all of the aforementioned compounds was evaluated in parallel on treated-uninfected cells.
Table 2.
Antiviral effect of N6-substituted adenosines on the replication of the EV71 strain BrCr in RD cells.
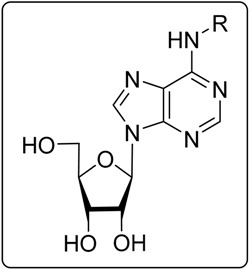
| No. | Compound Name | Substituent (R) | CC50 ± SD a,b | EC50 ± SD a,b | SI с |
|---|---|---|---|---|---|
| 1 |
N6-benzyladenosine (BAPR) |
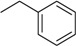 |
4.3 ± 1.6 | 0.28 ± 0.05 | 15 |
| 2 |
N6-isopenthenyladenosine (iPR) |
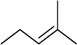 |
6.0 ± 0.6 | 1.0 ± 0.2 | 6.0 |
| 3 |
N6-furfuryladenosine (KINR) |
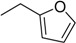 |
7.8 ± 3.4 | 1.4 ± 0.3 | 5.6 |
| 5 | N6-(2-fluorobenzyl) adenosine | 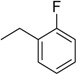 |
13.3 ± 3.7 | 0.30 ± 0.05 | 44 |
| 6 | N6-(3-fluorobenzyl) adenosine | 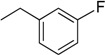 |
6.2 ± 1.8 | 0.24 ± 0.09 | 26 |
| 7 | N6-(4-fluorobenzyl) adenosine | 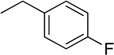 |
2.7 ± 0.9 | 0.14 ± 0.05 | 19 |
| 8 | N6-(2,6-difluorobenzyl) adenosine | 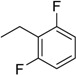 |
>254 | 0.21 ± 0.01 | >1210 |
| 9 | N6-(2-trifluoromethylbenzyl) adenosine | 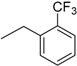 |
>235 | 1.0 ± 0.1 | >235 |
| 10 | N6-(3-trifluoromethylbenzyl) adenosine | 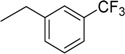 |
>235 | 0.068 ± 0.001 | >3456 |
| 11 | N6-(4-trifluoromethylbenzyl) adenosine | 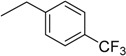 |
>235 | 1.0 ± 0.1 | >235 |
a All values are in μM and are based on at least three independent dose-response curves; b On rhabdomyosarcoma (RD) cells; c Selectivity Index (SI); SI = CC50/EC50; SD, standard deviation.
Overall, the incorporation of fluoro- and trifluoromethyl groups significantly improved the selectivity index of BAPR (Table 2). In particular, the monofluorination of the phenyl group (compounds 5–7) slightly changed the SI because of the concomitant cytotoxicity of such compounds. Surprisingly, the incorporation of a second fluorine atom resulted in a substantial improvement over the selectivity. In particular, compound 8 presented an EC50 comparable to BAPR with a dramatic reduction of cell toxcity: from a CC50 of 13.3 ± 3.7 μM for the monofluorinated analogue to a CC50 higher than 250 μM for the difluorinate counterpart. We wanted also to assess the effect of a trifluoromethyl group on the phenyl ring of BAPR on EV71 replication. Compounds 9, 10 and 11 did not show any cytotoxicity at the highest concentration tested, and the analogue N6-(3-trifluoromethylbenzyl)-adenosine (compound 10) exhibited also a four-fold improvement in potency as compared to BAPR.
Previous reports showed that the halogenation (and, in particular, the addition of I or Cl atoms) on a BAPR scaffold increased its selectivity by reducing the cell toxicity in cancer cell lines [26,30]. In line with these findings, we observed that the gain in selectivity in our model was mostly due to a decreased cell toxicity. In particular, only the analogues containing two fluorine or a trifluoromethyl group dramatically improved the cytotoxicity. In spite of our interest in understanding compound-driven cell toxicity, addressing this question was beyond our scientific scope. Future works on the optimization of this class of analogues may shed light on the mechanism of action and their metabolization within an infected cell.
Altogether, our data revealed that the introduction of at least two fluorine atoms or a trifluoromethyl group on the phenyl ring of BAPR dramatically improved its selectivity by reducing the cytotoxicity, and in case of compound 10, also by increasing the potency.
3. Materials and Methods
3.1. General
The solvents and materials were reagent grade and were used without additional purification. Column chromatography was performed on silica gel (Kieselgel 60 Merck, Germany, 0.063–0.200 mm). TLC was performed on an Alugram SIL G/UV254 (Macherey-Nagel, Düren, Germany) with UV visualization. The melting points were determined with Electrothermal Melting Point Apparatus IA6301 and are uncorrected. The 1H and 13C (with complete proton decoupling) NMR spectra were recorded on a Bruker (Karlsruhe, Germany) AMX 400 NMR instrument at 303 K. The 1H-NMR-spectra were recorded at 400 MHz and the 13C-NMR-spectra at 100 MHz. The chemical shifts in ppm were measured relative to the residual solvent signals as internal standards (CDCl3, 1H: 7.26 ppm, 13C: 77.1 ppm; DMSO-d6, 1H: 2.50 ppm, 13C: 39.5 ppm). Spin-spin coupling constants (J) are given in Hz. The high resolution mass spectra (HRMS) were registered on a Bruker Daltonics (Manning Park, Billerica, MA, USA) micrOTOF-Q II instrument using electrospray ionization (ESI). The measurements were done in positive ion mode. Interface capillary voltage: 4500 V; mass range from m/z 50 to 3000; external calibration (Electrospray Calibrant Solution, Fluka); nebulizer pressure: 0.4 Bar; flow rate: 3 µL/min; dry gas: nitrogen (4 L/min); interface temperature: 200 °C. Samples were injected into the mass spectrometer chamber from the Agilent 1260 HPLC system equipped with an Agilent (Santa Clara, CA, USA) Poroshell 120 EC-C18 (3.0 × 50 mm; 2.7 µm) column: the flow rate was 200 µL/min; and the samples were injected from the acetonitrile–water (1:1) solution and eluted in a linear gradient of acetonitrile concentrations (50→100%).
3.2. 9-(2,3,5-Tri-O-isobutyroyl-β-d-ribofuranosyl)-6-chloropurine (4)
Isobutyric anhydride (5.6 mL, 33.8 mmol) was added in portions to a suspension of 3 g (11.2 mmol) of inosine in 18 mL of dry pyridine. The reaction mixture was stirred for 24 h at room temperature, and then evaporated in vacuum. The residue was diluted with the mixture ethanol:water (50 mL), and the suspension was filtered. The resulting powder was washed with the mixture ethanol:water (50 mL) and dried in a vacuum dessicator over phosphorous pentoxide for 2 days. The yield was 5 g (93%) of 2′,3′,5′-tri-O-isobutyroylinosine as a white powder. Rf 0.39 (CH2Cl2:EtOH—98:2). 1H-NMR (400 MHz, CDCl3): δ = 1.23–1.12 (m, 18H, Me-i-Bu), 2.66–2.50 (m, 3H, CH-i-Bu), 4.40 (d, 2H, J5′4′ = 3.7 Hz, H5′), 4.45 (dt, 1H, J4′5′ = 3.7 Hz, J4′3′ = 4.6 Hz, H4′), 5.60 (dd, 1H, J3′4′ = 4.6 Hz, J3′2′ = 5.4 Hz, H3′), 5.84 (dd, 1H, J2′3′ = 5.4 Hz, J2′1′ = 5.3 Hz, H2′), 6.18 (d, 1H, J1′2′ = 5.3 Hz, H1′), 8.21 (br s, 2H, H2, H8 Hyp), 12.99 (br s, 1H, NH Hyp).
The compound 2′,3′,5′-tri-O-isobutyroylinosine (2.9 g, 6.05 mmol) was then dissolved in 30 mL of a DMF:dichloroethane (1:15) mixture, and thionyl chloride (1.14 mL, 15.7 mmol) was added dropwise to the mixture under intensive stirring. After stirring at 65 °C for 15 min, the reaction mixture was diluted with dichloromethane (60 mL) and washed successively with 10% sodium bicarbonate (4 × 50 mL) and water (2 × 50 mL). The organic layer was separated, dried over anhydrous Na2SO4, filtered, and evaporated in a vacuum. The residue was purified by column chromatography on silica gel (200 mL). The column was washed with dichloromethane (200 mL). The product was eluted with the system CH2Cl2:EtOH—96:4. The yield was 2.8 g (93%) as a slightly yellow syrup. Rf 0.52 (CH2Cl2:EtOH—98:2). 1H-NMR (400 MHz, CDCl3): δ = 1.09–1.23 (m, 18H, Me-i-Bu), 2.49–2.69 (m, 3H, CH-i-Bu), 4.40 (d, 2H, J5′,4′ = 3.7 Hz, H5′), 4.27 (dd, 1H, J4′,5′ = 3.7 Hz, J4′,3′ = 4.5 Hz, H4′), 5.63 (dd, 1H, J3′,2′ = 5.3 Hz, J4′,3′ = 4.5 Hz, H3′), 5.89 (t, 1H, J2′,3′ = 5.3 Hz, J2′,1′ = 5.3 Hz, H2′), 6.22 (d, 1H, J1′,2′ = 5.3 Hz, H1′), 8.30 (s, 1H, H2), 8.76 (s, 1H, H8). 13C-NMR (100 MHz, CDCl3): δ = 18.77, 18.85, 18.93, 18.99, 19.01, 19.11 (CH3-i-Bu), 33.80, 33.92, 34.05 (CH-i-Bu), 63.05 (C5′), 70.55 (C3′), 73.46 (C2′), 81.12 (C4′), 87.10 (C1′), 132.40 (C6), 143.57 (C5), 151.42 (C8), 151.73 (C4), 152.44 (C2), 175.51, 175.71, 176.57 (C=O). HRMS: m/z [M + H]+ calculated C22H30ClN4O7+ 497.1798, found 497.1798.
3.3. N6-(2-Fluorobenzyl)adenosine (5)
A mixture of 4 (200 mg, 0.4 mmol) and 2-fluorobenzylamine (0.091 mL, 0.8 mmol) was dissolved in MeCN (3 mL), and then DIPEA (0.14 mL, 0.8 mmol) was added in one portion. The solution was stirred at 70 °C. The reaction was monitored by TLC (CH2Cl2:EtOH—99.5:0.5). After 22 h, the reaction mixture was evaporated in a vacuum and the residue was diluted with methylene chloride (30 mL) and washed with water (2 × 15 mL). The organic layer was separated, dried over anhydrous Na2SO4, and evaporated in a vacuum. The residue was purified by column chromatography on silica gel. The product was eluted with CH2Cl2:EtOH—99:1. The yield was 208 mg (89%) of N6-(2-fluorobenzyl)-2′,3′,5′-tri-O-isobutyroyladenosine as a syrup. Rf 0.6 (CH2Cl2:EtOH—99.5:0.5). 1H-NMR (400 MHz, DMSO-d6): δ = 1.0–1.2 (m, 18H, Me-i-Bu), 2.51–2.66 (m, 3H, CH-i-Bu), 4.35–4.41 (m, 2H, H5′), 4.27 (ddd, 1H, J4′,5′ = 6.1 Hz, H4′), 4.76 (br s, 2H, CH2), 5.75 (dd, 1H, J3′,2′ = 5.4 Hz, H3′), 6.03 (dd, 1H, J2′,3′ = 5.4 Hz, J2′,1′ = 4.9 Hz, H2′), 6.21 (d, 1H, J1′,2′ = 4.9 Hz, H1′), 7.11 (td, 1H, 3J5-6 = 7.5 Hz, 3J5-4 = 7.5 Hz, 4J5-3 = 1.0 Hz, 5H-2-F-Ph), 7.16 (ddd, 1H, 3JH-F = 8.2 Hz, 3J3-4 = 8.8 Hz, 4J3-5 = 1.0 Hz, 3H-2-F-Ph), 7.28 (dddd, 1H, 4JH-F = 5.5 Hz, 3J4-3 = 8.8 Hz, 3J4-5= 7.5 Hz, 4J4-6 = 1.6 Hz, 4H-2-F-Ph), 7.32 (ddd, 1H, 4JH-F = 7.5 Hz, 3J6-5 = 7.5 Hz, 4J6-4 = 1.6 Hz, 6H-2-F-Ph), 7.23 (s, 1H, H2 Ade), 8.36 (s, 1H, H8 Ade), 8.42 (1H, NH).
The resulting N6-(2-fluorobenzyl)-2′,3′,5′-tri-O-isobutyroyladenosine (206 mg, 0.352 mmol) was treated with 8 M MeNH2 in EtOH solution (4.5 mL). After 2 days, the mixture was evaporated in a vacuum and the residue was purified by column chromatography on silica gel. The column was washed with CH2Cl2:EtOH—95:5, and then eluted with CH2Cl2:EtOH—90:10 to give 5 as a white powder. The yield was 118 mg (79% for two steps). Rf 0.15 (CH2Cl2:EtOH—95:5). m.p. 194–195 °C. 1H-NMR (400 MHz, DMSO-d6): δ = 3.55 (ddd, 1H, J5′b,5′a = −12.0 Hz, J5′b,4′ = 3.4 Hz, J5′b,OH = 6.9 Hz, H5′b), 3.68 (ddd, 1H, J5′a,5′b = −12.0 Hz, J5′a,4′ = 3.4 Hz, J5′a,OH = 4.7 Hz, H5′a), 3.96 (q, 1H, J4′,5′b = 3.4, J4′,5′a = 3.4 Hz, J4′,3′ = 3.4 Hz, H4′), 4.15 (ddd, 1H, J3′,4′ = 3.4 Hz, J3′,2′ = 4.9 Hz, J3′,OH = 4.7 Hz, H3′), 4.61 (ddd, 1H, J2′,3′ = 4.9 Hz, J2′,1′ = 6.1 Hz, J2′,OH = 6.2 Hz, H2′), 4.77 (br s, 2H, CH2), 5.15 (d, 1H, JOH-3′ = 4.7 Hz, 3′OH), 5.31 (dd, 1H, JOH-5′b = 6.9 Hz, JOH-5′a = 4.7 Hz, 5′OH), 5.41 (d, 1H, JOH-2′ = 6.2 Hz, 2′OH), 5.90 (d, 1H, J1′,2′ = 6.1 Hz, H1′), 7.11 (td, 1H, 3J5-6 = 7.5 Hz, 3J5-4 = 7.5 Hz, 4J5-3 = 1.0 Hz, 5JH-F < 1.0 Hz, 5H-2-F-Ph), 7.16 (ddd, 1H, 3JH-F = 9.4 Hz, 3J3-4 = 8.2 Hz, 4J3-5 = 1.0 Hz, 3H-2-F-Ph), 7.26 (dddd, 1H, 4JH-F = 5.5 Hz, 3J4-3 = 8.2 Hz, 3J4-5 = 7.5 Hz, 4J4-6 = 1.7 Hz, 4H-2-F-Ph), 7.32 (ddd, 1H, 4JH-F = 6.7 Hz, 3J6-5 = 7.5 Hz, 4J6-4 = 1.7 Hz, 6H-2-F-Ph), 8.21 (s, 2H, H2, NH), 8.39 (s, 1H, H8). 13C-NMR (100 MHz, DMSO-d6): δ = 36.78 (CH2), 61.62 (C5′), 70.60 (C3′), 73.49 (C2′), 85.86 (C4′), 87.92 (C1′), 114.95 (d, 2JC-F = 21.1 Hz, C3-Ph), 119.77 (C5), 124.18 (C5-Ph), 126.52 (d, 2JC-F = 12.6 Hz, C1-Ph), 128.52 (d, 3JC-F = 7.4 Hz, C4-Ph), 128.85 (br s, C6-Ph), 140.00 (C8), 148.59 (C4), 152.29 (C2), 154.49 (C6), 160.00 (d, 1JC-F = 244.0 Hz, C2-Ph). HRMS: m/z [M + H]+ calculated C17H19FN5O4+ 376.1416, found 376.1417; m/z [M + Na]+ calculated C17H18FN5O4Na+ 398.1235, found 398.1238.
3.4. N6-(3-Fluorobenzyl)adenosine (6)
Following the procedure for the preparation of 5, the condensation of 4 (200 mg, 0.4 mmol) with 3-fluorobenzylamine (0.091 mL, 0.8 mmol) in the presence of DIPEA (0.14 mL, 0.8 mmol) in MeCN (3 mL) for 22 h at 70 °C with a subsequent deblocking in 8 M MeNH2 in EtOH solution (4.5 mL) at room temperature gave 6 as a white powder. The overall yield was 111 mg (74%). Rf 0.15 (CH2Cl2:EtOH—95:5). m.p. 159–160 °C. 1H-NMR (400 MHz, DMSO-d6): δ = 3.56 (ddd, 1H, J5′b,5′a = −12.0 Hz, J5′b,4′ = 3.4 Hz, J5′b,OH = 6.9 Hz, H5′b), 3.67 (ddd, 1H, J5′a,5′b = −12.0 Hz, J5′a,4′ = 3.4 Hz, J5′a,OH = 4.7 Hz, H5′a), 3.96 (q, 1H, J4′,5′b = 3.4 Hz, J4′,5′a = 3.4 Hz, J4′,3′ = 3.4 Hz, H4′), 4.15 (td, 1H, J3′,4′ = 3.4 Hz, J3′,2′ = 4.7 Hz, J3′,OH = 4.7 Hz, H3′), 4.61 (ddd, 1H, J2′,3′ = 4.7 Hz, J2′,1′ = 6.1 Hz, J2′,OH = 6.2 Hz, H2′), 4.77 (br s, 2H, CH2), 5.15 (d, 1H, JOH-3′ = 4.7 Hz, 3′OH), 5.32 (dd, 1H, JOH-5′b = 6.9 Hz, JOH-5′a = 4.7 Hz, 5′OH), 5.41 (d, 1H, JOH-2′ = 6.2 Hz, 2′OH), 5.91 (d, 1H, J1′,2′ = 6.1 Hz, H1′), 7.03 (dd, 1H, 3JH-F = 8.9 Hz, 3J4-5 = 8.2 Hz, 4H-3-F-Ph), 7.13 (d, 1H, 3JH-F = 8.9 Hz, 2H-3-F-Ph), 7.18 (dd, 1H, 3J6-5 = 8.2 Hz, 5JH-F = 2.2 Hz, 6H-3-F-Ph), 7.33 (td, 1H, 4JH-F = 6.3 Hz, 3J5-4 = 8.2 Hz, 3J5-6 = 8.2 Hz, 5H-3-F-Ph), 8.21 (s, 1H, H2 Ade), 8.39 (s, 1H, H8 Ade), 8.46 (br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6): δ = 42.51 (CH2), 61.61 (C5′), 70.60 (C3′), 73.49 (C2′), 85.86 (C4′), 87.93 (C1′), 113.29 (d, 2JC-F = 21.0 Hz, C2-Ph), 113.71 (d, 2JC-F = 21.5 Hz, C4-Ph), 119.76 (C5), 123.06 (C6-Ph), 130.10 (d, 3JC-F = 7.5 Hz, C5-Ph), 139.99 (C8), 143.09 (br s, C1-Ph), 148.54 (C4), 152.30 (C2), 154.44 (C6), 162.16 (d, 1JC-F = 243.1 Hz, C3-Ph). HRMS: m/z [M + H]+ calculated C17H19FN5O4+ 376.1416, found 376.1418; m/z [M + Na]+ calculated C17H18FN5O4Na+ 398.1235, found 398.1239.
3.5. N6-(4-Fluorobenzyl)adenosine (7)
Following the procedure for the preparation of 5, the condensation of 4 (200 mg, 0.4 mmol) with 4-fluorobenzylamine (0.091 mL, 0.8 mmol) in the presence of DIPEA (0.14 mL, 0.8 mmol) in MeCN (3 mL) for 22 h at 70 °C with a subsequent deblocking in 8 M MeNH2 in EtOH solution (4.5 mL) at room temperature gave 7 as a white powder. The overall yield was 81 mg (54%). Rf 0.15 (CH2Cl2:EtOH—95:5). mp 181–182 °C. 1H-NMR (400 MHz, DMSO-d6): δ = 3.55 (ddd, 1H, J5′b,5′a = −12.0 Hz, J5′b,4′ = 3.4 Hz, J5′b,OH = 6.9 Hz, H5′b), 3.68 (ddd, 1H, J5′a,5′b = −12.0 Hz, J5′a,4′ = 3.4 Hz, J5′a,OH = 4.6 Hz, H5′a), 3.96 (q, 1H, J4′,5′b = 3.4 Hz, J4′,5′a = 3.4 Hz, J4′,3′ = 3.4 Hz, H4′), 4.15 (ddd, 1H, J3′,4′ = 3.4 Hz, J3′,2′ = 4.7 Hz, J3′,OH = 4.7 Hz, H3′), 4.62 (ddd, 1H, J2′,3′ = 4.7 Hz, J2′,1′ = 6.1 Hz, J2′,OH = 6.2 Hz, H2′), 4.69 (br s, 2H, CH2), 5.15 (d, 1H, JOH-3′ = 4.7 Hz, 3′OH), 5.32 (dd, 1H, JOH-5′b = 6.9 Hz, JOH-5′a = 4.6 Hz, 5′OH), 5.41 (d, 1H, JOH-2′ = 6.2 Hz, 2′OH), 5.89 (d, 1H, J1′,2′ = 6.1 Hz, H1′), 7.11 (t, 2H, 3JH-F = 8.9 Hz, 3J3-2 = 8.9 Hz, 3H-4-F-Ph, 5H-4-F-Ph), 7.37 (dd, 2H, 3J2-3 = 8.9, 4JH-F = 6.0 Hz, 2H-4-F-Ph, 6H-4-F-Ph), 8.21 (s, 1H, H2 Ade), 8.37 (s, 1H, H8 Ade), 8.43 (br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6): δ = 42.21 (CH2), 61.62 (C5′), 70.61 (C3′), 73.48 (C2′), 85.87 (C4′), 87.93 (C1′), 114.87 (d, 2JC-F = 21.2 Hz, C3-Ph, C5-Ph), 119.74 (C5), 129.05 (d, 3JC-F = 7.5 Hz, C2-Ph, C6-Ph), 136.15 (C1-Ph), 139.91 (C8), 148.52 (C4), 152.30 (C2), 154.43 (C6), 161.07 (d, 1JC-F = 241.9 Hz, C4-Ph). HRMS: m/z [M + H]+ calculated C17H19FN5O4+ 376.1416, found 376.1407; m/z [M + Na]+ calculated C17H18FN5O4Na+ 398.1235, found 398.1227.
3.6. N6-(2,6-Difluorobenzyl)adenosine (8)
Following the procedure for the preparation of 5, the condensation of 4 (191 mg, 0.384 mmol) with 2,6-difluorobenzylamine (0.092 mL, 0.77 mmol) in the presence of DIPEA (0.134 mL, 0.77 mmol) in MeCN (2.5 mL) for 11 h at 70 °C with a subsequent deblocking in 8 M MeNH2 in EtOH solution (2 mL) at room temperature gave 8 as a white powder. The overall yield was 129 mg (85%). Rf 0.15 (CH2Cl2:EtOH—95:5). m.p. 192–194 °C. 1H-NMR (400 MHz, DMSO-d6): δ = 3.56 (ddd, 1H, J5′b,5′a = −12.0 Hz, J5′b,4′ = 3.5 Hz, J5′b,OH = 6.8 Hz, H5′b), 3.68 (ddd, 1H, J5′a,5′b = −12.0 Hz, J5′a,4′ = 3.5 Hz, J5′a,OH = 4.7 Hz, H5′a), 3.97 (td, 1H, J4′,5′b = 3.5 Hz, J4′,5′a = 3.5 Hz, J4′,3′ = 3.3 Hz, H4′), 4.15 (ddd, 1H, J3′,4′ = 3.3 Hz, J3′,2′ = 5.2 Hz, J3′,OH = 4.6 Hz, H3′), 4.61 (td, 1H, J2′,3′ = 5.2 Hz, J2′,1′ = 6.1 Hz, J2′,OH = 6.1 Hz, H2′), 4.80 (br s, 2H, CH2), 5.15 (d, 1H, JOH-3′ = 4.6 Hz, 3′OH), 5.31 (dd, 1H, JOH-5′b = 7.0 Hz, JOH-5′a = 4.7 Hz, 5′OH), 5.41 (d, 1H, JOH-2′ = 6.1 Hz, 2′OH), 5.89 (d, 1H, J1′,2′ = 6.1 Hz, H1′), 7.05 (dd, 2H, 3JH-F =7.6 Hz, Jm-H-p-H = 8.9 Hz, m-H-2,6-di-F-Ph), 7.37 (tt, 1H, Jp-H-m-H =8.9 Hz, 4JH-F = 6.0 Hz, p-H-2,6-di-F-Ph), 8.19 (br s, 1H, NH), 8.24 (s, 1H, H8 Ade), 8.36 (s, 1H, H2 Ade). 13C-NMR (100 MHz, DMSO-d6): δ = 32.17 (CH2), 61.61 (C5′), 70.59 (C3′), 73.50 (C2′), 85.83 (C4′), 87.93 (C1′), 111.38 (d, 2JC-F = 23.6 Hz, C3-Ph, C5-Ph), 114.44 (t, 2JC-F = 17.6 Hz, C1-Ph), 119.70 (C5), 129.60 (C4-Ph), 139.85 (C8), 148.64 (C4), 152.17 (C2), 154.16 (C6), 161.23 (d, 1JC-F = 248.0 Hz, C2-Ph, C6-Ph). HRMS: m/z [M + H]+ calculated C17H18F2N5O4+ 394.1321, found 394.1325; m/z [M + Na]+ calculated C17H17F2N5O4Na+ 416.1141, found 416.1143.
3.7. N6-(2-Trifluoromethylbenzyl)adenosine (9)
Following the procedure for the preparation of 5, the condensation of 4 (213 mg, 0.43 mmol) with 2-trifluoromethylbenzylamine (0.12 mL, 0.86 mmol) in the presence of DIPEA (0.15 mL, 0.86 mmol) in MeCN (3 mL) for 10 h at 70 °C with a subsequent deblocking in 8 M MeNH2 in EtOH solution (3 mL) at room temperature gave 9 as a white powder. The overall yield was 168 mg (92%). Rf 0.13 (CH2Cl2:EtOH—95:5). m.p. 204–205 °C. 1H-NMR (400 MHz, DMSO-d6): δ = 3.56 (ddd, 1H, J5′b,5′a = −12.0 Hz, J5′b,4′ = 3.5 Hz, J5′b,OH = 6.9 Hz, H5′b), 3.68 (ddd, 1H, J5′a,5′b = −12.0 Hz, J5′a,4′ = 3.5 Hz, J5′a,OH = 4.7 Hz, H5′a), 3.97 (q, 1H, J4′,5′b = 3.5 Hz, J4′,5′a = 3.5 Hz, J4′,3′ = 3.5 Hz, H4′), 4.16 (ddd, 1H, J3′,4′ = 3.5 Hz, J3′,2′ = 5.2 Hz, J3′,OH = 4.7 Hz, H3′), 4.63 (ddd, 1H, J2′,3′ = 5.2 Hz, J2′,1′ = 6.1 Hz, J2′,OH = 6.2 Hz, H2′), 4.93 (br s, 2H, CH2), 5.15 (d, 1H, JOH-3′ = 4.7 Hz, 3′OH), 5.29 (dd, 1H, JOH-5′b = 6.9 Hz, JOH-5′a = 4.7 Hz, 5′OH), 5.41 (d, 1H, JOH-2′ = 6.2 Hz, 2′OH), 5.91 (d, 1H, J1′,2′ = 6.1 Hz, H1′), 7.40–7.75 (m, 4H, Ph), 8.19 (s, 1H, H8), 8.42 (s, 1H, H2), 8.45 (br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6): δ = 39.52 (CH2, overlapping with the solvent peak), 61.62 (C5′), 70.62 (C3′), 73.51 (C2′), 85.89 (C4′), 87.92 (C1′), 119.83 (C5), 123.24 (C2 Ph), 125.77 (C6 Ph), 125.95 (q, JC-F = 30.7 Hz, CF3), 127.03 (C3 Ph), 127.54 (C4 Ph), 132.55 (C5 Ph), 138.10 (C1 Ph), 140.17 (C8), 146.68 (C4), 152.38 (C2), 154.52 (C6). HRMS: m/z [M + H]+ calculated C18H19F3N5O4+ 426.1384, found 426.1387; m/z [M + Na]+ calculated C18H18F3N5O4Na+ 448.1203, found 448.1210.
3.8. N6-(3-Trifluoromethylbenzyl)adenosine (10)
Following the procedure for the preparation of 5, the condensation of 4 (137 mg, 0.276 mmol) with 3-trifluoromethylbenzylamine (0.08 mL, 0.55 mmol) in the presence of DIPEA (0.096 mL, 0.55 mmol) in MeCN (2 mL) for 20 h at 70 °C with a subsequent deblocking in 8 M MeNH2 in EtOH solution (2 mL) at room temperature gave 10 as a white powder. The overall yield was 59 mg (50%). Rf 0.11 (CH2Cl2:EtOH—95:5). m.p. 139–140 °C. 1H-NMR (400 MHz, DMSO-d6): δ = 3.55 (ddd, 1H, J5′b,5′a = −12.0 Hz, J5′b,4′ = 3.6 Hz, J5′b,OH = 7.0 Hz, H5′b), 3.68 (ddd, 1H, J5′a,5′b = −12.0 Hz, J5′a,4′ = 3.6 Hz, J5′a,OH = 4.7 Hz, H5′a), 3.97 (q, 1H, J4′,5′b = 3.6 Hz, J4′,5′a = 3.6 Hz, J4′,3′ = 3.6 Hz, H4′), 4.16 (ddd, 1H, J3′,4′ = 3.6 Hz, J3′,2′ = 5.2 Hz, J3′,OH = 4.7 Hz, H3′), 4.62 (ddd, 1H, J2′,3′ = 5.2 Hz, J2′,1′ = 6.1 Hz, J2′,OH = 6.2 Hz, H2′), 4.80 (br s, 2H, CH2), 5.14 (d, 1H, JOH-3′ = 4.7 Hz, 3′OH), 5.30 (dd, 1H, JOH-5′b = 7.0 Hz, JOH-5′a = 4.7 Hz, 5′OH), 5.40 (d, 1H, JOH-2′ = 6.2 Hz, 2′OH), 5.90 (d, 1H, J1′,2′ = 6.1 Hz, H1′), 7.5–7.75 (m, 4H, Ph), 8.21 (s, 1H, H8), 8.39 (s, 1H, H2), 8.53 (br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6): δ = 42.60 (CH2), 61.63 (C5′), 70.63 (C3′), 73.54 (C2′), 85.89 (C4′), 87.97 (C1′), 119.79 (C5), 123.41 (C4 Ph), 123.69 (C2 Ph), 125.64 (C6 Ph), 128.96 (q, JC-F = 31.4 Hz, CF3), 129.30 (C5 Ph), 131.32 (C3 Ph), 140.08 (C8), 141.54 (C1 Ph), 148.58 (C4), 152.33 (C2), 154.43 (C6). HRMS: m/z [M + H]+ calculated C18H19F3N5O4+ 426.1384, found 426.1384; m/z [M + Na]+ calculated C18H18F3N5O4Na+ 448.1203, found 448.1204
3.9. N6-(4-Trifluoromethylbenzyl)adenosine (11)
Following the procedure for the preparation of 5, the condensation of 4 (190 mg, 0.382 mmol) with 4-trifluoromethylbenzylamine (0.11 mL, 0.76 mmol) in the presence of DIPEA (0.132 mL, 0.76 mmol) in MeCN (3 mL) for 10 h at 70 °C with a subsequent deblocking in 8 M MeNH2 in EtOH solution (3 mL) at room temperature gave 11 as a white powder. The overall yield was 160 mg (98%). Rf 0.09 (CH2Cl2:EtOH—95:5). m.p. 152–153 °C. 1H-NMR (400 MHz, DMSO-d6): δ = 3.55 (ddd, 1H, J5′b,5′a = −12.0 Hz, J5′b,4′ = 3.6 Hz, J5′b,OH = 6.9 Hz, H5′b), 3.68 (ddd, 1H, J5′a,5′b = −12.0 Hz, J5′a,4′ = 3.6 Hz, J5′a,OH = 4.7 Hz, H5′a), 3.97 (q, 1H, J4′,5′b = 3.6, J4′,5′a = 3.6 Hz, J4′,3′ = 3.6 Hz, H4′), 4.16 (ddd, 1H, J3′,4′ = 3.6 Hz, J3′,2′ = 5.2 Hz, J3′,OH = 4.7 Hz, H3′), 4.62 (ddd, 1H, J2′,3′ = 5.2 Hz, J2′,1′ = 6.1 Hz, J2′,OH = 6.2 Hz, H2′), 4.80 (br s, 2H, CH2), 5.14 (d, 1H, JOH-3′ = 4.7 Hz, 3′OH), 5.30 (dd, 1H, JOH-5′b = 6.9 Hz, JOH-5′a = 4.7 Hz, 5′OH), 5.40 (d, 1H, JOH-2′ = 6.2 Hz, 2′OH), 5.90 (d, 1H, J1′,2′ = 6.1 Hz, H1′), 7.5–7.75 (m, 4H, Ph), 8.20 (s, 1H, H8), 8.39 (s, 1H, H2), 8.51 (br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6): δ = 42.65 (CH2), 61.60 (C5′), 70.59 (C3′), 73.49 (C2′), 85.86 (C4′), 87.91 (C1′), 119.79 (C5), 122.98 (C4 Ph), 125.07 (C3 Ph, C5 Ph), 127.48 (q, JC-F = 30.7 Hz, CF3), 127.67 (C2 Ph, C6 Ph), 140.02 (C8), 144.92 (C1 Ph), 148.59 (C4), 152.29 (C2), 154.42 (C6). HRMS: m/z [M + H]+ calculated C18H19F3N5O4+ 426.1384, found 426.1383; m/z [M + Na]+ calculated C18H18F3N5O4Na+ 448.1203, found 448.1203.
3.10. Antiviral Assay Against EV71 in RD Cells
An EV71 BrCr laboratory-adapted strain was used at a low multiplicity of infection (MOI) in a standardized antiviral assay [31]. Briefly, freshly harvested rhadbdosarcoma (RD) cells were seeded in a 96-well plate (2 × 104 cells/well) and incubated at 37 °C in 5% CO2. The next day, a serial dilution of the compounds was prepared in assay medium and added to the RD cells. Then, the cells were supplemented with the viral suspension. The assay plates were incubated until full virus–induced cell death was observed in the untreated, infected controls (3–4 days post-infection). Subsequently, the antiviral effect was quantified using a colorimetric readout with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium/phenazine methosulfate (MTS/PMS method), and the concentration of compound at which a 50% inhibition of virus-induced cell death would be observed (EC50) was calculated from the antiviral dose-response curves. A similar assay setup was used to determine the adverse effect of the compound on uninfected, treated cells for the calculation of the CC50 (concentration of compound that reduces overall cell health with 50% as determined by the MTS/PMS method). The selectivity index (SI) was calculated as a ratio of EC50/CC50.
4. Conclusions
We reported here the antiviral profile of a class of analogues of the cytokinin nucleoside BAPR, previously described by our groups as a potent and selective inhibitor of EV71 replication. Interestingly, we showed that the replacement of hydrogen with fluorine atoms or a trifluoromethyl group in the aromatic moiety of BAPR overall increased its SI. In particular, the least successful analogue (compound 7) with the addition of one fluorine atom at position 4 of the BAPR phenyl showed a 1.25-times increase of SI, whereas the best analogue of the class (compound 10) with a trifluoromethyl at position 3 exhibited an SI that was 230 times higher than BAPR. Fluorinated analogues of natural substances are of interest, since often the biological activity of the parent compounds is improved. However, the tremendous gain in selectivity reported here represents one of the most spectacular examples of structure optimization of a lead natural compound by introducing fluoro- or trifluoromethyl groups into an aromatic moiety.
Acknowledgments
This work was supported by the Russian Foundation for Basic Research (16-04-01594 and 17-04-01939) and Russian Academy of Sciences (Program ‘Molecular and Cell Biology’) and by Russian Federation President Program for young scientists (MK-8496.2016.4). Liang Sun is supported by a CSC: Project of Europe grant from the China Scholarship Council (CSC No. 201403250056). Kim Donckers and Caroline Collard are acknowledged for their excellent assistance in the acquisition of the antiviral data.
Supplementary Materials
Supplementary Materials are available online.
Author Contributions
Liang Sun, Carmen Mirabelli and Sergey N. Mikhailov conceived and designed the experiments; in particular, Liang Sun, Mikhail S. Drenichev, Vladimir E. Oslovsky, Nikolay N. Kurochkin and Vladislav E. Kunetsky performed the experiments; Johan Neyts contributed reagents/materials/analysis tools; Carmen Mirabelli, Pieter Leyssen and Sergey N. Mikhailov wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors.
References
- 1.Butler M.S., Robertson A.A., Cooper M.A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014;31:1612–1661. doi: 10.1039/C4NP00064A. [DOI] [PubMed] [Google Scholar]
- 2.Maier M.E. Design and synthesis of analogues of natural products. Org. Biomol. Chem. 2015;13:5302–5343. doi: 10.1039/C5OB00169B. [DOI] [PubMed] [Google Scholar]
- 3.Drenichev M.S., Oslovsky V.E., Mikhailov S.N. Cytokinin nucleosides-natural compounds with a unique spectrum of biological activity. Curr. Top. Med. Chem. 2016;16:2562–2576. doi: 10.2174/1568026616666160414123717. [DOI] [PubMed] [Google Scholar]
- 4.Mikhailov S.N., Scotti L., Singla R.K., Scotti M.T. Perspectives in Medicinal Chemistry. Curr. Top. Med. Chem. 2016;16:2725–2726. doi: 10.2174/156802661625160816180839. [DOI] [PubMed] [Google Scholar]
- 5.De Clercq E., Li G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rozenski J., Crain P.F., McCloskey J.A. The RNA Modification Database: 1999 update. Nucleic Acids Res. 1999;27:196–197. doi: 10.1093/nar/27.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agris P., Crain P.F., Rozenski J., Fabris D., Vendeix F.A.P. The RNA Institute; [(accessed on 23 June 2017)]. The RNA Modification Database. Available online: http://mods.rna.albany.edu. [Google Scholar]
- 8.Ernst D., Scháfer W., Oesterhelt D. Isolation and identification of a new, naturally occurring cytokinin (6-benzylaminopurineriboside) from an anise cell culture (Pimpinella anisum L.) Planta. 1983;159:222–225. doi: 10.1007/BF00397528. [DOI] [PubMed] [Google Scholar]
- 9.Horgan R., Hewett E.W., Horgan J.M., Purse J., Wareing P.F. A new cytokinin from Populus x robusta. Phytochemistry. 1975;14:1005–1008. doi: 10.1016/0031-9422(75)85176-4. [DOI] [Google Scholar]
- 10.Ge L., Yong J.W.H., Goh N.K., Chia L.S., Tan S.N., Ong E.S. Identification of kinetin and kinetin riboside in coconut (Cocos nucifera L.) water using a combined approach of liquid chromatography-tandem mass spectrometry, high performance liquid chromatography and gel electrophoresis. J. Chromatogr. B. 2005;829:26–34. doi: 10.1016/j.jchromb.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 11.Young D.C., Layre E., Pan S.-J., Tapley A., Adamson J., Seshadri C., Wu Z., Buter J., Minnaard A.J., Coscolla M., et al. In vivo biosynthesis of terpene nucleosides provides unique chemical markers of Mycobacterium tuberculosis infection. Chem. Biol. 2015;22:516–526. doi: 10.1016/j.chembiol.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arita M., Wakita T., Shimizu H. Characterization of pharmacologically active compounds that inhibit poliovirus and enterovirus 71 infectivity. J. Gen. Virol. 2008;89:2518–2530. doi: 10.1099/vir.0.2008/002915-0. [DOI] [PubMed] [Google Scholar]
- 13.Chang L.Y., Lin T.Y., Hsu K.H., Huang Y.C., Lin K.L., Hsueh C., Shih S.R., Ning H.C., Hwang M.S., Wang H.S., et al. Clinical features and risk factors of pulmonary oedema after enterovirus-71-related hand, foot, and mouth disease. Lancet. 1999;354:1682–1686. doi: 10.1016/S0140-6736(99)04434-7. [DOI] [PubMed] [Google Scholar]
- 14.Wong K.T., Munisamy B., Ong K.C., Kojima H., Noriyo N., Chua K.B., Ong B.B., Nagashima K. The distribution of inflammation and virus in human enterovirus 71 encephalomyelitis suggests possible viral spread by neural pathways. J. Neuropathol. Exp. Neurol. 2008;67:162–169. doi: 10.1097/nen.0b013e318163a990. [DOI] [PubMed] [Google Scholar]
- 15.Solomon T., Lewthwaite P., Perera D., Cardosa M.J., McMinn P., Ooi M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 2010;10:778–790. doi: 10.1016/S1473-3099(10)70194-8. [DOI] [PubMed] [Google Scholar]
- 16.Xing W., Liao Q., Viboud C., Zhang J., Sun J., Wu J.T., Chang Z., Liu F., Fang V.J., Zheng Y., et al. Hand, foot, and mouth disease in China, 2008-12: An epidemiological study. Lancet Infect. Dis. 2014;14:308–318. doi: 10.1016/S1473-3099(13)70342-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yip C.C.Y., Lau S.K.P., Woo P.C.Y., Yuen K.-Y. Human enterovirus 71 epidemics: What’s next? Emerg. Health Threats J. 2013;6:e19780. doi: 10.3402/ehtj.v6i0.19780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunaseelan S., Chu J.J.H. Identifying novel antiviral targets against enterovirus 71: Where are we? Future Virol. 2017;12:171–191. doi: 10.2217/fvl-2016-0144. [DOI] [Google Scholar]
- 19.Bauer L., Lyoo H., van der Schaar H.M., Strating J.R., van Kuppeveld F.J. Direct-acting antivirals and host-targeting strategies to combat enterovirus infections. Curr. Opin. Virol. 2017;24:25–30. doi: 10.1016/j.coviro.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tararov V.I., Tijsma A., Kolyachkina S.V., Oslovsky V.E., Neyts J., Drenichev M.S., Leyssen P., Mikhailov S.N. Chemical modification of the plant isoprenoid cytokinin N6-isopentenyladenosine yields a selective inhibitor of human enterovirus 71 replication. Eur. J. Med. Chem. 2015;90:406–413. doi: 10.1016/j.ejmech.2014.11.048. [DOI] [PubMed] [Google Scholar]
- 21.Drenichev M.S., Oslovsky V.E., Sun L., Tijsma A., Kurochkin N.N., Tararov V.I., Chizhov A.O., Neyts J., Pannecouque C., Leyssen P., et al. The length and structure of the linker in N6-benzyladenosine derivatives affects the antiviral potency against enterovirus 71. Eur. J. Med. Chem. 2016;111:84–94. doi: 10.1016/j.ejmech.2016.01.036. [DOI] [PubMed] [Google Scholar]
- 22.Müller K., Faeh C., Diederich F. Fluorine in pharmaceuticals: Looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 23.Wang J., Sánchez-Roselló M., Aceña J.L., del Pozo C., Sorochinsky A.E., Fustero S., Soloshonok V.A., Liu H. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011) Chem. Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 24.Tararov V.I., Kolyachkina S.V., Alexeev C.S., Mikhailov S.N. N6-acetyl-2′,3′,5′-tri-O-acetyladenosine, a convenient, missed out substrate for regioselective N6-alkylations. Synthesis. 2011:2483–2489. doi: 10.1055/s-0030-1260090. [DOI] [Google Scholar]
- 25.Kolyachkina S.V., Tararov V.I., Alexeev C.S., Krivosheev D.M., Romanov G.A., Stepanova E.V., Solomko E.S., Inshakov A.N., Mikhailov S.N. N6-Substituted adenosines. Cytokinin and antitumor activities. Collect. Czech. Chem. Commun. 2011;76:1361–1378. doi: 10.1135/cccc2011114. [DOI] [Google Scholar]
- 26.Doležal K., Popa I., Hauserová E., Sṕıchal L., Chakrabarty K., Novák O., Kryštof V., Voller J., Holub J., Strnad M. Preparation, biological activity and endogenous occurrence of N6-benzyladenosines. Bioorg. Med. Chem. 2007;15:3737–3747. doi: 10.1016/j.bmc.2007.03.038. [DOI] [PubMed] [Google Scholar]
- 27.Fleysher M.H., Bloch A., Hakala M.T., Nichol C.A. Synthesis and biological activity of some new N6-substituted purine nucleosides. J. Med. Chem. 1969;12:1056–1061. doi: 10.1021/jm00306a021. [DOI] [PubMed] [Google Scholar]
- 28.Žemlička J., Owens J. 6-Chloro-9-β-d-ribofuranosylpurine. A versatile intermediate in the synthesis of purine ribonucleosides. In: Townsend L.B., Tipson R.S., editors. Nucleic Acid Chemistry, Part 2. Wiley; New York, NY, USA: 1978. pp. 611–614. [Google Scholar]
- 29.Marek R., Sklenàř V. NMR studies of purines. Annu. Rep. NMR Spectrosc. 2004;54:201–242. [Google Scholar]
- 30.Dolezel P., Koudelkova P., Mlejnek P. Halogenation of N6-benzyladenosine decreases its cytotoxicity in human leukemia cells. Toxicol. In Vitro. 2010;24:2079–2083. doi: 10.1016/j.tiv.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Tijsma A., Franco D., Tucker S., Hilgenfeld R., Froeyen M., Leyssen P., Neyts J. The capsid binder vapendavir and the novel protease inhibitor SG85 inhibit enterovirus 71 replication. Antimicrob. Agents Chemother. 2014;58:6990–6992. doi: 10.1128/AAC.03328-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.