

ABSTRACT
Recently, accumulating evidences have indicated miRNAs play critical roles in the progression and development of various human complex diseases, which pointed out that identifying miRNA-disease association could enable us to understand diseases at miRNA level. Thus, revealing more and more potential miRNA-disease associations is a vital topic in biomedical domain. However, it will be extremely expensive and time-consuming if we examine all the possible miRNA-disease pairs. Therefore, more accurate and efficient methods are being highly requested to detect potential miRNA-disease associations. In this study, we developed a computational model of Ensemble Learning and Link Prediction for miRNA-Disease Association prediction (ELLPMDA) to achieve this goal. By integrating miRNA functional similarity, disease semantic similarity, miRNA-disease association and Gaussian profile kernel similarity for miRNAs and diseases, we constructed a similarity network and utilized ensemble learning to combine rank results given by three classic similarity-based algorithms. To evaluate the performance of ELLPMDA, we exploited global and local Leave-One-Out Cross Validation (LOOCV), 5-fold Cross Validation (CV) and three kinds of case studies. As a result, the AUCs of ELLPMDA is 0.9181, 0.8181 and 0.9193+/−0.0002 in global LOOCV, local LOOCV and 5-fold CV, respectively, which significantly exceed almost all the previous methods. Moreover, in three distinct kinds of case studies for Kidney Neoplasms, Lymphoma, Prostate Neoplasms, Colon Neoplasms and Esophageal Neoplasms, 88%, 92%, 86%, 98% and 98% out of the top 50 predicted miRNAs has been confirmed, respectively. Besides, ELLPMDA is based on global similarity measure and applicable to new diseases without any known related miRNAs.
KEYWORDS: association prediction, disease, ensemble learning, link prediction, microRNA
Introduction
MicroRNAs (miRNAs) are a family of small non-coding RNAs (containing about 22 nucleotides) that play a significant regulatory roles in animals and plants by targeting mRNAs for cleavage or translational repression [1]. Currently, a great many of studies have indicated that miRNA is one of the most important component in cell, which makes a vital contribution in multiple fundamental biological processes, including cell development, proliferation, signal transduction, differentiation, apoptosis, viral infection, metabolism, aging and so on [2–8]. Apparently, taking all the above functions into account, seeking for comprehensive information about miRNA might be a superior way to understand creatures at cell level. Twenty four years after the discovery of the first two miRNAs (Caenorhabditis elegans lin-4 and let-7), due to various experimental methods and computational models, thousands of miRNAs have been discovered recently [9–12]. In the latest version of miRBase [12] (see http://www.mirbase.org/), there are 28645 entries and more than 1000 human miRNAs. Furthermore, up to now, hundreds of miRNAs with different sequences and expression patterns have been discovered in diverse animals [7,13,14].
In recent years, the relationship between miRNAs and human diseases has attracted the most attention. Accumulating evidences have indicated that miRNA mutations or mis-expression correlate with various human cancers which implies miRNAs can function as tumor suppressors and oncogenes [15]. For instance, the dysregulation of the miRNAs has been confirmed as a main reason of aberrant cell behavior by many studies [11]. Furthermore, Single-nucleotide polymorphisms (SNPs) located at miRNA-binding sites (miRNA-binding SNPs) are likely to affect the expression of the miRNA target and may contribute to the susceptibility of humans to common diseases [16]. Moreover, downregulation of miR-101 is involved in cyclooxygenase-2 overexpression in human colon cancer cells. Also, miR-214 has been confirmed to be related to human ovarian cancer by inducing cell survival and cisplatin resistance [17,18]. Another example is that biological studies pointed out miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44 [19]. Besides, researches had pointed out that the miR-15a–miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities [20].
Therefore, considering the close relationship between miRNAs and human diseases, identifying more potential associations between miRNAs and diseases is currently an important goal in biomedical domain because it might contribute a lot to the research about diseases. However, it will be extremely expensive and time-consuming if we test all the miRNA-disease pairs by biological experiment. Thus, in order to avoid the waste of money and time, it is necessary to predict the most potential miRNA-disease pairs. As more and more databases are available, developing computational model would be an effective way to reveal potential association between miRNAs and diseases [21–30].
Based on the assumption that miRNAs with similar functions are more likely to have connections with diseases which share similar phenotypes, various computational methods have been established to predict the potential associations between miRNAs and diseases [31–34]. Jiang et al. [30] prioritized the entire human microRNAome for diseases of interest by integrating predicted miRNA-target associations, disease phenotype similarities, and known miRNA-disease associations. However, this computational model could not reach a satisfactory predictive accuracy because it strongly relies on the predicted miRNA-target interactions with high false-positive and false-negative results. Moreover, Shi et al. [35] advanced a model which mapped disease genes and miRNA targets on the protein-protein interaction (PPI) network by integrating the information of miRNA-target interactions, disease-gene associations and PPIs. Base on the assumption that a miRNA tends to be related with the diseases whose genes are correlated with the target of this miRNA, they paid attention to the aforementioned information to identify miRNA-disease associations. Xu et al. [36] made use of a miRNA prioritization method which exploited the similarity between the miRNAs targets and disease genes instead of exploiting the known miRNA-disease association. They constructed the miRNA target–dysregulated network (MTDN) and trained a support vector machine (SVM) on their own-defined gold standard data set to reveal novel miRNA-disease associations. Although it is a creative challenge, this method could not achieve a satisfactory accuracy because of the aforementioned reason that the miRNA-target interaction is not accurate enough. Besides, by integrating miRNA-protein association scores and protein-disease association scores, Mork et al. [37] presented a miRPD method to create scoring schemes that enable them to rank candidate miRNA–disease pairs. Furthermore, they obtained high-confidence and medium-confidence sets of miRNA-disease associations. However, because of the similar reason that miRNA-target interactions were not reliable enough, the predictive performance is not very satisfactory.
Considering the apparent limitation of miRNA-target interactions, several networks such as miRNA functional similarity network, disease semantic similarity network, disease phenotype similarity network, and miRNA-disease associations network were constructed, after that, multiple methods depending on them were developed. For example, Xuan et al. [29] presented a method names HDMP relied on weighted k most similar neighbors to predict disease-related miRNAs. To increase the accuracy of miRNA functional similarity calculated by the classic methods, they introduced information content of disease terms and disease phenotype similarity. HDMP did get a higher predictive accuracy than most of previous methods. However, it is based on a local similarity measure rather than a global measure which is obviously better. Furthermore, HDMP could not be used on new diseases without any known related miRNAs.
Moreover, to overcome the limitation of local similarity measure, Chen et al. [25] presented the model of Random Walk with Restart for MiRNA–Disease Association (RWRMDA) to infer potential miRNA–disease association. Based on global network information, random walk with restart was implemented on the miRNA functional similarity network. Moreover, RWRMDA was capable of simultaneously ranking all the miRNA-disease pairs. Although the predictive accuracy had been improved a lot, this model could not be exploited for diseases without any known associated miRNAs. In order to solve this problem, Chen et al. [28] further introduced another method called WBSMDA, which is based on miRNA functional similarity, disease semantic similarity, miRNA-disease associations, and Gaussian interaction profile kernel similarity for miRNAs and diseases. Comparing with RWRMDA, WBSMDA could be applied to diseases without any related miRNAs, which is a significant breakthrough in miRNA-disease association prediction. However, the performance of WBSMDA was not very satisfactory and they did not find a way reasonable enough to combine the Within-Score and Between-Score. What's more, to take the prior information regarding the network nodes and the respective local topological structures of the different categories of nodes into account. Xuan et al. [38] introduced another method named MIDP, which took advantage of the characteristics of the nodes and the various ranges of topologies. MIDP was also based on random walk, and it effectively relieved the negative effect of noisy data.
Also, more and more machine learning-based computational models were introduced in miRNA-disease association prediction. For instance, according to the assumption that miRNAs implicated in a specific tumor phenotype will show aberrant regulation of their target genes, Xu et al. [36] established a heterogeneous miRNA-target dysregulated network, extracted four network topological features, and developed Support Vector Machine (SVM)-based Supervised classifier to distinguish positive disease related miRNAs from negative ones. Although SVM is a theoretically accurate method, collecting known negative associations is a very difficult and even impossible task. Thus, this method did not give a good performance. Chen et al. [24] proposed another machine learning method which made use of semi-supervised learning to predict potential disease-related miRNAs (RLSMDA). Though RLSMDA is applicable for diseases without any related miRNAs, there are also limitations in combination of two classifiers in the different spaces and the selection of parameter values.
Considering that both similarity-based algorithms and machine learning methods achieve good results in previous researched [24,27,36], in this study, by combining the advantages of both methods, we advanced a novel computational model of Ensemble Learning and Link Prediction for miRNA-Disease Association prediction (ELLPMDA) to predict potential miRNA-disease associations. Instead of combining the result of a few classifiers, we output the weighted combination of the ranks given by three classic similarity-based algorithms, Common Neighbors, Jaccard index and Katz index. Unlike some methods mentioned above, ELLPMDA based on known miRNA-disease associations, miRNA functional similarity, disease semantic similarity and Gaussian interaction profile kernel similarity for diseases and miRNAs rather than disease-gene associations and miRNA-target interaction which are incomplete and inaccurate. Besides, ELLPMDA is based on global similar measure and is applicable to diseases without any known related miRNAs. Leave-One-Out Cross-Validation (LOOCV) and 5-fold Cross Validation were implemented for ELLPMDA based on the experimentally confirmed associations between miRNAs and diseases. Significantly better than six classic methods (HGIMDA, RLSMDA, HDMP, WBSMDA, RWRMDA and MCMDA) [21,22,24,25,28,29], the AUCs of global and local LOOCV were 0.9181 and 0.8181, respectively. Furthermore, ELLPMDA was evaluated in three distinct kinds of case studies. In the first case study, 44, 46 and 43 out of the top 50 predictive miRNAs for Colon Neoplasms, Esophageal Neoplasms and Kidney Neoplasms were confirmed in dbDEMC [39] and miR2Disease [40] databases. Secondly, to test ELLPMDA's predictive ability for new diseases without any known related miRNAs, we set up a novel case study for Lung Neoplasms. In this case, we removed all the experimentally confirmed miRNA-disease associations which including Lung Neoplasms from the training samples. At this time, Lung Neoplasms could be treated as a new disease without any known related miRNAs. After implementing ELLPMDA, we examined the top 50 miRNAs which were predicted to be associated with lung neoplasms in dbDEMC [39], HMDD v2.0 [41] and miR2Disease [40] databases, and 49 out of top 50 miRNAs have been confirmed. Finally, we utilized the old version of HMDD, a database that only include 1395 associations between 271 miRNAs and 137 diseases to be the training set. In our case study of Breast Neoplasms, 49 out of top 50 miRNAs were affirmed in dbDEMC [39], HMDD v2.0 [41] and miR2Disease [40] databases. Therefore, according to the cross validation and case studies, ELLPMDA is a highly reliable method, which is obviously superior to the aforementioned classic algorithms.
Results
Performance
Based on experimentally verified associations between miRNAs and diseases, we implemented global LOOCV, local LOOCV and 5-fold CV to evaluate the predictive accuracy of ELLPMDA. After that, we compared the evaluation result with six previous methods (HGIMDA, RLSMDA, HDMP, WBSMDA, RWRMDA and MCMDA) [21,22,24,25,28,29]. In LOOCV evaluation, every confirmed association was regard as a test sample in turn while the rest associations were treated as training samples. All the miRNA-disease pairs that had not been confirmed by experimental studies were regard as candidate samples. After executing ELLPMDA, every miRNA-disease pair will obtain a association score. A higher score means a link is more likely to exist between this pair. The difference between global LOOCV and local LOOCV lies in whether we simultaneously inspected all the diseases or not. In global LOOCV, we compared the score of the test sample with all the candidate samples. In local LOOCV, considering that every test sample was a pair consisting of particular disease and miRNA, we merely compared the test sample with candidate samples which included this particular disease. Furthermore, we drew Receiver operating characteristics (ROC) curve by plotting the true positive rate (TPR, sensitivity) against the false positive rate (FPR, 1-specificity) at different thresholds. Sensitivity denotes the percentage of miRNA-disease test samples whose ranks exceeded the given threshold while specificity represents the percentage of negative miRNA-disease associations whose ranks were lower than the threshold [42]. After plotting, we could calculate the area under ROC curve (AUC). In general, AUC = 1 means this method gives a perfect prediction performance while AUC = 0.5 indicates the performance of this method is same as random selection. As a result, the performance comparison of ELLPMDA to (HGIMDA, RLSMDA, HDMP, WBSMDA, RWRMDA and MCMDA) [21,22,24,25,28,29] have been shown in Fig. 1. This figure straightly indicate that for global LOOCV, the AUCs of HGIMDA, RLSMDA, HDMP, WBSMDA and MCMDA is respectively 0.8781, 0.8426, 0.8366, 0.8030 and 0.8759, while the AUC of ELLPMDA is 0.9181. The AUCs of HGIMDA, RLSMDA, HDMP, WBSMDA, RWRMDA and MCMDA in local LOOCV is respectively 0.8077, 0.6953, 0.7702, 0.8031, 0.7891 and 0.7718, as contract, the AUC of ELLPMDA is 0.8181.
Figure 1.
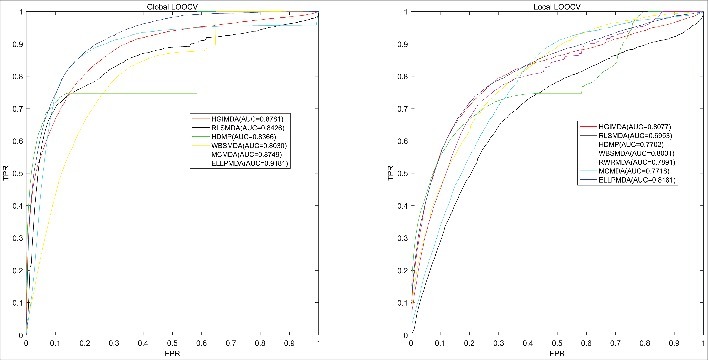
AUC of global LOOCV compared with HGIMDA, RLSMDA, HDMP, WBSMDA and MCMDA; AUC of local LOOCV compared with HGIMDA, RLSMDA, HDMP, WBSMDA, RMRMDA and MCMDA. As a result, ELLPMDA obtained AUCs of 0.9181 and 0.8181 in global and local LOOCV, which significantly exceed previous classic methods.
Moreover, we exploited 5-fold CV to further examine the predictive accuracy. Similar to LOOCV, in 5-fold CV, we randomly divided all the experimentally confirmed associations between miRNAs and diseases into 5 equal-size parts, and then we treated one part as test samples in turn and the other 4 parts as training samples. After executing ELLPMDA, the score of each test sample was compared with the scores of all the candidate samples, respectively. At this time, we could obtain the rank of every association in test samples. All the associations would obtain a rank after the above 5 parts were regard as test samples separately. In order to avoid random error, we took advantage of 5-fold CV for 100 times. As a result, the AUCs of MCMDA, HDMP and WBSMDA were respective 0.8767+/−0.0011, 0.8342+/−0.0010 and 0.8185+/−0.0009, while the AUC of ELLPMDA is 0.9193+/−0.0002.
Case study
Furthermore, we designed three kinds of case studies to comprehensively evaluate the predictive accuracy of ELLPMDA. Firstly, we took experimentally confirmed miRNA-disease associations captured from HMDD v2.0 [41] as the training set and did case studies for three common human cancers: Colon Neoplasms, Esophageal Neoplasms and Kidney Neoplasms. In this part, after implementing ELLPMDA, we obtained the scores of all the miRNA-disease pairs. Similar to cross-validation, we set experimentally verified associations as training samples and other miRNA-disease pairs were considered as candidate samples. Then for every disease mentioned above, we examined the top 10 and top 50 of predictive potential miRNA-disease pairs in dbDEMC [39] and miR2Disease [40] databases.
Colon Neoplasms is a big threaten of people's live with low detection rate in early stage, moreover, studies showed that about half of the Colon Neoplasms patients die of metastatic disease within 5 years from diagnosis [43]. Obviously, it is really urgent for us to achieve more information about it, which might contribute a lot to improve the accuracy of detection [44,45]. After a great many of biological experiments, lots of miRNAs had been proved to be associated with Colon Neoplasms. For example, miR-126 suppresses the growth of neoplastic cells by targeting phosphatidylinositol 3‐kinase signaling and is frequently lost in colon neoplasms [46]. Hsa-miR-181b and hsa-miR-200c were over-expressed in colon tumor tissues compared to normal tissues [47]. Also, hsa-miR-145 can inhabits the growth of Colon Neoplasms cells by targeting the insulin receptor substrate-1 [48]. Taking Colon Neoplasms as a case study, we implemented ELLPMDA and 9 out of top 10 and 44 out of top 50 potential related miRNAs given by ELLPMDA had been confirmed in miR2Disease and dbDEMC (See Table 1). Taking the top 5 predicted miRNAs as examples, miR-155(2nd) and miR-20a (5th) were confirmed to be up-regulated in colon neoplasms [49]. MiR-21(1st) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion in colon neoplasms [50]. What's more, miR-221(3rd), a miRNA directly amplified from plasma, is a potential diagnostic and prognostic marker of colon neoplasms [51]. Also, studies showed that miR-125b (4th) is straightly involved in cancer progression and is linked with poor prognosis in human colon neoplasms [52].
Table 1.
Prediction of the top 50 predicted miRNAs associated with Colon Neoplasms based on known associations in HMDD v2.0 database. The first column records top 1–25 related miRNAs. The second column records the top 26–50 related miRNAs.
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-21 | miR2Disease:dbDEMC: | hsa-mir-141 | miR2Disease:dbDEMC: |
| hsa-mir-155 | miR2Disease:dbDEMC: | hsa-mir-31 | miR2Disease:dbDEMC: |
| hsa-mir-221 | miR2Disease:dbDEMC: | hsa-mir-34c | miR2Disease: |
| hsa-mir-125b | dbDEMC: | hsa-let-7e | dbDEMC: |
| hsa-mir-20a | miR2Disease:dbDEMC: | hsa-mir-101 | unconfirmed |
| hsa-mir-34a | miR2Disease:dbDEMC: | hsa-mir-142 | unconfirmed |
| hsa-mir-16 | dbDEMC: | hsa-mir-15a | dbDEMC: |
| hsa-mir-222 | dbDEMC: | hsa-let-7f | miR2Disease:dbDEMC: |
| hsa-mir-199a | unconfirmed | hsa-mir-29b | miR2Disease:dbDEMC: |
| hsa-mir-200b | dbDEMC: | hsa-let-7i | dbDEMC: |
| hsa-mir-18a | miR2Disease:dbDEMC: | hsa-mir-205 | dbDEMC: |
| hsa-let-7a | miR2Disease:dbDEMC: | hsa-let-7d | dbDEMC: |
| hsa-mir-19a | miR2Disease:dbDEMC: | hsa-mir-122 | unconfirmed |
| hsa-mir-143 | miR2Disease:dbDEMC: | hsa-mir-196a | miR2Disease:dbDEMC: |
| hsa-mir-29a | miR2Disease:dbDEMC: | hsa-mir-106b | miR2Disease:dbDEMC: |
| hsa-mir-146a | dbDEMC: | hsa-mir-210 | dbDEMC: |
| hsa-mir-19b | miR2Disease:dbDEMC: | hsa-let-7g | miR2Disease:dbDEMC: |
| hsa-mir-200c | miR2Disease:dbDEMC: | hsa-mir-34b | miR2Disease:dbDEMC: |
| hsa-let-7b | miR2Disease:dbDEMC: | hsa-mir-214 | dbDEMC: |
| hsa-mir-92a | unconfirmed | hsa-mir-125a | miR2Disease:dbDEMC: |
| hsa-mir-223 | miR2Disease:dbDEMC: | hsa-mir-10b | miR2Disease:dbDEMC: |
| hsa-mir-200a | unconfirmed | hsa-mir-182 | miR2Disease:dbDEMC: |
| hsa-mir-1 | miR2Disease:dbDEMC: | hsa-mir-93 | miR2Disease:dbDEMC: |
| hsa-mir-9 | miR2Disease:dbDEMC: | hsa-mir-181b | miR2Disease:dbDEMC: |
| hsa-let-7c | dbDEMC: | hsa-mir-25 | miR2Disease:dbDEMC: |
Esophageal Neoplasms is reported as the eighth most common cancer worldwide and the sixth leading cause of deaths related with cancers based on the pathological characteristics and it affect males more times than females [45,53]. Because of the potential characteristics of invasion and metastasis in esophageal carcinoma cells, the overall 5-year survival rate is poor despite of advanced treatment [45,53,54]. Previous studies indicated that miRNAs is important in tumorigenesis of esophageal neoplasms [53]. For instance, miR-373 post-transcriptionally regulates large tumor suppressor in human esophageal neoplasms [55]. Down-regulation of miR-27a might reverse multidrug resistance of esophageal squamous cell carcinoma [56]. Also, research pointed out that miR-203 inhibits the proliferation and self-renewal of esophageal neoplasms stem-like cells by suppressing stem renewal factor Bmi-1 [57]. In this case, as a result, 9 out of top 10 and 46 out of top 50 predictive miRNAs given by ELLPMDA had been verified in miR2Disease and dbDEMC (See Table 2).
Table 2.
Prediction of the top 50 predicted miRNAs associated with Esophageal Neoplasms based on known associations in HMDD v2.0 database. The first column records top 1–25 related miRNAs. The second column records the top 26–50 related miRNAs.
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-200b | dbDEMC: | hsa-mir-29b | dbDEMC: |
| hsa-let-7e | dbDEMC: | hsa-mir-146b | dbDEMC: |
| hsa-let-7d | dbDEMC: | hsa-mir-191 | dbDEMC: |
| hsa-let-7f | unconfirmed | hsa-mir-106a | dbDEMC: |
| hsa-let-7i | dbDEMC: | hsa-mir-18b | dbDEMC: |
| hsa-mir-18a | dbDEMC: | hsa-mir-194 | miR2Disease:dbDEMC: |
| hsa-mir-125a | dbDEMC: | hsa-mir-302b | dbDEMC: |
| hsa-mir-17 | dbDEMC: | hsa-mir-24 | dbDEMC: |
| hsa-mir-19b | dbDEMC: | hsa-mir-7 | dbDEMC: |
| hsa-mir-429 | dbDEMC: | hsa-mir-199b | dbDEMC: |
| hsa-let-7g | dbDEMC: | hsa-mir-181b | dbDEMC: |
| hsa-mir-218 | unconfirmed | hsa-mir-30d | dbDEMC: |
| hsa-mir-132 | dbDEMC: | hsa-mir-20b | dbDEMC: |
| hsa-mir-125b | dbDEMC: | hsa-mir-302c | dbDEMC: |
| hsa-mir-127 | dbDEMC: | hsa-mir-195 | dbDEMC: |
| hsa-mir-30c | dbDEMC: | hsa-mir-30a | dbDEMC: |
| hsa-mir-106b | dbDEMC: | hsa-mir-181a | dbDEMC: |
| hsa-mir-9 | dbDEMC: | hsa-mir-107 | miR2Disease:dbDEMC: |
| hsa-mir-10b | dbDEMC: | hsa-mir-142 | dbDEMC: |
| hsa-mir-16 | dbDEMC: | hsa-mir-182 | dbDEMC: |
| hsa-mir-29a | dbDEMC: | hsa-mir-373 | miR2Disease:dbDEMC: |
| hsa-mir-222 | dbDEMC: | hsa-mir-92b | dbDEMC: |
| hsa-mir-1 | dbDEMC: | hsa-mir-30e | unconfirmed |
| hsa-mir-221 | dbDEMC: | hsa-mir-204 | unconfirmed |
| hsa-mir-93 | dbDEMC: | hsa-mir-367 | dbDEMC: |
Kidney Neoplasm, also known as renal cancer, is a cancer starting in the cells of kidney that includes many different types. It accounts for 3% of adult malignancies [58,59]. Renal cell carcinoma (RCC) and transitional cell carcinoma (TCC, also known as urothelial cell carcinoma) are the most common types of kidney neoplasms [60]. According to previous studies, various miRNAs had been examined to be linked with kidney neoplasms. For example, evidence had pointed out that miR-519 suppresses tumor growth in human kidney neoplasms by reducing HuR levels [61]. Also, VHL-regulated miR-204 is able to suppress tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma [62]. Besides, Overexpression of miR-210 causes centrosome amplification in renal carcinoma cells [63]. In this case, 8 out of top 10 and 43 out of top 50 predicted potential miRNAs had been confirmed (See Table 3). For instance, the miR-17-92(2nd) cluster is overexpressed in renal cell carcinoma and has an oncogenic effect on it [64]. Studies also indicated that miR-145 (3rd) functions as tumor suppressor and targets two oncogenes, ANGPT2 and NEDD9, in renal cell carcinoma [65]. What's more, miR-34a (4th) promote renal senescence by suppressing mitochondrial antioxidative enzymes [66] while miR-155(1st) expression was absent in nonlymphoid organs such as lung, heart and kidney [67].
Table 3.
Prediction of the top 50 predicted miRNAs associated with Kidney Neoplasms based on known associations in HMDD v2.0 database. The first column records top 1–25 related miRNAs. The second column records the top 26–50 related miRNAs.
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-155 | dbDEMC: | hsa-mir-29b | miR2Disease:dbDEMC: |
| hsa-mir-17 | miR2Disease: | hsa-let-7c | dbDEMC: |
| hsa-mir-145 | dbDEMC: | hsa-mir-1 | dbDEMC: |
| hsa-mir-34a | dbDEMC: | hsa-mir-429 | dbDEMC: |
| hsa-mir-125b | unconfirmed | hsa-mir-210 | miR2Disease:dbDEMC: |
| hsa-mir-200b | miR2Disease:dbDEMC: | hsa-mir-34b | dbDEMC: |
| hsa-mir-221 | unconfirmed | hsa-let-7f | miR2Disease:dbDEMC: |
| hsa-mir-20a | miR2Disease:dbDEMC: | hsa-mir-106b | miR2Disease:dbDEMC: |
| hsa-mir-199a | miR2Disease:dbDEMC: | hsa-mir-143 | dbDEMC: |
| hsa-mir-146a | dbDEMC: | hsa-mir-93 | dbDEMC: |
| hsa-mir-126 | miR2Disease:dbDEMC: | hsa-let-7e | unconfirmed |
| hsa-mir-200a | dbDEMC: | hsa-mir-218 | dbDEMC: |
| hsa-let-7a | dbDEMC: | hsa-mir-27a | miR2Disease:dbDEMC: |
| hsa-mir-19a | dbDEMC: | hsa-mir-223 | dbDEMC: |
| hsa-mir-18a | dbDEMC: | hsa-mir-182 | miR2Disease:dbDEMC: |
| hsa-mir-16 | dbDEMC: | hsa-mir-10b | dbDEMC: |
| hsa-mir-19b | miR2Disease:dbDEMC: | hsa-let-7i | dbDEMC: |
| hsa-mir-222 | dbDEMC: | hsa-mir-196a | dbDEMC: |
| hsa-mir-29a | miR2Disease:dbDEMC: | hsa-mir-101 | miR2Disease:dbDEMC: |
| hsa-mir-92a | unconfirmed | hsa-mir-133a | unconfirmed |
| hsa-mir-205 | unconfirmed | hsa-mir-29c | miR2Disease:dbDEMC: |
| hsa-let-7b | unconfirmed | hsa-mir-214 | miR2Disease:dbDEMC: |
| hsa-mir-9 | dbDEMC: | hsa-mir-146b | dbDEMC: |
| hsa-mir-34c | dbDEMC: | hsa-mir-181a | dbDEMC: |
| hsa-let-7d | dbDEMC: | hsa-let-7g | dbDEMC: |
Besides the aforementioned disease, we took advantage of ELLPMDA to rank all the miRNA-disease pairs between 383 diseases and 495 miRNAs in HMDD v2.0 [41] database (See Supplementary Table 1). We hope future biological experiments can confirm the prediction of ELLPMDA.
Secondly, in order to validate the predictive ability of ELLPMDA in new diseases without any known linked miRNAs, we set up a special case study. In this case, we examined ELLPMDA on Lung Neoplasms, a common human cancer which has a lot of experimentally verified related miRNAs. Similar to case study 1, we utilized the experimentally verified miRNA-disease associations achieved from HMDD v2.0 database [41] as the initial training set, however, by this time, we removed all the associations including lung neoplasms from the training set. Hence, lung neoplasms could be regard as a disease without any known related miRNAs. After executing ELLPMDA base on the brand new training set, we chose the top 50 of predicted miRNAs and examined them in dbDEMC [39], HMDD v2.0 [41] and miR2Disease [40] databases. As a result, 10 out of top 10 and 49 out of top 50 (See Table 4) potential associations were confirmed. For example, focus on the top 5 predicted miRNAs. High expression of miR-21(1st) and miR-155 (2nd) could predict recurrence and unfavorable survival in non-small cell lung neoplasms [68]. Also, studies indicated that circulating miR-125b (5th) is a novel biomarker for screening non-small-cell lung neoplasms [69]. Besides, miR-17-92 (4th) is overexpressed in human lung neoplasms cell and evidences had proved that it plays a key role in lung neoplasms development [70,71]. What's more, the reduction of miR-221 (3rd) inhibited cell proliferation and induced mitochondrial-mediated apoptosis in human lung neoplasms cells [72].
Table 4.
Prediction of the top 50 predicted miRNAs associated with Lung Neoplasms based on known associations in HMDD v2.0 database. In this case study, we eliminate all the known associations which including Lung Neoplasms to consider Lung Neoplasms as a new disease without any known related miRNAs. The first column records top 1–25 related miRNAs. The second column records the top 26–50 related miRNAs.
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-21 | miR2Disease:dbDEMC:HMDD: | hsa-mir-205 | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-155 | miR2Disease:dbDEMC:HMDD: | hsa-mir-223 | HMDD: |
| hsa-mir-221 | dbDEMC:HMDD: | hsa-mir-200c | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-17 | miR2Disease:HMDD: | hsa-mir-34b | dbDEMC:HMDD: |
| hsa-mir-125b | miR2Disease:HMDD: | hsa-mir-29b | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-222 | dbDEMC:HMDD: | hsa-mir-93 | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-34a | dbDEMC:HMDD: | hsa-mir-210 | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-199a | miR2Disease:dbDEMC:HMDD: | hsa-mir-200a | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-16 | miR2Disease:dbDEMC: | hsa-mir-106b | dbDEMC: |
| hsa-mir-20a | miR2Disease:dbDEMC:HMDD: | hsa-mir-182 | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-146a | miR2Disease:dbDEMC:HMDD: | hsa-mir-214 | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-145 | miR2Disease:dbDEMC:HMDD: | hsa-let-7c | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-18a | miR2Disease:dbDEMC:HMDD: | hsa-let-7d | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-126 | miR2Disease:dbDEMC:HMDD: | hsa-mir-181a | dbDEMC:HMDD: |
| hsa-mir-29a | miR2Disease:dbDEMC:HMDD: | hsa-mir-143 | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-19b | dbDEMC:HMDD: | hsa-mir-133a | dbDEMC:HMDD: |
| hsa-mir-19a | miR2Disease:dbDEMC:HMDD: | hsa-mir-122 | unconfirmed |
| hsa-let-7a | miR2Disease:dbDEMC:HMDD: | hsa-mir-196a | dbDEMC:HMDD: |
| hsa-mir-1 | miR2Disease:dbDEMC:HMDD: | hsa-mir-141 | miR2Disease:dbDEMC: |
| hsa-mir-200b | miR2Disease:dbDEMC:HMDD: | hsa-let-7e | miR2Disease:HMDD: |
| hsa-mir-15a | dbDEMC: | hsa-mir-101 | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-34c | dbDEMC:HMDD: | hsa-mir-31 | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-9 | miR2Disease:HMDD: | hsa-mir-29c | miR2Disease:dbDEMC:HMDD: |
| hsa-let-7b | miR2Disease:HMDD: | hsa-let-7f | miR2Disease:HMDD: |
| hsa-mir-92a | HMDD: | hsa-mir-27a | dbDEMC:HMDD: |
Finally, in order to examine the robustness of ELLPMDA's predictive accuracy in different databases, we utilized old version of HMDD, a database which only include 1395 experimentally verified associations between 271 miRNAs and 137 diseases, and these associations were treated as training samples. In this case, we tested ELLPMDA on Breast Neoplasms, a popular human disease which threating female's health a lot. Breast Neoplasms is now considered as the most leading type of invasive cancer in women with 1,384,155 estimated new cases worldwide and with nearly 459,000 related deaths each year [73]. It has been predicted that the worldwide incidence of female breast cancer will reach approximately 3.2 million new cases per year by 2050 [73]. These scary numbers indicate that it is extremely urgent for us to get more information about breast neoplasms to further interpret it and develop more effective methods for disease detection and treatment. In fact, lots of evidences had proved that various miRNAs were linked with breast neoplasms. For example, the overexpression of miR-21 in human breast neoplasms is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis [74]. MiR-17-5p regulates breast neoplasms cell proliferation by inhibiting translation of AIB1 mRNA [75] while miR-125b acts as a marker predicting chemoresistance in breast neoplasms [76]. Besides, biological researches had convinced that miR-210 is an independent prognostic factor in breast neoplasms [77] .We took advantage of old version of HMDD as training set and implemented ELLPMDA. As a result, 10 out of top 10 and 49 out of top 50 predicted miRNAs had been confirmed in dbDEMC [39], HMDD v2.0 [41] and miR2Disease [40] databases (see Table 5).
Table 5.
Prediction of the top 50 predicted miRNAs associated with Breast Neoplasms based on known associations in old version of HMDD database. The first column records top 1–25 related miRNAs. The second column records the top 26–50 related miRNAs.
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-let-7b | dbDEMC:HMDD: | hsa-mir-335 | miR2Disease:dbDEMC:HMDD: |
| hsa-let-7i | miR2Disease:dbDEMC:HMDD: | hsa-mir-106a | dbDEMC: |
| hsa-let-7e | dbDEMC:HMDD: | hsa-mir-26a | miR2Disease:dbDEMC:HMDD: |
| hsa-let-7c | dbDEMC:HMDD: | hsa-mir-128b | miR2Disease: |
| hsa-let-7g | dbDEMC:HMDD: | hsa-mir-203 | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-191 | miR2Disease:dbDEMC:HMDD: | hsa-mir-181a | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-92b | dbDEMC: | hsa-mir-135a | dbDEMC:HMDD: |
| hsa-mir-101 | miR2Disease:dbDEMC:HMDD: | hsa-mir-199b | dbDEMC:HMDD: |
| hsa-mir-126 | miR2Disease:dbDEMC:HMDD: | hsa-mir-532 | dbDEMC: |
| hsa-mir-520b | dbDEMC:HMDD: | hsa-mir-130b | dbDEMC: |
| hsa-mir-30e | unconfirmed | hsa-mir-24 | dbDEMC:HMDD: |
| hsa-mir-130a | dbDEMC: | hsa-mir-99a | dbDEMC: |
| hsa-mir-223 | dbDEMC:HMDD: | hsa-mir-95 | dbDEMC: |
| hsa-mir-18b | dbDEMC:HMDD: | hsa-mir-186 | dbDEMC: |
| hsa-mir-27a | miR2Disease:dbDEMC:HMDD: | hsa-mir-520c | miR2Disease:HMDD: |
| hsa-mir-373 | miR2Disease:dbDEMC:HMDD: | hsa-mir-22 | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-98 | miR2Disease:dbDEMC: | hsa-mir-196b | dbDEMC: |
| hsa-mir-100 | dbDEMC:HMDD: | hsa-mir-491 | dbDEMC: |
| hsa-mir-99b | dbDEMC: | hsa-mir-455 | dbDEMC: |
| hsa-mir-372 | dbDEMC: | hsa-mir-193b | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-192 | dbDEMC: | hsa-mir-29c | miR2Disease:dbDEMC:HMDD: |
| hsa-mir-32 | dbDEMC: | hsa-mir-23b | dbDEMC:HMDD: |
| hsa-mir-16 | dbDEMC:HMDD: | hsa-mir-30a | miR2Disease:HMDD: |
| hsa-mir-92a | HMDD: | hsa-mir-224 | dbDEMC:HMDD: |
| hsa-mir-182 | miR2Disease:dbDEMC:HMDD: | hsa-mir-340 | dbDEMC:HMDD: |
In conclusion, based on the evaluation in all cross validation (global LOOCV, local LOOCV and 5-fold CV) and three kinds of case studies, ELLPMDA achieved an excellent predictive accuracy which is significantly better than most of previous methods. What's more, ELLPMDA was based on global similar network and could be applied to new diseases without any know linked miRNAs.
Discussions
With the rapid development of human society, human health is currently one of the most concerned topics worldwide, furthermore, how to overcome more and more human diseases is an international problem which has received a great many of attentions. Considering that miRNAs play a critical role in multiple biological processes as well as the developments and progressions of various human diseases, identifying potential miRNA-disease associations is currently a vital research topic which might contribute a lot in the protection, detection and treatment of complex human diseases. However, it will be very expensive and time-consuming if we test all the miRNA-disease pairs using biological experiment. Therefore, lots of computational models have been proposed to predict novel associations between miRNAs and diseases. Although these previous methods have successfully predicted a great many of miRNA-disease associations, there were always limitations in these methods, such as applicable scope and predictive accuracy. To solve these problems, in this study, we developed a novel predictive method ELLPMDA, which utilized ensemble learning to combine results given by three classic algorithms to reveal potential miRNA-disease associations. As mentioned above, in LOOCV, the AUCs of ELLPMDA is 0.9181 and 0.8181 in global case and local case, respectively. Also, in 5-fold CV, the AUC of ELLPMDA is 0.9193+/−0.0002, which means the predictive accuracy of ELLPMDA in different cases is obviously better than most of previous methods. Furthermore, case studies indicated that ELLPMDA could be implemented in all kinds of diseases, whatever related miRNAs exist or not, and ELLPMDA gave excellent performances in various diseases.
The success of ELLPMDA can be mainly attributed to several factories. First and the most important factory is that we exploited ensemble learning into novel miRNA-disease association prediction. In this study, we developed three models depending on classic similarity-based algorithms and utilized these models into prediction works. Every model gave ranks of all the miRNA-disease pairs and we obtained the overall predictive result by exploiting ensemble learning to weightedly combine the sorted results given by these separate models. Concrete weight was calculated according to the predictive accuracies of these methods in case study 1. By combining results from three models, we achieved an overall result which was better than all the three results given by single model. Secondly, integrated similarity for miRNAs and diseases, which obtained by integrating Gaussian interaction profile kernel similarity, miRNA functional similarity and disease semantic similarity gave us precise information about the similarities between every miRNA-miRNA pair and disease-disease pair. Such information enabled us to take advantage of similarity-based algorithms on miRNA-disease network. Besides, experimentally verified associations between miRNA-disease given by HMDD v2.0 database [41] helped a lot, because it is the foundation to construct the miRNA-disease networks.
Although ELLPMDA achieved a great performance, there are still a few ways to further improve it. First, the current miRNA-disease association is insufficient. Thus, more information about experimentally confirmed miRNA-disease associations could help a lot by completing the miRNA-disease association network and further improving the accuracy of all the existing similarity networks. Apparently, ELLPMDA will perform better when more information is available. Besides, there is not a powerful method to choose the optimal parameter for ELLPMDA. What's more, the weight of three methods might be calculated in a more reasonable way.
For future work, we would like to pay attention to the following two aspects. First, as discussed in [78], biological systems are seen as a network of interconnected components, which indicates the importance of taking the interconnection into account. In our approach, after obtaining the predicted similarities between all the miRNAs and diseases given by ELLPMDA, examining the existence of certain regular loops between these similarities is a wise practice to further improve the performance of our method. For example, we would expect an association exists between disease and miRNA if all the diseases which are associated with miRNA and are associated with miRNA, and disease is associated with both miRNA and . In addition, [79] offers us a method to modeling existing data, which invites us countless inspiration for further study.
Materials and methods
Human miRNA-disease associations
Information about human miRNA-disease associations is available in HMDD v2.0 database [41], which includes 5430 distinct experimentally confirmed miRNA-disease associations about 495 miRNAs and 383 diseases. For convenience, we constructed an adjacency matrix to better describe these associations. The element equals to 1 if there is an experimentally confirmed association between miRNA and disease. Otherwise, equals to 0.
MiRNA functional similarity
Based on the assumption that functionally similar miRNAs tend to be associated with phenotypically similar diseases, the miRNA functional similarity was calculated in previous work [32]. Thanks to these excellent works, we can straightly download the miRNA functional similarity data from http://www.cuilab.cn/files/images/cuilab/misim.zip. Similar to adjacency matrix , the matrix was constructed, in which the entity represented the value of similarity between the miRNAs and .
Disease semantic similarity model 1
It is reasonable to use a distinct Directed Acyclic Graph (DAG) to describe each disease. To state it more clearly, we exploited a to represent a disease D, in which every disease is regard as a node and there are directed edges from parent nodes to child nodes. is a node set which contains disease D itself and its parent nodes and is a edge set including all the directed edges. Then we defined the contribution of disease d in DAG (D) to the semantic value of disease D as follows:
| (1) |
where is the semantic contribution factor. The contribution score for disease d is inversely proportional to the distance from d to D. The semantic value of disease D can be defined as follows:
| (2) |
According to the observation that two diseases will have larger similarity score if they have larger shared part of their DAGs, the semantic similarity value between diseases and can be defind as follows:
| (3) |
Where is the disease semantic similarity matrix 1.
Disease semantic similarity model 2
What's more, we further constructed another disease semantic similarity matrix by considering the difference between the contributions of different disease terms in the same layer of . Apparently, in the SS1 defined above, the disease terms in the same layer of have the same contribution to the semantic value of disease D. However, it might be unfair if we give two diseases the same contribution when they appear different times in disease DAGs. To state it more clearly, if two diseases lie in the same layer of and the first disease appears much more times in disease DAGs than the second disease, we believed that the second disease is more specific to disease D. Thus, it is reasonable to give the second disease a higher weight. Thus, the contribution of disease term d in to the semantic value of disease can be defined as follows:
| (4) |
Combining the weighted contributions and the similar ways in Disease semantic similarity model 1, the disease semantic similarity value 2 between diseases and can be calculated as follows:
| (5) |
Where is the disease semantic similarity matrix 2.
Gaussian interaction profile kernel similarity
According to the assumption that functionally similar miRNAs tend to be associated with similar diseases, Gaussian interaction profile kernel similarity for diseases are calculated by considering the topologic information of known miRNA–disease association network. We respectively used binary vector and to denote the interaction profiles of disease and miRNA . Because the adjacency matrix contains the information about the associations between diseases and miRNAs, we use the and to represent the i-th row and j-th column in the adjacency matrix which was defined above. Thus, the Gaussian interaction profile kernel similarity of diseases and miRNAs can be defined as follows:
| (6) |
| (7) |
where parameter and were used to control the kernel bandwidth, which can be obtained by normalizing the new bandwidth parameter and (Both were set as 1 based on previous work (van Laarhoven, T. et al.. (2011)) [80]) which can be denoted as follows:
| (8) |
| (9) |
Yet we got two matrix and , in which the entity represent the Gaussian interaction profile kernel similarity between disease and diseases , and represent the Gaussian interaction profile kernel similarity between miRNAs and .
Integrated similarity for miRNAs and diseases
After the above processes, we now have two matrices and including similarity value between miRNAs, and three matrices , and including similarity value between diseases. By combining these matrices, the integrated similarity for miRNAs and diseases were defined as follows:
| (10) |
| (11) |
Where the represents the integrated similarity value between diseases and represent the integrated similarity value between miRNAs.
Ellpmda
In this study, we exploited ensemble learning into novel miRNA-disease association prediction. In short, ensemble learning is a common machine learning method which could improve the classification accuracy by combining classified results given by different classifiers. Based on this method, we weightedly combined the rank results given by Common Neighbors, Jaccard index and Katz index to achieve a better prediction result (see Fig. 2).
Figure 2.
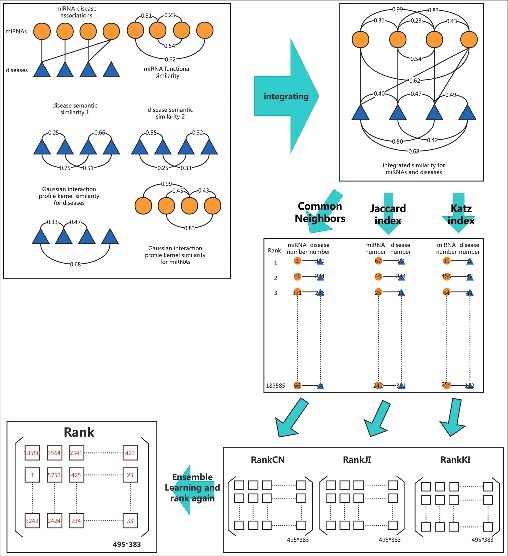
Flow chart of ELLPMDA in novel miRNA-disease association prediction by integrating known miRNA-disease associations, miRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity.
Common neighbors
For a note x, let donates the neighbor nodes set of node x. Intuitively speaking, if two nodes x and y share more neighbor nodes, a link is more likely to exist between x and y. In general, Common Neighbors score between node x and node y can be defined as follows:
| (12) |
Obviously, this equation directly count the number of nodes which are the neighbor of both x and y. In our study, considering the structure of our integrated network, which was obtained by combining human miRNA-disease associations and integrated similarity for miRNAs and diseases, we defined as the neighbor nodes set of x which only contains disease nodes while the donates the neighbors nodes set of x which only contain miRNA nodes. We can then redefine CN score between disease and miRNA as:
| (13) |
After calculating the scores of all the miRNA-disease pairs (between 495 miRNAs and 383 diseases), we were able to rank all the candidate samples based on CN score and further obtain an rank matrix , where the element represents the rank of pair. For example, if the CN score between miRNA 35 and disease 46 is highest and miRNA 78 and disease 365 achieved the second highest CN score, equals to 1 and equals to 2.
Jaccard index
The Jaccrad index [81] could be regard as the normalized Common Neighbors. Considering that if a node x have only 5 neighbor nodes and a node y have 200 neighbor nodes, however, they both have 5 common neighbor nodes with node z. It is reasonable to believe x is more ‘valuable’ to node z. As a result, to eliminate the bias to nodes with a lot of neighbor nodes, we defined JI score between disease and miRNA as follows:
| (14) |
where
| (15) |
| (16) |
In general, we ought to normalize CN score between disease and miRNA by dividing the number of neighbor nodes of them. However, to take the integrated similarity for miRNAs and diseases into account, we used the similarity between two nodes to replace the existence of a link between them. Thus, equation (16) is easy to understand. Similar to Common Neighbors, we obtained a rank matrix to save the ranks for all the miRNA-disease pairs.
Katz index
The Katz index [82] is a path-dependent global measure which directly sums all the possible paths between two nodes in a network and is exponentially damped to give the shorter paths more weight [83] .In order to take all the aforementioned materials into account, the matrix , a biadjacency matrix consist of human miRNA-disease associations matrix and integrated similarity for miRNAs and diseases, has been constructed. It can be represented as follow:
| (17) |
In this way, we could construct a network, in which every miRNA and disease represents a node. Undirected edges exist between two miRNAs or two diseases, and the weight of edge equals to the similarity between them (An edge won't exist if the similarity between two nodes is 0). Moreover, every experimentally confirmed miRNA-disease association corresponds an edge with a weight of 1. After establishing this network, path-depended algorithm could be applied on our network.
In our work for potential miRNA-disease association prediction, calculating the similarity between diseases and miRNAs could be transform to the problem of counting the walks from one miRNA nodes to a disease node. Considering that shorter walks tend to contribute more, we introduced a nonnegative parameter to control the weight of walks with different lengths. A smaller value of means the weights of longer walks is smaller. Thus, the Katz index could be calculated as follows:
| (18) |
where denotes the number of walks with length l which links node and node . Then, we further let and replaced the , the whole Katz index could be written in matrix form as:
| (19) |
The records the similarities between all the nodes. Specifically, it can be represented by a partitioned matrix:
| (20) |
Being aware of it was generated based on matrix , it could be easily detected that S3 record the association probability between every miRNA-disease pair. Also, to ensure that the Katz index converge, the value ofmust be less than the reciprocal of the largest eigenvalue of . Considering that longer walks tend to be meaningless for our predictive work, we set k to 1, 2 and 3 respectively to evaluate the influence of this parameter. Based on equation (18), the can be writing as follows:
| (21) |
Here, means the probability of going from node x to node y in i steps which could be represented by matrix A, SD and SR as follows:
| (22) |
| (23) |
| (24) |
According to previous research [84] and our own test, we chose β as 0.01. Similar to Common Neighbors, we obtained a rank matrix to save the ranks for all the miRNA-disease pairs.
Ensemble learning
As mentioned above, we would achieve a rank matrix of every method which enabled us to examine these methods in case study 1. We chose 14 popular human diseases and predicted the top 50 potential associations for them. Therefore, there were 700 predictive results and we confirmed these results in miR2Disease and dbDEMC [39,40]. Let denoted as the predicted error rate of every method respectively. Base on classic ensemble learning method, we calculated the weight of methods as follows:
| (25) |
As a result, the final weights of Common Neighbor, Jaccard index and Katz index are 0.1725, 0.3992 and 0.4283. By this time, we could take advantage of these weights to weightedly combine , and to obtain the overall rank matrix. As mentioned above, every miRNA-disease pair could obtain a rank based on Common Neighbor, Jaccard index and Katz index. To accurately combine these ranks, we exploited reciprocal ranking method, which means a pair ranks t will achieve a score . Hence, every miRNA-disease pair could achieve three scores depending on its ranks in the aforementioned three methods. Exploiting the final weight to weightedly combine the three scores of all the miRNA-disease pairs and taking advantage of the same sorting method used in Common Neighbor, we finally obtained the overall rank matrix Rank, which records the ranks of every miRNA-disease pair given by ELLPMDA.
Supplementary Material
Funding Statement
XC was supported by the National Natural Science Foundation of China under Grant Nos. 61772531 and 11631014.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
XC was supported by National Natural Science Foundation of China under Grant Nos. 61772531 and 11631014.
References
- [1].Bartel DP. MicroRNAs.: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. [DOI] [PubMed] [Google Scholar]
- [2].Cheng AM, Byrom MW, Shelton J, et al... Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005;33(4):1290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Karp X, Ambros V. Encountering microRNAs in cell fate signaling. Science. 2005;310(5752):1288–9. [DOI] [PubMed] [Google Scholar]
- [4].Miska EA. How microRNAs control cell division, differentiation and death. Curr Opin Genetics Dev. 2005;15(5):563–8. [DOI] [PubMed] [Google Scholar]
- [5].Xu P, Guo M, Hay BA. MicroRNAs and the regulation of cell death. Trends Genetics. 2004;20(12):617–24. [DOI] [PubMed] [Google Scholar]
- [6].Alshalalfa M, Alhajj R. Using context-specific effect of miRNAs to identify functional associations between miRNAs and gene signatures. BMC Bioinformatics. 2013;14(12):S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cui Q, Yu Z, Purisima EO, et al... Principles of microRNA regulation of a human cellular signaling network. Mol Systems Biol. 2006;2(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–54. [DOI] [PubMed] [Google Scholar]
- [10].Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75(5):855–62. [DOI] [PubMed] [Google Scholar]
- [11].Griffiths-Jones S, Grocock RJ, Van Dongen S, Bateman A, et al... miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34(suppl 1):D140–D4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2010;39(Database issue):gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5. [DOI] [PubMed] [Google Scholar]
- [14].He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genetics. 2004;5(7):522–31. [DOI] [PubMed] [Google Scholar]
- [15].Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–69. [DOI] [PubMed] [Google Scholar]
- [16].Yu Z, Li Z, Jolicoeur N, et al... Aberrant allele frequencies of the SNPs located in microRNA target sites are potentially associated with human cancers. Nucleic Acids Res. 2007;35(13):4535–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Strillacci A, Griffoni C, Sansone P, et al... MiR-101 downregulation is involved in cyclooxygenase-2 overexpression in human colon cancer cells. Exp Cell Res. 2009;315(8):1439–47. [DOI] [PubMed] [Google Scholar]
- [18].Yang H, Kong W, He L, et al... MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res. 2008;68(2):425–33. [DOI] [PubMed] [Google Scholar]
- [19].Liu C, Kelnar K, Liu B, et al... The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17(2):211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bonci D, Coppola V, Musumeci M, et al... The miR-15a–miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14(11):1271–7. [DOI] [PubMed] [Google Scholar]
- [21].Chen X, Yan CC, Zhang X, et al... HGIMDA: Heterogeneous graph inference for miRNA-disease association prediction. Oncotarget. 2016;7(40):65257–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li J-Q, Rong Z-H, Chen X, et al... MCMDA: Matrix completion for MiRNA-disease association prediction. Oncotarget. 2017;8(13):21187–21199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen X, Yan CC, Zhang X, et al... RBMMMDA: predicting multiple types of disease-microRNA associations. Scientific Reports. 2015;5:13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen X, Yan G-Y. Semi-supervised learning for potential human microRNA-disease associations inference. Scientific Reports. 2014;4:5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen X, Liu M-X, Yan G-Y. RWRMDA: predicting novel human microRNA–disease associations. Mol BioSystems. 2012;8(10):2792–8. [DOI] [PubMed] [Google Scholar]
- [26].Chen X, Liu M-X, Cui Q-H, et al... Prediction of disease-related interactions between microRNAs and environmental factors based on a semi-supervised classifier. PloS One. 2012;7(8):e43425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen X. KATZLDA: KATZ measure for the lncRNA-disease association prediction. Scientific Reports. 2015;5:16840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen X, Yan CC, Zhang X, et al... WBSMDA: within and between score for MiRNA-disease association prediction. Scientific Reports. 2016;6:21106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xuan P, Han K, Guo M, et al... Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PloS One. 2013;8(8):e70204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jiang Q, Hao Y, Wang G, et al... Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Sys Biol. 2010;4(1):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bandyopadhyay S, Mitra R, Maulik U, et al... Development of the human cancer microRNA network. Silence. 2010;1(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang D, Wang J, Lu M, et al... Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics. 2010;26(13):1644–50. [DOI] [PubMed] [Google Scholar]
- [33].Goh K-I, Cusick ME, Valle D, et al... The human disease network. Proc Natl Acad Sci. 2007;104(21):8685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pasquier C, Gardès J. Prediction of miRNA-disease associations with a vector space model. Scientific Reports. 2016;6:27036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shi H, Xu J, Zhang G, et al... Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Systems Biol. 2013;7(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Xu C, Ping Y, Li X, et al... Prioritizing candidate disease miRNAs by integrating phenotype associations of multiple diseases with matched miRNA and mRNA expression profiles. Mol BioSystems. 2014;10(11):2800–9. [DOI] [PubMed] [Google Scholar]
- [37].Mørk S, Pletscher-Frankild S, Caro AP, et al... Protein-driven inference of miRNA–disease associations. Bioinformatics. 2013;30(3):btt677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xuan P, Han K, Guo Y, et al... Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics. 2015;31(11):btv039. [DOI] [PubMed] [Google Scholar]
- [39].Yang Z, Ren F, Liu C, et al... dbDEMC: a database of differentially expressed miRNAs in human cancers. BMC Genomics. 2010;11(4):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jiang Q, Wang Y, Hao Y, et al... miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37(suppl 1):D98–D104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li Y, Qiu C, Tu J, et al... HMDD v2. 0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014;42(D1):D1070–D4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Oved K, Morag A, Pasmanik-Chor M, et al... Genome-wide miRNA expression profiling of human lymphoblastoid cell lines identifies tentative SSRI antidepressant response biomarkers. Pharmacogenomics. 2012;13(10):1129–39. [DOI] [PubMed] [Google Scholar]
- [43].Drusco A, Nuovo GJ, Zanesi N, et al... MicroRNA profiles discriminate among colon cancer metastasis. PloS One. 2014;9(6):e96670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ogata-Kawata H, Izumiya M, Kurioka D, et al... Circulating exosomal microRNAs as biomarkers of colon cancer. PloS One. 2014;9(4):e92921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jemal A, Bray F, Center MM, et al... Global cancer statistics. CA: Cancer J Clin. 2011;61(2):69–90. [DOI] [PubMed] [Google Scholar]
- [46].Guo C, Sah JF, Beard L, et al... The noncoding RNA, miR‐126, suppresses the growth of neoplastic cells by targeting phosphatidylinositol 3‐kinase signaling and is frequently lost in colon cancers. Genes Chromosomes Cancer. 2008;47(11):939–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nakajima G, Hayashi K, Xi Y, et al... Non-coding MicroRNAs hsa-let-7g and hsa-miR-181b are associated with Chemoresponse to S-1 in colon cancer. Cancer Genomics-Proteomics. 2006;3(5):317–24. [PMC free article] [PubMed] [Google Scholar]
- [48].Shi B, Sepp-Lorenzino L, Prisco M, et al... Micro RNA 145 targets the insulin receptor substrate-1 and inhibits the growth of colon cancer cells. J BiolChem. 2007;282(45):32582–90. [DOI] [PubMed] [Google Scholar]
- [49].Volinia S, Calin GA, Liu C-G, et al... A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natal Acad Sci U S A. 2006;103(7):2257–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Asangani I, Rasheed S, Nikolova D, et al... MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27(15):2128–36. [DOI] [PubMed] [Google Scholar]
- [51].Pu Xx, Huang GL, Guo HQ, et al... Circulating miR‐221 directly amplified from plasma is a potential diagnostic and prognostic marker of colorectal cancer and is correlated with p53 expression. J Gastroenterol Hepatol. 2010;25(10):1674–80. [DOI] [PubMed] [Google Scholar]
- [52].Nishida N, Yokobori T, Mimori K, et al... MicroRNA miR-125b is a prognostic marker in human colorectal cancer. Int J Oncol. 2011;38(5):1437. [DOI] [PubMed] [Google Scholar]
- [53].He B, Yin B, Wang B, et al... microRNAs in esophageal cancer (Review). Mol Med Reports. 2012;6(3):459–65. [DOI] [PubMed] [Google Scholar]
- [54].Enzinger PC, Mayer RJ. Esophageal cancer. N Eng J Med. 2003;349(23):2241–52. [DOI] [PubMed] [Google Scholar]
- [55].Lee K-H, Goan Y-G, Hsiao M, et al... MicroRNA-373 (miR-373) post-transcriptionally regulates large tumor suppressor, homolog 2 (LATS2) and stimulates proliferation in human esophageal cancer. Exp Cell Res. 2009;315(15):2529–38. [DOI] [PubMed] [Google Scholar]
- [56].Zhang H, Li M, Han Y, et al... Down-regulation of miR-27a might reverse multidrug resistance of esophageal squamous cell carcinoma. Digestive Dis Sci. 2010;55(9):2545–51. [DOI] [PubMed] [Google Scholar]
- [57].Yu X, Jiang X, Li H, et al... miR-203 inhibits the proliferation and self-renewal of esophageal cancer stem-like cells by suppressing stem renewal factor Bmi-1. Stem Cells Dev. 2013;23(6):576–85. [DOI] [PubMed] [Google Scholar]
- [58].Jemal A, Siegel R, Ward E, et al... Cancer statistics, 2006. CA: Cancer J Clin. 2006;56(2):106–30. [DOI] [PubMed] [Google Scholar]
- [59].Manojlović D, Elaković D, Mladenović L. Therapeutic value of transcatheter embolization in malignant tumors of the renal parenchyma. Srpski Arhiv Za Celokupno Lekarstvo. 1986;114(7):631–7. [PubMed] [Google Scholar]
- [60].Takehara K, Nishikido M, Koga S, et al... Multifocal transitional cell carcinoma associated with renal cell carcinoma in a patient on long‐term haemodialysis. Nephrol Dialysis Transplantation. 2002;17(9):1692–4. [DOI] [PubMed] [Google Scholar]
- [61].Abdelmohsen K, Kim MM, Srikantan S, et al... miR-519 suppresses tumor growth by reducing HuR levels. Cell Cycle. 2010;9(7):1354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mikhaylova O, Stratton Y, Hall D, et al... VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell. 2012;21(4):532–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nakada C, Tsukamoto Y, Matsuura K, et al... Overexpression of miR‐210, a downstream target of HIF1α, causes centrosome amplification in renal carcinoma cells. J Pathol. 2011;224(2):280–8. [DOI] [PubMed] [Google Scholar]
- [64].Tsz-fung FC, Mankaruos M, Scorilas A, et al... The miR-17-92 cluster is over expressed in and has an oncogenic effect on renal cell carcinoma. J Urol. 2010;183(2):743–51. [DOI] [PubMed] [Google Scholar]
- [65].Lu R, Ji Z, Li X, et al... miR-145 functions as tumor suppressor and targets two oncogenes, ANGPT2 and NEDD9, in renal cell carcinoma. J Cancer Res Clin Oncol. 2014;140(3):387–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bai X-Y, Ma Y, Ding R, et al... miR-335 and miR-34a Promote renal senescence by suppressing mitochondrial antioxidative enzymes. J Am Soc Nephrol. 2011;22(7):1252–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Thai T-H, Calado DP, Casola S, et al... Regulation of the germinal center response by microRNA-155. Science. 2007;316(5824):604–8. [DOI] [PubMed] [Google Scholar]
- [68].Yang M, Shen H, Qiu C, et al... High expression of miR-21 and miR-155 predicts recurrence and unfavourable survival in non-small cell lung cancer. Eur J Cancer. 2013;49(3):604–15. [DOI] [PubMed] [Google Scholar]
- [69].Yuxia M, Zhennan T, Wei Z. Circulating miR-125b is a novel biomarker for screening non-small-cell lung cancer and predicts poor prognosis. J Cancer Res Clin Oncol. 2012;138(12):2045–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Osada H, Takahashi T. let‐7 and miR‐17‐92: Small‐sized major players in lung cancer development. Cancer Sci. 2011;102(1):9–17. [DOI] [PubMed] [Google Scholar]
- [71].Hayashita Y, Osada H, Tatematsu Y, et al... A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005;65(21):9628–32. [DOI] [PubMed] [Google Scholar]
- [72].Zhang C, Zhang J, Zhang A, et al... PUMA is a novel target of miR-221/222 in human epithelial cancers. Int J Oncol. 2010;37(6):1621. [DOI] [PubMed] [Google Scholar]
- [73].Tao Z, Shi A, Lu C, et al... Breast cancer: epidemiology and etiology. Cell Biochem Biophys. 2015;72(2):333–8. [DOI] [PubMed] [Google Scholar]
- [74].Yan L-X, Huang X-F, Shao Q, et al... MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. Rna. 2008;14(11):2348–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hossain A, Kuo MT, Saunders GF. Mir-17-5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol Cell Biol. 2006;26(21):8191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang H, Tan G, Dong L, et al... Circulating MiR-125b as a marker predicting chemoresistance in breast cancer. PloS One. 2012;7(4):e34210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Camps C, Buffa FM, Colella S, et al... hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res. 2008;14(5):1340–8. [DOI] [PubMed] [Google Scholar]
- [78].Cloutier M, Wang E. Dynamic modeling and analysis of cancer cellular network motifs. Integrative Biol. 2011;3(7):724–32. [DOI] [PubMed] [Google Scholar]
- [79].Wang E, Zaman N, Mcgee S, et al... editors Predictive genomics: a cancer hallmark network framework for predicting tumor clinical phenotypes using genome sequencing data. Semin Cancer Biol. 2015;30:4–12. Elsevier [DOI] [PubMed] [Google Scholar]
- [80].van Laarhoven T, Nabuurs SB, Marchiori E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics. 2011;27(21):3036–43. [DOI] [PubMed] [Google Scholar]
- [81].Jaccard P. Etude de la distribution florale dans une portion des Alpes et du Jura. Bulletin De La Societe Vaudoise Des Sciences Naturelles. 1901;37(142):547–79. [Google Scholar]
- [82].Katz L. A new status index derived from sociometric analysis. Psychometrika. 1953;18(1):39–43. [Google Scholar]
- [83].Lü L, Zhou T. Link prediction in complex networks: A survey. Physica A: Statistical Mechan Applications. 2011;390(6):1150–70. [Google Scholar]
- [84].Chen X, Huang Y-A, You Z-H, et al... A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics. 2016;33(5):btw715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.