

ABSTRACT
In mammals, transposable elements are largely silenced, but under fortuitous circumstances may be co-opted to play a functional role. Here, we show that when Alu elements are inserted within or nearby genes in sense orientation, they may contribute to the transcriptome diversity by forming new cleavage and polyadenylation sites. We mapped these new gene ends in human onto the Alu sequence and identified three hotspots of cleavage and polyadenylation site formation. Interestingly, the native Alu sequence does not contain any canonical polyadenylation signal. We therefore studied what evolutionary processes might explain the formation of these specific hotspots of novel gene ends. We show that two of the three hotspots might have emerged from mutational processes that turned sequences that resemble polyadenylation signals into full-blown canonical signals, whereas one hotspot is tightly linked to the process of Alu insertion into the genome. Overall, Alu elements may lie behind the formation of 302 new gene end variants, affecting a total of 243 genes. Intergenic Alu elements may elongate genes by creating a downstream cleavage site, intronic Alu elements may lead to gene variants which code for truncated proteins, and 3′UTR Alu elements may result in gene variants with alternative 3′UTR.
KEYWORDS: Alu elements, exaptation, polyadenylation signals, nicking signals, gene-end, transcriptome repertoire
Introduction
Alu elements are primate-specific non-autonomous retrotransposons of the SINE (Short INterspersed Element) family [1–3]. They are the most abundant transposable elements in human, appearing in more than one million copies and covering more than 10% of the genome [2]. The length of a typical Alu element is about 300 nucleotides and it is made of two arms constructed as left arm-linker-right arm-tail, where the linker and the tail are A-rich sequences [3]. Alu elements can be classified into three large families that slightly differ in their consensus sequence and reflect their evolutionary history, from the most ancient AluJ elements, through the AluS elements, and to the youngest AluY elements [4,5]. In addition, our genome harbors an older family of Alu-like elements, called fossil antique monomers (FAMs), that is 100–200 nucleotides in length. This family arose from a 7SL RNA sequence and gave birth to the free left arm monomer (FLAM) and the free right arm monomer (FRAM) families [6]. Most Alu elements reside in genomic regions where their impact on human biology is negligible. However, there are cases where an Alu element is inserted in the vicinity of a functional region and affects the function of this region. In many cases, this effect is disruptive, and leads to disease [7]. Sometimes, however, the insertion of an Alu element enlarges transcriptome diversity. It had been shown that some intronic Alu elements in antisense orientation triggered the creation of a novel splice junction, leading to the birth of an alternative exon [8–11].
Here, we show how Alu elements that are inserted within or next to a gene in sense orientation, may trigger the formation of a new cleavage and polyadenylation site (PAS), thus generating new transcript variants. Specific examples are given towards the end of the paper. Transcription termination is a two-step process comprising the cleavage of the pre-mRNA followed by polyadenylation, which is the enzymatic addition of a long sequence of adenines to its 3′-end, called the poly(A) tail. This process is aided by several cis regulatory elements at both sides of the cleavage position that direct the factors that participate in the termination process [12-16]. In mammals, the two main regulatory elements are AAUAAA and AUUAAA, located within 40 bases upstream of the cleavage point (though most of them are found 10 to 30 bases upstream of it). These two elements are usually referred to as the canonical polyadenylation signals, and are found in 53–58% (AAUAAA) and 15–17% (AUUAAA) of human polyadenylation sites [17]. Other, weaker, polyadenylation signals have been reported, most of them a single substitution away from the canonical signals [18]. They are called non-canonical polyadenylation signals, and are found in about 10–20% of the polyadenylation sites. In addition to these polyadenylation signals, other auxiliary regulatory elements have been suggested to reside in other locations with respect to the cleavage site. The UpStream Element (USE) is a U-rich region that tend to be associated with UGUA or UAUA elements, and is located within 20 bases upstream of the polyadenylation signal. The DownStream Element (DSE) is a GU- or U-rich element, including sequences such as UGUG and GUGU. DSEs are located within the 40 bases downstream of the cleavage site. The cleavage point itself shows some tendency to be right after a CA dinucleotide, but its position is probably not fixed, and the actual site may vary by up to tens of nucleotides for the same pre-mRNA [12,13].
Previous research already suggested that several types of transposable elements, including Alu sequences, may facilitate the formation of novel cleavage sites [19–23]. In Alu elements, the proximity of the cleavage site to the A-rich parts of the Alu sequence has been pointed out, suggesting that formation of polyadenylation signals in these parts gave rise to the novel cleavage sites [22]. Here, we thoroughly examine human cleavage sites, identify their relation to Alu elements, and analyze the molecular processes that may have led PAS-devoid Alu elements to promote the introduction of new cleavage and polyadenylation sites. We determine the order of evolutionary events that might underlie the creation of these new cleavage sites, and show how they may vary between the different Alu families. We identify three hotspots of cleavage sites within Alu elements, and show that two of them are related to the A-rich regions of the Alu elements, whereas a third is related to the mechanism that inserts Alu elements into the DNA sequence. Finally, we demonstrate the impact of this process on the human genome by examining the expression levels of the newly formed transcripts in comparison with the original transcripts.
Results and disscussion
Alu elements that overlap gene ends tend to be in sense orientation
We have downloaded human genome assembly hg38 annotations, and retrieved 59,413 protein-coding transcripts, as well as 1,238,897 Alu elements (of families AluJ, AluS, AluY, FLAM_A, FLAM_C, FRAM, and FAM). Of these, we focused on the 579,747 Alu elements (46.8%) that overlap gene bodies, and divided them into three groups based on their position relative to the gene: Alu elements that overlap a gene end (a cleavage site of any transcript of the gene, EoG-Alu); Alu elements that overlap an intron-exon junction (Junc-Alu); and Alu elements that reside totally within introns (intronic Alu). Each Alu element was marked as sense or antisense according to its orientation with respect to the gene (Table S1, Fig. 1). Comparing these groups of Alu elements to a control group of intronic Alu elements, we replicated the previous observation [9] that Junc-Alu elements tend to be in antisense orientation (86.1% versus 54.9%; , -test). We also observed, as was noticed previously [22, that EoG-Alu elements tend to be in sense orientation (77.7% versus 45.1%; , -test).
Figure 1.
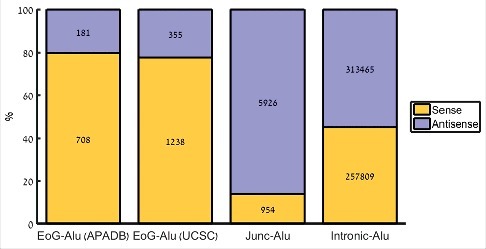
Distribution of Alu element orientation in different genomic positions. EoG-Alu – Alu elements that overlap gene ends; Junc-Alu – Alu elements that overlap splicing junctions; intronic-Alu – Alu elements that reside within introns to their full length. APADB and UCSC refer to the source of gene ends.
Gene end positions, namely the positions of the mRNA cleavage sites of any of the gene's transcripts, show high variability and are difficult to precisely determine, leading to partial and sometimes inaccurate annotations in current databases [24–26]. One source of error is the stochastic nature of the actual position of the cleavage site, which may vary by up to tens of nucleotides [12,13]. Another major source of error is internal priming, whereby during the generation of the cDNA the reverse transcriptase binds to an internal poly(A) stretch rather than to the poly(A) tail, leading to an appearance of a gene end in the middle of the gene. Alu elements are particularly prone to be targets of internal priming, as they harbor two A-rich stretches: in the linker at the middle of the element, and in the tail at its 3′-end. To reduce the level of falsely annotated gene ends, we have used a second list of gene ends, called APADB. APADB is a database that links cleavage and polyadenylation sites to nearby genes. It contains 71,829 human cleavage and polyadenylation sites that were experimentally measured in human using 3′-end sequencing method that is less sensitive to internal priming [27]. Using these data, we observed a similar enrichment of sense EoG-Alu elements (79.6% versus 45.1%; , -test; Fig. 1).
Alu elements harbor three cleavage site hotspots
In order to test whether gene ends tend to form in specific locations along the Alu sequence, we aligned all hg38 EoG-Alu elements to the Alu consensus sequence (AluJo) [5], and mapped the location of the gene ends onto the consensus sequence. This revealed six major hotspots, at positions 6, 118, 132, 141, 281, and 300 with respect to the Alu consensus sequence (Fig. 2A). Curiously, the hotspots at positions 118, 281 and 300 occur immediately upstream of A-rich stretches, raising the possibility that they might be an artifact of internal priming rather than true gene ends. To further examine this, we re-mapped gene ends that come from the APADB database onto the consensus sequence of EoG-Alu elements. This yielded five hotspots, at positions 6, 119, 134, 141, and 283 (Fig. 2B). The three hotspots at positions 6, 132/134, and 141 are apparent in both analyses. The two hotspots just upstream of the Alu tail were either dramatically reduced (position 281/283) or completely erased (position 300) for the APADB data, suggesting that they are indeed a result of internal priming. The hotspot upstream of the A-rich linker (positions 118 and 119 in Fig. 2A & B, respectively) is even stronger when using the APADB data. We hypothesize that this may be an artifact of the APADB annotation protocol that wrongly points at position 119 as the cleavage site in transcripts whose true cleavage site is at position 134. Such errors may stem from the fact that the entire Alu sequence between positions 119 and 134 is A-rich and might be wrongly considered as part of the poly(A) tail (Fig. S1).
Figure 2.
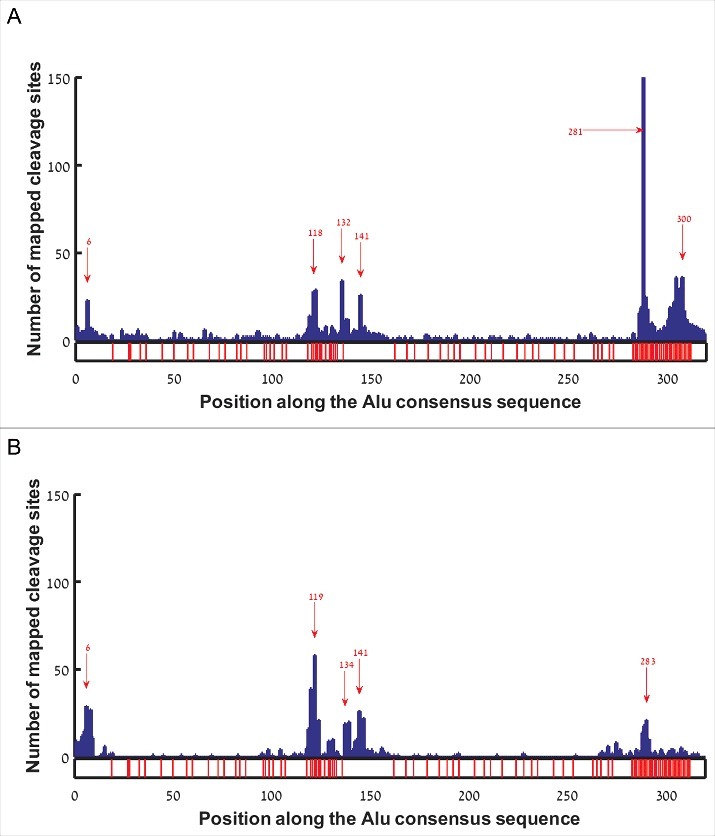
Histogram of gene end positions along the reference Alu sequence in sense orientation. Red arrows mark hotspots. Positions with adenine in the reference Alu sequence are marked by red bars at the bottom bar. (A) EoG-Alu elements based on the hg38 human genome annotations. (B) EoG-Alu elements based on the APADB database.
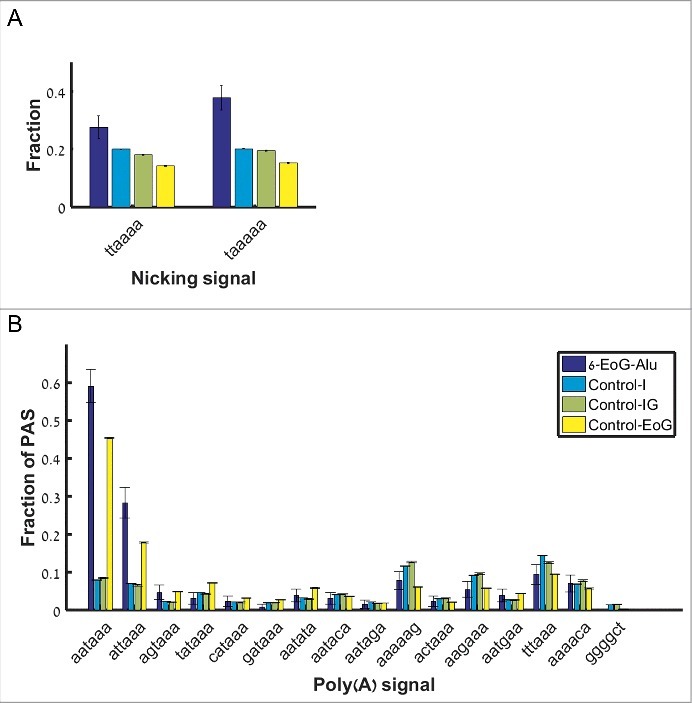
To further authenticate the cleavage site hotspots, we used the fact that genuine gene ends are significantly enriched with the canonical cleavage and polyadenylation signals AAUAAA and AUUAAA within the 40 nucleotides upstream of the cleavage point [17]. From the APADB EoG-Alu elements we isolated the five subgroups of elements where the cleavage point is mapped to the vicinity of one of the hotspots (Table S2), and named them 6-EoG-Alu elements, 119-EoG-Alu elements, etc. As a negative control, we used Alu elements that totally reside within introns (Control-I), and Alu elements that reside in intergenic regions (Control-IG). In each group, we measured the frequency of the canonical polyadenylation signals upstream of the relevant hotspot. As a positive control, we also measured this frequency upstream of genuine gene ends that do not overlap Alu elements (Control-EoG). We found a significant enrichment of canonical polyadenylation signals at hotspots 6, 134, and 141 (Fig. 3A, , one sided Fisher exact test). Interestingly, canonical polyadenylation signals are enriched in these EoG-Alu elements even when compared to Control-EoG ( , 0.028, 0.015 respectively, one sided proportion test). We provide a mechanistic explanation to this observation later in the paper.
Figure 3.
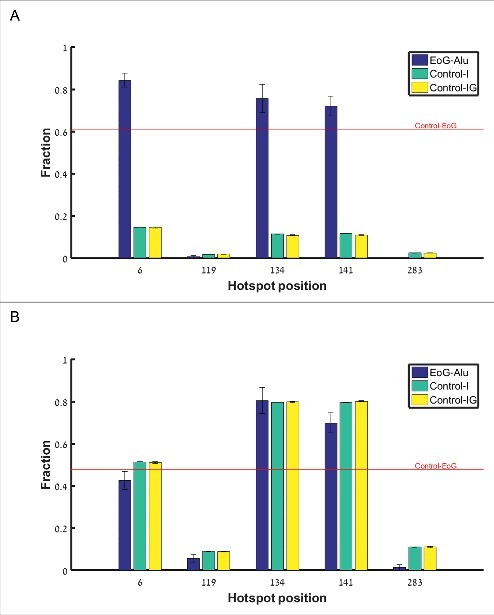
Fraction of polyadenylation signals upstream of gene ends mapped to the different hotspot positions along the reference Alu sequence (i.e., the number of Alu sequences where polyadenylation signals were found divided by the total number of Alu sequences). Blue –EoG-Alu elements; green –intronic Alu elements (Control-I); yellow –intergenic Alu elements (Control-IG). Red horizontal line represents the fraction of polyadenylation signals within the control set of gene ends (Control-EoG). (A) Canonical polyadenylation signals only, UUAUUU and UAAUUU. (B) Fourteen additional non-canonical polyadenylation signals.
For the hotspots at positions 119 and 283, the fraction of upstream canonical polyadenylation signals is very low, and there is no enrichment relative to Control-I and Control-IG (P = 0.94 and 0.999, respectively, one sided Fisher exact test). To test the possibility that these hotspots represent genuine cleavage points that use non-canonical polyadenylation signals (Table S3), we computed also the fraction of non-canonical polyadenylation signals and again found no enrichment (Fig. 3B).
We conclude that the gene ends that map to positions 6, 134, and 141 along the Alu sequence are genuine cleavage sites, whereas those that map to positions 119 and 283 may contain spurious cleavage sites. Here, we wished to apply strict criteria and therefore excluded from further analysis Alu elements where the cleave site was mapped to positions 119 and 283, or where it was mapped outside of any hotspot. We also conclude that gene ends based on APADB are less sensitive to internal priming, and hereinafter we will use APADB data for the analysis.
In total, we found 708 EoG-Alu elements, of them 127 within 6-EoG-Alu, 41 in 134-EoG-Alu and 93 in 141-EoG-Alu (Table S2). Next, we turn into investigating the molecular mechanisms behind the formation of the hotspots at positions 6, 134, and 141.
Cleave sites at 6-EoG-Alu elements arise from the Alu insertion mechanism
Nicking signals are co-opted to serve as polyadenylation signals
The canonical polyadenylation signal associated with cleavage sites at position 6 must reside outside and just upstream of the Alu sequence. Integration of new Alu elements into the genome starts when the L1 endonuclease recognizes the motif TTAAAA, and nicks the DNA in the opposite strand at the position corresponding to the junction between the T and the A. Then, a second nick occurs in the original strand 15–16 nucleotides downstream. Following these DNA breaks, an Alu RNA binds to the stretch of T's, and the DNA is complemented [28,29]. Hence, we expect to see the hexamer TTAAAA, known as the nicking signal, ∼18 bases upstream of the Alu insertion site. Notably, the nicking signal shows great similarity to the canonical polyadenylation signal ATTAAA, and is located in just the right distance from the cleavage point at the sixth base of the Alu sequence. To test whether indeed the nicking and the polyadenylation signals overlap, we identified both for the 6-EoG-Alu elements and for the Control-I Alu elements. As expected, we found that the spatial distributions of the two signals strongly overlap, with their modes located about 20 nucleotides upstream of the Alu insertion site (Fig. S2).
The sequence similarity between the nicking signal and the polyadenylation signal, combined with their co-localization with respect to the Alu element, suggest that a nicking signal that recruited an Alu element bears the potential to be later recognized as a polyadenylation signal, inducing a gene end at position 6 of the Alu element.
There are many ways by which the nicking signal TTAAAA can turn into a polyadenylation signal following mutations, insertions or deletions. However, the simplest way is if the nicking signal comes right after an A. Clearly, the sequence ATTAAAA serves as both a nicking signal and a canonical (ATTAAA) polyadenylation signal. To test whether we see evidence for such a scenario, we compared the fraction of nicking signals preceded by an A in 6-EoG-Alu elements to Control-I Alu elements, as well as to Alu elements that present gene ends in other positions (134- and 141-EoG-Alu elements). In support of our model, we found a significant enrichment in nicking signals preceded by an A only in 6-EoG-Alu elements (Fig. 4A, , when comparing 6-,134-, and 141-EoG-Alu elements to Control-I Alu elements, Fisher exact test, Table S4).
Figure 4.
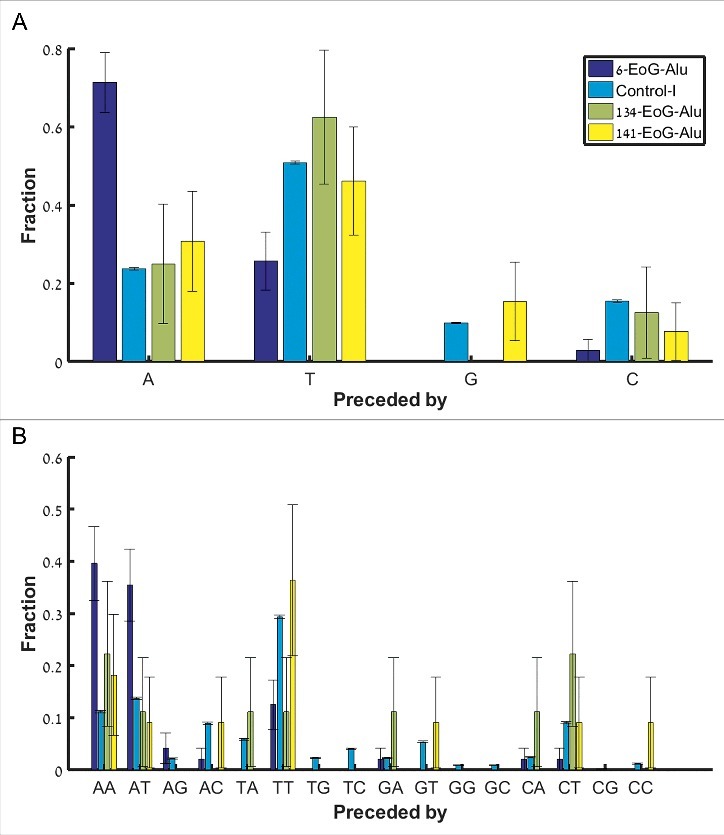
Fraction of nicking signals upstream of the Alu insertion site, split into separate groups (along the x-axis) based on the preceding nucleotide(s). (A) The canonical nicking signal TTAAAA, preceded by either A, C, G, and T. (B) The secondary nicking signal TAAAAA, preceded by each the 16 possible dinucleotides.
In addition to the primary nicking signal, a second variant TAAAAA may be in use. In this case, it may also be a canonical polyadenylation signal if preceded by the dinucleotides AA or AT. Indeed, we found a significant enrichment in these two cases within the 6-EoG-Alu elements, but not in other EoG-Alu elements (Fig. 4B, for AA and for AT, when comparing 6-, 134-, and 141-EoG-Alu elements to Control-I Alu elements, Fisher exact test; Table S5). In addition to fortuitous genomic context of the nicking signal, there are many possible ways by which a nicking signal can be mutated into a polyadenylation one, although the precise succession of these events is difficult to infer for any given Alu sequence (Table S6).
Nicking signals co-opted to be polyadenylation signals are more conserved
We prepared sequence logos of the region 40 bases upstream of the Alu element for the 6-EoG-, Control-I and Control-IG Alu elements (Fig. 5). The AT-rich nicking signal is well apparent in all groups, but is clearly more prominent for the 6-EoG-Alu elements. This is compatible with the notion that nicking signals that had been co-opted to serve as polyadenylation signals, especially those that did so by means of their genomic context, would be under stronger pressure to conserve their sequence. To test this we computed, for the different groups of Alu elements, the fraction of the two canonical nicking signals upstream of the Alu insertion site. Indeed, we saw that the two nicking signals are significantly enriched in 6-EoG-Alu elements ( for TTAAAA and for TAAAAA, compared to Control-I, Control-IG, and Control-EoG, respectively; Fisher exact test, Fig. 6A).
Figure 5.
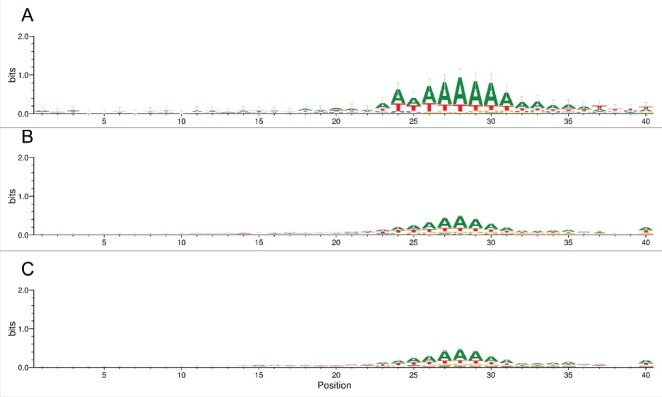
Sequence logo for the 40 bases upstream of the Alu element (The Alu sequence starts at position 41). (A) 6-EoG-Alu elements. (B) Control-I Alu elements. (C) Control-IG Alu elements.
Figure 6.
The frequency of signals upstream of the Alu insertion site, for different groups of Alu elements. (A) The two nicking signals (along the x-axis). (B) The 16 polyadenylation signals (along the x-axis).
Similarly, the leading mechanism that turns a nicking signal into a polyadenylation one (the right genomic context) creates one of the two canonical polyadenylation signals (ATTAAA and AATAAA) or one non-canonical signal (TTTAAA). Hence, we expected to see an enrichment in the use of these polyadenylation signals in 6-EoG-Alu elements even compared to normal gene ends. Indeed, we saw an increased use of the two canonical polyadenylation signals upstream of 6-EoG-Alu elements (for ATTAAA, and for AATAAA, compared to Control-I, Control-IG, and Control-EoG, respectively; Fisher exact test, Fig. 6B), but no preferential use of any of the 14 non-canonical signals. Notably, we do not observe preferential use of the non-canonical signal TTTAAA, probably as a result of the fact that this signal is weakly associated with polyadenylation.
We do not expect to see similar results in EoG-Alu elements where gene ends map to other hotspots, as we indeed confirm (Fig. S3, Table S7). For example, out of the 93 141-EoG-Alu elements only 11 (11.8%) harbor canonical polyadenylation signals upstream of their insertion site ( compared to 61% in Control-EoG; Fisher exact test), and 17 (18.3%) harbor canonical nicking signals ( compared to 15% in Control-I; Fisher exact test).
Polyadenylation signals in 134- and 141-EoG Alu elements arise from mutational events
Polyadenylation signals form spontaneously at the Alu linker and tail A-rich regions
Unlike gene ends at position 6, where the polyadenylation signals reside outside the Alu element, gene ends at positions 134 and 141 have their associated polyadenylation signals within the Alu sequence. However, canonical polyadenylation signals are not present along the reference sequence of any of the Alu families. Therefore, the emergence of canonical polyadenylation signals along Alu sequences must be a result of mutational processes. Spontaneous formation of polyadenylation signals is most likely within the A-rich linker in the middle of the Alu sequence, as well as within the A-rich tail. Indeed, when we scan Control-I Alu elements for polyadenylation signals, we find them almost exclusively in these two regions (Fig. S4).
Mutations that beget a polyadenylation signal at the linker region lead to the gene end hotspots at positions 134 and 141. We hypothesize that polyadenylation signals spontaneously formed at the tail similarly form novel gene ends, but these are not present in our data because they are located downstream of the Alu sequence rather than within it. To test it, we looked at Alu elements that totally reside inside 3′UTRs. As expected, these elements have significantly more polyadenylation signals within their tail when their distance to the gene end is shorter than ∼30 bases (, Fisher exact test, Fig. S5).
EoG-Alu elements share similar mutations
With time, each individual Alu element accumulates mutations that distinguish it from its family reference. To identify mutations that are critical for the formation of polyadenylation signals, we aligned all the 134-EoG- and 141-EoG-Alu elements to the Alu reference sequence (AluJo), and kept track of all mutations (Fig. S6). We applied a two-step alignment procedure, by which each Alu element is first aligned to its family reference, and only then the family references are mapped to the AluJo sequence (see Methods). This procedure ensures that we do not count mutations that underlie the formation of the Alu families themselves. We performed a separate analysis for substitutions, deletions and insertions.
For each position along the reference Alu sequence we counted how many times any possible substitution had occurred, and displayed the result as a sequence logo (Fig. S7). C>T substitutions in the context of a CpG dinucleotide are the predominant mutations, likely promoted by cytosine methylation [30] and are frequently observed in Alu elements [31, 32]. This explains the many apparent CG>TA mutations (C>T mutations in both strands in consecutive positions).
To find the critical substitutions that are over-represented in the 134-EoG and 141-EoG-Alu elements, we calculated P-values using the Fisher exact test for each mutation in every position compared to Control-IG, and corrected for multiple comparisons using Benjamini false discovery rate (FDR) correction. We found a single substitution that is significantly over-represented in 134-EoG-Alu elements, a C>A at position 119 (FDR corrected P-value = , Table S8A). The A-rich linker in most Alu families stretches from position 118 to position 133, and has the consensus sequence ACTAAAAATACAAAAA. A C>T or C>A substitutions at position 119 will generally form a canonical polyadenylation signal and will support a cleavage at position 134, hence their over-representation among the 134-EoG-Alu elements (Fig. 7A, B).
Figure 7.
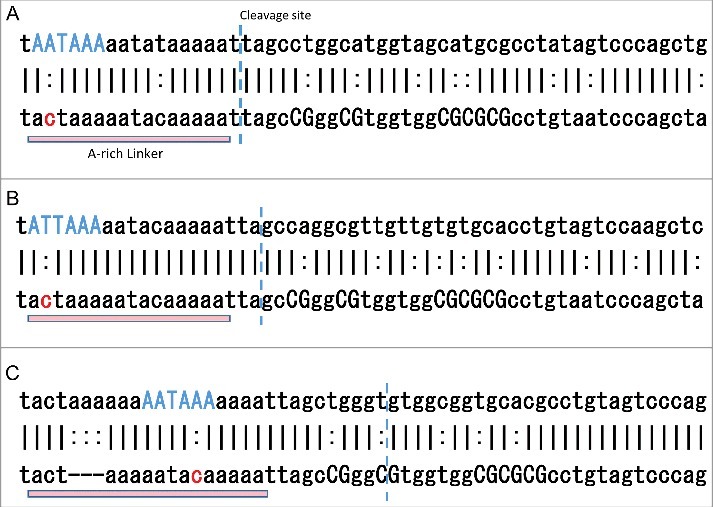
Examples of mutations in specific Alu elements that may underlie the emergence of polyadenylation signals. In each alignment, the top bar shows positions 117–172 of the specific Alu sequence, the bottom bar shows the same positions along its family reference, and the middle bar shows positions that are identical (|) or different (:). The positions of significantly over-represented mutations are marked in red. The canonical polyadenylation signal formed by the mutations is marked in blue. This signal, as well dinucleotide CG positions, are marked with capital letters. The cleavage position is marked by a vertical dashed blue line. (A) An AluSz6 element (chr1:160289488-160289990(−)), member of the 134-EoG-Alu set. A C>A mutation in position 119 creates the canonical polyadenylation signal AATAAA. (B) An AluSx element (chr1:27001322-27001784(+)), member of the 134-EoG-Alu set. A C>T mutation in position 119 creates the canonical polyadenylation signal ATTAAA. (C) An AluSc element (chr6:149590912-149591403(+)), member of the 141-EoG-Alu set. A C>A mutation in position 128 creates the canonical polyadenylation signal AATAAA.
141-EoG-Alu elements are characterized by 17 significantly over-represented substitutions (Table S8B). Of them, seven are upstream of the cleavage point, and 10 are downstream of it. The upstream substitutions usually generate a polyadenylation signal directly. The most enriched substitution is C>A at position 128, which generally creates a canonical polyadenylation signal that support cleavage at position 141 (Fig. 7C). The downstream substitutions are mostly CG>TG. This increases the abundance of TG's downstream of the cleavage site, in agreement with the suggestion that TG-rich downstream elements assist the main polyadenylation signal in promoting polyadenylation [17]. Similarly, significant CG>TG substitutions are found downstream of the cleavage point of 6-EoG-Alu elements probably for the same reason (Table S8C).
We have similarly mapped insertions and deletions onto the Alu reference sequence. Deletions are scarce, and no single deletion was found to significantly characterize EoG-Alu elements (Fig. S8A, B). In contrast, Alu elements tend to have many insertions. Interestingly, many single-nucleotide insertions may create polyadenylation signals within the A-rich linker segment. We could identify several positions upstream of the cleavage point, in which insertions are significantly over-represented in 134-EoG- and 141-EoG-Alu elements (Fig. S8A, B and Table S8D, E).
The fundamental difference between 6-EoG-Alu and 134-/141-EoG-Alu elements in the mechanisms behind gene end formation, suggests that gene ends at position 6 of the Alu sequence should form in equal rates in all Alu families, as all share the same insertion mechanism. In contrast, the mutational processes leading to the formation of polyadenylation signals in 134- and 141-EoG-Alu elements depend on the sequence and on the time available for mutations to accumulate. Therefore, 134- and 141-EoG-Alu elements are expected to show relative frequencies of the Alu families that are different from the relative frequencies within the set of control Alu elements. Indeed, we have validated these expectations (Fig. S9).
Contribution to the transcript repertoire
We identified a total of 708 EoG-Alu elements, of them 127 are 6-EoG-Alu elements, 41 are 134-EoG-Alu elements, and 93 are 141-EoG-Alu elements ( Table S2) (All other 215 EoG-Alu elements are mapped outside of a hotspot and were ignored in this work). To estimate the contribution of EoG-Alu elements to the transcriptome repertoire, we compared the level of expression of the new transcript variants they formed to that of other transcripts from the same gene.
The APADB database links cleavage and polyadenylation sites to nearby genes. To carry out a conservative analysis, we further filtered these data by crossing the positions of the cleavage sites with UCSC annotations, and removing all cleavage sites that were more than 40 bases away from an annotated gene end. This left 32 genes linked to 6-EoG-Alu elements, 13 genes linked to 134-EoG-Alu elements, and 24 genes linked to 141-EoG-Alu elements (Table S9). We downloaded transcript expression values for these genes in 53 tissues from the GTEx website [33]. Each transcript was assigned with a single RPKM value, taken as the maximum across all tissues. For each transcript formed by an EoG-Alu element, we calculated the ratio of its RPKM to that of the maximal RPKM across all the transcripts of the gene. A histogram of these values (Fig. S10A) shows that 28 (41.2%) of the EoG-Alu elements lead to transcripts whose expression is >50% of the maximal expression level of their gene. Notably, many of these Alu-promoted new transcripts show substantial RPKM values (Fig. S10B).
Our analysis was very strict, covering only Alu-elements whose cleavage site resides within one of the three hotspots, ignoring hotspots that might include high fraction of false positives. As a result, the above numbers are likely an underestimate of the true number of new transcripts formed by the introduction of cleavage and polyadenylation sites by Alu elements.
The exaptation of Alu elements, therefore, enriches the repertoire of the human transcriptome by generating new transcript variants. If the PAS-bearing Alu element resides just downstream of a 3′UTR of an existing gene, it may form a longer transcript variant. If the PAS-bearing Alu element resides in an intron, it may form a shorter transcript variant. Here, we describe a few specific examples. The transcript ENST00000411731 of the gene ERBB3 has an AluSz element overlapping its end. This transcript variant may have arisen following the insertion of the Alu element downstream of the 3′-end of the 3rd exon, making it the last exon of the new transcript variant. This change led to elongation of the coding region, and to the creation of a new 3′UTR (Fig. 8A). The RPKM value of this transcript (6.9) is 11.4% of the maximal RPKM in this gene. A shorter transcript may also be a result of Alu insertion within an existing 3′UTR. For example, in the gene THUMPD3, the insertion of an AluSq2 element into the 3′UTR generated two transcripts (ENST00000515662 and ENST00000464045) with shorter 3′UTR (Fig. 8B). In this case, the RPKM value of ENST00000464045 (a non-coding transcript, RPKM 47.7), is the maximal across all transcripts of the gene, whereas the RPKM of ENST00000515662 (2.4) is low, but still considerable.
Figure 8.
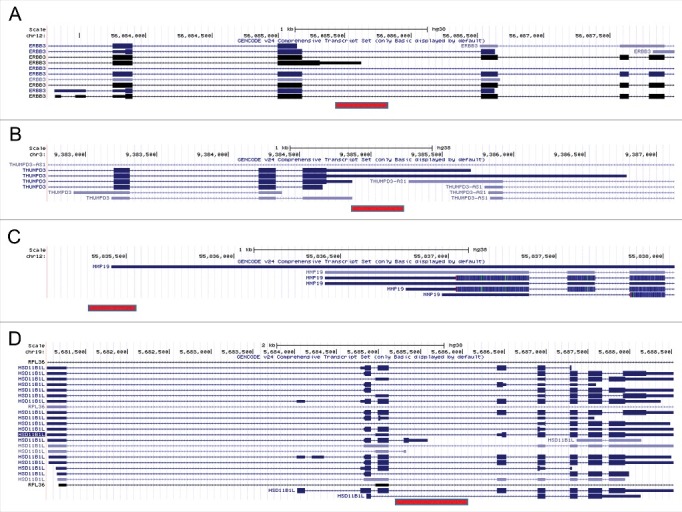
Examples of new transcript variants formed by Alu insertion (images were prepared using UCSC table browser). Alu elements are marked by a red bar. (A) AluJo insertion into an intron of the gene ERBB3 may have created a shorter transcript. (B) Insertion of an AluSq2 element into the 3′UTR of the gene THUMPD3 may have generated two transcripts with shorter 3′UTR. (C) The longest transcript of the gene MMP19 may have formed by the insertion of an AluJb element just downstream of its 3′UTR. (D) Insertion of an AluSx1 element within the intron of the gene HSD11B1L may have created shorter transcript with new exon and 3′UTR.
Alu elements inserted just downstream of an existing gene end may lead to 3′UTR elongation. For example, the longest transcript of the MMP19 gene, ENST00000548882, may have been formed by the insertion of an AluJb element downstream of its 3′UTR, leading to elongation of the 3′UTR (Fig. 8C). The RPKM value of this transcript (20.2) is the maximal across all transcripts of the gene.
Alu element insertion within a gene may also lead to completely new last exons. For example, the insertion of an AluSx1 element within an intron of the gene HSD11B1L was followed by the creation of an additional exon, and a new 3′UTR (Fig. 8D). The RPKM value of this transcript (ENST00000422535, RPKM 22.6) is maximal across all transcripts of this gene.
Summary
Within Alu elements, we have identified three positions that are hotspots for formation of new gene ends. While there are no gene-end databases that specifically account for mobile elements, we have crossed UCSC annotations with the APADB database to remove additional hotspots that may have arisen due to experimental artefacts. We showed that the hotspot at the beginning of the Alu element (position 6) is formed by exaptation of the nicking signal, used for the insertion of the Alu element into the genome, to serve as a polyadenylation signal. The two other hotspots in the middle of the Alu element are located downstream of the A-rich linker sequence. We showed that these hotspots likely formed as a result of a mutational process on the A-rich sequence.
We should stress out that the creation of a polyadenylation signal does not guarantee that it triggers the formation of novel gene end. The efficiency of any individual polyadenylation signal is difficult to predict, and it is believed that in many cases a true gene end would require additional auxiliary signals, both upstream and downstream of the cleavage point [12,13,15]. Indeed, we see many Alu elements among our control groups that present polyadenylation signals, and yet do not seem to promote a novel gene end (see, e.g., Fig. 3). We found that genuine EoG-Alu elements show enrichment in mutations that create TG's downstream of the cleavage site, which was already suggested to be an auxiliary polyadenylation signal [14,17].
Material and methods
Annotations of Alu elements and gene ends
Annotations of genes and Alu elements from human genome assembly hg38 were downloaded from UCSC genome browser using the Galaxy interface [34]. Noncoding genes and genes lacking 3′UTR annotation (3′UTR length < 4 nt) were excluded from the analysis. Experimentally measured gene ends were downloaded from the APADB database [27]. Alu families reference sequences were downloaded from Repbase [35] (www.girinst.org).
Control groups of Alu elements
In order to test how the properties of certain Alu elements allow them to create an alternative polyadenylation site, we mapped gene ends to the human genome, and looked for Alu elements that overlap them (EoG-Alu). As control, we wished to take Alu elements that do not contain gene ends. To properly construct this control, we annotated the genomic context of the EoG-Alu elements insertion point (exon, intron, 3′UTR, 5′UTR or intergenic region; Fig. S11). We found that most of the EoG-Alu elements were inserted within introns (35.2%) and intergenic regions (34.9%). In addition, we measured the distance of the EoG-Alu elements from the 3′-end of the nearest upstream exon, and found that most of them are located within 3000 nt from this exon (Fig. S12). Following these findings, we constructed two control groups of Alu elements that do not contain gene ends: a) Control-I. Alu elements that reside within introns in sense orientation to the gene, and their 5′ end is located no more than 3000 bases from the 3′-end of an upstream exon. If such intronic Alu elements start displaying polyadenylation signals, they would give rise to a new, shorter, transcript of the gene. b) Control-IG. Alu elements that reside in intergenic regions in sense orientation to the upstream gene, and their 5′ end is located no more than 3000 bases from the 3′-end of that gene. If such intergenic Alu elements start displaying polyadenylation signals, they would give rise to a new, longer, transcript of the gene.
To test additional aspects of EoG-Alu elements we constructed a third control group, consists of 80nt-long sequences that harbor a gene end in their center, and that do not overlap Alu elements (Control-EoG).
Sequence composition around Alu insertion sites
For analysis of sequence composition around Alu insertion sites, we fetched the 200 nt genomic sequence that flanks each Alu element (100 nt to each side). Nucleotide logo figures were prepared using WebLogo [36].
Mapping onto the Alu reference sequence
Sequence analysis of multiple Alu elements from different Alu families requires a definition of a common reference. We selected the sequence of the AluJo family as a common reference as it is the ancestral Alu form [5]. Mapping of a position along an Alu element to the reference sequence was performed using two consecutive pairwise alignments: First, the Alu element was aligned to the reference sequence of its own family. Second, the reference sequence of the family was aligned to the common AluJo reference. Alignments were performed using Needleman-Wunsch global alignment algorithm, and reference sequences for all Alu families were downloaded from Repbase [35]. Positions along Alu elements, whether EoG-Alu elements or control, are always given with respect to the equivalent position along the Alu reference sequence.
Alignment of the Alu elements to their family reference was also used to list the specific differences between each Alu element and its family reference. These differences were divided into single nucleotide substitutions (where we recorded the position, the consensus nucleotide and the mutated nucleotide); deletions (where we recorded the position and the deleted nucleotides); and insertions (where we recorded the position).
Gene end hotspots
All EoG-Alu elements whose gene end had been mapped to an interval (see Table S2) around a hotspot were considered as belonging to this hotspot.
Transcript expression data
Transcript RPKM values for 53 tissues in 8555 samples were downloaded from the GTEx portal [33] v6 (dbGaP Accession phs000424.v6.p1). RPKM value for each tissue was calculated as the median over the relevant samples. Each transcript was assigned with a single RPKM value, taken as the maximum across all tissues. RPKM values were computed for transcripts taken from the comprehensive gene annotations list of GENCODE v24 (Ensembl 83).
Supplementary Material
Funding Statement
This research was supported by the Israel Science Foundation [grant no. 1431/13].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Mighell AJ, Markham AF, Robinson PA. Alu sequences. FEBS Lett. 1997;417:1–5. doi: 10.1016/S0014-5793(97)01259-3 [DOI] [PubMed] [Google Scholar]
- [2].Deininger P. Alu elements: know the SINEs. Genome Biol. 2011;12:236. doi: 10.1186/gb-2011-12-12-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rowold DJ, Herrera RJ. Alu elements and the human genome. Genetica. 2000;108:57–72. doi: 10.1023/A:1004099605261 [DOI] [PubMed] [Google Scholar]
- [4].Jurka J, Smith T. A fundamental division in the Alu family of repeated sequences. Proc Natl Acad Sci U S A. 1988;85:4775–8. doi: 10.1073/pnas.85.13.4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Price AL, Eskin E, Pevzner PA. Whole-genome analysis of Alu repeat elements reveals complex evolutionary history. Genome research. 2004;14:2245–52. doi: 10.1101/gr.2693004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Quentin Y. Origin of the Alu family: a family of Alu-like monomers gave birth to the left and the right arms of the Alu elements. Nucleic Acids Res. 1992;20:3397–401. doi: 10.1093/nar/20.13.3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ule J. Alu elements: at the crossroads between disease and evolution. Biochemical Society transactions. 2013;41:1532–5. doi: 10.1042/BST20130157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sela N, Mersch B, Gal-Mark N, et al.. Comparative analysis of transposed element insertion within human and mouse genomes reveals Alu's unique role in shaping the human transcriptome. Genome Biol. 2007;8:R127. doi: 10.1186/gb-2007-8-6-r127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lev-Maor G, Sorek R, Shomron N, et al.. The birth of an alternatively spliced exon: 3′ splice-site selection in Alu exons. Science. 2003;300:1288–91. doi: 10.1126/science.1082588 [DOI] [PubMed] [Google Scholar]
- [10].Shen S, Lin L, Cai JJ, et al.. Widespread establishment and regulatory impact of Alu exons in human genes. Proc Natl Acad Sci U S A. 2011;108:2837–42. doi: 10.1073/pnas.1012834108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gal-Mark N, Schwartz S, Ast G. Alternative splicing of Alu exons–two arms are better than one. Nucleic Acids Res. 2008;36:2012–23. doi: 10.1093/nar/gkn024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Proudfoot NJ. Ending the message: poly(A) signals then and now. Genes & development. 2011;25:1770–82. doi: 10.1101/gad.17268411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14:496–506. doi: 10.1038/nrg3482 [DOI] [PubMed] [Google Scholar]
- [14].Millevoi S, Vagner S. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 2010;38:2757–74. doi: 10.1093/nar/gkp1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shi Y, Manley JL. The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015;29:889–97. doi: 10.1101/gad.261974.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tian B, Manley JL. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci. 2013;38:312–20. doi: 10.1016/j.tibs.2013.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tian B, Graber JH. Signals for pre-mRNA cleavage and polyadenylation. Wiley interdisciplinary reviews RNA. 2012;3:385–96. doi: 10.1002/wrna.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Beaudoing E, Freier S, Wyatt JR, et al.. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000;10:1001–10. doi: 10.1101/gr.10.7.1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hu J, Lutz CS, Wilusz J, et al.. Bioinformatic identification of candidate cis-regulatory elements involved in human mRNA polyadenylation. RNA. 2005;11:1485–93. doi: 10.1261/rna.2107305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Perepelitsa-Belancio V, Deininger P. RNA truncation by premature polyadenylation attenuates human mobile element activity. Nat Genet. 2003;35:363–6. doi: 10.1038/ng1269 [DOI] [PubMed] [Google Scholar]
- [21].Roy-Engel AM, El-Sawy M, Farooq L, et al.. Human retroelements may introduce intragenic polyadenylation signals. Cytogenet Genome Res. 2005;110:365–71. doi: 10.1159/000084968 [DOI] [PubMed] [Google Scholar]
- [22].Chen C, Ara T, Gautheret D. Using Alu elements as polyadenylation sites: A case of retroposon exaptation. Mol Biol Evol. 2009;26:327–34. doi: 10.1093/molbev/msn249 [DOI] [PubMed] [Google Scholar]
- [23].Lee JY, Ji Z, Tian B. Phylogenetic analysis of mRNA polyadenylation sites reveals a role of transposable elements in evolution of the 3′-end of genes. Nucleic Acids Res. 2008;36:5581–90. doi: 10.1093/nar/gkn540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Derti A, Garrett-Engele P, Macisaac KD, et al.. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22:1173–83. doi: 10.1101/gr.132563.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fujita PA, Rhead B, Zweig AS, et al.. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39:D876–82. doi: 10.1093/nar/gkq963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Proudfoot NJ, Furger A, Dye MJ. Integrating mRNA processing with transcription. Cell. 2002;108:501–12. doi: 10.1016/S0092-8674(02)00617-7 [DOI] [PubMed] [Google Scholar]
- [27].Muller S, Rycak L, Afonso-Grunz F, et al.. APADB: a database for alternative polyadenylation and microRNA regulation events. Database (Oxford). 2014;2014. doi: 10.1093/database/bau076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci U S A. 1997;94:1872–7. doi: 10.1073/pnas.94.5.1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Levy A, Schwartz S, Ast G. Large-scale discovery of insertion hotspots and preferential integration sites of human transposed elements. Nucleic Acids Res. 2010;38:1515–30. doi: 10.1093/nar/gkp1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pfeifer GP. Mutagenesis at methylated CpG sequences. Current topics in microbiology and immunology. 2006;301:259–81 [DOI] [PubMed] [Google Scholar]
- [31].Yang AS, Gonzalgo ML, Zingg JM, et al.. The rate of CpG mutation in Alu repetitive elements within the p53 tumor suppressor gene in the primate germline. J Mol Biol. 1996;258:240–50. doi: 10.1006/jmbi.1996.0246 [DOI] [PubMed] [Google Scholar]
- [32].Xing J, Hedges DJ, Han K, et al.. Alu element mutation spectra: molecular clocks and the effect of DNA methylation. J Mol Biol. 2004;344:675–82. doi: 10.1016/j.jmb.2004.09.058 [DOI] [PubMed] [Google Scholar]
- [33].Carithers LJ, Moore HM. The Genotype-Tissue Expression (GTEx) Project. Biopreserv Biobank. 2015;13:307–8. doi: 10.1089/bio.2015.29031.hmm [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Goecks J, Nekrutenko A, Taylor J, et al.. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA. 2015;6:11. doi: 10.1186/s13100-015-0041-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Crooks GE, Hon G, Chandonia JM, et al.. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90. doi: 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.