

ABSTRACT
CASP4/caspase-11-dependent inflammasome activation is important for the clearance of various Gram-negative bacteria entering the host cytosol. Additionally, CASP4 modulates the actin cytoskeleton to promote the maturation of phagosomes harboring intracellular pathogens such as Legionella pneumophila but not those enclosing nonpathogenic bacteria. Nevertheless, this non-inflammatory role of CASP4 regarding the trafficking of vacuolar bacteria remains poorly understood. Macroautophagy/autophagy, a catabolic process within eukaryotic cells, is also implicated in the elimination of intracellular pathogens such as Burkholderia cenocepacia. Here we show that CASP4-deficient macrophages exhibit a defect in autophagosome formation in response to B. cenocepacia infection. The absence of CASP4 causes an accumulation of the small GTPase RAB7, reduced colocalization of B. cenocepacia with LC3 and acidic compartments accompanied by increased bacterial replication in vitro and in vivo. Together, our data reveal a novel role of CASP4 in regulating autophagy in response to B. cenocepacia infection.
KEYWORDS: autophagy, Burkholderia cenocepacia, caspase-1 (CASP1), caspase-11 (CASP4), lysosome, macrophages
Introduction
Autophagy, an intracellular catabolic pathway, is a highly regulated stress response that ensures cellular homeostasis and provides protection against harmful conditions. Typical stimuli known to induce autophagy are nutrient deprivation, increased production of reactive oxygen species, and the accumulation of protein aggregates. The breakdown of damaged organelles and proteins via autophagy leads to the recycling of nutrients and sustains cellular energy levels. In addition, autophagy targets various intracellular pathogens such as Mycobacterium tuberculosis [1], Salmonella enterica serovar Typhimurium [2], Legionella pneumophila [3], Streptococcus pyogenes [4], and Burkholderia cenocepacia [5]. This bacteria-specific autophagic process, designated xenophagy, promotes bacterial clearance and prevents cellular escape. Bacteria such as Listeria monocytogenes [6,7] or Burkholderia pseudomallei [8], although recognized by members of the autophagic pathway, have developed mechanisms to avoid lysosomal degradation and xenophagy.
B. cenocepacia, commonly found in soil and water, is a Gram-negative facultative intracellular pathogen that causes severe infections among immune-deficient individuals, particularly patients with cystic fibrosis (CF) or chronic granulomatous disease [9–11]. We and others found that defective autophagy is responsible for the persistence of B. cenocepacia within eukaryotic cells. Maturation of the B. cenocepacia-containing vacuole (BCV) is delayed in murine RAW 264.7 macrophages, yet delivery to lysosomes still occurs and infection is restricted [12]. In contrast, CFTR (cystic fibrosis transmembrane conductance regulator) ∆F508 macrophages from human and mice are permissive to B. cenocepacia due to inefficient fusion of the BCV with lysosomes. Therefore, it can be concluded that defective autophagy promotes survival of B. cenocepacia in professional phagocytes. However, pharmacological stimulation of autophagy prior to infection restores bacterial delivery to acidic lysosomal compartments, thereby improving degradation of B. cenocepacia within murine CF macrophages [5]. Human macrophages derived either from patients with CF or chronic granulomatous disease also show improved restriction of B. cenocepacia after treatment with the autophagy-inducing agent rapamycin [13,14].
Alongside autophagy, inflammasome assembly is a crucial mechanism for innate immune cell defense against invading pathogens. Activation of CASP1/caspase-1 within a molecular platform comprising cytosolic NOD-like receptors (NLRs) and various adaptor proteins drives the secretion of pro-inflammatory cytokines such as IL1B/IL-1β (interleukin 1 beta) and IL18, which can be accompanied by plasma membrane disruption and host cell death (pyroptosis). As part of the non-canonical inflammasome, CASP4/caspase-11 contributes to cytokine release and bacterial restriction either upstream or independently of CASP1 [15–18]. B. cenocepacia activates CASP1 via NLRP3 in murine macrophages [19] and via MEFV/PYRIN in human monocytes [20] in a type VI secretion system (T6SS)-dependent manner, resulting in pronounced inflammation. A potential role for CASP4 during B. cenocepacia infection, however, has not yet been investigated.
Whereas cross-talk between autophagy and inflammasomes has been reported by several groups, these studies mostly report that autophagy negatively regulates inflammasome signaling to prevent excessive inflammation [21–23]. However, during L. pneumophila infection, inflammasome activation promotes autophagy [24]. Specifically, CASP4 activity modulates actin polymerization in L. pneumophila-infected macrophages, and hence facilitates the trafficking of phagosomes to lysosomal compartments for bacterial clearance [16,25]. Interestingly, transportation of phagosomes containing nonpathogenic Escherichia coli is not influenced by CASP4, suggesting differential regulation of endocytic pathways depending on the captured cargo [16]. Nonetheless, it remains unclear if CASP4 plays a role for the trafficking of other pathogenic bacteria such as B. cenocepacia.
Here we demonstrate that CASP4 positively regulates autophagosome formation and trafficking to lysosomes in murine macrophages. CASP4 deficiency leads to increased replication of B. cenocepacia in murine macrophages in vitro and infected lungs accompanied by elevated bacterial dissemination to the spleen in vivo. Furthermore, transport of B. cenocepacia to phagophores, the precursors to autophagosomes, and subsequent delivery to lysosomes is compromised in casp4−/- macrophages resulting in the accumulation of RAB7, a small GTPase that is involved in phagosome maturation and acidification [26]. Our findings provide new insights into non-pyroptotic functions of CASP4 by facilitating autophagosome biogenesis and trafficking in response to intracellular pathogens.
Results
CASP4 contributes to the restriction of B. cenocepacia and the release of cytokines in vitro
CASP4 restricts the growth of L. pneumophila, an intracellular pathogen that resides in autophagosomes in WT macrophages [3,16,25]. Notably, B. cenocepacia is also cleared by autophagosomes in healthy macrophages [5], but the role of CASP4 in intracellular trafficking of this bacterium is not known. In order to determine whether CASP4 is involved in the restriction of B. cenocepacia, we infected bone marrow-derived macrophages from C57BL/6 wild-type (WT) and casp4−/- mice and examined intracellular bacterial growth. Compared to WT macrophages, we found an approximately 1.9-fold increase in colony forming units (CFUs) at 6 h post infection in the absence of CASP4 (Figure 1a and S1A). When the numbers of intracellular bacteria was analyzed via confocal microscopy, a higher proportion of casp4−/- macrophages contained > 5–10 bacteria/cell compared to WT cells at 6 h post infection (Figure 1b and c). Accordingly, significantly more WT macrophages contained < 5 bacteria/cell (Figure 1c), suggesting that bacterial clearance is more effective in WT macrophages. In order to determine the amount of cell death in response to B. cenocepacia infection, we measured the release of LDH (lactate dehydrogenase) from WT and casp4−/- macrophages at 6 h post infection. Single casp1−/- macrophages that are defective in pyroptosis, were included as a control. We found lower levels of LDH in the supernatants from casp4−/- and casp1−/- macrophages, but overall cell death was low in all 3 genotypes (Figure 1d).
Figure 1.
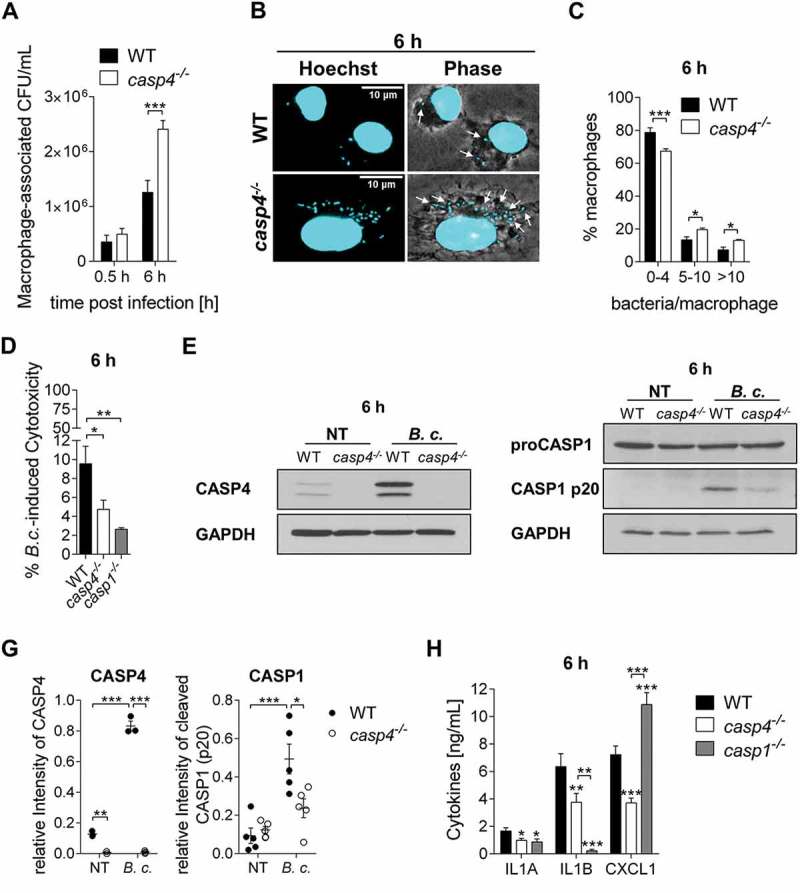
CASP4/caspase-11 contributes to the restriction of B. cenocepacia in vitro. (a) Intracellular survival of B. cenocepacia MH1K (B.c.) in WT and casp4−/- macrophages. Data represent mean ± SEM (n = 9). Statistical analysis was performed using two-way ANOVA. (b) Immunofluorescence assay of intracellular B. cenocepacia in WT and casp4−/- macrophages at 6 h post infection. White arrows point to B. cenocepacia. (c) Quantification of intracellular B. cenocepacia in WT and casp4−/- macrophages at 6 h post infection. Values are mean percentage ± SEM calculated by scoring 60 randomly chosen fields of view from 3 experiments. Statistical analyses were performed using a linear mixed effects model. (d) B. cenocepacia-induced cytotoxicity was calculated by measuring LDH release in supernatants from WT and casp4−/- macrophages at 6 h post infection. Statistical analyses were performed using one-way ANOVA. (e) Immunoblot analysis of CASP4 from WT and casp4−/- macrophages infected with B. cenocepacia MH1K at 6 h post infection. (f) CASP1 immunoblot analysis of whole cell lysates from WT and casp4−/- macrophages infected with B. cenocepacia MH1K at 6 h post infection. (g) Densitometry analysis of CASP4 (n = 3) and cleaved CASP1 (n = 5) at 6 h post infection. Shown are mean ± SEM. Statistical analysis was performed using two-way ANOVA. (h) Cytokine release at 6 h from WT, casp4−/- and casp1−/- macrophages infected with B. cenocepacia (n = 9). Shown are mean ± SEM. Statistical analysis was performed using one-way ANOVA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. NT, no treatment.
Basal expression of CASP4 is low in resting cells and its expression and activity are triggered by bacterial factors such as intracellular lipopolysaccharide [27,28]. To determine if B. cenocepacia infection of WT macrophages induces CASP4 protein expression, macrophages were infected with bacteria for 2 h or 6 h and the expression of CASP4 was evaluated by immunoblot. Like other Gram-negative bacteria, B. cenocepacia induced the expression of CASP4 in WT macrophages with an ~ 1.7-fold increase at 2 h (Fig. S1B-C) and a 6-fold increase at 6 h compared to non-infected control cells (Figure 1e and g). Because CASP4 stimulates non-canonical NLRP3 inflammasome activation in response to Gram-negative bacteria [15], we further analyzed if CASP4 contributes to B. cenocepacia-induced CASP1 activation. The processing of CASP1 in WT and casp4−/- macrophages was compared via immunoblot. As shown in Figure 1f-g and S1D, proteolytic cleavage of CASP1 after B. cenocepacia infection is significantly decreased in casp4−/- macrophages, indicating a crucial role for CASP4 in B. cenocepacia-mediated inflammasome activity.
To determine if CASP4 also modulates the secretion of IL1B in response to B. cenocepacia, we measured the amount of released IL1B from macrophages at 6 h post infection using casp1−/- macrophages as a control. Figure 1h and S1E show that CASP4 deficiency resulted in significantly decreased production of IL1B. Interestingly, casp4−/- macrophages also produced lower levels of the inflammasome-independent cytokine IL1A (interleukin 1 alpha) and the chemokine CXCL1/KC (analog of human CXCL8/IL8) (Figure 1h). In contrast, the absence of CASP1 was not associated with decreased CXCL1/KC secretion.
Because CASP7 (caspase 7) is a known substrate of CASP1 [29,30], we further evaluated if CASP4 affected CASP7 activation. Indeed, B. cenocepacia induced cleavage of CASP7 in WT macrophages, which was significantly reduced in casp4−/- macrophages (Fig. S1F-G). Therefore, these results show that CASP4 restricts the growth of B. cenocepacia and modulates the release of cytokines and the activation of CASP1 and CASP7 in macrophages.
CASP4 is expressed in mefv−/-/pyrin−/- macrophages with reduced activation of CASP1 in response to B. cenocepacia
Because the PYRIN inflammasome also plays a role in the detection of B. cenocepacia in human and mouse macrophages [20,31,32], we next compared B. cenocepacia-induced CASP4 expression and the activation of CASP1 in WT, mefv−/-, and casp4−/- macrophages at 6 h post infection via immunoblot. Expression of CASP4 in response to B. cenocepacia was markedly induced in WT and mefv−/- macrophages (Fig. S2A). With respect to B. cenocepacia-induced CASP1 activation, proteolytic processing of CASP1 was comparably decreased in both mefv−/- and casp4−/- macrophages compared to the corresponding WT cells (Fig. S2B). These data indicate that both MEFV/PYRIN and CASP4 contribute to CASP1 activation in response to B. cenocepacia infection.
CASP4 promotes the restriction of B. cenocepacia and the release of cytokines in vivo
Next, to examine if CASP4 deficiency also contributes to B. cenocepacia persistence in vivo, we infected WT and casp4−/- mice intratracheally with sublethal and lethal doses of B. cenocepacia and followed the course of infection. After sublethal infection, bacterial loads in casp4−/- mice were increased by 3.5-fold in the lung and by 2.5-fold in the spleen after 48 h when compared to the corresponding WT mice (Figure 2a). We also found that lethal doses of B. cenocepacia led to the death of all WT mice, whereas 75% of casp4−/- mice survived at the end of the experiment at day 8 post infection (Figure 2b). Notably, casp4−/- mice exhibited significantly lower amounts of the inflammatory cytokines IL1A, IL1B, and CXCL1/KC in their bronchoalveolar lavage fluid (BALF) (Figure 2c) despite increased bacterial persistence in their lungs (Figure 2a). Histological sections of lung tissue from WT mice were characterized by marked alveolitis, multifocal vasculitis and fibrin exudation (Figure 2d and S3A). Compared to their casp4−/- counterparts, WT mice showed marked to severe inflammatory cell infiltration (Figure 2d) with increased levels of the inflammatory marker MPO (myeloperoxidase) in the BALF (Figure 2e). Notably, B. cenocepacia was still present in the lungs of surviving casp4−/- mice, indicating that the mice did not clear the infection (Fig. S3B). Together, these results indicate that expression of CASP4 restricts survival and dissemination of B. cenocepacia during infection, yet is associated with increased inflammatory cytokine production and the biomarker MPO, and decreased host survival.
Figure 2.
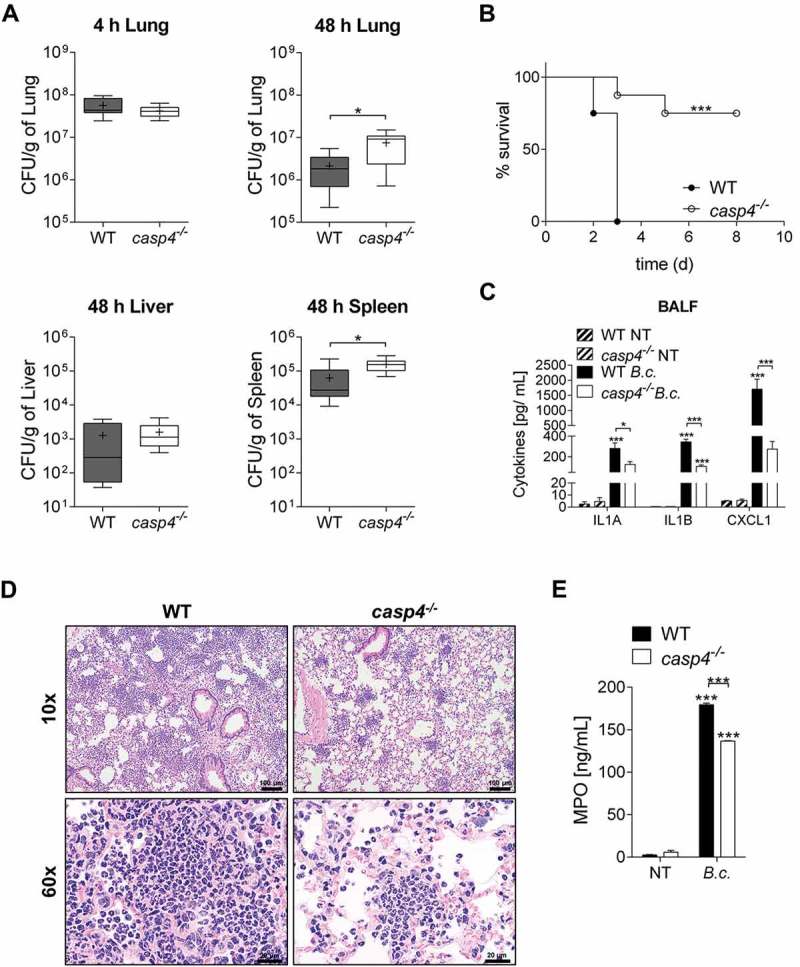
CASP4/caspase-11 contributes to the restriction of B. cenocepacia in vivo. (a) In vivo CFU from lung, liver and spleen of WT and casp4−/- mice infected with B. cenocepacia K-56 (B.c.) at 4 h (n = 9) and 48 h (n = 10). Pooled data from 2 independent experiments are shown as mean ± SEM. Statistical analysis was performed using two-tailed Student’s t-test. (b) Survival of WT and casp4−/- mice after intratracheal infection with 30 × 106 CFU. Pooled data from 2 independent experiments (n = 8). Statistical analysis was performed using Log-rank (Mantel-Cox) test. (c) Cytokine levels at 24 h in the BALF of WT (n = 7) and casp4−/- (n = 6) mice after intratracheal infection with 30 × 106 CFU. Statistical analysis was performed using two-way ANOVA. (d) Representative 10x (upper panel) and 60x (lower panel) magnification of H&E-stained lung sections from WT and casp4−/- mice showing increased inflammatory infiltrate within the WT sections. (e) MPO levels at 24 h in the BALF of WT and casp4−/- mice treated as in (C). Statistical analysis was performed using two-way ANOVA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
CASP4 promotes the formation of the B. cenocepacia-containing autophagosome
Because functional autophagy is important to clear intracellular B. cenocepacia in WT macrophages and epithelial cells [5], we next determined if the interaction between B. cenocepacia and the cellular autophagy system is affected by the lack of CASP4. We quantified the colocalization of B. cenocepacia with the autophagy marker LC3 in macrophages, and found fewer colocalization events at 0.5 h and 2 h post infection in the absence of CASP4 compared to WT cells (Figure 3a and b). Because stimulation of autophagy is characterized by the formation of autophagosomes, which can be identified as puncta using confocal microscopy, we next determined if puncta are formed in casp4−/- macrophages in response to infection. Notably, CASP4 deficiency led to reduced LC3 puncta formation in response to B. cenocepacia infection (Figure 3c). In contrast, no difference in LC3 colocalization between WT and casp4−/- macrophages was detected after infection with nonpathogenic E. coli (Figure 3d). Accordingly, restriction of intracellular E. coli was comparable in WT and casp4−/- macrophages (Fig. S4A). Therefore, these data indicate that CASP4 is required for the restriction of B. cenocepacia and its enclosure within autophagosomes but not for E. coli.
Figure 3.
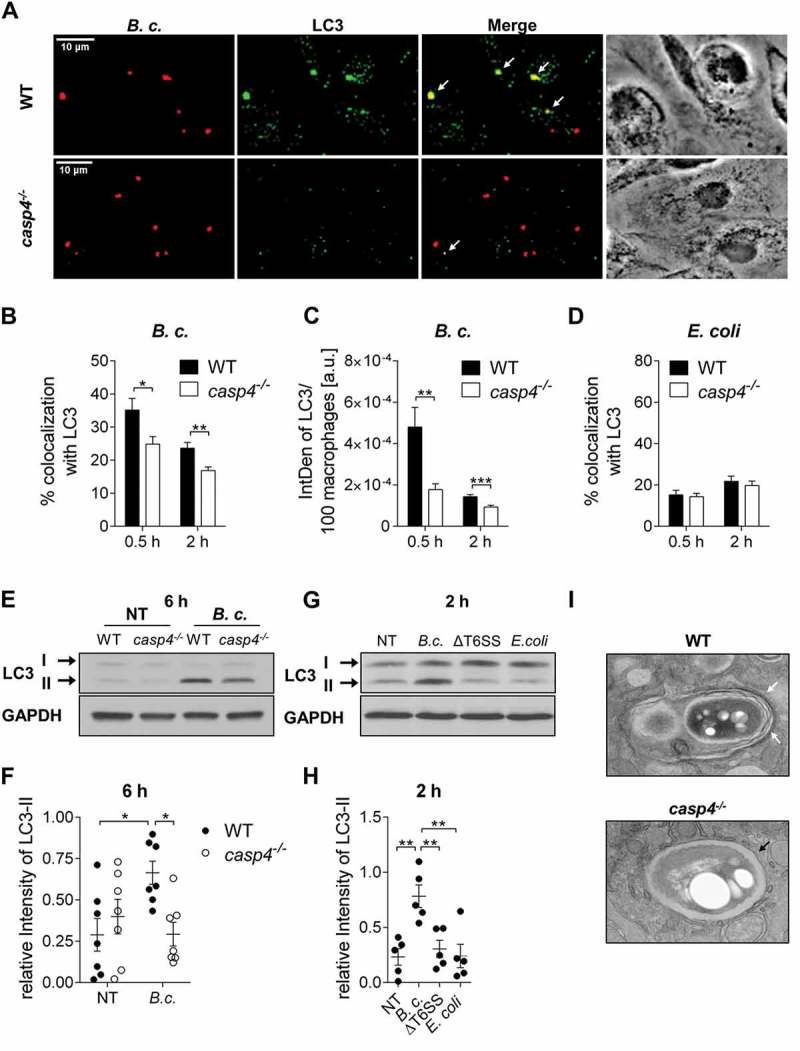
CASP4/caspase-11 promotes autophagosome formation in response to pathogenic bacteria. (a) LC3 immunofluorescence assay of B. cenocepacia (B.c.)-infected WT and casp4−/- macrophages at 30 min post infection. White arrows point to B. cenocepacia-LC3 colocalization. (b) Quantification of B. cenocepacia colocalized with LC3. (c) Quantification of total LC3 puncta in B. cenocepacia-infected WT and casp4−/- macrophages using ImageJ Software. a.u., arbitrary units. (d) Quantification of E. coli colocalized with LC3 in WT and casp4−/- macrophages. (A-D) Values are mean percentage ± SEM calculated by scoring 24 randomly chosen fields of view from at least 3 experiments. Statistical analyses were performed using Student’s t-test. (e) LC3-II immunoblot analysis from B. cenocepacia-infected WT and casp4−/- macrophages at 6 h post infection. (f) Densitometry analysis of B. cenocepacia-induced LC3-II in WT and casp4−/- macrophages at 6 h post infection. Shown are mean ± SEM of 7 independent experiments (n = 7). Statistical analysis was performed using two-way ANOVA. (g) LC3 immunoblot analysis in WT macrophages at 2 h after infection with B. cenocepacia wild-type, B. cenocepacia T6SS mutant and E. coli. (h) Densitometry analysis of LC3-II in WT macrophages treated as in (G). Shown are mean ± SEM of 5 independent experiments (n = 5). Statistical analysis was performed using one-way ANOVA. (i) Qualitative transmission electron microscopy images of B. cenocepacia-infected WT and casp4−/- macrophages at 2 h post infection. White arrows indicate a multilamellar membrane characteristic for autophagosomes. Black arrow indicates a single-membrane vacuole. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. No treatment (NT).
Another approach to detect the formation of autophagosomes is to determine the amount of cleavage and lipidation of LC3-I into LC3-II by western blot. To determine if B. cenocepacia infection was accompanied by the formation of autophagosomes, B. cenocepacia-induced LC3-II conversion was examined via immunoblot. The production of LC3-II upon B. cenocepacia infection was significantly lower within casp4−/- macrophages compared to their WT counterparts (Figure 3e and f). In WT macrophages, autophagy was clearly induced in response to B. cenocepacia, whereas nonpathogenic E. coli did not promote LC3-II conversion (Figure 3e-h and S4D-E).
The T6SS of B. cenocepacia is essential for intracellular pathogenicity [19,32]. Accordingly, T6SS deficiency led to enhanced phagocytosis and the impairment of bacterial replication in WT macrophages (Fig S4B and C). In addition, B. cenocepacia ΔT6SS did not induce autophagy within WT or casp4−/- macrophages. Figure 3g-h and S4F demonstrate that there was no significant increase in LC3-II formation upon infection with B. cenocepacia ΔT6SS when compared to non-infected control cells. To further visualize B. cenocepacia and its surrounding compartment in WT and casp4−/- macrophages, we used qualitative transmission electron microscopy (TEM). As shown in Figure 3i, in WT macrophages, B. cenocepacia was present in a multilamellar membrane structure typical of autophagosomes, whereas bacteria were enclosed by a single membrane in casp4−/- macrophages. Together, these results indicate that formation of autophagosomes in response to pathogenic B. cenocepacia is compromised in casp4−/- macrophages and requires a functional bacterial T6SS.
The absence of CASP4 impairs the delivery of B. cenocepacia to the lysosome
Degradation of bacteria such as L. pneumophila and B. cenocepacia via autophagy requires the fusion of the cargo-transporting autophagosomes with lysosomal compartments, leading to the formation of autolysosomes. Yet, a possible role of CASP4 in the trafficking of autophagosomes has not been examined. To determine if CASP4 contributes to the trafficking of compartments containing B. cenocepacia to lysosomes, we examined the uptake of LysoTracker Green by phagosomes containing B. cenocepacia via confocal microscopy in WT and casp4−/- macrophages. No difference in the number of bacteria colocalized with LysoTracker Green could be found between WT and casp4−/- macrophages during the early stages of infection (0.5 h). However, at later time points, WT macrophages exhibited significantly more bacteria colocalized with LysoTracker Green-labeled compartments than was seen with CASP4-deficient cells (Figure 4a and b). In addition, total LysoTracker Green fluorescence was decreased at 2 h post infection in both cell types, yet more pronounced in casp4−/- macrophages (Figure 4c). To further determine, if B. cenocepacia requires the T6SS to reduce acidic organelles, we compared total LysoTracker Green fluorescence in WT macrophages upon infection with B. cenocepacia and its corresponding T6SS mutant, or nonpathogenic E. coli. B. cenocepacia significantly reduced LysoTracker Green fluorescence in WT macrophages in a multiplicity of infection (MOI)-dependent manner (Figure 4d). In contrast, macrophages infected with B. cenocepacia ΔT6SS or E. coli, exhibited a similar level of LysoTracker Green fluorescence as measured in non-treated cells (Figure 4d). These results indicate that whereas CASP4 mediates the fusion of B. cenocepacia-containing compartments with lysosomes, the pathogen uses its T6SS to reduce the number of acidic compartments within macrophages.
Figure 4.
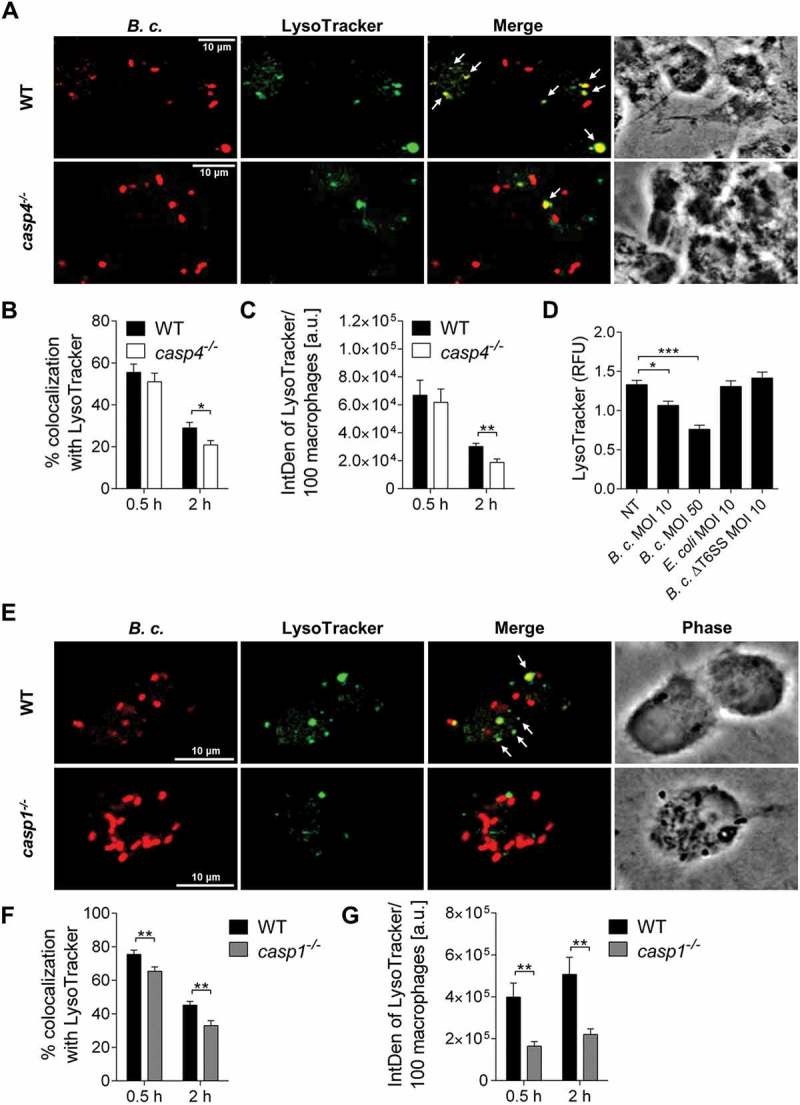
CASP4/caspase-11 and CASP1 modulate trafficking of the B. cenocepacia-containing vacuole. (a) LysoTracker Green immunofluorescence assay of B. cenocepacia (B.c.)-infected WT and casp4−/- macrophages at 2 h post infection. White arrows point to B. cenocepacia-LysoTracker colocalization. (b) Quantification of B. cenocepacia colocalized with LysoTracker Green in WT and casp4−/- macrophages. Statistical analyses were performed using Student’s t-test. (c) Quantification of total LysoTracker Green in B. cenocepacia-infected WT and casp4−/- macrophages shown in A-B using ImageJ Software. Values are mean ± SEM calculated by scoring 24 randomly chosen fields of view from at least 3 independent experiments. Statistical analyses were performed using Student’s t-test. a.u., arbitrary units. (d) Total LysoTracker Green fluorescence in WT macrophages. Shown are mean ± SEM of 15 independent experiments (n = 15). Statistical analysis was performed using one-way ANOVA. (e) LysoTracker Green immunofluorescence assay of B. cenocepacia-infected WT and casp1−/- macrophages at 2 h post infection. White arrows point to B. cenocepacia-LysoTracker colocalization. (f) Quantification of B. cenocepacia colocalized with LysoTracker Green WT and casp1−/- macrophages. Statistical analyses were performed using Student’s t-test. (g) Quantification of total LysoTracker Green in B. cenocepacia-infected WT and casp1−/- macrophages shown in E-F using ImageJ Software. Values are mean ± SEM calculated by scoring 24 randomly chosen fields of view from at least 3 experiments. Statistical analyses were performed using Student’s t-test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. NT, no treatment.
B. cenocepacia-containing autophagosomes require the enzymatic activity of CASP1 and CASP4 to fuse with lysosomes
Because CASP4 is partially required for CASP1 activity in response to B. cenocepacia (Figure 1f-g and S2B), we next examined if casp1−/- macrophages display impaired trafficking of B. cenocepacia to acidic vesicles as well. Indeed, CASP1 deficiency decreased colocalization of B. cenocepacia with LysoTracker Green (Figure 4e-g). These data show that inflammasome-dependent fusion of the B. cenocepacia-containing vacuoles with lysosomes is mediated by both CASP4 and CASP1. To further determine if CASP1 also plays a role for the trafficking of B. cenocepacia to phagophores, we quantified the colocalization of B. cenocepacia with LC3 in WT and casp1−/- macrophages at 0.5 h and 2 h post infection. In the absence of CASP1, fewer bacteria colocalized with LC3 (Figure 5a-c). Furthermore, to test if the enzymatic activities of CASP4 and CASP1 are required for autophagosome formation in response to B. cenocepacia, WT macrophages were treated with the inhibitor Ac-YVAD-CMK, which inhibits the activity of both CASP1 and CASP4. Then, the production of LC3-II was examined via immunoblot. Figure 5d-e show that Ac-YVAD-CMK treatment reduced the production of LC3-II at 6 h post infection. Thus, the autophagy response to B. cenocepacia requires enzymatically active CASP1 and CASP4.
Figure 5.

Autophagosome formation in response to B. cenocepacia requires active CASP1. (a) LC3 immunofluorescence assay of B. cenocepacia (B.c.)-infected WT and casp1−/- macrophages at 2 h post infection. White arrows point to B. cenocepacia-LC3 colocalization. (b) Quantification of B. cenocepacia colocalized with LC3. (c) Quantification of total LC3 puncta in B. cenocepacia-infected WT and casp1−/- macrophages using ImageJ Software. Values are mean percentage ± SEM calculated by scoring 24 randomly chosen fields of view from at least 3 experiments. Statistical analyses were performed using Student’s t-test. a.u., arbitrary units. (d) LC3-II immunoblot analysis from B. cenocepacia-infected WT macrophages treated with Ac-YVAD-CMK (50 µM) at 6 h post infection. (e) Densitometry analysis of B. cenocepacia-induced LC3-II in WT macrophages treated with Ac-YVAD-CMK at 6 h post infection. Shown are mean ± SEM of 6 independent experiments (n = 6). Statistical analysis was performed using two-way ANOVA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. NT, no treatment.
CASP4 deficiency is accompanied by the accumulation of RAB7 during B. cenocepacia infection
RAB proteins are small GTPases that, when activated in their GTP-bound form, regulate vesicle trafficking and fusion events. RAB7, usually present on late endosomes, regulates the transport of endocytic membranes [33,34], and promotes the maturation of autophagic compartments [35]. To determine the mechanism by which CASP4 mediates B. cenocepacia-phagosome maturation, we examined if the recruitment of RAB7 to the B. cenocepacia-containing vacuoles is altered in casp4−/- macrophages. Notably, there was increased colocalization of B. cenocepacia with RAB7 (Figure 6a-b) accompanied by an accumulation of RAB7-positive compartments in the absence of CASP4 (Figure 6c). An increase in RAB7 expression in casp4−/- macrophages after infection with B. cenocepacia was also observed by immunoblotting (Figure 6d-e). Interestingly, RAB7 activity assessed by immunoprecipitation of the GTP bound form revealed a higher proportion of GTP-bound RAB7 in B. cenocepacia-infected casp4−/- macrophages compared to the corresponding WT cells (Figure 6f-g). Thus, CASP4 deficiency is characterized by an accumulation of RAB7 around the B. cenocepacia-containing vacuole.
Figure 6.
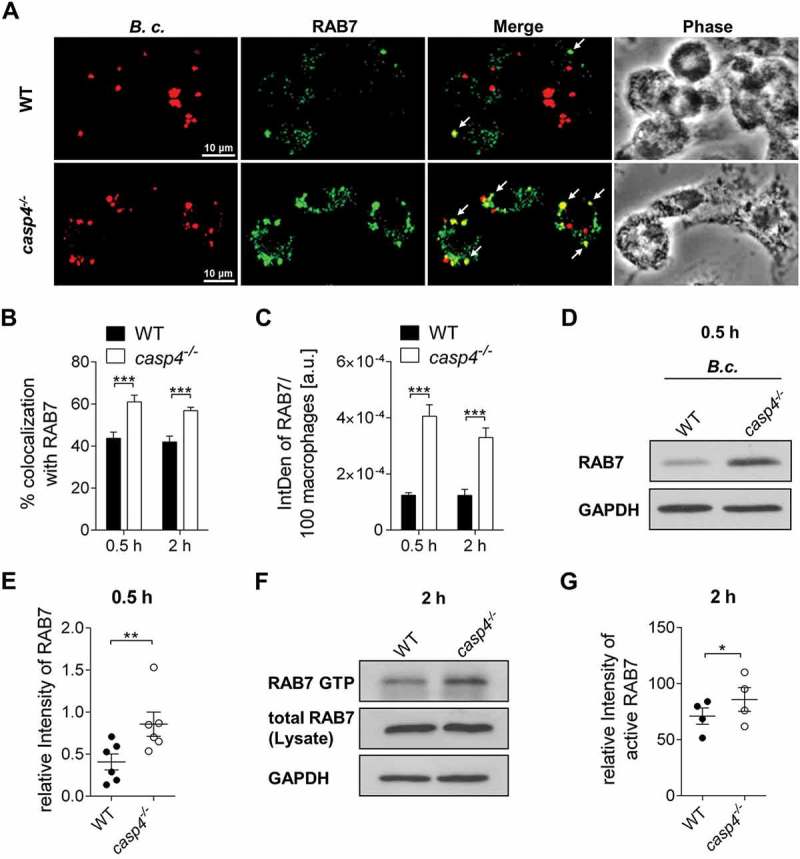
casp4−/-/caspase-11−/- macrophages accumulate RAB7 around the B. cenocepacia-containing vacuole. (a) RAB7 immunofluorescence assay of B. cenocepacia (B.c.)-infected WT and casp4−/- macrophages at 30 min post infection. White arrows point to B. cenocepacia-RAB7 colocalization. (b) Quantification of B. cenocepacia colocalized with RAB7. (c) Quantification of RAB7 in B. cenocepacia-infected WT and casp4−/-macrophages using ImageJ Software. (A-C) Values are mean percentage ± SEM calculated by scoring 24 randomly chosen fields of view from at least 3 experiments. Statistical analyses were performed using Student’s t-test. a.u., arbitrary units. (d) RAB7 immunoblot analysis from WT and casp4−/- macrophages infected with B. cenocepacia at 30 min post infection. (e) Densitometry analysis of RAB7 in B. cenocepacia-infected WT and casp4−/- macrophages. Shown are mean ± SEM of 6 independent experiments (n = 6). Statistical analyses were performed using paired t-test. (f) Immunoprecipitation of GTP-bound RAB7 in B. cenocepacia-infected WT and casp4−/- macrophages at 2 h post infection. (g) Densitometry analysis of active RAB7 in B. cenocepacia-infected WT and casp4−/- macrophages. Shown are mean ± SEM of 4 independent experiments (n = 4). Statistical analyses were performed using paired t-test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Less B. cenocepacia-containing vacuoles associate with CFL1 (cofilin 1, non-muscle) in casp4−/- macrophages
Microtubules and actin filaments provide a scaffold to allow vesicular trafficking, and remodeling of the actin cytoskeleton involves CASP4 [16,25,36]. Depolymerization of actin filaments specifically inhibits starvation-induced autophagosome formation, indicating a critical role for actin during the early stages of autophagy [37]. The actin-binding protein CFL1 (cofilin 1, non-muscle) is a key molecule that modulates actin-remodeling. To investigate the effect of CASP4 on CFL1 during B. cenocepacia infection and whether it is affected by CASP4, we followed the distribution of CFL1 in relation to the B. cenocepacia-containing vacuoles. Using confocal microscopy, we found that CFL1 was localized in proximity to phagosomes containing B. cenocepacia in WT macrophages (Figure 7a). Notably, colocalization of B. cenocepacia with CFL1 was reduced in the absence of CASP4 (Figure 7b). However, with regard to phagosomes harboring nonpathogenic E. coli, there was no difference in colocalization between WT and casp4−/- macrophages (Figure 7c). Together, these results indicate that CASP4 affects the recruitment of CFL1 in response to B. cenocepacia infection.
Figure 7.

CFL1 associates with the B. cenocepacia-containing phagosome. (a) CFL1 immunofluorescence assay of B. cenocepacia (B.c.)-infected WT and casp4−/-macrophages at 30 min post infection. White arrows point to B. cenocepacia-CFL1 colocalization. (b) Quantification of B. cenocepacia and E. coli colocalized with CFL1 in WT and casp4−/- macrophages. Values are mean percentage ± SEM calculated by scoring 24 randomly chosen fields of view from at least 3 experiments. Statistical analyses were performed using Student’s t-test. **p ≤ 0.01, ***p ≤ 0.001.
Discussion
Our study shows that CASP4 promotes the restriction of B. cenocepacia in vitro and in vivo. Although, CASP4 has been implicated in the restriction of L. pneumophila and the closely related Burkholderia species B. pseudomallei and B. thailandensis, the exact mechanism is still unclear [16,38]. Recognition of intracellular B. cenocepacia in murine macrophages is mediated through NOD-like receptors including NLRP3 and MEFV/PYRIN [19,20]. Non-canonical NLRP3-PYCARD/ASC-dependent inflammasome activation in response to Gram-negative pathogens requires CASP4 [15]. Here, we report that both CASP4 and MEFV/PYRIN are required for B. cenocepacia-induced CASP1 activation. Accordingly, cleavage of CASP7 downstream of CASP1 was also diminished in the absence of CASP4. Cleaved CASP7, as a result of CASP1 activation, has been described for S. Typhimurium [29], L. pneumophila [30] and B. pseudomallei [39] infection, all of which are pathogens predominantly sensed by the NLRC4 inflammasome [16]. Thus, our results demonstrate that CASP1-dependent CASP7 activation is not restricted to the NLRC4 inflammasome. Inflammasome activation via MEFV/PYRIN might very well account for the residual CASP7 activation observed in our study. However, putative CASP7 activity downstream of MEFV/PYRIN has yet to be determined.
The lack of CASP4, rather than CASP1, protects against lipopolysaccharide-induced septic shock due to reduced secretion of the pro-inflammatory cytokines IL1A and IL1B [15,40]. Yet, casp4−/- mice are highly susceptible to infection with virulent B. pseudomallei and avirulent B. thailandensis resulting in increased mortality [18]. In contrast, in our present study, we report for the first time that CASP4 deficiency mediates resistance to a lethal dose of B. cenocepacia even though casp4−/- mice showed that bacteria persisted within the lung at 8 days post infection. Notably, despite an increased pulmonary burden of B. cenocepacia, casp4−/- mice produced less IL1A and IL1B compared to WT in their BALF. In addition, we found reduced levels of the chemokine CXCL1/KC in the BALF of B. cenocepacia-infected casp4−/- mice, which could account for the reduced inflammatory infiltrate observed in these animals. In support of these findings, the levels of IL1A, IL1B, and CXCL1/KC released by casp4−/- macrophages were significantly reduced in vitro compared to WT cells. In this context, it should be noted, that mice deficient in MEFV/PYRIN exhibit reduced lung inflammation in response to B. cenocepacia infection as well [31]. Furthermore, mefv−/- macrophages stimulated with B. cenocepacia are characterized by decreased CASP1 activation and IL1B production [31], suggesting a possible link or cooperation between CASP4 and MEFV/PYRIN that orchestrates inflammation during B. cenocepacia infection.
The transcription of genes coding for major autophagy proteins is downregulated upon B. cenocepacia infection in both WT and CFTR∆F508 (CF) macrophages, suggesting that B. cenocepacia actively interferes with the autophagy pathway [5]. Nevertheless, WT macrophages successfully restrict B. cenocepacia growth, as functional autophagy machinery is restored in WT but not in CF cells [5,41]. Reduced autophagy activity has been implicated in dysfunctional cellular immunity to intracellular pathogens including the bacteria L. pneumophila, M. tuberculosis, L. monocytogenes, and parasites including Toxoplasma gondii [7,42,43].
Because colocalization of B. cenocepacia with lysosomes is reduced in casp4−/- macrophages, this raises the question of whether CASP4 takes part in the formation and maturation of autophagosomes. Here, we demonstrate that casp4−/- macrophages exhibit less LC3-stained autophagosomes (puncta) upon B. cenocepacia infection compared to WT cells, suggesting that CASP4 contributes to the autophagic response against bacterial pathogens (xenophagy). In fact, inflammasome components play a role in autophagosome formation [23]. Accordingly, inflammatory CASP1 has been described to coordinate autophagy in response to L. pneumophila [24]. However, this is the first report of CASP4 deficiency negatively affecting autophagy in macrophages, which may explain the ability of other autophagy-interacting bacteria to be cleared in the presence of CASP4 [2,8,16]. In contrast, clearance of nonpathogenic E. coli does not depend on CASP1 or CASP4 [16,25]. Notably, B. cenocepacia lacking a functional T6SS fails to induce autophagy in macrophages. This finding is corroborated by other reports demonstrating that P. aeruginosa and V. parahaemolyticus T6SS contribute to the activation of autophagy [44,45].
In addition, we found that casp4−/- macrophages infected with B. cenocepacia exhibit increased levels of total RAB7 and active GTP-bound RAB7, indicating that RAB7 activity is differentially modulated in the absence of CASP4. RAB7 has been implicated in lysosome biogenesis [46] and controls the interaction and fusion of late endosomes with lysosomes [47]. Active GTP-bound RAB7 on the surface of phagosomes promotes membrane motility leading to phagolysosome formation [26]. In addition, RAB7 regulates the final maturation of late autophagic vacuoles during starvation-induced autophagy [48]. Interestingly, B. cenocepacia inactivates RAB7 [12]. Because active RAB7 accumulates in casp4−/- macrophages, these data suggest that the inactivation of RAB7 by B. cenocepacia requires CASP4. Yet, it is also plausible to hypothesize that RAB7 accumulation on the B. cenocepacia-containing vacuoles is a result rather than a cause of arrested maturation. Recent reports demonstrate that RAB7 interacts with ATG4B, a key cysteine protease that cleaves LC3, and hence RAB7 seems to act as a negative regulator of autophagy which corroborates our findings in casp4−/- macrophages [49].
Likewise, the absence of PIK3C3/VPS34 which plays a crucial role in autophagosome formation and autophagic flux is characterized by RAB7 hyperactivation [50–52]. PIK3C3/VPS34 deficiency prevents recruitment of TBC1D2/Armus (the RAB7-specific GTPase activation protein) to endosomal membranes. TBC1D2 interacts with LC3 and regulates RAB7-GTP hydrolysis selectively on autophagosomes to enable fusion with lysosomes and subsequent degradation of the transported cargo [53]. Thus, higher amounts of RAB7-GTP in B. cenocepacia-infected casp4−/- macrophages may be attributed to decreased GTPase activity around the B. cenocepacia-containing vacuoles.
RAB7 on the surface of phagosomes was further reported to promote membrane motility leading to phagolysosome formation via recruitment of the effector protein RILP, which mediates the contact of phagosomes with microtubule-associated motor proteins [26]. Microtubules organize the formation of autophagosomes, their transport, and subsequent fusion events [54–56] by electrostatic interactions originating from the N-terminal domain of LC3 thereby allowing the attachment of autophagosomes to the microtubule network [57,58]. Chemical disruption of microtubules reduces the formation of mature autophagosomes [55]. Concurrently, actin-depolymerizing agents, such as latrunculin B or cytochalasins, reduce starvation-induced autophagosome formation without affecting general acidification of the autophagosome [37,59]. Nonetheless, details about the role of actin during autophagy of bacteria (xenophagy) are limited.
Here, we propose a role for CASP4 in autophagy through the modulation of actin dynamics during bacterial infection. CASP4 facilitates cell migration by directly interacting with the actin interacting protein WDR1/AIP1, thereby allowing CFL1-dependent actin depolymerization in the context of chemotaxis [36]. During phagocytosis, F-actin assembly around phagosomes is crucial to guarantee efficient phagosome-lysosome fusion [60]. However, the role of CASP4-actin interaction in intracellular autophagosome trafficking is still unclear. Notably, CASP4-dependent degradation of L. pneumophila in restrictive macrophages is driven by F-actin polymerization around the periphery of the L. pneumophila-containing vacuole. Likewise, we found reduced amounts of CFL1 adjacent to the B. cenocepacia-containing vacuole in CASP4-deficient cells. casp4−/- macrophages also exhibit significantly lower amounts of active RHOA and inactive CFL1, even under basal conditions [25]. In addition, the B. cenocepacia T6SS effector TecA mediates the deamidation of RHOA in murine cells [32]. RHOA is well known for its ability to induce actin-derived stress fibers and focal adhesions [61], and it also promotes the activation of LIM kinases, which inhibit CFL1 via phosphorylation [62,63].
Compartments in WT macrophages harboring L. pneumophila or B. cenocepacia have the characteristics of autophagosomes and proceed to fuse with lysosomes. However, in permissive casp4−/- and casp1−/- cells, the recruitment of lysosomes to vacuoles containing these pathogens is decreased. Notably, the number of lysosomes is reduced in casp4−/- and casp1−/- macrophages. Together, it is plausible to conclude that dysfunctional regulation of actin dynamics via CFL1 in the absence of CASP4 compromises the formation and trafficking of autophagosomes leading to accumulation of RAB7 during bacterial infection. In summary, our data present an intricate and novel relation between inflammatory caspases and the autophagy machinery.
Materials and methods
Bacterial strains
B. cenocepacia K56–2 is a clinical isolate obtained from a CF patient. Its derivate MH1K is a gentamicin-sensitive strain [64]. B. cenocepacia K56–2 or nonpathogenic E. coli BL21 used in immunofluorescence experiments are transformed with a plasmid for red fluorescent protein (Ds-Red). B. cenocepacia ΔT6SS was kindly provided by Dr. Valvano at Queen’s University, Belfast, UK. All bacterial strains were grown overnight in LB medium at 37°C and 200 rpm as previously described [5,14,41,65].
Mice
C57BL/6 WT mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). casp4−/-/caspase-11−/- mice were generously provided by Dr. Junying Yuan (Harvard Medical School, Boston, MA, USA) [40]. casp1−/− Casp4/Casp-11Tg mice were a gift from Dr. Vishva Dixit (Genentech) [15]. mefv−/-/Pyrin−/- mice were a generous gift from Dr. Thirumala -Devi Kanneganti (St. Jude Children’s Research Hospital). All mice are in the C57BL/6 background. All mice were housed in a pathogen-free facility and experiments were conducted with approval from the Animal Care and Use Committee at The Ohio State University (Columbus, OH, USA).
In vivo infection
For intratracheal infection, mice were anesthetized with Isoflurane and inoculated with 100 µl of phosphate-buffered saline (PBS; Thermo Fisher Scientific, 14,190,144) containing 10 × 106 (organ CFU, histology) or 30 × 106 (survival study) bacteria. To determine the bacterial load in organs, mice were sacrificed at 4 h and 24 h post infection to collect lung, liver and spleen for homogenization in PBS as previously described [5,16]. Data are presented as CFU per gram of organ. For survival experiments, animals were monitored daily for 8 days in total.
Bone marrow-derived macrophages
For the generation of primary bone marrow-derived macrophages from mice, tibias and femurs were flushed with IMDM medium (Thermo Fisher Scientific, 12,440,053) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, 16,000,044), 50% L cell-conditioned medium [66,67], 0.6x MEM non-essential amino acids (Thermo Fisher Scientific, 11,140,050) and 0.1% penicillin and streptomycin (Thermo Fisher Scientific, 15,140,122) as previously described [5,16,25,30,41,65]. Cells were cultivated at least 6 days at 37°C in a humidified atmosphere containing 5% CO2.
Cell culture and infection of primary macrophages
Prior to experiments, macrophages were cultivated in IMDM medium supplemented with 10% fetal bovine serum for at least 1 h. To stimulate autophagy, cells were incubated with 5 μg/mL rapamycin (Sigma Aldrich, R0395) 1 h prior to infection and continuously until the end of the experiment. To inhibit CASP1 and CASP4/caspase-11 activity, macrophages were incubated with 50 μM Ac-YVAD-CMK (Enzo Life Scienes, ALX-260–028) 1 h prior to infection and continuously until the end of the experiment. In vitro infections were performed with an MOI 10:1, including centrifugation for 5 min at 30xg to synchronize the infection. To determine cleavage of CASP1 via immunoblotting, macrophages were treated with an MOI 50:1. To analyze intracellular bacterial replication in WT and casp4−/- macrophages, cells were infected with B. cenocepacia MH1K for 1 h followed by 30 min incubation with 50 µg/mL gentamicin (Thermo Fisher Scientific, 15,750–060) to avoid extracellular bacterial replication. In contrast, to determine intracellular CFUs of the gentamicin-resistant B. cenocepacia T6SS mutant strain, macrophages were extensively washed 3x with PBS after the initial infection for 1 h. At the indicated time points, macrophages were lysed using 0.1% Triton X-100 (Fisher Scientific, BP151) in PBS, and serial dilutions of the lysates were incubated on LB agar for 48 h.
Confocal microscopy
Macrophages were cultured on glass coverslips in 24-well plates and fixed with 4% paraformaldehyde for 30 min at the indicated time points. For permeabilization, cells were treated with ice cold methanol for 10 sec (LC3, CFL1) or 0.1% Triton X-100 for 20 min (RAB7) followed by blocking with 5% goat serum (Thermo Fisher Scientific, 16,210,064) in PBS. LC3A/B (Cell Signaling Technology, 4108), CFL1 (Cell Signaling Technology, 5175), and RAB7 (Cell Signaling Technology, 9367,) were visualized using goat anti-rabbit IgG secondary antibody conjugated to Alexa Fluor® 488 (Molecular Probes, A-11,008,). LysoTracker™ Green (Molecular Probes, L7526) was used to stain acidic compartments of infected macrophages. Nuclei were stained with 1 µg/mL 4ʹ,6ʹ-diamino-2-phenylindole (DAPI; Molecular Probes, D1306,) in PBS-5% goat serum for 5 min. To stain intracellular B. cenocepacia strain MH1K, live macrophages were incubated with 1 μg/mL Hoechst in PBS for 20 min and subsequently fixed with 4% paraformaldehyde. Images were captured using a laser scanning confocal fluorescence microscope with a 60X objective (Olympus Fluoview FV10i) as previously described [25,41,65]. Intensities of target proteins were measured using ImageJ Software. Graphs depict integrated density (IntDen), which is defined as the ‘product of area and Mean Gray Value’ of the pixel values in the selected area, normalized to the cell number.
Quantification of lysotracker green using a microplate reader
Macrophages were cultured in flat clear bottom black 96-well plates (Corning, 3603). LysoTracker Green (1 µM) and 1 µg/mL Hoechst dye were added to the cells 1 h before the measurement. Then, cells were washed 1x with PBS and growth media was replaced with Hanks balanced salt solution (Corning, 21–022-CV). Fluorescence was read using a SpectraMax i3x microplate reader at 480/511 nm (LysoTracker Green) and 361/497 nm (Hoechst). Hoechst fluorescence was used to normalize LysoTracker Green signals.
Cytokine and MPO analysis
Cytokines in BALF were measured by the Analytical & Development Lab within the Center for Clinical and Translational Science (CCTS) at The Ohio State University using the V-PLEX Proinflammatory Panel 1 (mouse) Kit (MSD, K15048G) according to the manufacturer’s instructions. MPO levels in BALF were measured using a mouse MPO ELISA kit (Hycult Biotech, HK210).
Histological analysis
Lungs were removed from infected mice, inflated with PBS and fixed in 10% formalin at room temperature. Sample preparation, processing, hematoxylin & eosin staining, and semi-quantitative slide evaluation using ordinal grading scales was performed by the Histology laboratory within the Department of Veterinary Biosciences at The Ohio State University as previously described [5].
Immunoblotting
Protein extraction from macrophages was performed using TRIzol reagent (Thermo Fisher Scientific, 15,596,026) according to the manufacturer’s instructions. Briefly, after phase separation using chloroform, 100% ethanol was added to the interphase/phenol-chloroform layer to precipitate genomic DNA. Subsequently, the phenol-ethanol supernatant was mixed with isopropanol to isolate proteins. The Bradford method was used to determine protein concentrations. Equal amounts of protein were separated by 13.5% SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane. Membranes were incubated overnight with antibodies against CASP4 (Sigma Aldrich, C1354), CASP1 (Genentech), cleaved CASP7 (Cell Signaling Technology, 9491), LC3 (Sigma Aldrich, L8918,), RAB7 (Cell Signaling Technology, 9367), p-CFL1 (Cell Signaling Technology, 3313), CFL1 (Cell Signaling Technology, 5175) and GAPDH (Cell Signaling Technology, 2118). Corresponding secondary antibodies conjugated with horseradish peroxidase (Cell Signaling Technology, 7074; Santa Cruz Biotechnology, sc-2006) and in combination with enhanced chemiluminescence reagent (Amersham, RPN2209,) were used to visualize protein bands. Densitometry analyses were performed by normalizing target protein bands to their respective loading control (GAPDH) using ImageJ software as previously described [16,25,41]. For CASP4, graphs display average values of the upper and lower band.
Immunoprecipitation of RAB7
GTP-bound RAB7 from WT and casp4−/- macrophages was immunoprecipitated using the RAB7 Activation Assay Kit (New East Bioscienes, 82,501,) according to the manufacturer’s instructions.
Transmission electron microscopy
Macrophages were cultured in 2-well Permanox Lab-Tek chamber slides (Nunc, 177,429) and fixed with 2.5% glutaraldehyde (Ted Pella, 18,426) in 0.1 M phosphate buffer, pH 7.4 (Fisher Scientific, S369 and S373) containing 0.1 M sucrose (Fisher Scientific, S2–500). Sample processing was performed by the Campus Microscopy & Imaging Facility at The Ohio State University as previously described [5]. Images were taken using a FEI Tecnai G2 Spirit transmission electron microscope plus AMT camera system.
Statistical analyses
Data were analyzed using GraphPad Prism 6.0 and SAS 9.4. All figures display mean and standard error of the mean (SEM) from at least 3 independent experiments as indicted in the figure legends. Comparisons between groups were conducted with Student’s t-test, one-way or two-way ANOVA, or linear mixed effects model (depending on the data structure) followed by Holm’s adjustment for multiple comparisons as indicated. Adjusted p-values < 0.05 were considered statistically significant.
Funding Statement
This work was supported by the | NIH | National Institute of Allergy and Infectious Diseases (NIAID) [R01AI24121];HHS | NIH | National Institute of Allergy and Infectious Diseases (NIAID) [R21AI113477]; | NIH | National Heart, Lung, and Blood Institute (NHBLI) [R01HL127651-01A1]
Abbreviations
- BALF
bronchoalveolar lavage fluid
- BCV
Burkholderia cenocepacia-containing vacuole
- CASP1
caspase 1
- CASP4/caspase-11
caspase 4, apoptosis-related cysteine peptidase
- CASP7
caspase 7
- CF
cystic fibrosis
- CFL1
cofilin 1, non-muscle
- CFU
colony-forming unit
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- Mefv/Pyrin
Mediterranean fever
- MPO
myeloperoxidase
- RAB7
RAB7, member RAS oncogene family
- T6SS
type six secretion system
- WT
wild-type
Acknowledgments
We thank M. Valvano at Queen’s University, Belfast, UK for providing red fluorescent B. cenocepacia and B. cenocepacia ΔT6SS strains. We thank Daniel M. Layman, Mohamed Quteifan, and Anup Vaidya for technical help. We are grateful to Dr. Sue E. Knoblaugh at the College of Veterinary Medicine/Department of Veterinary Biosciences at the Ohio State University, Columbus OH, USA for the histological evaluation of lung tissue. Comparative Pathology & Mouse Phenotyping Shared Resource, Department of Veterinary Biosciences and the Comprehensive Cancer Center, The Ohio State University, are supported in part by grant P30 CA016058. Studies in the Amer laboratory are supported by Ohio State University (OSU) Bridge funds, Public Health Preparedness for Infectious Diseases (PHPID), Center for Clinical and Translational Science (CCTS), R21 AI113477, R01 AI24121, and R01 HL127651-01A1. Cytokine analysis using the V-PLEX Proinflammatory Panel 1 (mouse) Kit was supported by Award Number Grant UL1TR001070 from the National Center For Advancing Translational Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Advancing Translational Sciences or the National Institutes of Health. KK is supported by Deutsche Forschungsgemeinschaft (DFG - German Research Foundation). KC is supported by a Cystic Fibrosis Foundation Postdoctoral Research Fellowship. AB is supported by funding from the Egyptian Government. The authors declare that they have no conflict of interests.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplementary data for this article can be accessed here
References
- [1].Gutierrez MG, Master SS, Singh SB, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119(6):753–766. [DOI] [PubMed] [Google Scholar]
- [2].Birmingham CL, Smith AC, Bakowski MA, et al. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281(16):11374–11383. [DOI] [PubMed] [Google Scholar]
- [3].Amer AO, Swanson MS.. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol. 2005;7(6):765–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nakagawa I, Amano A, Mizushima N, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306(5698):1037–1040. [DOI] [PubMed] [Google Scholar]
- [5].Abdulrahman BA, Khweek AA, Akhter A, et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. 2011;7(11):1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol. 2003;5(7):455–468. [DOI] [PubMed] [Google Scholar]
- [7].Py BF, Lipinski MM, Yuan J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy. 2007;3(2):117–125. [DOI] [PubMed] [Google Scholar]
- [8].Cullinane M, Gong L, Li X, et al. Stimulation of autophagy suppresses the intracellular survival of Burkholderia pseudomallei in mammalian cell lines. Autophagy. 2008;4(6):744–753. [DOI] [PubMed] [Google Scholar]
- [9].Bressler AM, Kaye KS, LiPuma JJ, et al Risk factors for Burkholderia cepacia complex bacteremia among intensive care unit patients without cystic fibrosis: a case-control study. Infect Control Hosp Epidemiol. 2007;28(8):951–958. [DOI] [PubMed] [Google Scholar]
- [10].Drevinek P, Mahenthiralingam E. Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin Microbiol Infect. 2010;16(7):821–830. [DOI] [PubMed] [Google Scholar]
- [11].Ganesan S, Sajjan US. Host evasion by Burkholderia cenocepacia. Front Cell Infect Microbiol. 2011;1:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lamothe J, Huynh KK, Grinstein S, et al. Intracellular survival of Burkholderia cenocepacia in macrophages is associated with a delay in the maturation of bacteria-containing vacuoles. Cell Microbiol. 2007;9(1):40–53. [DOI] [PubMed] [Google Scholar]
- [13].Al-Khodor S, Marshall-Batty K, Nair V, et al. Burkholderia cenocepacia J2315 escapes to the cytosol and actively subverts autophagy in human macrophages. Cell Microbiol. 2014;16(3):378–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Assani K, Tazi MF, Amer AO, et al. IFN-gamma stimulates autophagy-mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PLoS One. 2014;9(5):e96681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. [DOI] [PubMed] [Google Scholar]
- [16].Akhter A, Caution K, Abu Khweek A, et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity. 2012;37(1):35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Casson CN, Copenhaver AM, Zwack EE, et al. Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog. 2013;9(6):e1003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aachoui Y, Leaf IA, Hagar JA, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339(6122):975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rosales-Reyes R, Aubert DF, Tolman JS, et al. Burkholderia cenocepacia type VI secretion system mediates escape of type II secreted proteins into the cytoplasm of infected macrophages. PLoS One. 2012;7(7):e41726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gavrilin MA, Abdelaziz DH, Mostafa M, et al. Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J Immunol. 2012;188(7):3469–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–268. [DOI] [PubMed] [Google Scholar]
- [22].Lee J, Kim HR, Quinley C, et al. Autophagy suppresses interleukin-1β (IL-1β) signaling by activation of p62 degradation via lysosomal and proteasomal pathways. J Biol Chem. 2012;287(6):4033–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Byrne BG, Dubuisson JF, Joshi AD, et al. Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. MBio. 2013;4(1):e00620–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Caution K, Gavrilin MA, Tazi M, et al. Caspase-11 and caspase-1 differentially modulate actin polymerization via RhoA and Slingshot proteins to promote bacterial clearance. Sci Rep. 2015;5:18479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Harrison RE, Bucci C, Vieira OV, et al. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol. 2003;23(18):6494–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hagar JA, Powell DA, Aachoui Y, et al. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341(6151):1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shi J, Zhao Y, Wang Y, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514(7521):187–192. [DOI] [PubMed] [Google Scholar]
- [29].Lamkanfi M, Kanneganti TD, Van Damme P, et al. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics. 2008;7(12):2350–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Akhter A, Gavrilin MA, Frantz L, et al. Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog. 2009;5(4):e1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xu H, Yang J, Gao W, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the pyrin inflammasome. Nature. 2014;513(7517):237–241. [DOI] [PubMed] [Google Scholar]
- [32].Aubert DF, Xu H, Yang J, et al. A Burkholderia type VI effector deamidates Rho GTPases to activate the pyrin inflammasome and trigger inflammation. Cell Host Microbe. 2016;19(5):664–674. [DOI] [PubMed] [Google Scholar]
- [33].Feng Y, Press B, Wandinger-Ness A. Rab 7: an important regulator of late endocytic membrane traffic. J Cell Biol. 1995;131(6 Pt 1):1435–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Press B, Feng Y, Hoflack B, et al. Mutant Rab7 causes the accumulation of cathepsin D and cation-independent mannose 6-phosphate receptor in an early endocytic compartment. J Cell Biol. 1998;140(5):1075–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gutierrez MG, Munafo DB, Beron W, et al. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117((Pt13)):2687–2697. [DOI] [PubMed] [Google Scholar]
- [36].Li J, Brieher WM, Scimone ML, et al. Caspase-11 regulates cell migration by promoting Aip1-cofilin-mediated actin depolymerization. Nat Cell Biol. 2007;9(3):276–286. [DOI] [PubMed] [Google Scholar]
- [37].Aguilera MO, Beron W, Colombo MI. The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy. 2012;8(11):1590–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hagar JA, Miao EA. Detection of cytosolic bacteria by inflammatory caspases. Curr Opin Microbiol. 2014;17:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bast A, Krause K, Schmidt IH, et al. Caspase-1-dependent and -independent cell death pathways in Burkholderia pseudomallei infection of macrophages. PLoS Pathog. 2014;10(3):e1003986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang S, Miura M, Jung YK, et al. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92(4):501–509. [DOI] [PubMed] [Google Scholar]
- [41].Tazi MF, Dakhlallah DA, Caution K, et al. Elevated Mirc1/Mir17–92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis transmembrane conductance regulator) function in CF macrophages. Autophagy. 2016;12(11):2026–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhao Z, Fux B, Goodwin M, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4(5):458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Castillo EF, Dekonenko A, Arko-Mensah J, et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A. 2012;109(46):E3168–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jiang F, Wang X, Wang B, et al. The pseudomonas aeruginosa type VI secretion PGAP1-like effector induces host autophagy by activating endoplasmic reticulum stress. Cell Rep. 2016;16(6):1502–1509. [DOI] [PubMed] [Google Scholar]
- [45].Yu Y, Fang L, Zhang Y, et al. VgrG2 of type VI secretion system 2 of vibrio parahaemolyticus induces autophagy in macrophages. Front Microbiol. 2015;6:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bucci C, Thomsen P, Nicoziani P, et al. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11(2):467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vanlandingham PA, Ceresa BP. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J Biol Chem. 2009;284(18):12110–12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jager S, Bucci C, Tanida I, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117(Pt 20):4837–4848. [DOI] [PubMed] [Google Scholar]
- [49].Kjos I, Borg Distefano M, Saetre F, et al. Rab7b modulates autophagic flux by interacting with Atg4B. EMBO Rep. 2017;18(10):1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jaber N, Mohd-Naim N, Wang Z, et al. Vps34 regulates Rab7 and late endocytic trafficking through recruitment of the GTPase activating protein armus. J Cell Sci. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Byfield MP, Murray JT, Backer JM. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem. 2005;280(38):33076–33082. [DOI] [PubMed] [Google Scholar]
- [52].Nobukuni T, Joaquin M, Roccio M, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A. 2005;102(40):14238–14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Carroll B, Mohd-Naim N, Maximiano F, et al. The TBC/RabGAP armus coordinates Rac1 and Rab7 functions during autophagy. Dev Cell. 2013;25(1):15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kochl R, Hu XW, Chan EY, et al. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7(2):129–145. [DOI] [PubMed] [Google Scholar]
- [55].Fass E, Shvets E, Degani I, et al. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem. 2006;281(47):36303–36316. [DOI] [PubMed] [Google Scholar]
- [56].Jahreiss L, Menzies FM, Rubinsztein DC. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9(4):574–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mann SS, Hammarback JA. Molecular characterization of light chain 3. A microtubule binding subunit of MAP1A and MAP1B. J Biol Chem. 1994;269(15):11492–11497. [PubMed] [Google Scholar]
- [58].Kouno T, Mizuguchi M, Tanida I,et al. Solution structure of microtubule-associated protein light chain 3 and identification of its functional subdomains. J Biol Chem. 2005;280(26):24610–24617. [DOI] [PubMed] [Google Scholar]
- [59].Aplin A, Jasionowski T, Tuttle DL, et al. Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J Cell Physiol. 1992;152(3):458–466. [DOI] [PubMed] [Google Scholar]
- [60].Marion S, Hoffmann E, Holzer D, et al. Ezrin promotes actin assembly at the phagosome membrane and regulates phago-lysosomal fusion. Traffic. 2011;12(4):421–437. [DOI] [PubMed] [Google Scholar]
- [61].Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70(3):389–399. [DOI] [PubMed] [Google Scholar]
- [62].Mizuno K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013;25(2):457–469. [DOI] [PubMed] [Google Scholar]
- [63].Mi N, Chen Y, Wang S, et al. CapZ regulates autophagosomal membrane shaping by promoting actin assembly inside the isolation membrane. Nat Cell Biol. 2015;17(9):1112–1123. [DOI] [PubMed] [Google Scholar]
- [64].Hamad MA, Skeldon AM, Valvano MA. Construction of aminoglycoside-sensitive Burkholderia cenocepacia strains for use in studies of intracellular bacteria with the gentamicin protection assay. Appl Environ Microbiol. 2010;76(10):3170–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Abdulrahman BA, Khweek AA, Akhter A, et al. Depletion of the ubiquitin-binding adaptor molecule SQSTM1/p62 from macrophages harboring cftr deltaF508 mutation improves the delivery of Burkholderia cenocepacia to the autophagic machinery. J Biol Chem. 2013;288(3):2049–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Amer A, Franchi L, Kanneganti TD, et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem. 2006;281(46):35217–35223. [DOI] [PubMed] [Google Scholar]
- [67].Lamkanfi M, Amer A, Kanneganti TD, et al. The nod-like receptor family member naip5/birc1e restricts Legionella pneumophila growth independently of caspase-1 activation. J Immunol. 2007;178(12):8022–8027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.