

ABSTRACT
This multinational, randomized, double-blind trial, (ClinicalTrials.gov identifier NCT02149121) was designed to demonstrate equivalence in pharmacokinetics and efficacy between CT-P10 and innovator rituximab (RTX) in patients with rheumatoid arthritis (RA). Adults with active RA were treated with CT-P10, United States-sourced RTX (US-RTX; Rituxan®), or European Union-sourced RTX (EU-RTX; MabThera®) at weeks 0 and 2. The co-primary pharmacokinetic endpoints were area under the serum concentration–time curve (AUC) from time zero to last measurable concentration (AUC0–last), AUC from time zero to infinity (AUC0–∞), and maximum concentration (Cmax) after two infusions. The primary efficacy endpoint was change from baseline to week 24 in Disease Activity Score using 28 joints-C-reactive protein (DAS28-CRP). Pharmacodynamics, immunogenicity, and safety were also assessed. 372 patients were randomly assigned to CT-P10 (n = 161) or RTX (n = 211 [US-RTX, n = 151; EU-RTX, n = 60]). For the co-primary pharmacokinetic endpoints, 90% confidence intervals (CI) for ratios of geometric means (CT-P10/US-RTX, CT-P10/EU-RTX or EU-RTX/US-RTX) all fell within the equivalence margin of 80–125%. Adjusted least squares (LS) mean (standard error) change from baseline in DAS28-CRP at week 24 was −2.13 (0.175) for CT-P10 and −2.09 (0.176) for RTX. The 95% CI (−0.29, 0.21) of the estimated treatment difference between CT-P10 and RTX (−0.04) was entirely within the efficacy equivalence margin of ±0.5. Pharmacodynamics, immunogenicity, and safety profiles were similar for CT-P10 and RTX. The pharmacokinetics of CT-P10, US-RTX, and EU-RTX were equivalent. CT-P10 and RTX were also equivalent in terms of efficacy and displayed similar pharmacodynamic, immunogenicity, and safety profiles up to week 24.
Keywords: Rituximab, CT-P10, rheumatoid arthritis, equivalence, biosimilar
Introduction
Rituximab (RTX) is a monoclonal antibody against B cell surface protein CD20 that is used to treat various B cell-related disorders, including rheumatoid arthritis (RA).1 RTX depletes CD20-expressing B cells, which play a critical role in the pathogenesis of RA.2–4 The efficacy of RTX in combination with methotrexate (MTX) in patients with active RA was established in randomized controlled trials (RCTs) published over a decade ago.5–7
A biosimilar drug is a highly similar version of an already-licensed biological drug, or reference product.8,9 For a biosimilar to be approved, it must be shown that there are no clinically meaningful differences between the two products.8,9 The stepwise, ‘totality of evidence’ approach adopted by regulatory authorities for biosimilars means that the type of clinical studies needed may vary on a case-by-case basis. However, statistically proven equivalence between biosimilar and reference product in both pharmacokinetics (PK) and efficacy are usually required, as is a demonstration of acceptable safety and immunogenicity.8,9
CT-P10 (CELLTRION, Inc., Incheon, Republic of Korea) is a RTX biosimilar approved in Europe and South Korea for the same indications as ‘innovator’ RTX. CT-P10 and RTX share an identical primary structure and have highly similar higher-order structures, post-translational modifications, and in vitro activities.10 A Phase 1 study of CT-P10 in patients with RA demonstrated equivalent PK to European Union-sourced RTX (EU-RTX; MabThera®; Roche, Welwyn Garden City, UK) and comparable efficacy, pharmacodynamics (PD), immunogenicity, and safety up to week 72.10–14 CT-P10 and United States-sourced RTX (US-RTX; Rituxan®; Genentech, Inc., South San Francisco, USA) have also been shown to be similar in patients with follicular lymphoma, a B cell-related hematological malignancy for which RTX is approved.15
This Phase 3 RCT was divided into two parallel parts, each of which assessed different primary endpoints. The objective of Part 1 was to demonstrate PK equivalence of CT-P10, US-RTX, and EU-RTX over 24 weeks. Part 2 aimed to demonstrate efficacy equivalence of CT-P10 and RTX (US-RTX and EU-RTX combined) at week 24 in a larger patient group. PD, immunogenicity, and safety were also assessed.
Results
Patients
In total, 495 patients were screened for the study. The first patient was recruited in August 2014; the last week-24 visit for the final patient was in January 2016. A total of 372 patients were randomly assigned to treatment (CT-P10, n = 161; RTX, n = 211 [US-RTX, n = 151; EU-RTX, n = 60]) (Figure 1). Of these, 345 (92.7%) patients completed the course (CT-P10, n = 145 [90.1%] and RTX, n = 200 [94.8%; US-RTX, n = 142 (94.0%); EU-RTX n = 58 (96.7%)]). Among the 372 patients, 189 (CT-P10, n = 64; US-RTX, n = 65; EU-RTX, n = 60) participated in Part 1 of the study. All patients from Part 1 were included in Part 2 and underwent all assessments performed in Part 2. The most frequently reported reasons for discontinuation in both Parts 1 and 2 were patient withdrawal of consent and adverse events (AEs) (Figure 1). The number of patients included in each treatment group for the PK, efficacy and other study assessments are shown in Figure 1. Demographics and baseline disease characteristics were similar among treatment groups (Table 1).
Figure 1.
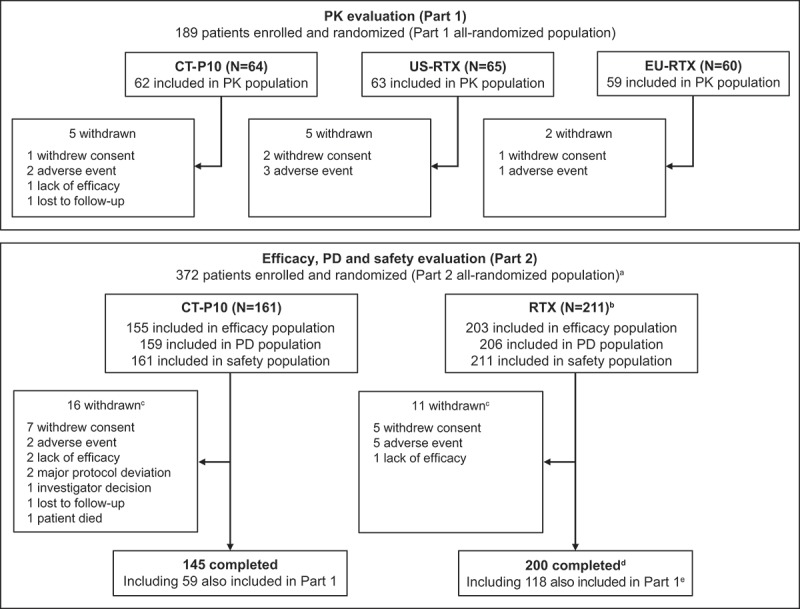
Patient flow and study analysis populations.
aIncludes all 189 patients from Part 1. The study comprised two parts that ran in parallel: Part 1 evaluated PK; Part 2 evaluated efficacy, PD and safety (plus immunogenicity). Patients included in Part 1 were randomly assigned (1:1:1) to CT-P10, US-RTX, or EU-RTX; these patients were also included in the Part 2 assessments. Part 2 also recruited additional patients (N = 183) who were randomly assigned (1:1) to either CT-P10 or US-RTX.b151 US-RTX, 60 EU-RTX. cIncludes withdrawals shown for Part 1. d142 US-RTX, 58 EU-RTX. e60 US-RTX, 58 EU-RTX. EU = European Union. PD = pharmacodynamics. PK = pharmacokinetics. RTX = rituximab. US = United States.
Table 1.
Baseline demographics and disease characteristics (all-randomized populations).
| Part 1 |
Part 2a |
||||
|---|---|---|---|---|---|
| Parameter | CT-P10 (n = 64) |
US-RTX (n = 65) |
EU-RTX (n = 60) |
CT-P10 (n = 161) |
RTXb (n = 211) |
| Age, years | |||||
| Mean (SD) | 52.4 (10.58) | 52.8 (11.84) | 50.8 (10.86) | 51.5 (11.54) | 51.8 (11.14) |
| Gender, n (%) | |||||
| Male | 10 (15.6%) | 14 (21.5%) | 10 (16.7%) | 23 (14.3%) | 31 (14.7%) |
| Female | 54 (84.4%) | 51 (78.5%) | 50 (83.3%) | 138 (85.7%) | 180 (85.3%) |
| Race, n (%) | |||||
| White | 48 (75.0%) | 53 (81.5%) | 41 (68.3%) | 91 (56.5%) | 138 (65.4%) |
| Asian | 4 (6.3%) | 3 (4.6%) | 5 (8.3%) | 12 (7.5%) | 12 (5.7%) |
| Other | 12 (18.8%) | 9 (13.8%) | 14 (23.3%) | 58 (36.0%) | 61 (28.9%) |
|
Height, cm Mean (SD) |
163.8 (9.79) |
165.4 (10.68) |
162.1 (7.55) |
162.1 (9.08) |
162.5 (9.08) |
|
Weight, kg Mean (SD) |
70.6 (17.70) |
76.3 (20.21) |
69.8 (18.12) |
70.6 (17.12) |
71.0 (16.91) |
| Region, n (%) | |||||
| EU | 28 (43.8%) | 31 (47.7%) | 21 (35.0%) | 38 (23.6%) | 65 (30.8%) |
| Non-EU | 36 (56.3%) | 34 (52.3%) | 39 (65.0%) | 123 (76.4%) | 146 (69.2%) |
| RF or anti-CCP status, n (%) | |||||
| RF positive | 53 (82.8%) | 55 (84.6%) | 49 (81.7%) | 127 (78.9%) | 174 (82.5%) |
| Anti-CCP positive | 53 (82.8%) | 55 (84.6%) | 53 (88.3%) | 131 (81.4%) | 178 (84.4%) |
|
SJC at baseline Mean (SD) |
16.3 (7.84) |
14.4 (6.87) |
15.2 (10.42) |
15.3 (7.99) |
14.3 (8.11) |
|
TJC at baseline Mean (SD) |
24.2 (14.06) |
23.4 (13.80) |
22.0 (12.89) |
22.4 (12.84) |
21.8 (12.77) |
| Prior anti-TNF blocker status, n (%) | |||||
| Inadequate response | 55 (85.9%) | 55 (84.6%) | 55 (91.7%) | 137 (85.1%) | 187 (88.6%) |
| Intolerant case | 9 (14.1%) | 10 (15.4%) | 5 (8.3%) | 22 (13.7%) | 24 (11.4%) |
| Duration of prior TNF-antagonist use, months | |||||
| Mean (SD) | 16.4 (22.06) | 13.7 (23.49) | 16.6 (18.74) | 15.5 (19.98)c | 17.0 (27.08) |
| Number of prior TNF-antagonist use, n (%) | |||||
| 0 | 0 | 0 | 0 | 2 (1.2%)d | 0 |
| 1 | 56 (87.5%) | 58 (89.2%) | 49 (81.7%) | 142 (88.2%) | 183 (86.7%) |
| 2 | 8 (12.5%) | 7 (10.8%) | 11 (18.3%) | 17 (10.6%) | 28 (13.3%) |
|
Prior TNF-antagonist used, n (%) |
|||||
| Adalimumab | 23 (35.9%) | 28 (43.1%) | 30 (50.0%) | 52 (32.3%) | 80 (37.9%) |
| Certolizumab | 2 (3.1%) | 5 (7.7%) | 2 (3.3%) | 5 (3.1%) | 11 (5.2%) |
| Etanercept | 21 (32.8%) | 17 (26.2%) | 15 (25.0%) | 55 (34.2%) | 55 (26.1%) |
| Golimumab | 5 (7.8%) | 6 (9.2%) | 7 (11.7%) | 17 (10.6%) | 26 (12.3%) |
| Infliximab | 18 (28.1%) | 15 (23.1%) | 16 (26.7%) | 44 (27.3%) | 65 (30.8%) |
| Unspecifiede | 1 (1.6%)e | 0 | 0 | 1 (0.6%)e | 0 |
| Investigational drug | 2 (3.1%) | 1 (1.5%) | 1 (1.7%) | 2 (1.2%) | 2 (1.0%) |
| Baseline CRP, mg/dL | |||||
| Mean (SD) | 2.2 (3.56) | 2.1 (2.79) | 3.4 (4.99) | 2.2 (3.22) | 2.6 (3.91) |
| Baseline ESR, mm/h | |||||
| Mean (SD) | 54.1 (26.35) | 53.2 (25.38) | 51.5 (20.54) | 54.7 (27.89) | 54.9 (26.67) |
| Baseline B cell count, cells/mcL | |||||
| Mean (SD)f | 200.5 (161.83) | 187.0 (115.92) | 159.7 (119.09) | 201.1 (140.46) | 192.4 (134.36) |
| DAS28-CRP | |||||
| Mean (SD) | 5.8 (0.86) | 5.8 (0.82) | 6.0 (0.86) | 5.8 (0.89) | 5.8 (0.91) |
| DAS28-ESR | |||||
| Mean (SD) | 6.8 (0.76) | 6.7 (0.77) | 6.8 (0.74) | 6.7 (0.82) | 6.7 (0.81) |
| Time since RA diagnosis, yearg | |||||
| Mean (SD) | 9.4 (6.83) | 8.2 (5.34) | 9.9 (7.39) | 10.7 (8.01) | 9.1 (7.41) |
| MTX dose, mg/weekh | |||||
| Mean (SD) | 15.2 (4.93) | 15.5 (5.21) | 15.6 (5.01) | 14.6 (4.34) | 15.0 (4.66) |
CCP = cyclic citrullinated peptide. CRP = C-reactive protein. DAS28 = Disease Activity Score using 28 joints. ESR = erythrocyte sedimentation rate. EU = European Union. MTX = methotrexate. PK = pharmacokinetics. RA = rheumatoid arthritis. RF = rheumatoid factor. RTX = rituximab. SD = standard deviation. SJC = swollen joint count. TJC = tender joint count. TNF = tumour necrosis factor. US = United States.
aAll patients in Part 1 were included in Part 2.
bThe RTX group consists of data from the combined US-RTX and EU-RTX treatment groups.
cn = 159.
dTwo patients had not received prior TNF-antagonist therapy and discontinued due to noncompliance with the inclusion criteria. Patients were excluded from the PK population for not providing at least one post-treatment PK concentration result and were excluded from the primary efficacy analysis for not having information regarding prior anti-TNF blocker status, which was one of the covariates for analysis.
eResults did not report whether the patient received adalimumab or certolizumab in the prior blinded clinical trial.
fPart 1: CT-P10, n = 61; US-RTX, n = 62; EU-RTX, n = 56. Part 2: CT-P10, n = 148; RTX, n = 194.
gCalculated as (date of randomization – date of diagnosis)/365.25.
hAt first infusion of first course.
Pharmacokinetics
Analysis of the co-primary PK endpoints showed that, for all comparisons, the 90% CIs of the ratio of geometric means for area under the serum concentration–time curve (AUC) from time zero to last measurable concentration (AUC0–last), AUC from time zero to infinity (AUC0–∞), and maximum concentration (Cmax) were entirely contained within the margin of 80–125%, indicating PK equivalence between CT-P10, US-RTX, and EU-RTX (Table 2). After two infusions on day 0 and day 14, mean serum concentrations of RTX in the PK population were similar for the CT-P10, US-RTX, and EU-RTX treatment groups (Figure 2). Co-primary and secondary PK endpoint values were similar for all three treatment groups (Table S1).
Table 2.
Analysis for the co-primary PK endpoints (PK population).
| Comparison | Parameter (unit) |
Treatment | na | Geometric LS Meanb | % Ratiob (T/R) |
90% CIb |
|---|---|---|---|---|---|---|
| CT-P10 (T) vs. US-RTX (R) |
AUC0–last (h*µg/mL) |
Test Reference |
62 60 |
162414.81 167309.07 |
97.07 | (88.08, 106.99) |
| AUC0–∞ (h*µg/mL) |
Test Reference |
59 60 |
162377.28 169480.80 |
95.81 | (87.39, 105.04) | |
| Cmax (µg/mL) |
Test Reference |
62 59 |
367.03 386.65 |
94.92 | (89.61, 100.55) | |
| CT-P10 (T) vs. EU-RTX (R) |
AUC0–last (h*µg/mL) |
Test Reference |
62 59 |
162414.81 172450.97 |
94.18 | (85.40, 103.86) |
| AUC0–∞ (h*µg/mL) |
Test Reference |
59 56 |
162377.28 180637.81 |
89.89 | (81.85, 98.72) | |
| Cmax (µg/mL) |
Test Reference |
62 59 |
367.03 412.40 |
89.00 | (84.01, 94.28) | |
| EU-RTX (T) vs. US-RTX (R) |
AUC0–last (h*µg/mL) |
Test Reference |
59 60 |
172450.97 167309.07 |
103.07 | (93.32, 113.85) |
| AUC0–∞ (h*µg/mL) |
Test Reference |
56 60 |
180637.81 169480.80 |
106.58 | (97.03, 117.08) | |
| Cmax (µg/mL) |
Test Reference |
59 59 |
412.40 386.65 |
106.66 | (100.56, 113.13) |
AUC0–last = area under the serum concentration–time curve from time 0 to the last measurable concentration. AUC0–∞ = AUC from time 0 extrapolated to infinity. CI = confidence interval. Cmax = maximum serum concentration after the second infusion. EU = European Union. LS = least squares. PK = pharmacokinetics. R = reference. RTX = rituximab. T = test. US = United States.
aOutliers were excluded from the statistical analysis (n = 3 in US-RTX for AUC0–last and Cmax and n = 2 in US-RTX for AUC0–∞). In addition, some patients were excluded from the statistical analysis for AUC0–∞ since R-square was lower than 0.8 in the terminal phase and the terminal slope could not be calculated (n = 3 in CT-P10; n = 1 in US-RTX; n = 3 in EU-RTX).
bPoint estimates and 90% CIs for differences on the log scale were exponentiated to obtain estimates for ratios of geometric LS means on the original scale.
Figure 2.
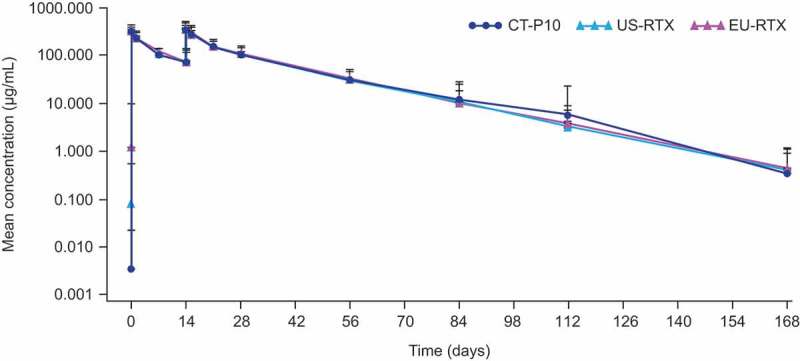
Mean (SD) serum concentration of study drug (PK populationa).
EU = European Union. PK = pharmacokinetics. RTX = rituximab. SD = standard deviation. US = United States.aCT-P10, N = 62; US-RTX, N = 63; EU-RTX, N = 59.
Efficacy
For the primary efficacy endpoint, the adjusted least squares (LS) mean (standard error [SE]) change from baseline in Disease Activity Score using 28 joints-C-reactive protein (DAS28-CRP) at week 24 in the efficacy population was −2.13 (0.175) for CT-P10 and −2.09 (0.176) for RTX (calculated without imputation of missing data, as for other reported efficacy data reported here). The 95% confidence intervals (CI; −0.29, 0.21) of the estimated treatment difference between CT-P10 and RTX (−0.04) were entirely within a systematic literature review-derived statistical equivalence margin of ±0.5, as well as the margin of ±0.6 derived from the pivotal REFLEX study5 and European League Against Rheumatism (EULAR) response criteria. The treatment difference (95% CI) was highly similar in the all-randomized population (−0.04 [−0.28, 0.21]). Mean decreases from baseline in DAS28-CRP over time were similar in the CT-P10 and RTX groups (Figure 3A).
Figure 3.
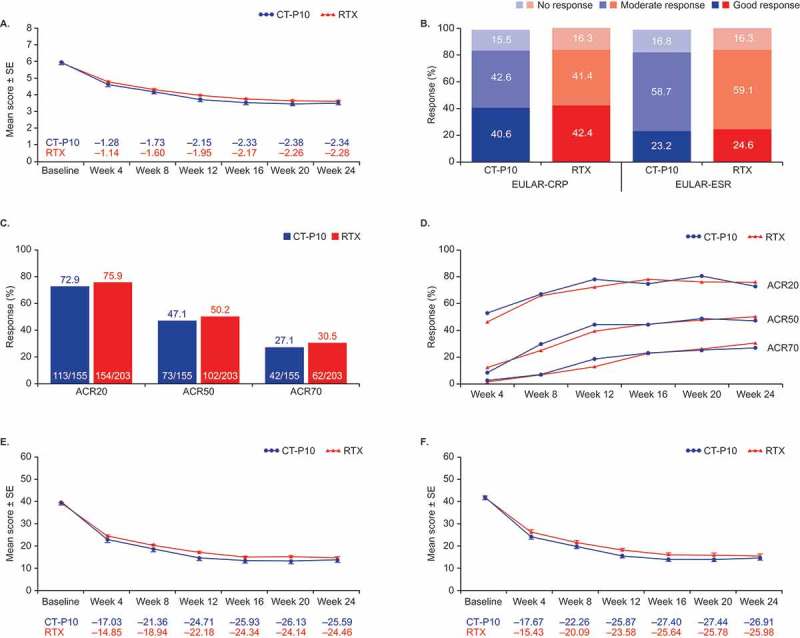
Efficacy outcomes (efficacy populationa). (A) Mean change from baselineb in DAS28-CRP. (B) Proportions of patients with good, moderate, or no EULAR response at week 24c. (C) Proportions of patients achieving clinical response at week 24 according to the ACR20, ACR50, and ACR70 criteria. (D) ACR response over time. (E) Mean change from baseline in clinical disease activity index. (F) Mean change from baseline in simplified disease activity index.
ACR = American College of Rheumatology. CI = confidence interval. CRP = C-reactive protein. DAS28 = Disease Activity Score using 28 joint counts. ESR = erythrocyte sedimentation rate. EULAR = European League Against Rheumatism. RTX = rituximab. SE = standard error. aCT-P10, N = 155; RTX, N = 203. bData shown are non-adjusted arithmetic means. cTwo patients in the CT-P10 group were non-evaluable at week 24 as they had undergone joint surgery during the study and were excluded.
Secondary efficacy outcomes were also similar between groups, including improvement in Disease Activity Score using 28 joints-erythrocyte sedimentation rate (DAS28-ESR) (Figure S1). In the efficacy population, the adjusted LS mean (SE) change from baseline in DAS28-ESR at week 24 was −2.40 (0.180) and −2.35 (0.181) for CT-P10 and RTX, respectively (estimated treatment difference for DAS28-ESR, −0.05; 95% CI, −0.31, 0.20). In the all-randomized population, estimated treatment difference (95% CI) was also −0.05 (−0.31, 0.20). The proportion of patients with a good/moderate EULAR response, or achieving an American College of Rheumatology (ACR) clinical response, up to 24 weeks was similar between the CT-P10 and RTX groups (Figure 3B–3D). The 95% CIs for the estimated treatment difference in ACR20, ACR50, and ACR70 response rates at week 24 demonstrated that there were no statistical differences between groups (Figure 3C). Mean change from baseline in Clinical Disease Activity Index and Simplified Disease Activity Index was comparable between CT-P10 and RTX (Figure 3E and F). The efficacy profile was also similar in a three treatment group comparison (Figure S2).
Pharmacodynamics
Median B cell counts decreased to below the lower limit of quantification (20 cells/μL) immediately after the first infusion and remained below this level up to 24 weeks in all treatment groups. Mean ESR and serum levels of CRP, anti-cyclic citrullinated peptide (CCP), and rheumatoid factor (RF) decreased from baseline at each time point measured in each treatment group. There were no significant differences in PD outcomes between groups (Table S2).
Immunogenicity
The number of patients with anti-drug antibodies (ADAs) at baseline was 19 (11.8%) and 20 (9.5%) in the CT-P10 and RTX groups, respectively. At week 24, ADAs were detected in 24 (14.9%) and 49 (23.2%) patients, in the CT-P10 and RTX groups, respectively. Fourteen patients (seven in each group) were ADA-positive at baseline and week 24. Two patients tested positive for neutralizing antibodies, one at baseline (CT-P10 group [0.6%]) and one at week 24 (US-RTX group [0.7%]).
Safety
A total of 421 AEs were reported up to week 24 in 205 (55.1%) patients, with a similar proportion of patients in each group experiencing AEs (Table 3; Table S3). Most AEs were grade 1 or grade 2 in intensity. The most frequently reported AEs were infusion-related reactions (IRRs) and upper respiratory tract infection. The number of patients considered to have treatment-related AEs was similar in both groups (50 [31.1%] and 60 [28.4%] patients in the CT-P10 and RTX groups, respectively). In total, 19 serious AEs were reported in 18 (4.8%) patients, with a similar proportion in each treatment group (Table 3; Table S4). Five patients from the RTX group, and none from the CT-P10 group, were considered to have experienced a treatment-related serious AE. AEs leading to permanent study drug discontinuation were reported for 3 (1.9%) and 5 (2.4%) patients in the CT-P10 and RTX groups, respectively. The most frequently reported AE leading to permanent study drug discontinuation was IRR, for 2 (1.2%) and 4 (1.9%) patients in the CT-P10 and RTX groups, respectively. Overall, IRRs were reported in 14.3% of patients in the CT-P10 group, 4.6% in the US-RTX group and 20.0% in the EU-RTX group after the first infusion, and 2.6%, 0.7% and 1.7%, respectively, after the second infusion. There were no cases of progressive multifocal leukoencephalopathy or malignancy up to week 24. One death was reported; this occurred in a patient in the CT-P10 group and was not considered related to study drug (Table 3).
Table 3.
Adverse events up to week 24 (safety population).
| Number of patients (%) | CT-P10 (n = 161) |
US-RTX (n = 151) |
EU-RTX (n = 60) |
RTXa (n = 211) |
|---|---|---|---|---|
| AE | 96 (59.6%) | 76 (50.3%) | 33 (55.0%) | 109 (51.7%) |
| Treatment-related | 50 (31.1%) | 38 (25.2%) | 22 (36.7%) | 60 (28.4%) |
| SAE | 9 (5.6%) | 9 (6.0%) | 0 | 9 (4.3%) |
| Treatment-related | 0 | 5 (3.3%) | 0 | 5 (2.4%) |
| IRR | 25 (15.5%) | 8 (5.3%) | 12 (20.0%) | 20 (9.5%) |
| Pruritus | 9 (5.6%) | 3 (2.0%) | 4 (6.7%) | 7 (3.3%) |
| Rash | 8 (5.0%) | 4 (2.6%) | 3 (5.0%) | 7 (3.3%) |
| Throat irritation | 5 (3.1%) | 1 (0.7%) | 3 (5.0%) | 4 (1.9%) |
| Infection | 40 (24.8%) | 36 (23.8%) | 11 (18.3%) | 47 (22.3%) |
| Upper respiratory tract infection | 17 (10.6%) | 18 (11.9%) | 4 (6.7%) | 22 (10.4%) |
| Urinary tract infection | 9 (5.6%) | 5 (3.3%) | 1 (1.7%) | 6 (2.8%) |
| Lower respiratory tract infection | 4 (2.5%) | 7 (4.6%) | 2 (3.3%) | 9 (4.3%) |
| Influenza | 1 (0.6%) | 0 | 2 (3.3%) | 2 (0.9%) |
| Malignancy | 0 | 0 | 0 | 0 |
| Death | 1 (0.6%)b | 0 | 0 | 0 |
| Discontinuation due to AEs | 3 (1.9%) | 4 (2.6%) | 1 (1.7%) | 5 (2.4%) |
AE = adverse event. EU = European Union. IRR = infusion-related reaction. RTX = rituximab. SAE = serious adverse event. US = United States.
Note: The total number of AEs included all patient events. At each level of summarization, a patient was counted only once if they reported one or more events. Only the most severe event was counted.
aThe RTX group consists of data from the combined US-RTX and EU-RTX treatment groups.
bOne patient, who had an ongoing medical history of hypertension, thrombocytosis, and anaemia of chronic disease, initially experienced cellulitis and thrombosis of the right brachial vein and was hospitalized approximately two months after receiving the first dose of study drug. The condition of the patient worsened and the patient died due to acute respiratory distress syndrome approximately three weeks after hospitalization. The death was considered unrelated to the study drug.
Discussion
Developing a biosimilar and obtaining market authorization is a complex process. CT-P10 was the first biosimilar to gain regulatory approval in the EU for RTX indications, and here we present the first full report of a Phase 3 study of CT-P10 in RA. PK equivalence was demonstrated between CT-P10 and both US-RTX and EU-RTX, and equivalent efficacy between CT-P10 and the combined RTX group with similar PD, immunogenicity, and safety profiles displayed up to week 24.
The strengths of this trial include its randomized design, objective and well-established outcome measures, high patient retention rates, and the three-way comparison of PK that included CT-P10 and both licensed reference products (US-RTX and EU-RTX). PK results in this study were similar to those observed in a Phase 1 study comparing CT-P10 to EU-RTX.10 Importantly, in this report the PK was also shown to be equivalent between CT-P10 and US-RTX. Considering the equivalence of EU-RTX and US-RTX shown in Part 1, it is appropriate to assess efficacy equivalence of CT-P10 against a combined RTX group, where both PK and efficacy of CT-P10 could be considered “as equivalent” to each form of RTX as each of these reference products was to each other. Because patients were randomized 1:1:1 for Part 1, and then 1:1 in Part 2, an overall 1:1 distribution of CT-P10- and RTX-treated patients was not created in the efficacy analysis. To address this possible limitation, study part was included as a covariate in the primary efficacy analysis. Results up to week 24 are reported here, but the study is ongoing and longer-term safety and efficacy will be assessed up to week 72.
While this study demonstrated PK equivalence, for the comparison between CT-P10 and EU-RTX, the upper and lower 90% CIs for both Cmax and AUC0–∞ fell below 100%, which may suggest that exposure to CT-P10 was lower than to EU-RTX. Visual inspection of PK data confirmed that the range of serum concentration distribution for CT-P10-treated patients substantially overlapped with that for EU-RTX and US-RTX-treated patients (Figure S3). A trend towards higher serum rituximab levels was observed in a small proportion of EU-RTX-treated patients at the earlier time-points. However, this difference did not result in any overt impact on PK similarity between the three treatment groups. PK similarity between CT-P10 and EU-RTX has previously been established in a Phase 1 study (NCT01534884). We are therefore satisfied that there is sufficient assurance that individual PK concentration levels seen with CT-P10 and reference products are likely due to random fluctuations, and not any intrinsic properties of either drug.10
Previous clinical trials established the efficacy of RTX in combination with MTX in reducing symptoms in patients with RA.5–7 These studies reported variable proportions of RTX-treated patients achieving ACR and EULAR responses at 24 weeks (ACR20, 51–73%; ACR50, 27–43%; ACR70, 12–23%; and EULAR moderate-to-good, 65–83%), and changes from baseline in DAS28-ESR at 24 weeks (−1.90 to −2.60).5–7 The findings of the present study demonstrate not only that the efficacy of CT-P10 is comparable to RTX, but that it is of similar magnitude to that reported in the pivotal RCTs of RTX in combination with MTX.5–7
Overall, 39 and 73 patients were ADA-positive at baseline and at week 24, respectively; 14 were positive at both timepoints. True ADA levels in patients treated with CD20-targeted biologic therapies are difficult to measure due to the potential for false positives in immunogenicity assays, thought to be caused by cell membrane fragments (CMF) expressing CD20.16 The cutoff point for this assay was determined statistically with a false positive rate of 5%, and so the ADA positive rate observed at screening in this study was within the range of false positivity anticipated by Amaravadi et al. (2–11%).17 In this study, confirmatory and titration methods were improved by using ofatumumab, another anti-CD20 antibody, which reduced potential interference of circulating CD20 CMFs by preventing the binding of CD20 in the sample to labelled rituximab reagents in the assay. However, further non-specific binding by other unknown factors remains possible. No difference was observed between groups in ADA-positivity at baseline, and neutralizing antibodies were observed in only two patients. Furthermore, we noted no noticeable trends in PK, PD, efficacy or safety according to baseline ADA-positivity in the current study.
Incidence of IRR in the CT-P10 treatment group after the first infusion (14.3%) was less than observed in the EU-RTX treatment group (20.0%) and greater than in the US-RTX treatment group (4.6%). After the second infusion the incidence of IRR was low in all groups (CT-P10, 2.6%; EU-RTX, 1.7% and US-RTX, 0.7%). The proportions observed in this study are slightly lower than historical studies of RTX where the incidence of IRRs following the first infusion has ranged from 23–32%, and declined following the second infusion (6–9%).5,7,18 However, the present results demonstrate a similar pattern of IRR predominantly occurring during the first infusion.
RTX therapy is an established treatment option for patients with RA with an inadequate response or intolerance to anti-tumour necrosis factor (TNF) biologics.19 Patient access to innovator biologics such as RTX can, however, be restricted by cost, particularly in lower income countries.20 Biosimilars offer financial savings and reductions in time for development and manufacture because the companies developing them can rely on the research efforts implemented for the reference product without jeopardizing efficacy and safety.21 The increased availability of these therapeutic agents due to lower cost may improve patient care and lower the burden on healthcare providers. As such, both patients with RA and healthcare systems are expected to benefit from the development of RTX biosimilars.22
In conclusion, this Phase 3 study demonstrated the PK and efficacy equivalence of CT-P10 and RTX in patients with RA. PD and immunogenicity findings were also comparable between groups, and no unexpected safety concerns were raised.
Patients and methods
Patients
Eligible patients were aged 18–75 years with active RA diagnosed per revised 1987 ACR classification criteria23 ≥6 months before randomization. Active disease was defined by ≥6 swollen joints and ≥6 tender joints, and serum CRP ≥1.5 mg/dL or an ESR ≥28 mm/hour. Patients had received MTX treatment (7.5 to 25 mg/week orally or parenterally) for the past ≥12 weeks, with the last four weeks at a stable dose before screening, and had experienced an inadequate response or were intolerant to anti-TNF agents. Full eligibility criteria are presented in Table S5 (supplementary information).
Study design
This was a randomized, double-blind, parallel-group, active-controlled Phase 3 study conducted in 76 centers in Europe, Asia Pacific, and Latin America (ClinicalTrials.gov identifier NCT02149121). A total of 361 patients were planned to be enrolled in the entire study. Of these, the first 189 study patients were randomly assigned to Part 1 in a 1:1:1 ratio to CT-P10, US-RTX, or EU-RTX for the PK evaluation. All patients in Part 1 were also included in Part 2 of the study. In Part 2, newly enrolled patients (n = 183) were randomly assigned (1:1) to either CT-P10 or US-RTX (Figure 1). Part 2 ran independently and in parallel with Part 1. Further details are provided in online supplementary material A. Patients received two intravenous (IV) infusions of 1,000 mg CT-P10, US-RTX, or EU-RTX separated by a 2-week interval. MTX (7.5–25 mg orally or parenterally, weekly) and folic acid (≥5 mg orally, weekly) were co-administered with study drug. Methylprednisolone (100 mg IV), an antipyretic (acetaminophen/paracetamol 500–1,000 mg, orally), and an antihistamine (chlorpheniramine 2–4 mg [or equivalent], orally) were administered 30–60 minutes before each infusion of study drug.
The study was performed according to the principles of the Declaration of Helsinki and International Conference on Harmonisation Good Clinical Practice guidelines. The study design was approved by an independent ethics committee for each investigational site. All patients provided written informed consent.
Assessments
Blood samples for PK and B cell kinetic analyses were collected from all patients within 15 minutes before and after, and one hour (±15 minutes) after study drug infusion. Additional samples were collected from patients in Part 1 for PK analysis and from all patients for B cell kinetic analysis 24 hours (±2 hours) after the start of each study drug infusion (first infusion only for B cell kinetics), and on days 7, 21, 28, 56, 84, 112, and 168 (see supplementary material B). DAS28 and ACR response were evaluated at baseline and at 4-weekly intervals. EULAR response criteria were derived from DAS28. Safety was monitored throughout the study. Immunogenicity was measured at baseline and at week 24 (see supplementary material C).
Endpoints
The two parts of the study had different primary endpoints. The co-primary endpoints for Part 1 were AUC from time zero to last measurable concentration over both doses of study drug (AUC0–last), AUC from time zero extrapolated to infinity over both doses of study drug (AUC0–∞), and observed maximum concentration after the second infusion of study drug (Cmax). The primary endpoint for Part 2 was the LS mean change from baseline in disease activity measured by DAS28-CRP at week 24. Secondary endpoints included additional PK parameters for Parts 1 and 2, and additional efficacy parameters plus PD, immunogenicity, and safety outcomes for Part 2. A full list is provided in supplementary material D.
Statistical analysis
Part 1 of the study was powered to demonstrate PK equivalence of CT-P10, US-RTX, and EU-RTX in AUC0–last, AUC0–∞, and Cmax. Equivalence was to be demonstrated if the 90% CI for the geometric mean ratio of CT-P10 to US-RTX and EU-RTX in these endpoints was within 80–125%. In Part 1, a sample size of 189 patients (PK population: all patients who received two full doses [at weeks 0 and 2] of study drug and provided at least one post-treatment PK concentration result) was based on overall 90% power, two one-sided α equal to 0.05, an interpatient coefficient of variation in AUC0–last of 30%, and an assumed drop-out rate of 20%. The target sample size for Part 2 of the study (n = 361 in the efficacy population [all patients who received at least one full dose of study drug and provided at least one post-treatment efficacy result]) allowed for a drop-out rate of 10% and provided 82% power for demonstration of similarity based on a 95% CI for the estimate of the difference in change in DAS28 score from baseline at week 24 between CT-P10 and RTX groups. The Part 2 power calculation assumed a clinical equivalence margin of ±0.60, which was sufficient to exclude clinically relevant effects and consistent with a change in DAS28 of ≤0.6 representing ‘no response’ according to EULAR response criteria.24 A tighter statistical equivalence margin of ±0.5 was used for evaluation of the primary efficacy endpoint. This statistical margin was derived from a systematic literature review of DAS28 responses to RTX in patients with RA who had an inadequate response to one or more TNF-antagonists. Using the observed standard deviations of the change in DAS28 with the efficacy population sample size, the post-hoc statistical power was about 95% for the equivalence margin of ±0.5 and the two, one-sided 2.5% α. Data from four RCTs identified in this review were used to determine an appropriate equivalence margin.5–7,18 All patients who received at least one full dose of study drug and provided at least one post-treatment PD result were included in the PD population. All patients that received study drug were included in the safety population.
Primary PK and efficacy endpoints, and PD endpoints, were analyzed using an analysis of covariance (ANCOVA) model with treatment group as a fixed effect and gender, region, race, study part (efficacy endpoints only), prior anti-TNF blocker status, and RF or CCP status fitted as covariates. Additional ANCOVAs of DAS28-ESR data were performed in the efficacy population and of DAS28-CRP and DAS28-ESR data in all patients randomly assigned to treatment. Exact binomial analyses were performed to assess any treatment differences in ACR response rates. Continuous data were described using descriptive statistics, and categorical data were summarized using patient counts and percentages, unless otherwise specified.
PK parameters were calculated by standard non-compartmental methods (linear trapezoidal rule) using Phoenix WinNonlin v6.4. All analyses were conducted using SAS software v9.1.3 (SAS Institute, Cary, NC, USA) or higher.
Supplementary Material
Funding Statement
This work was supported by CELLTRION, Inc. (Incheon, South Korea).
Acknowledgments
Medical writing support (writing assistance, assembling tables and figures, collating author comments, copyediting, grammatical editing, and referencing) was provided by Joanna Chapman PhD at Aspire Scientific Limited (Bollington, UK) and was funded by CELLTRION, Inc. (Incheon, Republic of Korea).
Disclosure of interest
WP received consulting fees from CELLTRION during the conduct of the study. SJL, SYL, and SHK are employees of CELLTRION. DHY is a scientific consultant and on the speaker’s bureau of CELLTRION, and has received research grants not related to this clinical study. PM received fees from CELLTRION for conducting the trial and has performed trials for CELLTRION outside the submitted work. LB-M and DM received fees from CELLTRION for conducting the trial. AK has received fees from CELLTRION for conducting the trial and grants and personal fees from Roche outside the submitted work. ABK, ECE-K, FI-P, FFCM, PS, FMR, PW, SJ, JC-C, OG, TL, DR, PH, NF, OP, MA-B, C-HS, and SCS declare no conflicts of interest.
Contributors
WP, C-HS, SCS, SJL, SYL, SHK, and DHY were involved in conception and design of the study, acquisition of data, and/or analysis and interpretation of data. LB-M, DM, ABK, ECE-K, FI-P, FFCM, PS, PM, FGM-R, PW, SJ, JC-C, OG, TL, DR, PH, AK, NF, OP, and MA-B were involved in the acquisition of data. All authors reviewed drafts of the manuscript and approved the final version.
Abbreviation
- ACR
American College of Rheumatology
- ADAs
anti-drug antibodies
- AEs
adverse events
- ANCOVA
analysis of covariance
- AUC
area under the serum concentration–time curve
- AUC0–last
area under the serum concentration–time curve from time zero to last measurable concentration
- AUC0–∞
area under the serum concentration–time curve from time zero to infinity
- CCP
cyclic citrullinated peptide
- CI
confidence interval
- Cmax
maximum concentration
- CMF
cell membrane fragments
- DAS28-CRP
Disease Activity Score using 28 joints-C-reactive protein
- DAS28-ESR
Disease Activity Score using 28 joints-erythrocyte sedimentation rate
- ESR
erythrocyte sedimentation rate
- EU
European Union
- EULAR
European League Against Rheumatism
- EU-RTX
European Union-sourced rituximab
- IRRs
infusion-related reactions
- IV
intravenous
- LS
least squares
- MTX
methotrexate
- PD
pharmacodynamics
- PK
pharmacokinetics
- RA
rheumatoid arthritis
- RCTs
randomized controlled trials
- RF
rheumatoid factor
- RTX
rituximab
- SE
standard error
- TNF
tumour necrosis factor
- US-RTX
United States-sourced rituximab
References
- 1.European Medicines Agency MabThera (rituximab) [Summary of Product Characteristics]. 2018. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000165/WC500025821.pdf
- 2.Marston B, Palanichamy A, Anolik JH.. B cells in the pathogenesis and treatment of rheumatoid arthritis. Curr Opin Rheumatol. 2010;22(3):307–315. doi: 10.1097/BOR.0b013e3283369cb8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panayi GS. B cells: a fundamental role in the pathogenesis of rheumatoid arthritis? Rheumatology (Oxford). 2005;44 (Suppl 2):ii3–ii7. doi: 10.1093/rheumatology/keh616. [DOI] [PubMed] [Google Scholar]
- 4.Pescovitz MD. Rituximab, an anti-CD20 monoclonal antibody: history and mechanism of action. Am Journal Transplant. 2006;6(5 Part 1):859–866. doi: 10.1111/j.1600-6143.2006.01288.x. [DOI] [PubMed] [Google Scholar]
- 5.Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester G-R, Cravets MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54(9):2793–2806. doi: 10.1002/art.22025. [DOI] [PubMed] [Google Scholar]
- 6.Edwards JCW, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350(25):2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 7.Emery P, Fleischmann R, Filipowicz-Sosnowska A, Schechtman J, Szczepanski L, Kavanaugh A, Racewicz AJ, van Vollehoven RF, Li NF, Agarwal S, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54(5):1390–1400. doi: 10.1002/art.21778. [DOI] [PubMed] [Google Scholar]
- 8.European Medicines Agency Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf
- 9.U.S. Food and Drugs Administration Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. 2015. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf
- 10.Yoo DH, Suh C-H, Shim SC, Jeka S, Cons Molina FF, Hrycaj P, Wiland P, Lee EY, Medina-Rodriguez FG, Shesternya P, et al. A multicentre randomised controlled trial to compare the pharmacokinetics, efficacy and safety of CT-P10 and innovator rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76(3):566–570. doi: 10.1136/annrheumdis-2016-209540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park W, Suh C-H, Shim SC, Cons Molina FF, Jeka S, Medina-Rodriguez FG, Hrycaj P, Wiland P, Lee EY, Shesternya P, et al. Efficacy and safety of switching from innovator rituximab to biosimilar CT-P10 compared with continued treatment with CT-P10: results of a 56-week open-label study in patients with rheumatoid arthritis BioDrugs. 2017;31(4):369–377. doi: 10.1007/s40259-017-0233-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoo DH, Suh C-H, Shim SC, Jeka S, Cons Molina FF, Hrycaj P, Wiland P, Lee EY, Medina-Rodriguez FG, Shesternya P, et al. Efficacy, safety and pharmacokinetics of up to two courses of the rituximab biosimilar CT-P10 versus innovator rituximab in patients with rheumatoid arthritis: results up to week 72 of a phase I randomized controlled trial. BioDrugs. 2017;31(4):357–367. doi: 10.1007/s40259-017-0232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoo DH, Park W, Suh C-H, Shim SC, Jeka S, Cons Molina FF, Hrycaj P, Spieler W, Wiland P, Brzezicki J, et al. Efficacy and safety of rituximab biosimilar candidate (CTP-10) and innovator rituximab in patients with rheumatoid arthritis: results from phase I randomized controlled trial over 72 weeks. 2015 ACR/ARHP annual meeting: abstract number 2058. http://acrabstracts.org/abstract/efficacy-and-safety-of-rituximab-biosimilar-candidate-ct-p10-and-innovator-rituximab-in-patients-with-rheumatoid-arthritis-results-from-phase-i-randomized-controlled-trial-over-72-weeks/
- 14.Yoo DH, Park W, Suh C-H, Shim SC, Cons Molina FF, Jeka S, Brzezicki J, Medina-Rodriguez FG, Hrycaj P, Wiland P, et al. Efficacy and safety of switched CT-P10 from innovator rituximab compared to those of maintained CT-P10 in patients with rheumatoid arthritis up to 56 weeks. 2015 ACR/ARHP annual meeting: abstract number 1675. http://acrabstracts.org/abstract/efficacy-and-safety-of-switched-ct-p10-from-innovator-rituximab-compared-to-those-of-maintained-ct-p10-in-patients-with-rheumatoid-arthritis-up-to-56-weeks/
- 15.Kim WS, Buske C, Ogura M, Jurczak W, Sancho JM, Zhavrid E, Kim JS, Hernández-Rivas JÁ, Prokharau A, Vasilica M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362–e373. doi: 10.1016/S2352-3026(17)30120-5. [DOI] [PubMed] [Google Scholar]
- 16.Chen K, Page JG, Schwartz AM, Lee TN, DeWall SL, Sikkema DJ, Wang C. False-positive immunogenicity responses are caused by CD20+ B cell membrane fragments in an anti-ofatumumab antibody bridging assay. J Immunol Methods. 2013;394(1–2):22–31. doi: 10.1016/j.jim.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 17.Amaravadi L, Song A, Myler H, Thway T, Kirshner S, Devanarayan V, Ni YG, Garofolo F, Birnboeck H, Richards S, et al. 2015 White Paper on recent issues in bioanalysis: focus on new technologies and biomarkers (Part 3–LBA, biomarkers and immunogenicity). Bioanalysis. 2015;7(24):3107–3124 doi: 10.4155/bio.15.226. [DOI] [PubMed] [Google Scholar]
- 18.Emery P, Deodhar A, Rigby WF, Isaacs JD, Combe B, Racewicz AJ, Latinis K, Abud-Mendoza C, Szczepanski LJ, Roschmann RA, et al. Efficacy and safety of different doses and retreatment of rituximab: a randomised, placebo-controlled trial in patients who are biological naive with active rheumatoid arthritis and an inadequate response to methotrexate (Study Evaluating Rituximab’s Efficacy in MTX iNadequate rEsponders (SERENE)). Ann Rheum Dis. 2010;69(9):1629–1635. doi: 10.1136/ard.2009.119933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh JA, Saag KG, Bridges SL, Jr., Akl EA, Bannuru RR, Sullivan MC, Vaysbrot E, McNaughton C, Osani M, Shmerling RH, et al. 2015 American College of Rheumatology Guideline for the treatment of rheumatoid arthritis. Arthritis Care Res. 2016. 2015;68(1):1–25. doi: 10.1002/acr.22783. [DOI] [PubMed] [Google Scholar]
- 20.Putrik P, Ramiro S, Kvien TK, Sokka T, Pavlova M, Uhlig T, Boonen A. Inequities in access to biologic and synthetic DMARDs across 46 European countries. Ann Rheum Dis. 2014;73(1):198–206. doi: 10.1136/annrheumdis-2012-202603. [DOI] [PubMed] [Google Scholar]
- 21.Schellekens H, Smolen JS, Dicato M, Rifkin RM. Safety and efficacy of biosimilars in oncology. Lancet Oncol. 2016;17(11):e502–e509. doi: 10.1016/S1470-2045(16)30374-6. [DOI] [PubMed] [Google Scholar]
- 22.Gulacsi L, Brodszky V, Baji P, Rencz F, Pentek M. The rituximab biosimilar CT-P10 in rheumatology and cancer: a budget impact analysis in 28 European countries. Adv Ther. 2017;34(5):1128–1144. doi: 10.1007/s12325-017-0522-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheumat. 1988;31(3):315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 24.Fransen J, van Riel PL. The Disease Activity Score and the EULAR response criteria. Clin Exp Rheumatol. 2005;23(5 Suppl 39):S93–S99. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.