

ABSTRACT
We recently identified a novel ribonuclease in Bacillus subtilis called Rae1 that cleaves mRNAs in a translation-dependent manner. Rae1 is a member of the NYN/PIN family of ribonucleases and is highly conserved in the Firmicutes, the Cyanobacteria and the chloroplasts of photosynthetic algae and plants. We have proposed a model in which Rae1 enters the A-site of ribosomes that are paused following translation of certain sequences that are still ill-defined. In the only case identified thus far, Rae1 cleaves between a conserved glutamate and lysine codon during translation of a short peptide called S1025. Certain other codons are also tolerated on either side of the cleavage site, but these are recognized less efficiently. The model of Rae1 docked in the A-site allows us to make predictions about which conserved residues may be important for recognition of mRNA, the tRNA in the adjacent P-site and binding to the 50S ribosome subunit.
KEYWORDS: A-site, ribosomes, co-translational mRNA decay, phylogeny, ribonuclease
In contrast to the transcription and translation machineries, which are highly conserved in the different forms of life, the enzymes involved in RNA maturation and degradation vary widely. Only a handful of ribonucleases (RNases) can be considered as pervasive in biology and no single RNA processing or decay enzyme is truly ubiquitous. For a long time, it was assumed that the RNA subunit of the tRNA 5’ processing enzyme RNase P was universally conserved, but it is now known that at least three different enzymes can catalyze this reaction, two of which are protein-only forms [1,2]. Some organisms have even found a way to survive without RNase P, by initiating transcription at the first nucleotide (nt) of the mature tRNA [3].
We have been intrigued for some time by the contrast between the RNA degrading machineries of the two best-studied bacteria, the Firmicute Bacillus subtilis and the γ–Proteobacterium Escherichia coli. These two organisms diverged somewhere between 1.5 and 3 billion years ago and are essentially on opposite sides of the bacterial phylogenetic tree as it is currently understood (Fig. 1). They each have about 20 known exo- or endoribonucleases, only 8 of which are clearly identifiable orthologs that are present in both organisms [4]. While is sometimes a useful exercise to distinguish the Gram-positive and Gram-negative RNA maturation/degradation apparatuses, in fact, if one looks at the bigger picture, RNases are globally arranged according to two gradients across the phylogenetic tree, one extending from the Firmicutes to the Proteobacteria and the other in the reverse orientation (Fig. 1). Organisms located between B. subtilis and E. coli on the tree have mixtures of these two sets of enzymes that are roughly correlated to their evolutionary distance from either species.
Figure 1.
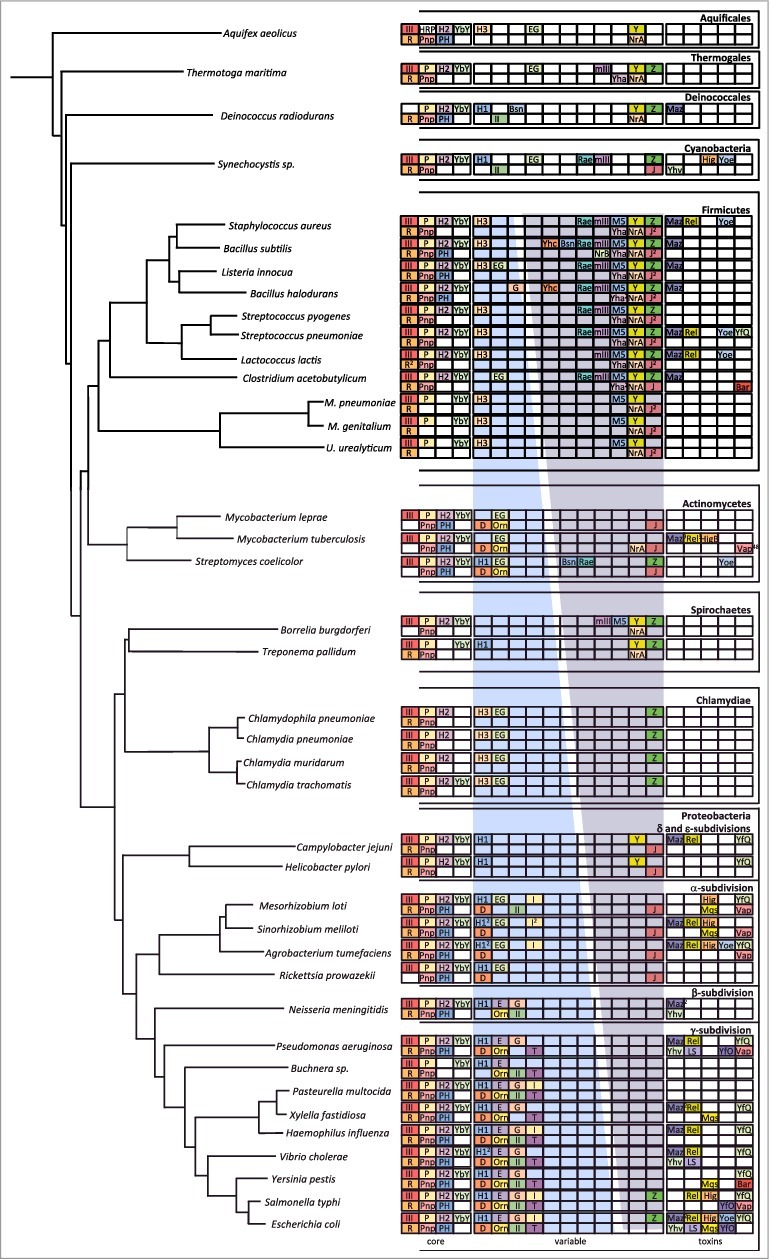
Phylogeny of ribonucleases in bacteria. The image is an update of Figure 1 in [ref. 18]. The most highly conserved RNases are in a subsection labelled ‘core’. More variable RNases are shown in a subsection labelled ‘variable’. For these two subsections, endoribonucleases are confined to the top line and exoribonucleases to the bottom line. Within the variable subsection, the blue gradient represents the distribution of RNases mostly identified with the E. coli degradation machinery; the purple gradient represents the distribution of RNases mostly identified in B. subtilis. Horizontally transferred toxin RNases (all endoribonucleases) are shown in a subsection labelled ‘toxins’. Abbreviations are as follows: Endondonucleases: III = RNase III; P = RNase P; H2 = RNase HII; H1 = RNase HI; H3 = RNase HIII; E = RNase E; G = RNase G; EG = RNase E/G; YbY = YbeY; I = RNase I; Z = RNase Z; M5 = RNase M5; Y = RNase Y; mIII = Mini-III; Rae = Rae1; Maz = MazF; Rel = RelE; Yoe = YoeB; Hig = HigB; YfQ = YafQ; Yhv = YhaV; LS = RNase LS, Mqs = MqsR; YfO = YafO; Bsn = RNase Bsn; Bar = Barnase; Exoribonucleases: R = RNase R; Pnp = polynucleotide phosphorylase; PH = RNase PH; D = RNase D; Orn = oligoribonuclease; II = RNase II; T = RNase T; J = RNase J; BN = RNase BN; Yha = RNase YhaM. Numbers in superscript refer to the number of orthologs present when identified.
A few RNases, first identified in the Firmicutes, have a particular distribution pattern and are basically only additionally found in the Cyanobacteria, perhaps indicative of ancient horizontal transfer events in the shared ecological niche of the soil. These RNases, which include the 23S ribosomal RNA (rRNA) processing enzyme Mini-III [5] and the recently discovered ribosome associated endoribonuclease Rae1 [6], can be traced from the Cyanobacteria to the chloroplasts of green algae and higher plants. In B. subtilis, the rae1 gene (previously known as yacP) is the last gene of an operon that encodes two subunits of a cysteinyl-tRNA synthetase (cysE-cysS), the Mini-III gene (mrnC) and the 23S rRNA (G2251) methylase encoded by rlmB/yacO. This synteny is highly conserved throughout the Firmicutes and was the first clue that Rae1 might play a role in translation [6]. Indeed, the rae1 gene is located downstream of the cysS-mrnC-rlmB triad >90% of the time in this bacterial clade. Its synteny in the Cyanobacteria and in chloroplasts is unrelated and is less well conserved, perhaps indicative of other functions.
RNA sequencing (RNAseq) experiments performed in a B. subtilis rae1 deletion mutant and plasmid complemented strains gave the first handle on the role of this enzyme in vivo. Although about 60 genes in total showed altered expression levels in the RNAseq experiment [6], two operons stood out because they showed strong up-effects in the deletion strain and strong down-effects in the plasmid complemented strain. These were the yrzI operon, encoding several small peptides of unknown function, and the bmrBCD operon, encoding two subunits of a multi-drug efflux pump and its regulatory region. Direct endonucleolytic cleavage of the mRNA encoding the 17-amino acid (aa) S1025 peptide just downstream of the yrzI open reading frame (ORF) accounted for the Rae1-mediated destabilization of this mRNA. So far, we have been unable to map a clear Rae1-dependent cleavage site in the bmrBCD mRNA (unpublished data), but this search is complicated by its large size (5 kb) and a difficulty in identifying degradation intermediates in strains lacking either 5’ or 3’ exoribonucleases.
Rae1 associates with the B. subtilis ribosome and its cleavage within the S1025 ORF is dependent on both its translation and reading frame [6]. Cleavage occurs precisely between a glutamate codon (GAG) in position 13 and a lysine codon (AAG) in position 14 in the highly conserved nucleotide sequence of the second half of this short reading frame. A resolution of the Rae1 crystal structure allowed us to make a model of the protein docked into the A-site of the ribosome, positioned to cleave between the P-site and A-site codons. In this model, Rae1 can only directly read the Lys codon in position 14. However, an extensive mutational analysis of both codons 13 and 14 showed that, while synonymous changes (GAA or AAA) in these two positions are perfectly tolerated by Rae1, non-synonymous changes in either position have deleterious effects ranging from partial to a total lack of cleavage. In sum, the data show that Lys codon 14 is most important for Rae1 cleavage, but that the Glu codon in position 13 clearly also makes a contribution.
Rae1 presumably only gains access to the A-site when there is a problem with translation. Under normal circumstances, the A-site is rapidly occupied by elongation factor EF-Tu bound to aminoacylated tRNA. However, a number of physiological situations are known in which translation stalls and the A-site becomes temporarily available for other factors to bind. These are: 1) during amino acid starvation, when there is no available amino-acylated tRNA for EF-Tu to bring to the ribosome. In this case, Rae1 would have to compete with uncharged tRNA and the RelA protein, whose role is to induce the stringent response by synthesizing ppGpp; 2) during translation of mRNAs lacking a stop-codon (through mutation, RNase cleavage or physical breakage). Ribosomes are unable to dissociate from such mRNAs via the normal release factor mechanism. In bacteria, ribosomes are rescued by tmRNA or other proteins (e.g. ArfA, ArfB in E. coli) that bind to the A-site to promote translation termination and ribosome release [7]; 3) during translation of a series of rare codons or strong secondary structure. This process is termed No-Go-Decay (NGD) in yeast [8]; 4) during strong ribosome pausing induced by particular, but ill-defined peptide sequences that get stuck in the ribosome exit channel [9]. In this case, the fate of the mRNA is likely to be varied. In a classical example, an erythromycin induced stall on the leader peptide of the ermC mRNA leads to its dramatic stabilization through its protection against the 5’-3’ exoribonuclease RNase J1 [10,11]. However, one could also imagine situations where RNase cleavage sites might be revealed by the lack of translation of the downstream mRNA, leading to destabilization.
The burning question is which of these situations applies to Rae1 and S1025? Currently, we have a preference for the scenario involving peptide induced pausing due to the following considerations: 1) given its apparent preference for lysine codons in the A-site, we asked whether lysine starvation would be a general mechanism for Rae1 cleavage of mRNAs in B. subtilis. While all four specific mRNAs tested indeed showed evidence of lysine starvation-dependent cleavage, the pattern was identical in a ∆rae1 strain, suggesting this phenomenon is Rae1-independent [6]; 2) with non-stop mRNAs, the ribosome runs all the way to the end of the transcript and, depending on the phase, the A-site will be either completely devoid of RNA, or only contain one or two nucleotides. Since Rae1 appears to have a preference for a lysine triplet in the A-site, it is unlikely to be employed in this case. Furthermore, its impact would be limited because it would at most only trim one or two extra nts from the already truncated mRNA; 3) examination of the S1025 ORF did not reveal anything out if the ordinary in terms of codon usage or RNA secondary structure. Thus, the ribosome pause signal does not appear to be rare codons or secondary structure. By process of elimination, therefore, we arrive at our working hypothesis 4) that the sequence of the first 13 amino acids of the S1025 peptide, though an interaction with the exit tunnel, causes the ribosome to pause long enough for Rae1 to gain access to Lys codon in the A-site. This scenario might account for the role of the Glu codon in position 13, either through a direct role in the pausing process per se or through contacts between Rae1 and the Glu-tRNA in the P-site that could facilitate optimal docking. We developed a coupled in vitro translation/Rae1 cleavage assay that will greatly facilitate the testing of this model, via a high throughput analysis of the roles of specific codons in the ribosome pausing and Rae1 cleavage processes.
Clearly, we need to identify more direct substrates of Rae1 and map its cleavage sites within these mRNAs to be able to draw general conclusions. The RNAseq data is a reservoir of other potential candidates waiting to be explored. One class of Rae1-sensitive RNAs (Class I) showed up-effects in the ∆rae1 deletion strain, but no effect in the plasmid complemented strain grown in the presence or absence of inducer [6]. We speculate that leaky expression in the absence of inducer may already provide sufficient Rae1 to fully cleave its substrates and that these represent high affinity targets. A striking feature of this class of mRNAs is that a large proportion encodes stress response proteins, regulated by the iron stress regulator Fur, the starvation response regulator AbrB or the membrane stress sigma factor SigW. No effect of Rae1 on fur [6], abrB or sigW (unpublished data) mRNA levels was observed, suggesting the effect occurs at the protein level. These data point to a general role for Rae1 in stress management that remains to be characterized.
There is an interesting parallel between the proposed Rae1 mechanism and that of the RelE family of toxins (which includes YoeB, HigB, YafQ; see Fig. 1) that are expressed during stress conditions and enter the ribosome A-site to provoke mRNA cleavage with particular codon specificity [12–15]. Furthermore, the PIN-domain structure of Rae1 is related to the VapC family of toxins, although these are not currently known to enter the A-site. Two features differentiate Rae1 from the RelE family of toxin RNases. The first is the lack of an obvious antitoxin coded in the same operon to keep its activity in check. In fact, as mentioned above, the rae1 gene is found in an operon encoding a number of house-keeping genes in the Firmicutes, suggesting that its stress related function has been fully integrated into the basic cell machinery for this whole bacterial clade. The second is that in contrast to the RelE family of toxins that promote ribosome-mediated cleavage of the mRNA [16], Rae1 has its own magnesium-dependent catalytic site that performs the cleavage reaction [6].
The model of Rae1 in the A-site of the ribosome [6] requires validation either through structural studies (crystallography or cryo-EM) or mutational analysis. Most of the highly conserved residues in the Firmicute Rae1 proteins have obvious roles in the structure or catalytic activity of the NYN/PIN domain. However, we would anticipate that a number of conserved residues also play roles in the recognition of the mRNA, the ribosomal RNA lining the A-site pocket or the P-site tRNA. Using ConSurf [17], we aligned 500 unique Firmicute Rae1 sequences to highlight the positions of conserved residues in the model of Rae1 docked in the A-site (Fig. 2). In this model, the highly conserved residues Tyr9 and Asp53 are close enough to potentially interact with the first and second nucleotides of the A-site codon, respectively, while Thr79 and Glu107 are the best candidates for the recognition of the third nucleotide. The mutational analysis of the different positions of the Lys14 codon (AAA/AAG) showed that the central A-residue was most important for Rae1 recognition, but that both the first and third positions contribute. Our model would suggest that these four residues in particular should be targets of future studies on A-site codon recognition. This analysis also highlighted something we had not anticipated; that is, the possibility that Rae1 may also be able to ‘read’ the next codon to be translated on the mRNA. Indeed, in addition to its potential role in recognizing the third nucleotide of the A-site codon, Glu107 lies close enough in the model to make contacts with the first position of the next codon, while the adjacent residue, the highly conserved Gln108 could simultaneously contact the bases of nucleotides 2 and 3. The potential role of this conserved Asp codon (GAC) in Rae1 cleavage within the S1025 ORF and its recognition by the EQ motif in Rae1 will a focus of future attention.
Figure 2.
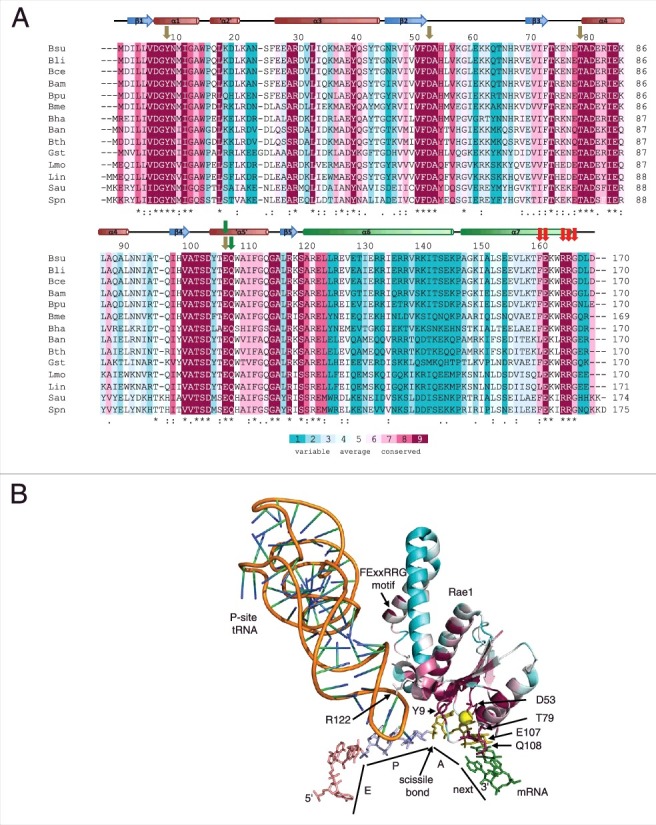
Conserved residues in Rae1. (A) Clustal Omega [19] alignment of 14 Firmicute Rae1 proteins. The alignment is color-coded according to an alignment of 500 non-identical sequences with Consurf [17]. The secondary structure of the Rae1 protein is shown above the alignment, with the two C-terminal helices shown in green. Abbreviations are as follows: Bsu, Bacillus subtilis; Bli, Bacillus lichiniformis; Bce, Bacillus cereus; Bam, Bacillus amyloliquifaciens; Bpu, Bacillus pumilus; Bme, Bacillus megaterium; Bha, Bacillus halodurans; Ban, Bacillus anthracis; Bth, Bacillus thuringiensis; Gst, Geobacillus stearothermophilus; Lmo, Listeria monocytogenes; Lin, Listeria innocua; Sau, Staphylococcus aureus; Spn, Streptococcus pneumoniae. Olive colored arrows show residues potentially involved in A-site codon recognition, green arrows point to amino acids potentially involved in interactions with the next codon and red arrows identify residues potentially involved in 50S subunit interactions. (B) Structure of Rae1 docked in A-site showing positions of conserved residues color-coded as in panel (A). The P-site tRNA is shown as an orange ribbon. The exit site (pink), peptidyl-transfer site (blue), acceptor site olive) and next (green) codons in the mRNA are labeled as E, P, A and next, respectively. The site of Rae1 cleavage is shown. Relevant amino acids for the potential A-site, next codon, P-site tRNA and 50S subunits interactions are labeled.
Although Rae1 lies close to the P-site tRNA in the docking model, there are no obvious highly conserved residues that we can point to as potential ligands; only Arg122 (65% conservation) points towards the phosphodiester backbone of the anticodon loop, but even this would not confer any obvious specificity for the Glu-tRNA in the P-site. Thus, whether Rae1 can indirectly read the P-site codon through specific interactions with the P-site tRNA is an open question. Indeed, the docking model does not allow us to rule out the possibility that Rae1 recognizes the Glu13-Lys14 codons of the S1025 ORF in the A-site and next codon positions, respectively, rather than in the P-site and A-site as currently proposed.
In wild-type Rae1, a tryptophan residue W109 (65% conserved) projects into the active site and would be predicted to interfere with catalytic activity [6]. Intriguingly, in the structure of a fortuitously isolated Rae1 (W164L) mutant, this Trp residue is flipped out of the catalytic site [6]. In the current docking model, W109 would point towards helix 34 of 16S rRNA in the flipped out orientation (not shown). A specific interaction between this residue and 16S rRNA upon binding to the A-site could provide a potential activation mechanism for Rae1. Again, it should be stressed that the roles we propose for specific residues of Rae1 in codon recognition and A-site binding are highly speculative and completely dependent on the accuracy of our docking model. The goal of this discussion is simply to establish a framework for future experiments to validate or invalidate some of the principal interactions proposed.
Despite the snug fitting of NYN/PIN domain of Rae1 in the A-site of the 30S ribosomal subunit, we identified a strong interaction with both 50S subunits and 70S ribosomes (in the absence of mRNA or tRNA) [6]. We propose that the two C-terminal α-helices of Rae1 may be important for the interaction with the 50S subunit. The conformation adopted by these helices in the crystal structure is clearly dictated by formation of a dimer in the crystal and they are almost certainly free to adopt other conformations in the monomeric form bound to the ribosome. The sequences in the first helix and the beginning of the second helix are not well conserved. However, a motif at the extreme C-terminus of Rae1 (FExxRRG; residues 161–167 in B. subtilis) shows a much higher level of conservation, suggesting these residues are more important for Rae1 function. The overall length of these two helices also varies very little, suggesting that their main purpose is to display the C-terminal motif at a certain distance from the catalytic site to interact with a specific domain 23S rRNA. Because of the intrinsic flexibility of the C-terminal helices, is not possible to predict which domain this may be.
In summary, our studies so far have led us to the working hypothesis that Rae1 enters the A-site of ribosome paused during the translation of certain amino acid sequences to cleave the mRNA. This would presumably initiate a ribosome rescue process, e.g. by tmRNA or equivalent system. No homologs of ArfA or ArfB are present in B. subtilis, but analogous systems presumably exist, since the tmRNA is not essential in this organism. It remains to be seen how widespread Rae1 cleavages are on Firmicute mRNAs in general and whether this can be influenced by environmental conditions.
Funding Statement
Agence Nationale de la Recherche (ARNr-QC), Agence Nationale de la Recherche (CACSICE), Agence Nationale de la Recherche (DYNAMO), Centre National de la Recheche Scientifique (UMR8261).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by funds from the CNRS (UMR 8261), Université Paris VII-Denis Diderot, the Agence Nationale de la Recherche (ARNr-QC). This work has been published under the framework of Equipex (Cacsice) and a LABEX program (Dynamo) that benefit from a state funding managed by the French National Research Agency as part of the Investments for the Future program.
References
- [1].Holzmann J, Frank P, Loffler E, et al.. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008;135:462–74. [DOI] [PubMed] [Google Scholar]
- [2].Nickel AI, Waber NB, Gossringer M, et al.. Minimal and RNA-free RNase P in Aquifex aeolicus. Proc Natl Acad Sci U S A. 2017;114:11121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Randau L, Schroder I, Soll D. Life without RNase P. Nature. 2008;453:120–3. [DOI] [PubMed] [Google Scholar]
- [4].Durand S, Tomasini A, Braun F, et al.. sRNA and mRNA turnover in Gram-positive bacteria. FEMS Microbiol Rev. 2015;39:316–330. [DOI] [PubMed] [Google Scholar]
- [5].Redko Y, Bechhofer DH, Condon C. Mini-III, an unusual member of the RNase III family of enzymes, catalyses 23S ribosomal RNA maturation in B. subtilis Mol Microbiol. 2008;68:1096–106. [DOI] [PubMed] [Google Scholar]
- [6].Leroy M, Piton J, Gilet L, et al.. Rae1/YacP, a new endoribonuclease involved in ribosome-dependent mRNA decay in Bacillus subtilis. EMBO J. 2017;36:1167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Himeno H, Nameki N, Kurita D, et al.. Ribosome rescue systems in bacteria. Biochimie. 2015;114:102–12. [DOI] [PubMed] [Google Scholar]
- [8].Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Woolstenhulme CJ, Parajuli S, Healey DW, et al.. Nascent peptides that block protein synthesis in bacteria. Proc Natl Acad Sci U S A. 2013;110:E878–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bechhofer DH, Zen KH. Mechanism of erythromycin-induced ermC mRNA stability in Bacillus subtilis. J Bacteriol. 1989;171:5803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yao S, Sharp JS, Bechhofer DH. Bacillus subtilis RNase J1 endonuclease and 5' exonuclease activities in the turnover of DeltaermC mRNA. Rna. 2009;15:2331–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pedersen K, Zavialov AV, Pavlov MY, et al.. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell. 2003;112:131–40. [DOI] [PubMed] [Google Scholar]
- [13].Hurley JM, Woychik NA. Bacterial toxin HigB associates with ribosomes and mediates translation-dependent mRNA cleavage at A-rich sites. J Biol Chem. 2009;284:18605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Prysak MH, Mozdzierz CJ, Cook AM, et al.. Bacterial toxin YafQ is an endoribonuclease that associates with the ribosome and blocks translation elongation through sequence-specific and frame-dependent mRNA cleavage. Mol Microbiol. 2009;71:1071–87. [DOI] [PubMed] [Google Scholar]
- [15].Hwang JY, Buskirk AR. A ribosome profiling study of mRNA cleavage by the endonuclease RelE. Nucleic Acids Res. 2017;45:327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Neubauer C, Gao YG, Andersen KR, et al.. The structural basis for mRNA recognition and cleavage by the ribosome-dependent endonuclease RelE. Cell. 2009;139:1084–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ashkenazy H, Abadi S, Martz E, et al.. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44:W344–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Condon C, Putzer H. The phylogenetic distribution of bacterial ribonucleases. Nucleic Acids Res. 2002;30:5339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sievers F, Wilm A, Dineen D, et al.. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular systems biology. 2011;7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]