

Abstract
Twenty-three new berberine (BBR) analogues defined on substituents of ring D were synthesized and evaluated for their activity for suppression of tumor necrosis factor (TNF)-α-induced nuclear factor (NF)-κB activation. Structure–activity relationship (SAR) analysis indicated that suitable tertiary/quaternary carbon substitutions at the 9-position or rigid fragment at position 10 might be beneficial for enhancing their anti-inflammatory potency. Among them, compounds 2d, 2e, 2i and 2j exhibited satisfactory inhibitory potency against NF-κB activation, with an inhibitory rate of around 90% (5 μM), much better than BBR. A preliminary mechanism study revealed that all of them could inhibit TNF-α-induced NF-κB activation via impairing IκB kinase (IKK) phosphorylation as well as cytokines interleukin (IL)-6 and IL-8 induced by TNF-α. Therefore, the results provided powerful information on further structural modifications and development of BBR derivatives into a new class of anti-inflammatory candidates for the treatment of inflammatory diseases.
Keywords: berberine, anti-inflammatory, NF-κB, IκB kinase, structure-activity relationship
1. Introduction
Inflammation represents a response to tissue injury induced by a wide variety of stimuli, such as pathogens, damaged cells, or irritants, and is a protective response involving immune cells, blood vessels, and molecular mediators [1,2,3,4]. As a consequence, diverse pathological conditions involve inflammatory processes including arthritis, atherosclerosis, the metabolic syndrome, sepsis, and cancer. For most of these conditions, no satisfactory treatment is available [5,6]. Initial stages of inflammation involve cytokine-mediated activation of the vascular endothelium leading to adhesion and transmigration of leukocytes into the site of inflammation. Many of the pro-inflammatory processes elicited at the endothelium and leukocytes are mediated by the transcription factor nuclear factor (NF)-κB. The most prevalent inducers of the NF-κB signaling pathway are cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1, various mitogens, and bacterial components such as lipopolysaccharides (LPS).
NF-κB is the name used for a family of homodimers and heterodimers, the NF-κB dimers are maintained in an inactive state in the cytoplasm bound to the inhibitor of κB (IκB) proteins, of which the prototypical member is IκB-α [7]. Upon a pro-inflammatory signal, such as binding of TNF-α to its membrane receptor, IκB-α becomes phosphorylated at Ser32/Ser36 by IκB kinase (IKK). The IKK is a multi-subunit kinase complex, most typically composed of IKK-α and IKK-β and two molecules of IKKγ/NF-kappa-B essential modulator (NEMO) [8]. The IKK-catalyzed phosphorylation triggers degradation of IκB-α leading to the release of NF-κB followed by its translocation to the nucleus where it regulates gene expression [9]. As NF-κB is a key regulator of many pro-inflammatory responses, inhibition of different mediators of the NF-κB signaling pathway, including IKK, has emerged as a promising approach for the development of anti-inflammatory candidates.
Natural isoquinoline alkaloid berberine (BBR, Figure 1), as a nonprescription anti-diarrhea drug, has been extensively used in China for decades with a confirmed safety. Recently, several other pharmacological and biological properties of BBR including anti-inflammatory and anti-carcinogenic activities have been identified [10,11,12,13,14,15,16]. It was reported that BBR could suppress NF-κB activation induced by various inflammatory agents or carcinogens with a mild potency [17]. In our study, BBR’s moderate anti-inflammatory potency was further confirmed by an inhibitory rate of 36% at the concentration of 5 μM. The unique isoquinoline skeleton of BBR provoked us to conduct structural modification and optimization to enhance the anti-inflammatory effect. Therefore, in our present study, taking BBR as the lead, a series of novel BBR analogues defined on the substituents of ring D (Figure 1) was achieved to elucidate the structure-activity relationship (SAR) and develop a novel class of anti-inflammatory agents. Specifically, novel ester, amide, and sulfonate BBR derivatives were prepared and evaluated for their effect to inhibit TNF-α-induced NF-κB activation, and the mechanism exploration of the key compounds was carried out as well.
Figure 1.

Chemical structures of berberine (BBR), and its structure modification strategy.
2. Results and Discussion
2.1. Chemistry
Firstly, taking commercial available BBR as the starting material, a demethylation reaction was conducted to afford the key intermediate 1 (Scheme 1) [18]. The esters 2a–k and sulfonates 3a–f were obtained by esterification and sulfonation of compound 1 with various acyl chloride and sulfonyl chloride, respectively, using acetonitrile as solvent and triethylamine as the base. Compounds 4 and 7 were prepared in the presence of corresponding amino compounds [19]. After acidification, compound 6 was obtained taking pyridine as the base.
Scheme 1.

Reagents and conditions: (a) 195 °C, 30–40 mmHg, 60 min; (b) R1COX/R1SO2Cl, triethylamine, CH3CN, reflux; (c) 2,4-Dimethoxybenzylamine, 120 °C; (d) HCl, CH3OH; (e) R2COX, pyridine, CH2Cl2, reflux; (f) R-NH2, 120 °C.
Secondly, compounds 16a and 16b were synthesized through a seven-step process (Scheme 2), using commercially available 2,3-dihydroxybenzaldehyde (8) as the starting materials with the methods reported previously [20,21]. Condensation was conducted between 2-methoxy-3-hydroxy benzaldehyde (9) and homopiperonylamine (10) after selective methylation of 8. In the intermolecular cyclization of the intermediate 12, the skeleton formed according to Pictet-Spengler cyclization and Friedel-Crafts alkylation rule in one step, and, subsequently, the key intermediate 13 was obtained with an ideal yield of 53%. After acidification, alkalization, and esterification, the final products 16a and 16b were purified via flash column chromatography using methanol/dichloromethane as the gradient eluent.
Scheme 2.

Reagents and conditions: (a) CH3I, room temperature (r.t.); (b) 100 °C, 8 h; (c) NaBH4, methanol, reflux, 5 h; (d) glyoxal, formic acid, CuSO4, HCl, 100 °C, 5 h; (e) methanol/H2O, CaO, r.t., 2 h; (f) ethanol, HCl, r.t., 0.5 h; (g) RCOX, triethylamine, CH3CN, reflux.
2.2. Biology
2.2.1. SAR for Suppressing TNF-α-induced NF-κB Activation
All the newly synthesized compounds were examined in 293T cells for their anti-inflammatory activities. Given the key role of NF-κB signaling in inflammation, we investigated these analogues for their anti-inflammatory abilities by using TNF-α-induced NF-κB-responsive promoter reporter assays. Structures of the analogues and their inhibitory rates on TNF-α-induced NF-κB-responsive promoter activity are shown in Table 1.
Table 1.
Structures and inhibition effect of target compounds on tumor necrosis factor (TNF)-induced nuclear factor (NF)-κB activities.

| No. | R | Inhibitory Rate% | No. | R | Inhibitory Rate% | No. | R | Inhibitory Rate% |
|---|---|---|---|---|---|---|---|---|
| 2a | 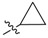 |
56 | 2i | 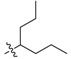 |
96 | 3f | 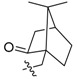 |
48 |
| 2b | 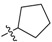 |
34 | 2j | 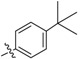 |
88 | 4 | 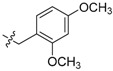 |
43 |
| 2c | 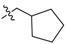 |
52 | 2k | 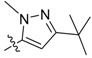 |
72 | 5 | H | 52 |
| 2d | 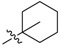 |
96 | 3a | 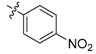 |
65 | 6 | 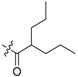 |
43 |
| 2e | 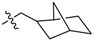 |
83 | 3b | 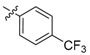 |
86 | 7 | 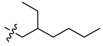 |
37 |
| 2f | 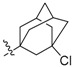 |
39 | 3c | 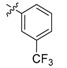 |
85 | 16a | 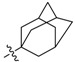 |
96 |
| 2g | 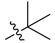 |
81 | 3d | 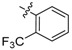 |
87 | 16b | 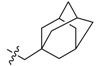 |
84 |
| 2h | 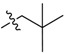 |
92 | 3e | 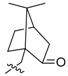 |
82 | BBR | - | 36 |
| PS1145 | - | 73 |
SAR analysis was first focused on the influence of substitutions on position 9 of ring D, by which eleven new ester derivatives (2a–k) were prepared and tested taking PS1145 as the positive control [22]. Different kinds of cyclic carboxylic acid were introduced on 9-hydroxyl by which cyclopropanecarboxylate (2a), cyclopentanecarboxylate (2b), 2-cyclopentylacetate (2c), and 1-methylcyclohexane-1-carboxylate (2d) derivatives were examined for their abilities to inhibit NF-κB activation. An inspiring potency with the inhibitory rate of 96% was given by compound 2d while compounds 2a–c only gave comparable inhibitory rates (34–56%) to BBR (36%). Then, two bridged-ring derivatives (2e,f) were created; norbornane substituted compound 2e exhibited an improved inhibitory effect on NF-κB activation with the rate of 83%, while the activity of chlorinated adamantane compound 2f was only comparable to that of BBR. Finally, three alkyl ring-opening analogues bearing tertiary carbon and quaternary carbon substitutions (2g–i) were prepared and tested, and they gave the improved activities with inhibitory rates of 81–96%. Based on the results above, it was speculated that the introduction of an ester group bearing a suitable tertiary/quaternary carbon substitution at the 9-position was beneficial for the activity. Moreover, the activity was retained when a benzene or unsaturated heterocycle ring (2j,k) was inserted between the ester bond and quaternary carbon substituent.
Meanwhile, 6 sulfonate derivatives (3a–f) were designed to explore further SAR on position 9. Compounds 3a–d, possessing substituted benzenesulfonate, were tested, and they could suppress NF-κB expression by 65–87%. It seemed that trifluoromethyl was a more favorable substitution than a nitro group for the ability to inhibit TNF-α-induced NF-κB activation. Then, 10′-camphorsulfonyl analogues 3e,f with different chiral configurations were generated, and apparently, compound 3e with a d-configuration showed obviously higher activity than its entantiomer 3f, which indicated the possible effect of chiral configuration.
Then, converting the sulfonyl linker to an amine or amide linker, and the generated compounds 4–7 did not show improved activities compared to the lead BBR, and the result might hint that amine and amide were not suitable to be applied as linkers for the inhibitory activity.
Next, the SAR study was conducted for the substituents on the 10-position of ring D. Introducing adamantate at position 10 in BBR, compounds 16a and 16b gave satisfactory potencies with inhibitory rates of 96% and 84%, respectively, which indicated that rigid structure on position 10 might be also favorable for the ability to inhibit TNF-α-induced NF-κB activation.
Based on the preliminary SAR analysis, the IC50 of representative compounds 2d, 2e, 2i and 2j on inhibition of TNF-α-induced NF-κB activation were tested, as listed in Table 2. To evaluate the effect of the novel BBR derivatives on cell viability, the cytotoxic effects of representative compounds 2d, 2e, 2i and 2j on 293T cells were determined by MTT assay. Our results revealed that 2d, 2e, 2i and 2j failed to affect cell viability for 24 h at concentrations up to 20 μM (Figure 2). These data demonstrated that 2d, 2e, 2i and 2j within 20 μM has little cytotoxic effects on 293T cells.
Table 2.
The IC50 of compounds 2d, 2e, 2i and 2j on inhibition of TNF-α-induced NF-κB activation.
| Compound | 2d | 2e | 2i | 2j | PS1145 |
|---|---|---|---|---|---|
| IC50 (ìM) | 1.01 ± 0.19 | 3.91 ± 1.04 | 0.55 ± 0.43 | 10.13 ± 1.40 | 2.493 ± 0.23 |
Figure 2.

Cytotoxic effects of compounds 2d, 2e, 2i and 2j on 293T cells. Following pretreatment with compounds 2d, 2e, 2i and 2j at the indicated concentrations for 24 h, the cell viability of 293T cells were determined by MTT assay.
2.2.2. Preliminary Mechanism Study
Nine key compounds (2d–j, 3b and 16a) with different types of structure were selected to investigate the preliminary mechanism of NF-κB inactivation. Considering that IKK plays a critical role in TNF-α-induced NF-κB activation, the experiment was carried out to verify if the 9 compounds suppressed NF-κB activation through the IKK pathway. The translocation of NF-κB to the nucleus is preceded by the phosphorylation, ubiquitination, and proteolytic degradation of IκBα [23]. To determine whether inhibition of TNF-α-induced NF-κB activation was due to inhibition of IκBα degradation through IKK, we pretreated 293T cells with our representative compounds and then exposed them to TNF-α for 2 h. We then examined the cells for IKK phosphorylation with antibodies specific for phospho-IKKα (Ser180), phosphor-IKKβ (Ser181), and IκBα degradation by Western blot. As shown in Figure 3, TNF-α induced IKKα/β phosphorylation continued to increase at 120 min but had no effect on compounds 2d, 2e, 2h, 2i, 2j and 16a-pretreated cells, this was not the result of the variation of IKK expression, as the total amount of IKK protein remained unchanged during the treatment (Figure 3). In addition, compounds 2d, 2e, 2i and 2j delayed TNF-α-induced degradation of IκBα (Figure 3). Moreover, 2d, 2e, 2i, 2j, as well as the negative control 2f inhibited TNF-α-induced expression of cytokines including IL-6 and IL-8, as depicted in Figure 4. These results demonstrated that compounds 2d, 2e, 2i and 2j inhibited both TNF-α-induced IKK phosphorylation and IκBα degradation, indicating these compounds had great potential as anti-inflammatory agents. IKK serves as a key node in inflammatory signaling, inhibition of which may represent a novel therapeutic target for inflammatory diseases such as rheumatoid arthritis and atherosclerosis. Identification of compounds 2d, 2e, 2i and 2j as novel IKK inhibitors will allow us to better define the potential of using BBR derivatives for the treatment of inflammatory diseases.
Figure 3.

BBR analogues inhibited TNF-α-induced NF-κB activation by impairing IκB kinase (IKK) phosphorylation. Following pretreatment with BBR analogs 2d–j, 3b and 16a (10 μM) for 2 h, 293T cells were treated with TNF-α (20 ng/mL) for 30 min. The phosphorylation of IKKα/β (Ser180/181) and expression levels of IKKα and IκBα were measured by Western blot using corresponding antibodies. GADPH = glyceraldehyde-3-phosphate dehydrogenase.
Figure 4.

Effects of BBR analogues on TNF-α-induced cytokine of interleukin (IL)-6 and IL-8. Following pretreatment with compounds 2d, 2e, 2f, 2i and 2j (10 μM) for 2 h, 293T cells were treated with TNF-α (20 ng/mL) for 6 h, and then the medium was collected for ELISA analysis.
3. Materials and Methods
3.1. Apparatus, Materials, and Analysis Reagents
Antibodies against IκBα, IKKα, phospho-IKKα/β (Ser180/181), and glyceraldehyde-3-phosphate dehydrogenase (GADPH) were purchased from Cell Signaling Technology (Danvers, MA, USA). TNF-α was obtained from R&D Systems (Minneapolis, MN, USA). The p5×nuclear factor (NF)-κB-luciferase reporter plasmid, the pRL-TK plasmid and Dual-luciferase reporter assay system were from Promega (Madison, WI, USA).
Melting point (mp) was obtained with CXM-300 melting point apparatus and was uncorrected. The 1H-NMR spectra was performed on a Varian Inova 500 or 600 MHz spectrometer (Varian, San Francisco, CA, USA) and 13C-NMR on a Bruker Avance III 500 or 600 spectrometer in Dimethyl sulfoxide (DMSO)-d6, or CDCl3, with Me4Si as the internal standard. Electrospray ionization (ESI) high-resolution mass spectra (HRMS) were recorded on an Autospec UItima-TOF mass spectrometer (Micromass UK Ltd., Manchester, UK). Flash chromatography was performed on a Combiflash Rf 200 (Teledyne, NE, USA), particle size 0.038 mm.
3.2. Synthesis
3.2.1. General Procedure for the Synthesis of 2a–k and 3a–f
BBR (3.71 g, 10 mmol) was heated at 195–210 °C for 10–15 min under vacuum (30–40 mmHg) to afford the black oil, which was acidified with ethanol/concentrated HCl (95:5). The solvent was removed by evaporation and the residue was collected and then purified by flash chromatography over silica gel using CH2Cl2/CH3OH as the gradient eluent, giving the title compound 1 (2.85 g, 80%) as an orange solid.
To a stirred solution of 1 (100 mg, 0.28 mmol) in anhydrous CH3CN, triethylamine (175 µL, 1.26 mmol) was added and heated to 70 °C. Then the RCOX/RSO2Cl (1.1–1.2 eq) was added and stirred for 5–6 h. The mixture was cooled to precipitate completely, filtrated and washed with CH2Cl2 to afford target compounds 2a–k and 3a–f.
2,3-Methylenedioxy-9-((cyclopropanecarbonyl)oxy)-10-methoxy protoberberine chloride (2a). Compound 1 (100 mg, 0.28 mmol) was treated with cyclopropanecarbonyl chloride (28 µL, 0.31 mmol) according to the general procedure to give the desired product 2a as a yellow solid, yield: 34%; M.p.: 193–195 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.89 (s, 1H, CHarom), 9.08 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.21 (d, J = 9.2 Hz, 1H, CHarom), 7.82 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.98 (t, J = 6.4 Hz, 2H, CH2), 4.04 (s, 3H, OCH3), 3.22 (t, J = 6.4 Hz, 2H, CH2), 2.13 (tt, J = 7.8, 4.7 Hz, 1H, CH), 1.26–1.16 (m, 4H, 2 × CH2); 13C-NMR (126 MHz, DMSO-d6) δ 172.3, 151.0, 150.6, 148.3, 144.9, 138.7, 134.2, 133.5, 131.5, 127.3, 126.5, 121.8, 121.3, 121.0, 109.1, 106.2, 102.8, 57.9, 56.7, 26.8, 13.4, 10.4(2); HRMS: calcd. for C23H20NO5Cl [M − Cl]+ 390.1336, found 390.1342.
2,3-Methylenedioxy-9-((cyclopentanecarbonyl)oxy)-10-methoxy protoberberine chloride (2b). Compound 1 (100 mg, 0.28 mmol) was treated with cyclopentanecarbonyl chloride (38 µL, 0.31 mmol) according to the general procedure to give the desired product 2b as a yellow solid, yield: 32%; M.p.: 207–209 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.89 (s, 1H, CHarom), 9.07 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.21 (d, J = 9.2 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.96 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.47–3.38 (m, 1H, CH), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.14–2.00 (m, 4H, 2 × CH2), 1.79–1.64 (m, 4H, 2 × CH2); 13C-NMR (126 MHz, DMSO-d6) δ 174.1, 150.9, 150.6, 148.3, 145.1, 138.7, 134.5, 133.5, 131.5, 127.2, 126.5, 121.8, 121.2, 121.0, 109.1, 106.2, 102.8, 57.9, 56.0, 43.6, 30.1(2), 26.8, 26.1(2); HRMS: calcd. for C25H24NO5Cl [M − Cl]+ 418.1649, found 418.1653.
2,3-Methylenedioxy-9-(2′-cyclopentylacetoxy)-10-methoxy protoberberine chloride (2c). Compound 1 (100 mg, 0.28 mmol) was treated with 2-cyclopentylacetyl chloride (42 µL, 0.31 mmol) according to the general procedure to give the desired product 2c as a yellow solid, yield: 38%; M.p.: 186–188 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.94 (s, 1H, CHarom), 9.06 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.21 (d, J = 9.2 Hz, 1H, CHarom), 7.82 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.95 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.88 (d, J = 7.3 Hz, 2H, CH2), 2.41–2.30 (m, 1H, CH), 1.96–1.88 (m, 2H, CH2), 1.74–1.63 (m, 2H, CH2), 1.63–1.53 (m, 2H, CH2), 1.40–1.27 (m, 2H, CH2); 13C-NMR (126 MHz, CDCl3) δ 171.5, 151.3, 151.0, 148.7, 147.3, 138.3, 136.3, 133.1, 131.0, 125.9, 125.5, 122.5, 120.2, 119.7, 108.9, 105.3, 102.4, 57.2, 55.3, 40.5, 36.7, 32.6(2), 27.8, 25.4(2); HRMS: calcd. for C26H26NO5Cl [M − Cl]+ 432.1805, found 432.1808.
2,3-Methylenedioxy-9-((1′-methylcyclohexane-1′-carbonyl)oxy)-10-methoxy protoberberine chloride (2d). Compound 1 (100 mg, 0.28 mmol) was treated with 1-methyl-1-cyclohexanecarboxylic acid (48 mg, 0.34 mmol) according to the general procedure to give the desired product 2d as a yellow solid, yield: 39%; M.p.: 215–217 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.42 (s, 1H, CHarom), 9.09 (s, 1H, CHarom), 8.30 (d, J = 9.2 Hz, 1H, CHarom), 8.22 (d, J = 9.2 Hz, 1H, CHarom), 7.84 (s, 1H, CHarom), 7.12 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.96 (t, J = 6.2 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.21 (t, J = 6.2 Hz, 2H, CH2), 2.24–2.15 (m, 2H, CH2), 1.70–1.34 (m, 11H, 4 × CH2 and CH3); 13C-NMR (126 MHz, DMSO-d6) δ 175.1, 150.8, 150.6, 148.4, 144.4, 138.7, 134.4, 133.7, 131.6, 127.3, 126.6, 121.5, 121.4, 121.0, 109.1, 106.2, 102.8, 57.8, 56.5, 44.0, 35.6(2), 26.8, 26.3, 25.9, 23.1(2); HRMS: calcd. for C27H28NO5Cl [M − Cl]+ 446.1962, found 446.1962.
2,3-Methylenedioxy-9′-(2-(bicyclo[2.2.1]heptan-2-yl)acetoxy)-10-methoxy protoberberine chloride (2e). Compound 1 (100 mg, 0.28 mmol) was treated with 2-norbornaneacetic acid (49 µL, 0.34 mmol) according to the general procedure to give the desired product 2e as a yellow solid, yield: 24%; M.p.: 189–191 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 10.01–9.94 (m, 1H, CHarom), 9.07 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.21 (d, J = 9.2 Hz, 1H, CHarom), 7.82 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.96 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.83 (dd, J = 16.0, 7.5 Hz, 1H, CH2), 2.72 (dd, J = 16.0, 7.5 Hz, 1H, CH2), 2.29–2.24 (m, 1H, CH), 2.18–2.14 (m, 1H, CH2), 2.05–1.97 (m, 1H, CH2), 1.62–1.44 (m, 4H, 2 × CH2), 1.29–1.12 (m, 4H, 2 × CH2); 13C-NMR (126 MHz, DMSO-d6) δ 170.5, 151.0, 150.6, 148.3, 145.1, 138.7, 134.2, 133.5, 131.5, 127.3, 126.5, 121.8, 121.2, 121.0, 109.1, 106.2, 102.8, 57.8, 55.9, 41.2, 40.8, 38.6, 38.0, 36.9, 35.5, 30.1, 28.9, 26.8; HRMS: calcd. for C28H28NO5Cl [M − Cl]+ 458.1962, found 458.1963.
2,3-Methylenedioxy-9-((3′-chloroadamantane-1′-carbonyl)oxy)-10-methoxy protoberberine chloride (2f). Compound 1 (100 mg, 0.28 mmol) was treated with 3-chloroadamantane-1-carboxylic acid (73 mg, 0.34 mmol) according to the general procedure to give the desired product 2f as a brown solid, yield: 33%; M.p.: 206–208 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.63 (s, 1H, CHarom), 9.08 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.22 (d, J = 9.2 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.12 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.99 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.56 (s, 2H CH2), 2.38–2.31 (m, 2H, CH2), 2.21–2.17 (m, 4H, 2 × CH2), 2.17–2.08 (m, 4H, 2 × CH2), 1.79–1.67 (m, 2H, 2 × CH); 13C-NMR (126 MHz, DMSO-d6) δ 173.2, 150.6, 150.6, 148.4, 144.6, 138.7, 134.3, 133.6, 131.56, 127.4, 126.5, 121.5, 121.4, 121.0, 109.1, 106.2, 102.8, 69.1, 58.0, 56.2, 48.0, 46.7(2), 45.4, 37.2(2), 34.3, 31.2(2), 26.8; HRMS: calcd. for C30H29NO5Cl2 [M − Cl]+ 518.1728, found 518.1733.
2,3-Methylenedioxy-9-pivaloyloxy-10-methoxy protoberberine chloride (2g). Compound 1 (100 mg, 0.28 mmol) was treated with pivaloyl chloride (38 µL, 0.31 mmol) according to the general procedure to give the desired product 2g as a yellow solid, yield: 32%; M.p.: 205–207 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.58 (s, 1H, CHarom), 9.09 (s, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 8.22 (d, J = 9.2 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.12 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.99 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.22 (t, J = 6.3 Hz, 2H, CH2), 1.47 (s, 9H,(CH3)3); 13C-NMR (151 MHz, CD3OD) δ 175.7, 150.9, 150.9, 148.5, 143.4, 138.8, 134.7, 133.7, 130.6, 126.4, 125.5, 121.7, 120.7, 120.3, 107.9, 105.2, 102.3, 56.3, 56.1, 39.3, 26.6, 26.2(3); HRMS: calcd. for C24H24NO5Cl [M − Cl]+ 406.1649, found 406.1653.
2,3-Methylenedioxy-9-((3′,3′-dimethylbutanoyl)oxy)-10-methoxy protoberberine chloride (2h). Compound 1 (100 mg, 0.28 mmol) was treated with 3,3-dimethylbutyryl chloride (43 µL, 0.31 mmol) according to the general procedure to give the desired product 2h as a yellow solid, yield: 27%; M.p.: 212–214 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.96 (s, 1H, CHarom), 9.07 (s, 1H, CHarom), 8.30 (d, J = 9.2 Hz, 1H, CHarom), 8.22 (d, J = 9.2 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.96 (t, J = 6.3 Hz, 2H, CH2), 4.04 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.75 (s, 2H, CH2), 1.18 (s, 9H, (CH3)3); 13C-NMR (126 MHz, DMSO-d6) δ 169.5, 151.0, 150.6, 148.3, 145.1, 138.7, 134.1, 133.6, 131.5, 127.3, 126.5, 121.8, 121.2, 121.0, 109.1, 106.2, 102.8, 57.7, 55.9, 47.6, 31.4, 30.0(3), 26.8; HRMS: calcd. for C25H26NO5Cl [M − Cl]+ 420.1805, found 420.1813.
2,3-Methylenedioxy-9-((2′-propylpentanoyl)oxy)-10-methoxy protoberberine chloride (2i). Compound 1 (100 mg, 0.28 mmol) was treated with 2-propylpentanoyl chloride (58 µL, 0.34 mmol) according to the general procedure to give the desired product 2i as a yellow solid, yield: 24%; M.p.: 199–201 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.68 (s, 1H, CHarom), 9.10 (s, 1H, CHarom), 8.30 (d, J = 9.2 Hz, 1H, CHarom), 8.23 (d, J = 9.2 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.95 (t, J = 6.3 Hz, 2H, CH2), 4.02 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.95 (p, J = 6.6 Hz, 1H, CH), 1.88–1.79 (m, 2H, CH2), 1.72–1.63 (m, 2H, CH2), 1.56–1.42 (m, 4H, 2 × CH2), 0.99 (t, J = 7.3 Hz, 6H, 2 × CH3); 13C-NMR (126 MHz, DMSO-d6) δ 173.4, 150.9, 150.6, 148.4, 144.6, 138.7, 134.1, 133.6, 131.5, 127.3, 126.5, 121.6, 121.3, 121.0, 109.1, 106.2, 102.8, 57.8, 56.2, 44.4, 33.6(2), 26.8, 20.3(2), 14.7(2); HRMS: calcd. for C27H30NO5Cl [M − Cl]+ 448.2118, found 448.2128.
2,3-Methylenedioxy-9-((p-tert-butylbenzoyl)oxy)-10-methoxy protoberberine chloride (2j). Compound 1 (100 mg, 0.28 mmol) was treated with 4-tert-butylbenzoyl chloride (61 µL, 0.31 mmol) according to the general procedure to give the desired product 2j as a brown solid, yield: 28%; M.p.: 205–207 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 10.00 (s, 1H, CHarom), 9.11 (s, 1H, CHarom), 8.35 (d, J = 9.2 Hz, 1H, , CHarom), 8.27 (d, J = 9.2 Hz, 1H, CHarom), 8.20 (d, J = 8.5 Hz, 2H, 2 × CHarom), 7.85 (s, 1H, CHarom), 7.72 (d, J = 8.5 Hz, 2H, 2 × CHarom), 7.10 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.91 (t, J = 6.3 Hz, 2H, CH2), 4.03 (s, 3H, OCH3), 3.20 (t, J = 6.3 Hz, 2H, CH2), 1.38 (s, 9H, (CH3)3); 13C-NMR (126 MHz, DMSO-d6) δ 164.0, 158.6, 151.1, 150.6, 148.4, 145.1, 138.8, 134.3, 133.6, 131.5(2), 131.1, 127.5, 126.6(2), 126.5, 125.9, 122.0, 121.3, 121.0, 109.1, 106.2, 102.8, 57.9, 55.8, 35.8, 31.5(3), 26.8; HRMS: calcd. for C30H28NO5Cl [M − Cl]+ 482.1962, found 482.1962.
2,3-Methylenedioxy-9-((3′-tert-butyl-1′-methyl-1H-pyrazole-5′-carbonyl)oxy)-10-methoxy protoberberine chloride (2k). Compound 1 (100 mg, 0.28 mmol) was treated with 5-tert-butyl-2-methylpyrazole-3-carbonyl chloride (59 µL, 0.34 mmol) according to the general procedure to give the desired product 2k as a yellow solid, yield: 42%; M.p.: 207–209 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 10.03 (s, 1H, CHarom), 9.12 (s, 1H, CHarom), 8.36 (d, J = 9.2 Hz, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 7.84 (s, 1H, CHarom), 7.15 (s, 1H, CHpyrazole), 7.11 (s, 1H, CHarom), 6.19 (s, 2H, OCH2O), 4.92 (t, J = 6.4 Hz, 2H, CH2), 4.13 (s, 3H, NCH3), 4.06 (s, 3H, OCH3), 3.22 (t, J = 6.4 Hz, 2H, CH2), 1.34 (s, 9H, (CH3)3); 13C-NMR (126 MHz, DMSO-d6) δ 160.5, 157.2, 151.1, 150.7, 148.4, 145.1, 138.9, 133.6, 133.1, 131.5, 131.2, 127.9, 126.5, 121.8, 121.3, 121.0, 109.6, 109.1, 106.2, 102.8, 58.0, 55.9, 40.0, 32.5, 31.0(3), 26.8; HRMS: calcd. for C28H28N3O5Cl [M − Cl]+ 486.2023, found 486.2027.
2,3-Methylenedioxy-9-((p-nitrophenylsulfonyl)oxy)-10-methoxy protoberberine chloride (3a). Compound 1 (100 mg, 0.28 mmol) was treated with 4-nitrobenzenesulfonyl chloride (75 mg, 0.34 mmol) according to the general procedure to give the desired product 3a as an orange solid, yield: 31%; M.p.: 146–148 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.69 (s, 1H, CHarom), 9.14 (s, 1H, CHarom), 8.52 (d, J = 8.6 Hz, 2H, 2 × CHarom), 8.30 (d, J = 9.3 Hz, 1H, CHarom), 8.27 (d, J = 8.6 Hz, 2H, 2 × CHarom), 8.22 (d, J = 9.3 Hz, 1H, CHarom), 7.83 (s, 1H, CHarom), 7.13 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 4.98 (t, J = 6.3 Hz, 2H, CH2), 3.69 (s, 3H, OCH3), 3.22 (t, J = 6.3 Hz, 2H, CH2); 13C-NMR (126 MHz, DMSO-d6) δ 151.8, 151.8, 150.7, 148.2, 144.2, 140.4, 139.3, 133.9, 131.6, 131.1, 130.8(2), 129.3, 126.5, 125.4(2), 122.1, 121.5, 120.6, 108.9, 106.1, 102.7, 57.3, 56.1, 26.6; HRMS: calcd. for C25H19N2O8SCl [M − Cl]+ 507.0856, found 507.0860.
2,3-Methylenedioxy-9-((p-trifluoromethylphenyl sulfonyl)oxy)-10-methoxy protoberberine chloride (3b). Compound 1 (100 mg, 0.28 mmol) was treated with 4-(trifluoromethyl)benzene-1-sulfonyl chloride (83 mg, 0.34 mmol) according to the general procedure to give the desired product 3b as a yellow solid, yield: 41%; M.p.: 190–191 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.70 (s, 1H, CHarom), 9.17 (s, 1H, CHarom), 8.30 (d, J = 9.2 Hz, 1H, CHarom), 8.24–8.20 (m, 4H, 4 × CHarom), 8.15 (d, J = 8.5 Hz, 2H, 2 × CHarom), 7.84 (s, 1H, CHarom), 7.13 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 4.98 (t, J = 6.3 Hz, 2H, CH2), 3.23 (t, J = 6.3 Hz, 2H, CH2); 13C-NMR (126 MHz, DMSO-d6) δ 152.0, 150.9, 148.4, 144.5, 139.5, 139.2, 135.3, 134.1, 131.7, 131.3, 130.4(2), 129.5, 127.6(2), 126.6, 123.8, 122.3, 121.7, 120.8, 109.1, 106.3, 102.9, 57.4, 56.3, 26.8; HRMS: calcd. for C26H19F3NO6SCl [M − Cl]+ 530.0879, found 530.0884.
2,3-Methylenedioxy-9-((m-trifluoromethylphenylsulfonyl)oxy)-10-methoxy protoberberine chloride (3c). Compound 1 (100 mg, 0.28 mmol) was treated with 3-(trifluoromethyl)benzenesulfonyl chloride (55 µL, 0.34 mmol) according to the general procedure to give the desired product 3c as an orange solid, yield: 37%; M.p.: 184–186 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.71 (s, 1H, CHarom), 9.18 (s, 1H, CHarom), 8.31–8.26 (m, 2H, 2 × CHarom), 8.17 (d, J = 9.4 Hz, 1H, CHarom), 8.13 (t, J = 7.7 Hz, 1H, CHarom), 8.07–8.03 (m, 1H, CHarom), 7.99–7.94 (m, 1H, CHarom), 7.13 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 5.04 (t, J = 6.4 Hz, 2H, CH2), 3.46 (s, 3H, OCH3), 3.24 (t, J = 6.4 Hz, 2H, CH2); 13C-NMR (126 MHz, DMSO-d6) δ 151.5, 150.7, 148.2, 144.2, 139.3, 136.4, 134.0, 133.8, 133.9, 133.0, 131.6, 131.4, 129.7, 129.3, 127.8, 126.3, 122.9, 122.4, 121.6, 120.6, 108.9, 106.1, 102.7, 57.2, 56.2, 26.6; HRMS: calcd. for C26H19F3NO6SCl [M − Cl]+ 530.0879, found 530.0883.
2,3-Methylenedioxy-9-((o-trifluoromethylphenylsulfonyl)oxy)-10-methoxy protoberberine chloride (3d). Compound 1 (100 mg, 0.28 mmol) was treated with 2-(trifluoromethyl)benzenesulfonyl chloride (52 µL, 0.34 mmol) according to the general procedure to give the desired product 3d as an orange solid, yield: 38%; M.p.: 183–185 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.73 (s, 1H, CHarom), 9.17 (s, 1H, CHarom), 8.36–8.29 (m, 3H, 3 × CHarom), 8.24 (d, J = 1.8 Hz, 1H, CHarom), 8.23 (d, J = 9.4 Hz, 1H, CHarom), 8.01 (t, J = 7.9 Hz, 1H, CHarom), 7.84 (s, 1H, CHarom), 7.13 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 4.98 (t, J = 6.3 Hz, 2H, CH2), 3.66 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2); 13C-NMR (126 MHz, DMSO-d6) δ 151.8, 150.7, 148.2, 144.4, 139.2, 136.3, 134.0, 133.2, 132.8, 132.0, 131.5, 131.1, 130.7, 129.3, 126.3, 125.6, 123.6, 122.2, 121.5, 120.6, 108.9, 106.1, 102.7, 57.3, 56.1, 26.6; HRMS: calcd. for C26H19F3NO6SCl [M − Cl]+ 530.0879, found 530.0880.
2,3-Methylenedioxy-9-((D-(+)-10′-camphorsulfonyl)oxy)-10-methoxy protoberberine chloride (3e). Compound 1 (100 mg, 0.28 mmol) was treated with D(+)-10-camphorsulfonyl chloride (85 mg, 0.34mmol) according to the general procedure to give the desired product 3e as a yellow solid, yield: 20%; M.p.: 193–195 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.77 (s, 1H, CHarom), 9.14 (s, 1H, CHarom), 8.37 (d, J = 9.2 Hz, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 7.84 (s, 1H, CHarom), 7.12 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 5.01 (t, J = 6.4 Hz, 2H, CH2), 4.20 (d, J = 15.0 Hz, 1H ,COCH2), 4.13 (s, 3H, OCH2), 3.98 (d, J = 15.0 Hz, 1H, COCH2), 3.23 (t, J = 6.4 Hz, 2H, CH2), 2.48–2.40 (m, 1H, CH2), 2.39–2.30 (m, 1H, CH2), 2.14 (t, J = 4.5 Hz, 1H, CH), 2.07–1.98 (m, 2H, CH2), 1.73–1.64 (m, 1H, CH2), 1.53–1.44 (m, 1H, CH2), 1.10 (s, 3H, CH3), 0.91 (s, 3H, CH3); 13C-NMR (126 MHz, DMSO-d6) δ 214.2, 152.3, 150.8, 148.4, 144.8, 139.1, 134.0, 132.1, 131.7, 128.6, 127.1, 122.5, 121.5, 120.8, 109.1, 106.2, 102.8, 58.5, 58.1, 56.4, 50.6, 48.8, 42.9, 42.6, 26.9, 26.8, 25.9, 20.0, 19.9; HRMS: calcd. for C29H30NO7SCl [M − Cl]+ 536.1737, found 536.1745.
2,3-Methylenedioxy-9-((L-(−)-10′-camphorsulfonyl)oxy)-10-methoxy protoberberine chloride (3f). Compound 1 (100 mg, 0.28 mmol) was treated with L(−)-10-camphorsulfonyl chloride (85 mg, 0.34 mmol) according to the general procedure to give the desired product 3f as a yellow solid, yield: 21%; M.p.: 201–203 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.76 (s, 1H, CHarom), 9.13 (s, 1H, CHarom), 8.37 (d, J = 9.2 Hz, 1H, CHarom), 8.29 (d, J = 9.2 Hz, 1H, CHarom), 7.84 (s, 1H, CHarom), 7.12 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 5.01 (t, J = 6.4 Hz, 2H, CH2), 4.20 (d, J = 15.0 Hz, 1H, COCH2), 3.97 (d, J = 15.0 Hz, 1H, COCH2), 3.23 (t, J = 6.4 Hz, 2H, CH2), 2.47–2.40 (m, 1H, CH2), 2.38–2.31 (m, 1H, CH2), 2.14 (t, J = 4.5 Hz, 1H, CH), 2.06–1.98 (m, 2H, CH2), 1.73–1.64 (m, 1H, CH2), 1.53–1.46 (m, 1H, CH2), 1.10 (s, 3H), 0.91 (s, 3H); 13C-NMR (126 MHz, DMSO-d6) δ 214.2, 152.3, 150.8, 148.4, 144.8, 139.3, 134.0, 132.1, 131.7, 128.6, 127.9, 122.5, 121.5, 120.8, 109.1, 106.2, 102.8, 58.5, 58.1, 56.4, 50.6, 48.8, 42.9, 42.6, 26.9, 26.8, 25.9, 20.0, 19.9; HRMS: calcd. for C29H30NO7SCl [M − Cl]+ 536.1737, found 536.1742.
3.2.2. Synthesis of 2,3-Methylenedioxy-9-(o,p-dimethoxybenzyl amino)-10-methoxy Protoberberine Chloride (4)
To a stirred solution of BBR (7.4 g, 20 mmol) in 2,4-dimethoxybenzylamine (15 mL, 78 mmol) at 120 °C for 6–8 h. The mixture was cooled to room temperature and washed with acetone (3 × 50 mL) to remove the remaining amine. The reside was purified by flash chromatography over silica gel using CH2Cl2/CH3OH (96.5:3.5) as the gradient eluent to afford red solid 4, yield: 37%; M.p.: 239–240 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 9.98 (s, 1H, CHarom), 8.73 (s, 1H, CHarom), 7.88 (d, J = 8.8, 1H, CHarom), 7.77 (s, 1H, CHarom), 7.52 (d, J = 8.8 Hz, 1H, CHarom), 7.14 (d, J = 8.3 Hz, 1H, CHarom), 7.09 (s, 1H, CHarom), 6.52 (d, J = 2.2 Hz, 1H, CHarom), 6.44 (t, J = 6.5 Hz, 1H, NH), 6.42–6.39 (m, 1H, CHarom), 6.17 (s, 2H, OCH2O), 4.80 (t, J = 6.2 Hz, 2H ,NCH2), 4.66 (d, J = 6.3 Hz, 2H, CH2), 3.87 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 3.71 (s, 3H, OCH3), 3.21 (t, J = 6.3 Hz, 2H, CH2); 13C-NMR (126 MHz, DMSO-d6) δ 160.5, 158.5, 150.1, 148.2, 148.1, 147.1, 137.5, 136.3, 133.5, 130.9, 130.3, 124.8, 121.2, 120.5, 120.3, 118.0, 117.7, 109.1, 105.9, 104.8, 102.6, 98.9, 57.5, 56.0, 55.8, 55.6, 47.0, 27.3; HRMS: calcd. for C28H27N2O5Cl [M − Cl]+ 471.1914, found 471.1919.
3.2.3. Synthesis of 2,3-Methylenedioxy-9-amino-10-methoxy Protoberberine Chloride (5)
Compound 4 (3 g, 6.4 mmol) was dissolved in CH3OH, and hydrochloric acid 3 mL was added. The mixture was stirred for 5–6 h, filtered, and washed with 80% ethanol to afford red solid 5, yield: 80%; M.p.: 212–214 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 10.19 (s, 1H, CHarom), 8.64 (s, 1H, CHarom), 7.84 (d, J = 8.6 Hz, 1H, CHarom), 7.76 (s, 1H, CHarom), 7.32 (d, J = 8.6 Hz, 1H, CHarom), 7.08 (s, 1H, CHarom), 6.89 (s, 2H, NH2), 6.16 (s, 2H, OCH2O), 4.70 (t, J = 6.3 Hz, 2H, CH2), 3.98 (s, 3H, OCH3), 3.20 (t, J = 6.3 Hz, 2H, CH2); 13C-NMR (126 MHz, DMSO-d6) δ 149.8, 148.0, 147.0, 143.9, 138.1, 135.4, 132.1, 130.4, 123.0, 121.2, 119.8, 113.7, 113.3, 108.9, 105.6, 102.4, 56.9, 55.1, 27.2; HRMS: calcd. for C19H17N2O3Cl [M − Cl]+ 321.1233, found 321.1235.
3.2.4. Synthesis of 2,3-Methylenedioxy-9-(2′-propylpentanamido)-10-methoxy Protoberberine Chloride (6)
To a stirred solution of 5 (100 mg, 0.28 mmol) and pyridine (100 µL, 1.24 mmol) in anhydrous CH2Cl2 (5 mL), the 2-propylpentanoyl chloride (143 µL, 0.84 mmol) was added and refluxed for 10–12 h. The solvent was removed by evaporation and purified by flash chromatography over silica gel using CH2Cl2/CH3OH (95:5) as the gradient eluent to give yellow solid 6, yield: 34%; M.p.: 249–251 °C (Dec.); 1H-NMR (600 MHz, DMSO-d6) δ 10.04 (s, 1H, CHarom), 9.26 (s, 1H, CHarom), 8.99 (s, 1H, CHarom), 8.25–8.13 (m, 2H, CHarom), 7.79 (s, 1H, CHarom), 7.09 (s, 1H, CHarom), 6.15 (s, 2H, CHarom), 4.87 (t, J = 6.4 Hz, 2H, OCH2O), 3.99 (s, 3H, OCH3), 3.19 (t, J = 6.4 Hz, 2H, CH2), 2.67 (s, 1H, CH), 1.70–1.59 (m, 2H, CH2), 1.48–1.36 (m, 6H, 3 × CH2), 0.99 – 0.90 (m, 6H, 2 × CH3); 13C-NMR (151 MHz, DMSO-d6) δ 176.0, 154.8, 150.3, 148.1, 146.1, 137.8, 133.7, 131.0, 127.8, 125.8, 124.2, 122.3, 121.2, 120.8, 108.9, 106.0, 102.5, 57.4, 56.4, 45.6, 34.9(2), 26.8, 20.6(2), 14.6(2); HRMS: calcd. for C27H31N2O4Cl [M − Cl]+ 447.2278, found 447.2284.
3.2.5. Synthesis of 2,3-Methylenedioxy-9-(2′-ethylhexyl amino)-10-methoxy Protoberberine Chloride (7)
To a stirred solution of BBR (2.5 g, 6.7 mmol) in 2,4-dimethoxybenzylamine (5 mL, 26 mmol) at 120 °C for 6–8 h. The mixture was cooled to room temperature and washed with acetone (3 × 20 mL) to remove the remaining amine. The reside was purified by flash chromatography over silica gel using CH2Cl2/CH3OH (95:5) as the gradient eluent to afford red solid 7, yield: 41%; M.p.: 219–211 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 10.29 (s, 1H, CHarom), 8.69 (s, 1H, CHarom), 7.88 (d, J = 8.7 Hz, 1H, CHarom), 7.76 (s, 1H, CHarom), 7.44 (d, J = 8.7 Hz, 1H, CHarom), 7.07 (s, 1H, CHarom), 6.54 (t, J = 6.0 Hz, 1H, NH), 6.16 (s, 2H, OCH2O), 4.82 (t, J = 6.3 Hz, 2H, CH2), 3.95 (s, 3H, OCH3), 3.50 (t, J = 6.4 Hz, 2H, NCH2), 3.20 (t, J = 6.3 Hz, 2H, CH2), 1.70–1.62 (m, 1H, CH), 1.45–1.15 (m, 7H, 2 × CH2 and CH3), 0.89–0.80 (m, 7H, 2 × CH2 and CH3); 13C-NMR (126 MHz, DMSO-d6) δ 150.1, 148.2, 147.0, 146.9, 138.1, 136.1, 133.6, 130.8, 124.9, 121.2, 120.2, 117.2, 116.2, 109.0, 105.8, 102.6, 57.4, 55.2, 51.0, 40.4, 30.9, 28.9, 27.3, 24.2, 23.2, 14.6, 11.3; HRMS: calcd. for C27H33N2O3Cl [M − Cl]+ 433.2485, found 433.2489.
3.2.6. Synthesis of 2,3-Methylenedioxy-9-methoxy-10-(adamantane-1′-carbonyl)oxy Protoberberine Chloride (16a)
Compound 15 was obtained according to the reported procedure as an orange solid [18]. Compound 15 (100 mg, 0.28 mmol) was treated with 1-adamantanecarbonyl chloride (61 mg, 0.31 mmol) according to the general procedure to give the desired product 16a as a brown solid, yield: 27%; M.p.: 144–146 °C (Dec.); 1H-NMR (500 MHz, DMSO-d6) δ 10.03 (s, 1H, CHarom), 9.07 (s, 1H, CHarom), 7.98 (d, J = 9.1 Hz, 1H, CHarom), 7.96 (d, J = 9.1 Hz, 1H, CHarom), 7.86 (s, 1H, CHarom), 7.12 (s, 1H, CHarom), 6.20 (s, 2H, OCH2O), 4.97 (t, J = 6.3 Hz, 2H, CH2), 4.10 (s, 3H, OCH3), 3.23 (t, J = 6.3 Hz, 2H, CH2), 2.14–2.07 (m, 9H, 3 × CH and 3 × CH2), 1.80–1.74 (m, 6H, 3 × CH2); 13C-NMR (126 MHz, DMSO-d6) δ 175.5, 151.0, 148.9, 148.4, 146.9, 141.6, 140.5, 138.0, 135.4, 132.0, 123.8, 122.2, 121.0, 120.8, 109.1, 106.4, 102.9, 63.8, 55.8, 41.3, 38.8(3), 36.4(3), 27.9(3), 26.8; HRMS: calcd. for C30H30NO5Cl [M − Cl]+ 484.2118, found 484.2122.
3.2.7. Synthesis of 2,3-Methylenedioxy-9-methoxy-10-(2′-(adamantan-1-yl)acetoxy) Protoberberine Chloride (16b)
Compound 15 (100 mg, 0.28 mmol) was treated with 2-(1-adamantyl)acetyl chloride (66 mg, 0.31 mmol) according to the general procedure to give the desired product 16b as an orange solid, yield: 28%; M.p.: 141–143 °C (Dec.); 1H-NMR (500 MHz, CDCl3) δ 10.85 (s, 1H, CHarom), 8.32 (s, 1H, CHarom), 7.78 (d, J = 8.8 Hz, 1H, CHarom), 7.74 (d, J = 8.8 Hz, 1H, CHarom), 7.39 (s, 1H, CHarom), 6.85 (s, 1H, CHarom), 6.12 (s, 2H, OCH2O), 5.42 (t, J = 6.2 Hz, 2H, CH2), 4.38 (s, 3H, OCH3), 3.32 (d, J = 6.2 Hz, 2H, COCH2), 2.45 (s, 2H, CH2), 2.09–2.04 (m, 3H, 3 × CH), 1.79 (d, J = 2.8 Hz, 6H, 3 × CH2), 1.78–1.70 (m, 6H, 3 × CH2); 13C-NMR (126 MHz, CDCl3) δ 169.4 151.4, 150.0, 148.7, 148.3, 141.6, 140.2, 137.7, 135.3, 131.5, 122.8, 122.1, 120.0, 119.5, 108.9, 105.5, 102.5, 64.2, 55.9, 48.5, 42.5(3), 36.8(3), 33.6, 28.7(3), 27.7; HRMS: calcd. for C31H32NO5Cl [M − Cl]+ 498.2275, found 498.2275.
3.3. Biology Assay
3.3.1. Cell Culture and NF-κB Luciferase Reporter Gene Assay
The 293T cell line was purchased from the American Type Culture Collection (ATCC, VA, USA). Cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) (Gibico, NY, USA) supplemented with 10% fetal bovine serum (Hyclone, UT, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin sulfate and incubated at 37 °C in a humidified atmosphere with 5% CO2.
The 293T cells were co-transfected with pNF-κB-Lucreporter plasmid plus the pRL-TK plasmid using the Vigofect transfection reagent (Vigorous Biotechnology, Beijing, China) as instructed by the manufacturers [23]. After 24 h of transfection, cells were pretreated with 5 μM of BBR analogues for 2 h and then stimulated with TNF-α (20 ng/mL) for 6 h. Following TNF-α treatment, the cells were lysed, and the luciferase activity was determined using the Dual-luciferase reporter assay system (Promega, Madison, CA, USA) according to the manufacture′s protocols. The luciferase activity values were normalized to the expression of the Renilla luciferase and presented as the percentages of luciferase activity measured without TNF-α or BBR analogues treatment.
3.3.2. Western Blot
Western blot was performed as described previously. Briefly, cells were washed with phosphate-buffered saline (PBS) and were lysed in lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM β-glycerophosphate, 5 mM egtazic acid (EGTA), 1 mM sodium pyrophosphate, 5 mM NaF, 1 mM Na3VO4, 0.5% Triton X-100, and 1 mM dithiothreitol (DTT)) supplemented with protease inhibitors (1 mM phenylmethylsulfonyl fluoride (PMSF), 5 μg/mL leupeptin, 5 μg/mL pepstatin A, and 5 μg/mL aprotinin). Proteins were separated by SDS-PAGE and were electrically transferred to a polyvinylidene difluoride membrane. The membranes were blocked for one hour at room temperature in TBS (150 mM NaCl, 20 Mm Tris, pH 7.5, 0.05% Tween-20) containing 5% milk and probed with specific first antibodies for one hour at room temperature. Blots were then incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit (7074, Cell Signaling) or anti-mouse (7076, Cell Signaling) secondary antibody. Immunoreactive proteins were visualized using the ECL detection system (Bio-Rad, Hercules, CA, USA).
3.3.3. Determination of Cytokines Production
The 293T cells cultured in 24-well plates were pretreated with compounds 2d, 2e, 2i and 2j for 2 h and then stimulated with TNF-α (20 ng/mL) for 6 h. Levels of IL-6 and IL-8 in the culture media were quantified using ELISA kits (R&D Systems Inc., MN, USA) in accordance with the manufacturer's instructions.
3.3.4. Cell Survival Assay
The cell survival was evaluated by MTT assay. Briefly, Cell suspensions (100 μL) of 293T cells at concentration of 50% confluence were seeded into the 96-well plates, and then were treated with various concentrations of BBR derivate. After 24 h of incubation, 10 μL of the MTT (1 mg/mL) solution was added into each plate and incubated for 2 h at 37 °C, 5% CO2. Subsequently, the culture supernatant was replaced with 100 μL DMSO to dissolve the formazan crystal made from succinic dehydrogenase in the mitochondria and its substrate MTT. The optical density (OD) at 550 and 630 nm were measured using a microplate reader. The net absorbance (OD630–OD550) indicates the enzymatic activity of mitochondria and provides information on cell viability.
4. Conclusions
To conclude, 23 new BBR analogues defined on substituents of the ring D were synthesized and evaluated for their effect to inhibit TNF-α-induced NF-κB activation. SAR analysis indicated that tertiary or quaternary carbon substitutions on position 9 or rigid fragment on position 10 might be beneficial for the activity. Among them, compounds 2d, 2e, 2i and 2j exhibited satisfactory potency with inhibitory rates of over 90% at the concentration of 5 μM compared with that of BBR. A preliminary mechanism study revealed that all of them could inhibit TNF-α-induced NF-κB activation via impairing IKK phosphorylation as well as TNF-α induced expression of cytokines including IL-6, and IL-8. Our current study supports the potential role of compounds 2d, 2e, 2i and 2j in the prevention and treatment of inflammatory diseases, and they have been selected for the further investigation.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81321004 and 81473248) and CAMS initiative for innovative medicine 2016-12M-1-011.
Supplementary Materials
Author Contributions
Y.-X.W. performed part of synthetic experiments and wrote the paper, L.L. performed the biological assay, Q.-X.Z. and T.-Y.F. performed part of synthetic experiments, J.-D.J., H.-B.D. and D.-Q.S. conceived and designed the experiments.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 2a–k, 3a–f, 4–7 and 16a–b are available from the authors.
References
- 1.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 2.McCulloch C.A., Downey G.P., El-Gabalawy H. Signaling platforms that modulate the inflammatory response: New targets for drug development. Nat. Rev. Drug Discov. 2006;5:864–876. doi: 10.1038/nrd2109. [DOI] [PubMed] [Google Scholar]
- 3.Ferrero-Miliani L., Nielsen O.H., Andersen P.S., Girardin S.E. Chronic inflammation: Importance of NOD2 and NALP3 in interleukin-1β generation. Clin. Exp. Immunol. 2007;147:227–235. doi: 10.1111/j.1365-2249.2006.03261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tilg H., Moschen A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006;6:772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 5.Woolbright B.L., Jaeschke H. Role of the inflammasome in acetaminophen-induced liver injury and acuteliver failure. J. Hepatol. 2017;66:836–848. doi: 10.1016/j.jhep.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fatkhullina A.R., Peshkova I.O., Koltsova E.K. The role of cytokines in the development of atherosclerosis. Biochemistry (Moscow) 2016;81:1358–1370. doi: 10.1134/S0006297916110134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixit V., Mak T.W. NF-κB signaling: Many roads lead tomadrid. Cell. 2002;111:615–619. doi: 10.1016/S0092-8674(02)01166-2. [DOI] [PubMed] [Google Scholar]
- 8.Hayden M.S., Ghosh S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vallabhapurapu S., Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 10.Lau C.W., Yao X.Q., Chen Z.Y., Ko W.H., Huang Y. Cardiovascular actions of berberine. Cardiovasc. Drug Rev. 2001;19:234–244. doi: 10.1111/j.1527-3466.2001.tb00068.x. [DOI] [PubMed] [Google Scholar]
- 11.Tang Q.L., Lai M.L., Zhong Y.F., Wang A.M., Su J.K., Zhang M.Q. Antinociceptive effect of berberine on visceral hypersensitivity in rats. World J. Gastroenterol. 2013;19:4582–4589. doi: 10.3748/wjg.v19.i28.4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang M., Wang C.M., Li J., Meng Z.J., Wei S.N., Li J., Bucala R., Li Y.L., Chen L. Berberine protects against palmitate-induced endothelial dysfunction: Involvements of up regulation of AMPK and eNOS and down regulation of NOX4. Med. Inflamm. 2013;2013:260464. doi: 10.1155/2013/260464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heidarian E., Rafieian-Kopaei M., Khoshdel A., Bakhshesh M. Metabolic effects of berberine on liver phosphatidate phosphohydrolase in rats fed on high lipogenic diet: An additional mechanism for the hypolipidemic effects of berberine. Asian Pac. J. Trop. Biomed. 2014;4:S429–S435. doi: 10.12980/APJTB.4.2014C474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ansari N., Khodagholi F. Natural products as promising drug candidates for the treatment of Alzheimer′s disease: Molecular mechanism aspect. Curr. Neuropharmacol. 2013;11:414–429. doi: 10.2174/1570159X11311040005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Q., Tang Z.H., Peng J., Liao L., Pan L.H., Wu C.Y., Jiang Z.S., Wang G.X., Liu L.S. The dual behavior of PCSK9 in the regulation of apoptosis is crucial in Alzheimer’s disease progression. Biomed. Rep. 2014;2:167–171. doi: 10.3892/br.2013.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X., Gu L., Li J., Shah N., He J., Yang L., Hu Q., Zhou M. Degradation of MDM2 by the interaction between berberine and DAXX leads to potent apoptosis in MDM2-overexpressing cancer cells. Cancer Res. 2010;70:9895–9904. doi: 10.1158/0008-5472.CAN-10-1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pandey M.K., Sung B., Kunnumakkara A.B., Sethi G., Chaturvedi M.M., Aggarwal B.B. Berberine modifies cysteine 179 of IκBα kinase, suppresses nuclear factor-κB–regulated antiapoptotic gene products, and potentiates apoptosis. Cancer Res. 2008;68:5370–5379. doi: 10.1158/0008-5472.CAN-08-0511. [DOI] [PubMed] [Google Scholar]
- 18.Li Y.H., Li Y., Yang P., Kong W.J., You X.F., Ren G., Deng H.B., Wang Y.M., Wang Y.X., Jiang J.D., et al. Design, synthesis, and cholesterol-lowering efficacy for prodrugs of berberrubine. Bioorg. Med. Chem. 2010;18:6422–6428. doi: 10.1016/j.bmc.2010.06.106. [DOI] [PubMed] [Google Scholar]
- 19.Shan W.J., Huang L., Zhou Q., Meng F.C., Li X.S. Synthesis, biological evaluation of 9-N-substituted berberine derivatives as multi-functional agents of antioxidant, inhibitors of acetylcholinesterase, butyrylcholinesterase and amyloid-β aggregation. Eur. J. Med. Chem. 2011;46:5885–5893. doi: 10.1016/j.ejmech.2011.09.051. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y.X., Kong W.J., Li Y.H., Tang S., Li Z., Li Y.B., Shan Y.Q., Bi C.W., Jiang J.D., Song D.Q. Synthesis and structure–activity relationship of berberine analogues in LDLR up-regulation and AMPK activation. Bioorg. Med. Chem. 2012;20:6552–6558. doi: 10.1016/j.bmc.2012.09.029. [DOI] [PubMed] [Google Scholar]
- 21.Boger D.L., Hong J.Y., Hikota M., Ishida M. Total synthesis of phomazarin. J. Am. Chem. Soc. 1999;121:2471–2477. doi: 10.1021/ja983631q. [DOI] [Google Scholar]
- 22.Yemelyanov A., Gasparian A., Lindholm P., Dang L., Pierce J.W., Kisseljov F., Karseladze A., Budunova I. Effects of IKK inhibitor PS1145 on NF-kappaB function, proliferation, apoptosis and invasion activity in prostate carcinoma cells. Oncogene. 2006;25:387–398. doi: 10.1038/sj.onc.1209066. [DOI] [PubMed] [Google Scholar]
- 23.Zhang N., Bi C.W., Liu L., Dou Y.Y., Tang S., Pang W.Q., Deng H.B., Song D.Q. IMB-6G, a novel N-substituted sophoridinic acid derivative, induces endoplasmic reticulum stress-mediated apoptosis via activation of IRE1α and PERK signaling. Oncotarget. 2016;7:23860–23873. doi: 10.18632/oncotarget.8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.